
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMicro-respirometry of whole cells and isolated mitochondria†
Yan Levitsky ab,
David J. Pegouskea,
Sandra S. Hammerb,
Nathan L. Frantza,
Kiera P. Fisherb,
Artem B. Muchnika,
Anand R. Saripallic,
Philip Kirschnerb,
Jason N. Bazilb,
Julia V. Busikb and
Denis A. Proshlyakov*a
ab,
David J. Pegouskea,
Sandra S. Hammerb,
Nathan L. Frantza,
Kiera P. Fisherb,
Artem B. Muchnika,
Anand R. Saripallic,
Philip Kirschnerb,
Jason N. Bazilb,
Julia V. Busikb and
Denis A. Proshlyakov*a
aDepartment of Chemistry, Michigan State University, 578 S Shaw Lane, East Lansing, MI 48824, USA. E-mail: dapro@chemistry.msu.edu
bDepartment of Physiology, Michigan State University, East Lansing, MI, USA
cREPID Program, Michigan State University, East Lansing, MI, USA
First published on 17th October 2019
Abstract
Oxygen consumption is a key metric of metabolism in aerobic organisms. Current respirometric methods led to seminal discoveries despite limitations such as high sample demand, exchange with atmospheric O2, and cumulative titration protocols leading to limited choice of useable tissue, complex data interpretation, and restricted experimental design. We developed a sensitive and customizable method of measuring O2 consumption rates by a variety of biological samples in microliter volumes without interference from the aerobic environment. We demonstrate that O2 permeability of the photopolymer, VeroClear, is comparable to that of polyetheretherketone (0.125 vs. 0.143 barrer, respectively) providing an efficient barrier to oxygen ingress. Optical transparency of VeroClear, combined with high resolution 3D printing, allows for optode-based oxygen detection in enclosed samples. These properties yield a microrespirometer with over 100× dynamic range for O2 consumption rates. Importantly, the enclosed respirometer configuration and very low oxygen permeability of materials makes it suitable, with resin pre-conditioning, for quantitative assessment of O2 consumption rates at any desired [O2], including hyperbaric, physiological or hypoxic conditions as necessary for each cell type. We characterized two configurations to study soluble enzymes, isolated mitochondria, cells in suspension, and adherent cells cultured on-chip. Improved sensitivity allows for routine quantitative detection of respiration by as few as several hundred cells. Specific activity of cell suspensions in the microrespirometer was in close agreement with that obtained by high-resolution polarographic respirometry. Adherent cell protocols allowed for physiologically relevant assessment of respiration in retinal pigment epithelial cells, ARPE-19, which displayed lower metabolic rates compared with those in suspension. By exchanging medium composition, we demonstrate that cells can be transiently inhibited by cyanide and that 99.6% of basal O2 uptake is recovered upon its removal. This approach is amenable to new experimental designs and precision measurements on limited sample quantities across basic research and applied fields.
Introduction
Oxidation is the most common means of transducing hydrocarbons into energy. Aerobic organisms oxidize molecules in a stepwise manner while synthesizing adenosine triphosphate (ATP), the energy currency of the cell. Mitochondria are the specialized organelles where oxidation is coupled to phosphorylation by capturing the energy of electron transport, via a chain of proteins to O2, to create a proton gradient across the inner mitochondrial membrane, which then fuels ADP phosphorylation.As the main ATP generation mechanism of the cell, mitochondria have been extensively studied for over a century. The rate of oxygen consumption, the final step of the mitochondrial electron transport chain, provides information about the activity of electron transport chain protein complexes, transporters and ATP synthase. Two major approaches, polarographic1 or fluorescence quenching,2,3 are used for the measurement of O2 concentration in solution, which provide the necessary data for calculation of oxygen consumption rates. Sophisticated titration protocols using varying substrate and inhibitor combinations were developed to glean information about specific segments of the oxidative phosphorylation machinery. Tremendous progress has been made in understanding mitochondrial function using these approaches. There are, however, several limitations of currently available methodology. As polarographic measurements are based on the current produced by reduction of O2 on an electrode, oxygen consumption by the sample must be significantly higher than that on the electrode, dictating the high tissue demand of this approach. Fluorescence quenching methods do not impose this demand, however existing approaches utilize open configurations that require compensation for ingress of atmospheric oxygen due to diffusion. Finally, both polarographic and fluorescence quenching measurements are performed with static samples that only allow for cumulative, non-reversible titration protocols.4,5
An enclosed flow-through cell respirometer has the potential of improving the currently available methodology by decreasing sample demand, greatly enhancing flexibility, and revolutionizing experimental approaches. The advantages of the flow-through approach were first demonstrated by Jekabsons and Nicholls.6 Using oxygen electrodes to determine pre- and post-sample differentials in oxygen tension in a continuous medium steam, they monitored the respiration of primary cerebellar granule neuron cultures and determined the ATP supply and demand, proton leak, and mitochondrial respiratory capacity during chronic glutamate exposure. Although revolutionary, this approach required complex custom assembly and was not amenable to automation and scaling up for high-throughput measurements.
Recent developments in microfluidics and 3D printing technology provide new opportunities in the development of custom instrumentation.7 In this study, we used O2-impermeable 3D printing plastics to manufacture microchannels. Optical transparency of the plastic allowed us to sample an oxygen-sensitive fluorescence-based thin film deposited on the inner surface of the channel without exposing the sample to atmospheric O2. We show that adherent cells can be cultured directly on-chip and sampled over prolonged periods of time using repetitive and reversible stimulation of a given sample for observation of metabolic response. In addition to adherent cells, the experimental protocol can be adapted to isolated mitochondria and cell suspensions. Ease of production, flexibility in protocol design, and direct quantitative reporting of O2 consumption rates make this system highly amenable to both precise individual measurements of traditional respirometry and parallelization as needed for drug discovery and testing.
Methods
Materials
Dulbecco's phosphate buffered saline (DPBS; D8662), bovine serum albumin (BSA), oligomycin, carbonyl cyanide m-chlorophenyl hydrazone (CCCP), polystyrene pellets (PS), KCN and general laboratory chemicals were of reagent or better grade from Sigma-Aldrich (St. Louis, MO) and were used as acquired. Platinum octaethylporphyrin (PtOEP) was from Frontier Scientific (Logan, Utah). Prefabricated materials were from McMaster-Carr (Elmhurst, IL).Micro-respirometric oxygen sampling
The microfluidic respirometer (MfR) was designed in Autodesk Inventor (Autodesk Inc, San Francisco, CA) and printed on an Objet Connex 350 or J750 (Objet Geometries Inc, Billerica, MA) 3D printer in VeroClear, a polymethylmethacrylate-like clear resin (Objet Geometries Inc). MfR consisted of three parts (Fig. 1A): the manifold (top), the sensing chip (middle), and the compression base (bottom). The manifold provided fluidic interface to the chip and positioned sampling optical fiber immediately above the optode (below). Continuity of the ports was achieved by compression of o-rings (size-001, Buna-N, shore durometer 70A) between the chip and the manifold. The entire assembly was compressed vertically using four bolts, hand tight. Pressure was transferred to the chip and the o-rings via a stacked wave disk spring for better alignment of contact surfaces of the chip and the manifold without pressure points.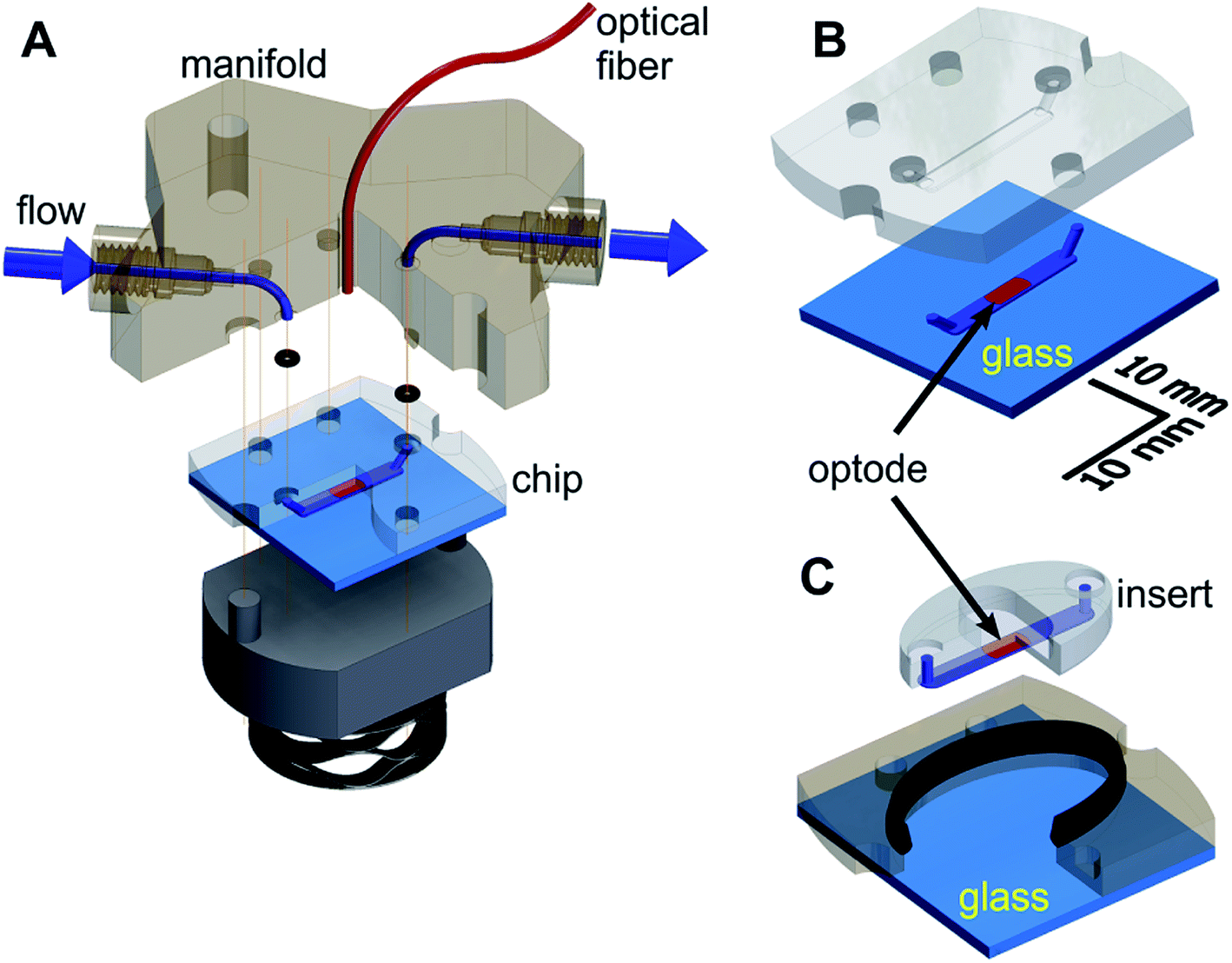 | ||
| Fig. 1 Schematic of the MfR for adherent and non-adherent samples. (A) An exploded view of MfR. Solution flow is shown in blue. Optical fiber and PS/PtOEP are shown in red. O-rings are shown in black. Vertical lines show alignment points. (B) Closed shell configuration for suspension measurements. A 3D-printed chip is fully adhered to glass (light blue). (C) Open shell configuration for adherent measurements. Integral seal is shown in black. | ||
Two interchangeable versions of the chip were produced: closed shell (Fig. 1B) and open shell (Fig. 1C) configurations for suspension and adherent samples, respectively. A 2.0 mm wide by 0.15 mm deep microchannel of the same geometry was manufactured by 3D printing in VeroClear in both cases.
In the closed-shell configuration, the channel was formed by permanent adhesion of the printed part to the flat glass substrate. Four parts of Loctite EA E-30CL epoxy mixture were thinned with 1-part chloroform and applied along the perimeter of the microchannel, ensuring no spillover into the channel. The chip was spun at 2100 rpm for 20 seconds and solvent evaporated for 5 minutes at room temperature. A glass cover, cleaned in acetone, was applied to the chip and cured overnight under mechanical pressure.
In the open-shell configuration, the 3D-printed part was split into two components (Fig. 1B). The outer section of the chip formed a well and was adhered to the glass the same way as for the closed-shell version. It ensured proper alignment on the central, oval section (insert) against the manifold. The microfluidic channel was formed by pressing the insert against the glass without additional adhesives. An elongated, 1.4 mm by 2.6 mm cross section soft seal (shore durometer 26–28A) was co-printed in Tango + 3D printing resin along the perimeter of the well to prevent leaks. The seal was positioned along the upper edge of the well (Fig. 1B) to provide simultaneous 2-way contact with the outer edge of the insert and the bottom surface of the manifold.
In both configurations, the microchannel-containing surface was oriented upwards on the printer bed to prevent support material deposition in the microchannel. Bulk supporting plastic was removed using a brush with medium-soft plastic bristles followed by overnight soaking in a stirred solution of 2 M potassium hydroxide in water saturated with sodium bicarbonate as a mild abrasive. Cleaned parts were washed with deionized water and air dried before further use. Glass-contacting surfaces of chips in both designs were polished on 5 μm grit silicon carbide sheet after optode deposition to improve flatness.
The O2 optodes were deposited by casting or spin coating methods. For drop casting, a ∼220 mg mL−1 stock solution of PS in bromobenzene or chloroform was prepared at room temperature overnight. The working PS/PtOEP solution was prepared by 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 (v/v) dilution of PS stock with a 2 mg mL−1 solution of PtOEP in the same solvent. One microliter of the PS/PtOEP mixture was deposited into the center of the microchannel and dried under a stream of warm air (T < 50 °C). For spin coating, PtOEP (∼1 mg mL−1) and 12.5–25% (m/m) PS in bromobenzene or chloroform solution was applied to the chip followed by spinning at 1000 rpm for 2 minutes. Remaining solvent was removed under reduced pressure (0.2 bar) overnight.
:4 (v/v) dilution of PS stock with a 2 mg mL−1 solution of PtOEP in the same solvent. One microliter of the PS/PtOEP mixture was deposited into the center of the microchannel and dried under a stream of warm air (T < 50 °C). For spin coating, PtOEP (∼1 mg mL−1) and 12.5–25% (m/m) PS in bromobenzene or chloroform solution was applied to the chip followed by spinning at 1000 rpm for 2 minutes. Remaining solvent was removed under reduced pressure (0.2 bar) overnight.
The measurements are based on the reversible quenching of the luminescence intensity and decay time of PtOEP by oxygen modelled by the Stern–Volmer equation.25 The measurements were performed using a NeoFox GT phase fluorimeter with NeoFox Viewer software (Ocean Optics, Dunedin, FL). Fluorescence lifetime of the PtOEP sensor was acquired through the MfR wall by an unterminated optical fiber (ThorLabs Inc., Newton, New Jersey) at a minimal sensor to fiber distance (Fig. 1A, see Results). The optical fiber was coupled to a bifurcated optical fiber of the fluorimeter by a bare fiber terminator (ThorLabs).
Oxygen permeability and solubility
Oxygen permeability was measured following ASTM D-3985 using a Mocon Oxtran 2/21 (Minneapolis, MN) as previously described.8,9 VeroClear discs were printed at 0.5 mm (N = 2) and 0.2 mm (N = 2) thicknesses in two separate batches. Measured thicknesses were within 8% of the nominal thicknesses (0.184 ± 0.008 mm; 0.185 ± 0.010 mm; 0.477 ± 0.003 mm; 0.479 ± 0.009 mm), thus nominal values were used for calculations. Polyetheretherketone (PEEK) film (76.2 μm thick) was cut to size and measured under identical conditions (N = 4). The thin film was conditioned for 1 h. Measurements were performed at 23 °C and 37 °C using dry 100% O2 at a flow rate of 20 sccm against 10 sccm flow of 98% N2, 2% H2 as a carrier gas.Assessment of oxygen solubility was performed using air-equilibrated and anaerobic water in ambient air and in a glovebox (Pas-Labs Inc., Lansing, Michigan) purged with nitrogen (<10 ppm O2). Anaerobic water was prepared using a Schlenk line over 7 cycles between 0.026 atm vacuum and 1 atm Ar gas with agitation. Solutions were delivered to the chip from a gas-tight syringe through a minimal length of PEEK tubing.
Cell culture and respiration assays
ARPE-19 cells were cultured, harvested, and counted according to standard methods (see ESI†). At 100% confluence, cells were trypsinized, counted and either plated in the MfR well or stored on ice prior to measurements.Glucose oxidase assays
Stock solutions of glucose oxidase (GOx) and glucose were prepared in 50 mM potassium phosphate buffer, pH 7.5, and diluted in the same buffer as necessary. Assays were prepared by mixing 1:1 (v/v) of glucose and GOx stock solutions to a final concentration of 75 mM glucose and varying concentrations of GOx as indicated. The microchannel was flushed with blank buffer between trials.
Non-adherent samples
ARPE-19 cell suspensions were stored in buffer containing 148 mM NaCl, 5 mM KCl, 0.81 mM MgSO4, 0.83 mM Na2HPO4, 0.14 mM KH2PO4, 1 mM CaCl2, 25 mM NaHCO3, 15 mM glucose at pH 7.5.10 Aliquots of the cell suspension stock were mixed with various buffers (vide infra) immediately prior to loading 2.0–33 × 103 cells per μL into the MfR using a 20 μL pipette. The microchannel was flushed with PBS between trials.A high-resolution respirometer, Oxygraph-2k (Oroboros Instruments Corp., Innsbruck, Austria), was used as a reference following standard protocols at 23 °C.5,11
Adherent samples
A removable, 3 mm in diameter by 3 mm in height seeding mask was used in preparation of the adherent cell respirometry. The seeding mask was printed in VeroClear and overcoated with polymethylmethacrylate (PMMA) by dip coating into a 5% (m/m) solution of PMMA in methylethylketone and drying overnight at 50 °C. The open-shell well (Fig. 1C) and the seeding mask were sterilized by washing with 70% ethanol in water followed by 15 min of UV irradiation prior to seeding.ARPE-19 cells were loaded into the mask at densities of 3.5–7.0 × 103 cells per mm2 and cultured overnight. Photomicrographs were acquired using an inverted light microscope equipped with an AmScope 0.3 megapixel color CMOS camera (United Scope, Irvine, CA) prior to measurements. Surface density was calculated using ImageJ software by converting the RGB photomicrograph into 16-bit greyscale and applying a median filter (2 pixel radius). Find maxima was used to segment cells (noise tolerance of 5–8) excluding edge particles.
Respiratory activity was measured under stationary medium conditions. Medium flow of 10–20 μL min−1 for re-oxygenation and reagent replenishment was controlled by a syringe infusion pump (Model 22, Harvard Apparatus, MA). Stationary conditions were assured by a manual diversion valve that isolated the chip from the pump.
The base cellular respirometry buffer (BBC) consisted of DPBS with calcium and magnesium (Sigma-Aldrich) supplemented with 0.2% BSA. Respiration buffer (RBC) consisted of BBC supplemented with 10 mM glucose, 10 mM lactate, and 1 mM pyruvate. Leak buffer (LBC) contained RBC with 2.5 μM oligomycin. Uncoupling buffer (UBC) contained LBC with 5 μM CCCP. Inhibition buffer (IBC) contained UBC with 5 mM KCN.12
Mitochondrial isolation and assay
Mitochondria were isolated from ARPE-19 cells using a standard differential centrifugation protocol (see ESI†).13 Isolated mitochondria were stored on ice prior to measurements.The base mitochondrial respiration buffer consisted of 130 mM KCl, 20 mM Tris, 10 mM EGTA, 1 mM sodium phosphate at pH 7.5. Mitochondrial leak buffer contained 5 mM pyruvate, 0.5 mM malate, 10 mM succinate in base buffer. Leak buffer was supplemented with 0.24 mM ADP for ADP-stimulated respiration. Inhibited rates were determined by addition of 5 mM KCN and ADP to leak buffer. Total mitochondrial protein was determined using Bradford assay (BioRad).
Calibration and data analysis
Calibration was performed daily using air equilibrated buffer as the aerobic standard (245 μM)14,15 and fresh ∼1 mM sodium dithionite solution in the same buffer as anaerobic standard. The Stern–Volmer relation was used to convert fluorescence lifetime to O2 concentration.16–18 Oxygen consumption rates (RO2) were determined by linear fitting of steady state phases. For enzyme assays, background activity was corrected by subtracting RO2 of the buffer blank. For mitochondrial or cellular measurements, the RO2 of inhibited samples was subtracted from all others. Polynomial background drift corrections were applied to O2 traces in some instances. Data analysis was performed using IgorPro (Wavemetrics Inc., Portland, OR) software.Results
Fig. 1 shows a schematic of the device consisting of 3D printed flat chip with O2 sensor film deposited in the microchannel. Optically transparent VeroClear allowed sampling of the sensor by the optical fiber (Fig. 1A). Patency of the microchannels was ensured by low-pressure perfusion prior to use. Distance between the optode and the optical fiber was 1.75 mm in the non-adherent configuration and 0.55 mm in the adherent configuration. An acceptance angle of 23° for the 1.0 mm fiber resulted in circular fields of view with diameters of 2.48 mm and 1.47 mm for the non-adherent and adherent configurations, respectively. Drop-coated optode, with a limiting area of 1.8 mm2, was probed by an oversized field of view in the non-adherent configuration. In the adherent configuration, a section of an oversized optode was sampled by a limiting 1.7 mm2 field of view.Nominal dimensions of non-adherent microchannels were 0.25 mm deep by 1.20 mm wide. Average printed microchannel width was 1.09 ± 0.117 mm (N = 3). Average channel depth was 0.235 ± 0.013 mm (N = 3) and 0.145 ± 0.017 mm (N = 3) before and after polishing, respectively. Nominal and measured dimensions of the adherent configuration were 0.150 mm vs. 0.131 ± 0.018 mm (N = 3) in depth and 2.00 mm vs. 2.23 ± 0.006 mm (N = 3) in width.
Sample demand for cellular respiration
ARPE-19 cells were chosen for their robust respiratory capacity and high rate of proliferation, yielding abundant sample for instrument characterization.A representative oxygen concentration trace using suspensions of ARPE-19 cells is shown in Fig. 2A and B. Buffer blanks were used for system equilibration (arrowheads) until stable baseline was observed. Aliquots of ARPE-19 cells were pre-mixed with respiration buffer (hollow arrows; 10 mM glucose, 10 mM lactate, 1 mM pyruvate) or inhibition buffer (solid arrows; respiration buffer + 5 mM KCN) and loaded into the microchannel. Separate aliquots were prepared from the same suspension stock and sequentially sampled. After acquiring cellular RO2 for 3–7 min, the microchannel was purged using blank buffer at 2.25 mL min−1 for 3–5 min, resulting in rapid re-oxygenation of the microchannel and no evidence of residual activity in the subsequent blank measurement. Cellular RO2 with inhibition buffer was not distinguishable from blank baseline. Injection of anaerobic standard (gray arrowhead) was used for calibration. Transient artifacts were seen immediately after sample loading, including O2 consumption during handling, and were excluded from the steady-state RO2 assessment.
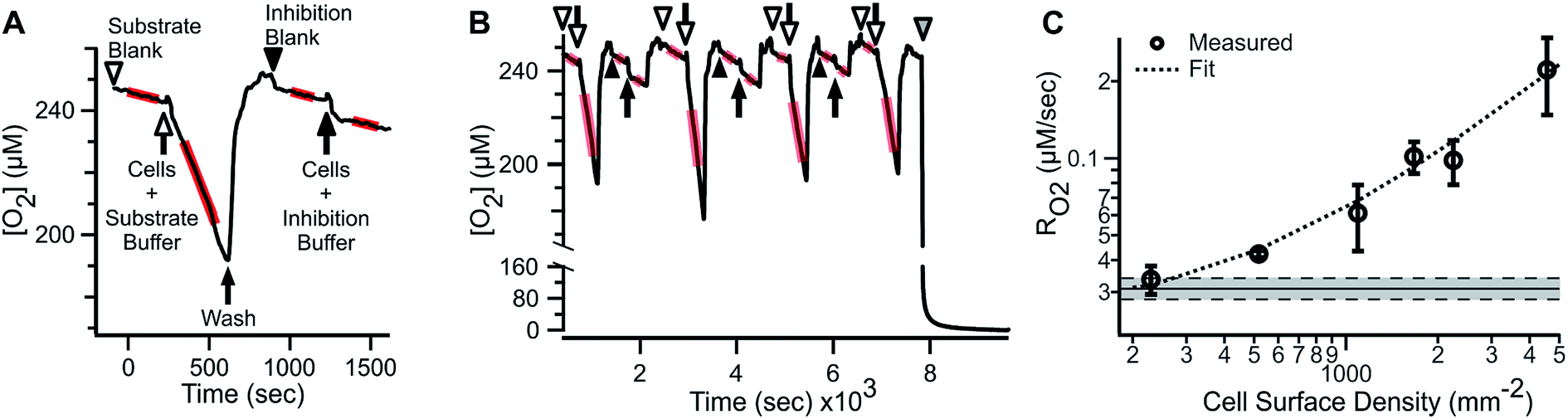 | ||
| Fig. 2 RO2 by ARPE-19 cell suspension in the microrespirometer. (A) Detailed view of ARPE-19 substrate supported and KCN inhibited respiration (12.5 × 103 cells per μL). (B) Reproducibility of alternating injections of respiring and inhibited cells. Arrows and arrowheads indicate cell suspension and blank measurements, respectively. Hollow and solid symbols indicate respiration buffer (RBC: 10 mM glucose, 10 mM lactate, 1 mM pyruvate) and inhibition (IBC: RBC + 5 mM KCN) buffer, respectively. Gray arrowhead shows anaerobic calibration. Regions used for determining RO2 are shown by red highlights. (C) Basal RO2 of ARPE-19 cells with various densities, including linear fit of data (dashed line, y = 0.02 + 4.22 × 10−5x, R2 = 0.98). Shaded area – mean of inhibited cells ± SEM (N = 5). RO2 are expressed as mean ± SEM (N = 3–5). | ||
The microfluidic respirometer sensitivity was characterized using varying cell suspension densities. Fig. 2C shows that RO2 scales linearly with cell density (R2 = 0.98, linear regression) across an order of magnitude change in RO2. Linear interpolation of observed RO2, and its errors, showed the lower limit of detection of approximately 500 cells. Variances increased with increasing cell density, effectively limiting the upper bound of meaningful RO2 determinations. Examination of cell suspension delivery into the MfR by light microscopy revealed non-homogeneous cell distribution at cell densities higher than 17 × 103 cells per μL, likely contributing to the increased variability in dense suspension.
Oxygen diffusion limits the sensitivity of respirometers and leads to ambiguity in data interpretation. As glass is impermeable to O2, diffusion of atmospheric O2 from the open ports along the major axis of the microchannel and diffusion through the VeroClear walls are the two remaining routes in the microchannel. The first mechanism is unlikely to contribute significantly to O2 ingress due to the diffusion distance (5.6 mm) and respiration by cells occupying the entire channel, effectively isolating cells in the sampled volume. Thus, O2 diffusion through VeroClear is the most probable source of O2 ingress. The O2 permeability of VeroClear at ambient and physiological temperatures is compared with that of commonly used polymers in Table S1 (see ESI†). The room temperature (23 °C) barrier properties of VeroClear (0.125 ± 0.007 barrer) were comparable to that of PEEK (0.143 ± 0.001 barrer). Permeability of VeroClear increased 1.8-fold to 0.218 ± 0.006 barrer at 37 °C. Since O2 permeability at both temperatures was comparable and this study focuses on the fitness of the current approach for respirometry over details such as metabolic response to temperature, further analysis was performed at ambient temperature.
Since the ingress of O2 into the sample during measurement may lead to a significant underestimation of RO2,11,19,20 we assessed the bi-directional mass transfer of dissolved O2 between the microchannel and the enclosing materials, including plastic and capillaries. Fig. 3 illustrates changes in dissolved O2 at the minimal and maximal concentration gradients in aerobically (left) and anaerobically (right) conditioned open-shell chip. To eliminate potential contribution of proteins adsorbed to the chip surfaces, measurement was performed on a fresh chip that was not used in other studies. Sequential sampling of aerobic water in the air-equilibrated chip (Fig. 3A) showed no distinguishable changes in the solution [O2], as expected for zero pO2 gradient between the plastic and the solution. Rapid replacement of aerobic water with anaerobic water to impose a maximal gradient on the same chip resulted in pronounced ingress of O2 from plastic into the solution (Fig. 3B). The observed ingress is attributed entirely to the immediate vicinity of the optode and not the upstream components because measurements were performed in the stationary solution. The average ingress rate decreased from 0.04 μM s−1 to 0.01 μM s−1 between the first and the third measurement, suggesting that while a substantial amount of O2 may be dissolved in the thin layer of air-equilibrated plastic, contribution of mass transfer from deeper layers is small, in agreement with direct permeability measurements.
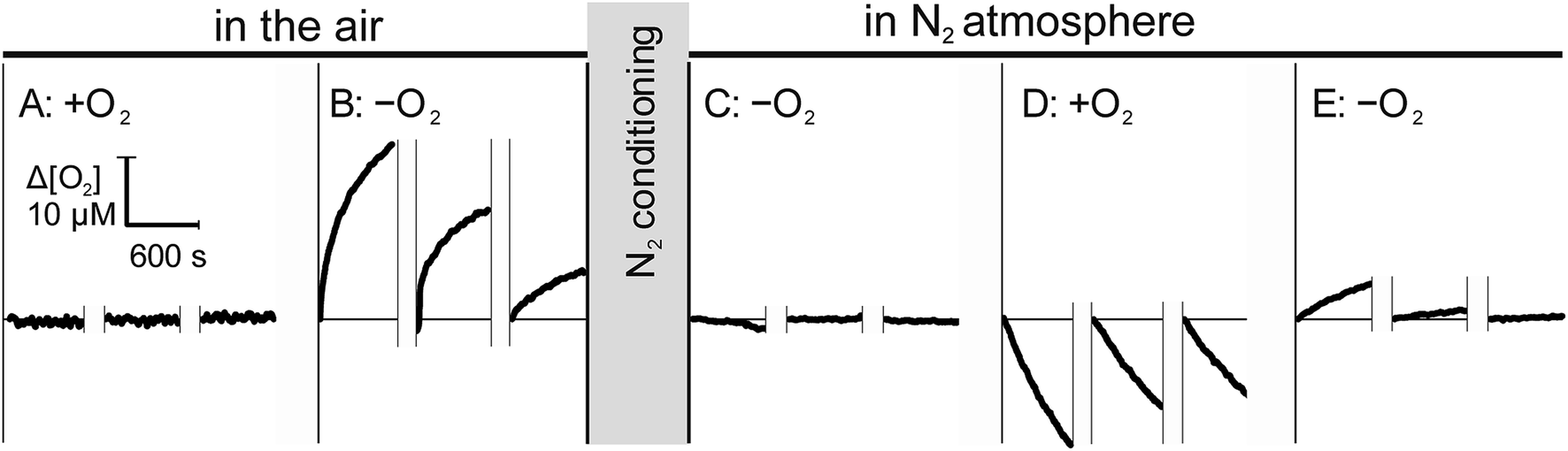 | ||
| Fig. 3 Interface mass transfer of O2 under zero and maximal gradients. Left: sequential measurements in a fresh, closed-shell chip that remained in the air since printing. Measurements using air-equilibrated (A) and anaerobic (B) water were performed in stationary solution and were alternated with 10 min wash and replenishment phases, shown by the breaks over time axis. Changeover from (A) to (B) included a 20min purge with anaerobic water. Right: the same chip was transferred into a glovebox and conditioned for 48 h with N2 stream over microchannel. Measurements were performed as in the left panel using anaerobic (C), air-equilibrated (D), and again anaerobic (E) water. In all cases, O2 concertation is expressed as a change from the start of each stationary measurement. Changes in the concentration indicate ingress (B and E) and egress (D) of O2 into and from the solution, respectively. | ||
This conclusion was further supported by the opposite trend that was observed when the same chip was pre-conditioned in anaerobic (N2) atmosphere for 48 hours. Initial measurement of anaerobic water in the N2-conditioned chip (Fig. 3C) showed no ingress of O2 into the sample. A noticeable loss of O2 was observed when air-equilibrated water was subsequently measured in the same N2-conditioned chip, attributable to the mass transfer of O2 from the solution into plastic. The O2 transferred into the plastic from the aerobic solution can be available for re-entry into the solution until it diffuses further from the interface. This was, indeed, observed when the perfusing solution was reverted to anaerobic water. Fig. 3E shows a noticeable, but rapidly diminishing, re-entry of O2 from transiently oxygenated plastic into the solution.
Isolated mitochondria
To demonstrate the suitability of MfR for metabolic studies, RO2 of mitochondria isolated from ARPE-19 cells was investigated under conditions mimicking standard respirometric measurements (Fig. 4). In the presence of saturating substrates (State II), mitochondrial respiration is limited by a proton motive force and the observed RO2 is controlled by ATP hydrolysis and proton leak. Addition of ADP activates ATP synthase, decreasing the proton motive force and increasing the observed RO2 (State III). Upon complete phosphorylation of ADP, the increase in proton motive force again limits the observed RO2 (State IV) to levels similar to the leak state. Background RO2 is determined from KCN-inhibited samples.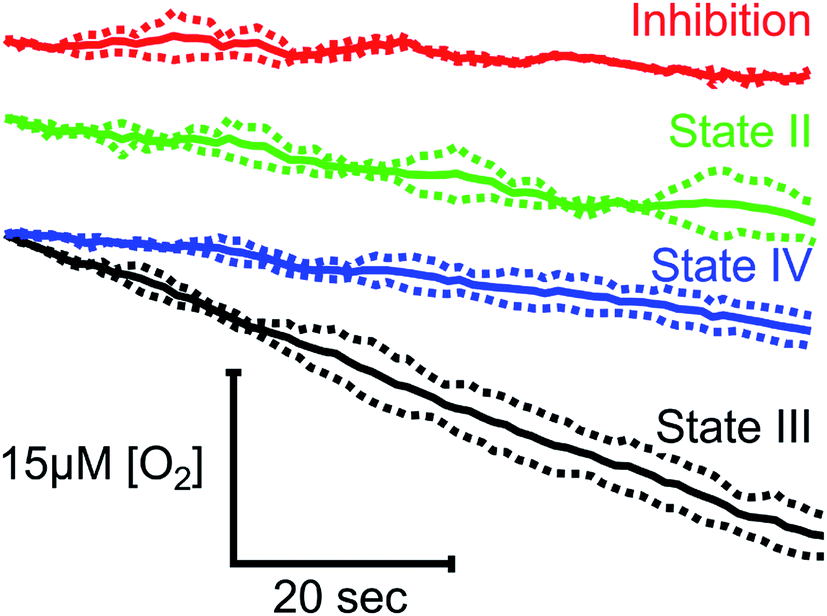 | ||
| Fig. 4 Classical states of mitochondrial RO2 in the MfR. Red trace indicates inhibition by KCN. Green – leak buffer, black – leak buffer + 0.24 mM ADP, blue – leak buffer + 0.24 mM ADP after complete ADP phosphorylation. Traces were overlaid at t = 0 and averaged over the steady state (solid lines). Dashed lines show ±SD. N = 2 for inhibition and leak, N = 4 for state III and IV. | ||
RO2 of cells in suspension
Similar substrate-inhibitor modulation of whole cell suspensions in the microchannel is shown in Fig. 5. Substrate-inhibitor modulation of RO2 by the same cell line in a high-resolution respirometer is shown for direct comparison. Inhibition of ATP synthase and a corresponding increase in the inner membrane potential by oligomycin caused the expected decrease in cell specific RO2 relative to basal RO2 in both approaches (leak). Maximal cell-specific RO2, achieved by dissipating the membrane potential using 5 μM CCCP (mitochondrial uncoupler) in the presence of substrates, were 0.025 ± 0.002 and 0.028 ± 0.004 nmolO2 per 106 cells per second in the oxygraph and in microfluidic respirometer, respectively (uncoupled). The increased variability of RO2 in the MfR was attributed to manual handling of high-density cellular suspensions.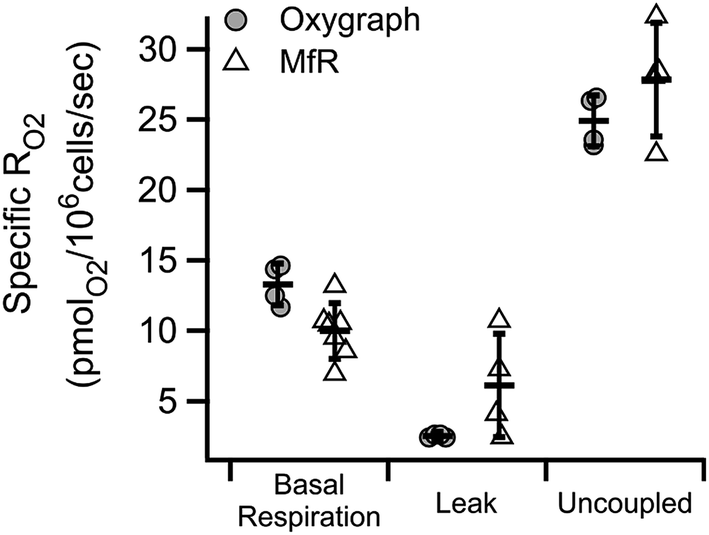 | ||
| Fig. 5 Characteristic RO2 states of whole cells in the MfR compared to traditional oxygraph. Specific RO2 are inhibition corrected, normalized to cell density and expressed as mean ± SD. N = 4 for oxygraph, N = 4–7 for MfR. | ||
Variability in homogeneous samples
To characterize the sensitivity and reproducibility of the MfR independent of the variability associated with suspensions, measurements were performed using a homogeneous glucose–glucose oxidase (Glu–GOx) enzyme system to mimic cellular RO2. Fig. 6A shows a representative trace of sequential Glu–GOx measurements. Inter-trial RO2 reproducibility was high and little residual Glu–GOx activity was detected following washes, which would appear as a loss of O2 during blank measurements. Fig. 6B shows averaged traces of multiplicate measurements of RO2 at [GOx] of 0, 0.09, 4.85 and 333.5 μg mL−1 with 75 mM Glu. At low [GOx] the RO2 (blue) is statistically indistinguishable from the blank (red). At intermediate [GOx], there is a linear decrease in [O2] (black), and at high [GOx] there is a loss of linearity in the [O2] decrease (green). The loss of linearity at the upper limit of MfR sensitivity corresponds to the sensor response time. Maximum apparent RO2 was 5.22 ± 0.43 μM s−1 (N = 3), observed following injection of anaerobic standard. RO2 between 0.03 and 2.5 μM s−1 showed a linear dependence on [GOx] (Fig. 6C).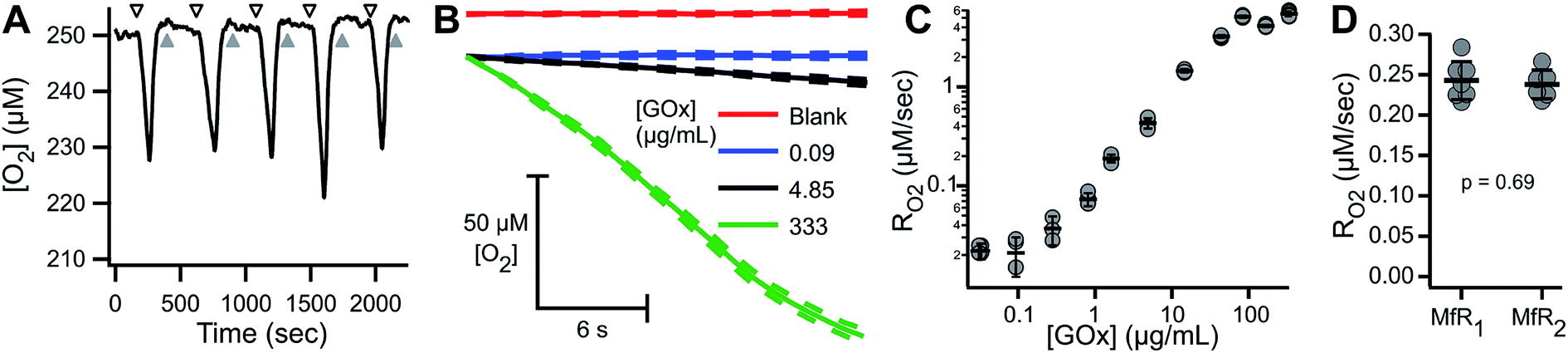 | ||
| Fig. 6 Characterization of MfR using homogeneous model reaction. (A) Raw trace of Glu–GOx RO2 assay. Gray and black arrows show buffer blank and 4.85 μg mL−1 GOx + 75 mM Glu in 50 mM KPi (pH 7.5), respectively. (B) O2 consumption traces by select GOx concentrations. Solid lines indicate overlaid and averaged blank and reaction trials. Dashed lines represent ±SD (N = 5–6). (C) Dynamic range of the MfR using Glu–GOx RO2 expressed as mean ± SD, N = 3–5. (D) Inter-MfR reproducibility using Glu–GOx expressed as mean ± SD, N = 6–7. | ||
The Glu–GOx model was also used to assess inter-instrument variability of the MfR (Fig. 6D). RO2 were measured in parallel, on two separate MfR and fluorimeters, using common Glu–GOx stocks. The RO2 values obtained on each instrument, 0.243 ± 0.024 and 0.238 ± 0.018 μM s−1 (N = 6–7), were not statistically distinguishable (p = 0.69, t-test).
RO2 of adherent cell samples
Adherent cell culture models were used to increase reproducibility, decrease sample demand, and permit transient sample stimulation using repetitive measurements of a given biological sample. As cell adhesion required extended incubation in proximity to a material with unknown biocompatibility, gross toxicity was assessed by culturing ARPE-19 cells on-chip, without a seeding mask, for several days (Fig. 7A and B). Photomicrographs, obtained daily, showed no visible differences between cells in the central and peripheral areas, indicating that proximity to polymer did not affect cell growth. After overnight incubation ARPE-19 cells exhibited an adherent cell morphology. Subsequent proliferation over the three-day incubation period resulted in the cobblestone appearance typical of this cell type.21 Cell surface density quantitation over time is presented in Fig. 7B, demonstrating an initial increase followed by a plateau at day two in culture. The biocompatibility of the MfR instrument was further confirmed by a long-term continuous measurement of adherent cells, which showed no changes in respiration over 24 hours (Fig. S1†).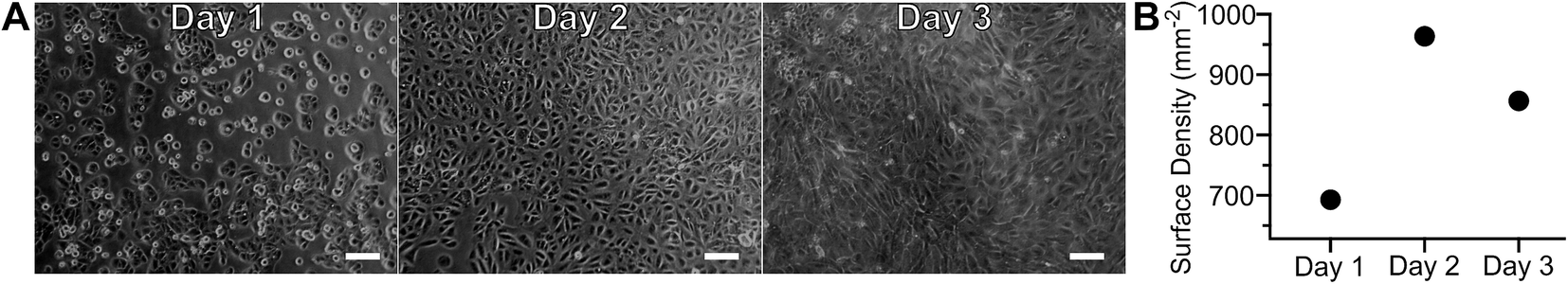 | ||
| Fig. 7 Adherent cell configuration of the MfR. Photomicrographs (A) and quantitation (B) of ARPE-19 cell growth and proliferation on cell culture well of the MfR. Scale bars = 100 μm. | ||
Following cell culture under standard conditions, the removable insert was used to form a microchannel immediately prior to RO2 measurements (Fig. S2†).
Fig. 8 shows an example of RO2 measurements in adherent ARPE-19 cells. Under a constant flow of buffer (Fig. 8A, arrows), the microchannel remains fully oxygenated as O2 delivery is faster than consumption. Assessment of RO2 was achieved by arresting buffer flow (Fig. 8A, arrowheads), resulting in a linear decrease of dissolved O2 over time. Resumption of flow (Fig. 8A, arrows), leads to re-oxygenation of the microchannel. Analysis showed a mean basal RO2 of 0.030 ± 0.002 (±8.4%) μM s−1 and a mean inhibited RO2 of 0.011 ± 0.002 (±14.6%) μM s−1 (N = 4) as shown in Fig. 8B.
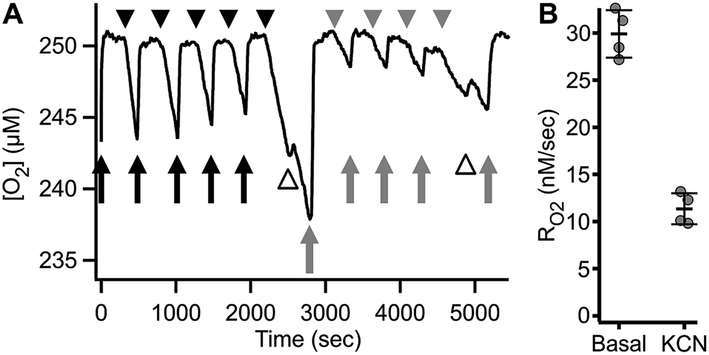 | ||
| Fig. 8 Repetitive RO2 assessment of adhered ARPE-19 cells. (A) Representative O2 trace showing RO2 of 2800 ARPE-19 cells. Arrowheads – no buffer flow, arrows – buffer flow, black – respiration buffer, gray – inhibition buffer, white arrowheads – buffer exchange artifact. (B) Quantitation of basal (RBC) and inhibited (IBC) RO2 obtained from (A), expressed as mean ± SD (N = 4). | ||
The continuous medium flow utilized in the microchannel allows for fast and complete exchange of buffer for reversible activation/inhibition cycles using a single cell sample. This capability is illustrated in Fig. 9A and B. Basal RO2 is inhibited in the presence of the reversible inhibitor KCN. Upon subsequent perfusion with KCN-free buffer (RBC), 99.6% of basal activity is recovered within 880 seconds at 10 μL min−1 flow.
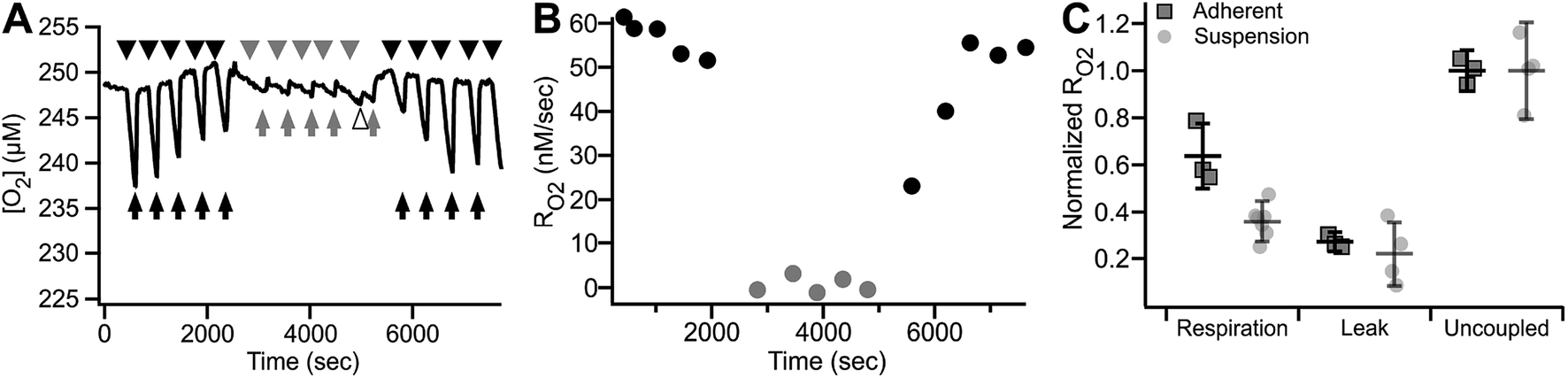 | ||
| Fig. 9 Reversible inhibition of respiration in the MfR. (A) Trace of reversible respiratory inhibition by KCN in adherent ARPE-19 cells cultured on MfR. Arrows – buffer flow, arrowheads – static buffer, black – RBC, gray – IBc, white arrowhead – buffer exchange artifact. (B) RO2 quantitation of the results shown in (A). Black circles – respiration buffer, gray circles – inhibition buffer. (C) Classical states of whole cell RO2 in the adherent compared to suspension MfR configuration. Respiration was in glucose, lactate, pyruvate (10, 10, 1 mM), leak was by addition of oligomycin (2.5 μM), and uncoupled RO2 was by addition of CCCP (5 μM). Squares – adherent cells, gray faded circles – suspension (from Fig. 5). RO2 were normalized to uncoupled respiration and expressed as mean ± SD (N = 3). | ||
Finally, RO2 of adherent ARPE-19 cells was examined using the same sequence of modulation as described above for cell suspensions (Fig. 5). Fig. 9C shows basal, oligomycin-inhibited, CCCP-uncoupled and KCN-inhibited RO2 obtained from 3.4 × 103 cells. The patterns of stimulation and inhibition were recapitulated with adherent cells and, importantly, showed significantly decreased variance of RO2 using the adherent configuration compared to cell suspensions. Significant and reproducible decreases were noted in RO2 after cell adhesion, as discussed below.
Discussion
Measurement of dissolved O2 content provides valuable information on metabolic activity of plant, bacterial, animal and human samples. Sophisticated systems are commercially available for respirometric measurements; however, the advantages of each are balanced by unique limitations such as high sample demand, operating cost, and limited adaptability to sample type.22–24 In this study we combined the advantages of high-resolution 3D printing with fluorometric O2 detection to produce a simple, versatile, and highly sensitive method for micro-scale repetitive respirometry.Measurements on cell suspensions demonstrated the high sensitivity of the MfR, detecting RO2 in as few as several hundred cells (Fig. 2). This represents three orders of magnitude higher sensitivity than large volume respirometers, and approximately 10 times the sensitivity of plate-based respirometry.22,23 Such sensitivities are afforded by the small volumes, tight control over O2 ingress, and short distances between the optode and respiring cells. Suspension measurements showed minimal gain from increasing cell density as higher absolute RO2 were offset by decreased reproducibility. Sensitivity of adherent cell measurements is proportional to surface cell density and inversely proportional to the channel depth, but is independent of channel width or length, assuming a uniform cell monolayer and sensor width greater than channel depth. Therefore, reduction in channel depth is beneficial until shear stress and O2 ingress become liming factors (below). The phase-based fluorescence lifetime detection of O2 by PtOEP in a polystyrene matrix39 is independent of film thickness and fluorophore concentration, enhancing reproducibility (Fig. 6D and 8).25
In Fig. 5 we showed RO2 modulation by respiratory effectors, measured using a traditional oxygraph and the MfR. The expected trends were recapitulated in the MfR with some notable exceptions. The magnitude of inhibition caused by oligomycin was smaller in the MfR whereas the magnitude of stimulation by CCCP was larger. Additionally, MfR cell suspensions showed high RO2 variances compared to polarographic RO2, where cell densities were 2–6-fold lower than in MfR. Both higher variances and changes in modulation efficiencies are likely to arise from the higher cell densities in the MfR. Though the chemical composition of assay buffers were the same, the lipophilic modulators, oligomycin and CCCP, require optimization of modulator to membrane ratios in order to achieve optimal membrane concentrations without side effects.26–29 Despite these differences, we observed excellent agreement of cell-specific uncoupled RO2 between the Oxygraph-2k and MfR, demonstrating the utility of microfluidic respirometry in classical respirometric assays. Uncoupled RO2 was chosen as a normalization between instruments because it represents near-maximal respiratory capacity without the limiting effects of metabolic load.
High variances in cell suspension measurements are attributed to micropipetting of concentrated cell stocks and cell sedimentation during sample loading, which can be minimized by incorporating hydrodynamic focusing.30,31 We reasoned, however, that the high variability between independent cell suspension trials can be remediated by immobilization of the sample, which also improves physiological relevance due cell–cell and cell–substrate contacts in natively adherent cell types. Repeated probing of a given sample in the adherent configuration not only improved variability of RO2 determination (Fig. 8) but further reduced sample demand relative to cell suspensions, requiring delivery of homogeneous medium between samplings. Importantly, ARPE-19 cells had lower metabolic rate in the native adherent state compared to the same cells in suspension. Such changes in metabolic rate upon resuspension of natively adherent cells is expected and highlight the versatility of the MfR, allowing for precise tuning of experimental design to the research question.
The interrupted-flow approach in the adherent MfR configuration enables development of novel measurement strategies.6 First, cell samples are kept at the desired [O2] (near saturation in this study) because cellular RO2 is measured for a short period before medium replenishment. This allows the MfR to sustain prolonged experiments without inducing metabolic changes associated with hypoxic responses (Fig. 8, 9A and S1†).37,38 Continuous buffer exchange can mimic classical titration-based protocols and is further amenable to addition and removal of metabolic stimuli to study reversibility of metabolic switches (Fig. 9). As an example, we demonstrated reversible respiratory inhibition by KCN (Fig. 9A and B) and observed the kinetics of RO2 recovery prior to reaching the near-complete pre-inhibition activity. These transient metabolic states are attributed either to partial washout of the inhibitor or, alternatively, to cellular recovery from the metabolic insult. Regardless of the cause, these observations open intriguing possibilities for the investigation of time-dependent metabolic changes affecting RO2 in real-time and the opportunity to resolve transient metabolic states during stimulation.
Isolation of the sample from the atmospheric environment is particularly important for microrespirometry due to the high surface area to volume ratios inherent in microfluidics. In such regimes, surface exchange of O2 can lead to relatively rapid changes in bulk O2 concentrations in the medium. This property is used widely for the development of microfluidic hypoxia incubators which focus on controlling and rapidly changing dissolved oxygen concentrations within microchannels.18,32 These devices, however, are fundamentally different from respirometers. Whereas control over dissolved oxygen concentration in a microchannel requires highly permeable polymers, microrespirometers require low permeability barriers because real-time detection of bulk analyte and the kinetic analysis of its dynamics are greatly hindered in the presence of extraneous analyte sources or sinks.18,33 Ingress of atmospheric O2 into the microchannel can adversely affect results, decreasing instrument sensitivities and causing non-linear responses due to the accumulation of concentration gradients and diffusion according to Fick's law.2,3,34 The non-linearity of steady-state RO2 is particularly problematic for multi-phasic processes, such as in the transition from ADP-dependent to ADP-limited respiratory states of isolated mitochondria (Fig. 4). For example, ADP-limited respiration could be under-estimated when samples reach lower [O2], affecting calculated parameters such as respiratory control and ADP:O ratios.11,35,36
Excellent barrier properties of VeroClear play a critical role in preventing interference from atmospheric O2. However, VeroClear has some O2 buffering capacity due to a limited solubility of O2 in the plastic. Oxygen transfer between the solution and the plastic results in small changes in the solution O2 following rapid imposition of concentration gradient of over 200 μM. Even at the maximal gradient, the observed mass transfer was significantly slower than the RO2 reported elsewhere in this study. Since mass transfer is directly proportional to the difference of concentration at the boundary of the sample and all biological measurements were performed with 5 to 10 times smaller gradients, effective contribution of mass transfer is negligible. It is important to note that the maximal gradient conditions were designed to test the limits of O2 mass transfer at the boundary and do not represent a biologically relevant model. A well-designed applied experiment should be performed at relatively constant [O2] at physiological pO2, including hypoxic conditions. In such cases, pre-conditioning of the MfR by gas stream or a flow of a solution is the best strategy.
The results shown in Fig. 3A clearly demonstrated that there is no chemical O2 scavenging by the resin itself. This shows that a small, but consistent, non-zero initial RO2 drift with static blank solutions (Fig. 2A, B and 6A) may originate from partial retention of proteins or organelles from previous measurements. Such background drifts are much smaller than mitochondrial RO2 and are corrected with standard respiratory inhibition controls in differential measurements.
While reduction in the depth of the channel enhances sensitivity in adherent cell measurements, it also increases shear stress on cells during perfusion. This physiologically relevant stimulus is lacking in traditional cell culture models.33,40,41 Physiological shear stress can range from 10−2 to 101 dyne per cm2.41,42 In the adherent MfR configuration, ARPE-19 cells experienced an estimated shear stress of 0.27 dyne per cm2, assuming a rectangular channel of nominal dimensions, flow rate of 10 μL min−1 and dynamic viscosity of 0.94 cP.33,41,43 As epithelial cells experience fluid flow velocities closer to that of the interstitium in vivo, the low, intermittent shear imposed by the MfR is likely comparable to physiologically relevant conditions40 and can be further adjusted as needed.
Optical transparency and O2 barrier properties of VeroClear are ideally suited for MfR, but its biocompatibility requires further studies. Cell suspension samples are exposed to VeroClear for ≤10 min in the MfR, in contrast to adherent cells which are cultured in the proximity of the polymer for many hours. We used several strategies to prevent or reduce potentially cytotoxic effects. First, the cells were seeded as a tight cluster in the center of the well with glass base and polymer walls, separated from the bulk media by a seeding mask. The seeding mask was coated with polymethylmethacrylate to act as a barrier film between VeroClear and cells. This impeded mass transfer from the perimeter of the well and sequestered the cell cluster from interaction with potentially toxic leachates.44 We have successfully cultured human epithelial (ARPE-19, Fig. 7A) and bovine retinal endothelial cells (Fig. S1†) in the well without the use of a mask, although further investigation of cytotoxicity is needed for printed materials. During adherent cell measurements, the polystyrene matrix of the PtOEP optode is unlikely to interfere with cellular metabolism, or suffer from bio-fouling, and provides an additional barrier against potential leaching of toxic compounds from the underlying printed polymer.44
3D printing technology now enables the production of micron scale features in an array of polymers. High resolution and flexibility permitted the manufacture of most components with adequate precision and reproducibility without specialized tooling or casting. An alternative to 3D printing, micro-milling, creates a rougher surface that is prone to trapping air bubbles, which can affect oxygen measurements. In contrast, 3D printed parts have smooth surfaces when properly oriented with respect to the supporting plastics, improving the reproducibility of the measurements. A more powerful and flexible open shell design (Fig. 1C) would be very difficult to implement using traditional manufacturing techniques.
While 3D printing has been a major enabling technology for this study, yielding hundreds of chambers of varying design, methodologies can vary widely from a single prototype chip to industrial production. Common to all methodologies is the concept of an isolated microchannel with an aspect ratio amenable to bulk analyte measurement without interference from mass transfer. The Z-dimension in 3D printing typically has higher resolution than the X- and Y-dimensions45,46 and limits microchannel depths to 10's of microns. This is sufficient for optimal MfR channel depths of 70–150 μm because shear stress scales linearly with flow rate and as the inverse square of the channel height. Further reduction of height would necessitate large reductions in flow rates to control shear of adherent cells. The increased cell aggregation would negatively affect reproducibility in suspension measurements.
Conclusion
This work describes the development of microfluidics-based respirometry for studies on biological energy transduction. Taking advantage of remote sensing in an isolated microchannel, this simple, yet versatile, method can detect O2 consumption by minute amounts of sample, ranging from soluble enzyme systems to cell or organelle suspensions and adherent samples. We demonstrated performance of this analytical tool in the context of animal respiration, although it can be employed for measurements of bacterial and plant metabolism. A combination of low oxygen permeability with flexible configuration allow for direct, uncompensated data acquisition, amenable for automation and development of fundamentally new experimental protocols for use in a wide array of basic and applied biomedical fields.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The research was supported by National Institutes of Health (NIH) grant EY028049 to D. A. P. and J. V. B. The authors thank Dr Dana Spence and Brian Wright, MSU, for support in microfluidic 3D printing and valuable discussions.References
- L. C. Clark, R. Wolf, D. Granger and Z. Taylor, J. Appl. Physiol., 1953, 6, 189–193 CrossRef CAS
.
- A. A. Gerencser, A. Neilson, S. W. Choi, U. Edman, N. Yadava, R. J. Oh, D. A. Ferrick, D. G. Nicholls and M. D. Brand, Anal. Chem., 2009, 81, 6868–6878 CrossRef CAS
- A. V. Kondrashina, D. B. Papkovsky and R. I. Dmitriev, Analyst, 2013, 138, 4915–4921 RSC
- G. W. Rogers, M. D. Brand, S. Petrosyan, D. Ashok, A. A. Elorza, D. A. Ferrick and A. N. Murphy, PLoS One, 2011, 6, e21746 CrossRef CAS
- E. Gnaiger, in Drug-Induced Mitochondrial Dysfunction, ed. J. A. Dykens and Y. Will, John Wiley and Sons, New Jersey, USA, 2008, ch. 12, pp. 327–352 Search PubMed
- M. B. Jekabsons and D. G. Nicholls, J. Biol. Chem., 2004, 279, 32989–33000 CrossRef CAS
- H. Zirath, M. Rothbauer, S. Spitz, B. Bachmann, C. Jordan, B. Muller, J. Ehgartner, E. Priglinger, S. Muhleder, H. Redl, W. Holnthoner, M. Harasek, T. Mayr and P. Ertl, Front. Physiol., 2018, 9, 815 CrossRef
- A. Kathuria, N. Brouwers, M. Buntinx, T. Harding and R. Auras, J. Appl. Polym. Sci., 2017, 53, 45690 Search PubMed
- R. A. Auras, S. P. Singh and J. J. Singh, Packag. Technol. Sci., 2005, 18, 207–216 CrossRef CAS
- M.-e. Harper and D. Brand, J. Biol. Chem., 1993, 268, 14850–14860 CAS
- E. Gnaiger, R. Steinlechner-Maran, G. Méndez, T. Eberl and R. Margreiter, J. Bioenerg. Biomembr., 1995, 27, 583–596 CrossRef CAS
- M. D. Brand and D. G. Nicholls, Biochem. J., 2011, 435, 297–312 CrossRef CAS
- C. Frezza, S. Cipolat and L. Scorrano, Nat. Protoc., 2007, 2, 287–295 CrossRef CAS
- E. J. Green and D. E. Carritt, Science, 1967, 157, 191–193 CrossRef CAS PubMed
- H. Forstner and E. Gnaiger, in Polarographic Oxygen Sensors: Aquatic and Physiological Applications, ed. H. Forstner and E. Gnaiger, Springer-Verlag Berlin Heidelberg, Heidelberg, 1st edn, 1983, ch. Appendix A, pp. 321–333 Search PubMed
- R. N. Gillanders, M. C. Tedford, P. J. Crilly and R. T. Bailey, Anal. Chim. Acta, 2004, 502, 1–6 CrossRef CAS
- V. Nock, R. J. Blaikie and T. David, Lab Chip, 2008, 8, 1300–1307 RSC
- P. C. Thomas, S. R. Raghavan and S. P. Forry, Anal. Chem., 2011, 83, 8821–8824 CrossRef CAS
- E. Gnaiger, B. Lassnig, a. Kuznetsov, G. Rieger and R. Margreiter, J. Exp. Biol., 1998, 201, 1129–1139 CAS
- K. Krab, H. Kempe and M. Wikström, Biochim. Biophys. Acta, Bioenerg., 2011, 1807, 348–358 CrossRef CAS
- K.-S. Vellonen, M. Malinen, E. Mannermaa, A. Subrizi, E. Toropainen, Y.-R. Lou, H. Kidron, M. Yliperttula and A. Urtti, J. Controlled Release, 2014, 190C, 94–114 CrossRef
- D. Pesta and E. Gnaiger, Methods in Molecular Biology, Humana Press, New Jersey, USA, 2012, ch. 3, pp. 25–58 Search PubMed
- G. W. Rogers, M. D. Brand, S. Petrosyan, D. Ashok, A. A. Elorza, D. A. Ferrick and A. N. Murphy, PLoS One, 2011, 6, e21746 CrossRef CAS
- M. P. Horan, N. Pichaud and J. W. O. Ballard, J. Gerontol. Ser. A Biol. Med. Sci., 2012, 67, 1022–1035 CrossRef PubMed
- P. E. Oomen, M. Skolimowski and S. Verpoorte, Lab Chip, 2016, 16, 3394–3414 RSC
- T. Arato-Oshima, H. Matsui, A. Wakizaka and H. Homareda, J. Biol. Chem., 1996, 271, 25604–25610 CrossRef CAS
- E. Glaser, B. Norling, J. Kopecky and L. Ernster, Eur. J. Biochem., 1982, 121, 525–531 CrossRef CAS
- B. S. Padman, M. Bach, G. Lucarelli, M. Prescott and G. Ramm, Autophagy, 2013, 9, 1862–1875 CrossRef CAS
- P. G. Heytler, Biochemistry, 1963, 2, 357–361 CrossRef CAS
- H. Yun, K. Kim and W. G. Lee, Biomicrofluidics, 2014, 8, 1–9 CrossRef
- M. Kolnik, L. S. Tsimring and J. Hasty, Lab Chip, 2012, 12, 4732–4737 RSC
- S. M. Grist, L. Chrostowski and K. C. Cheung, Sensors, 2010, 10, 9286–9316 CrossRef CAS
- H. E. Abaci, R. Devendra, Q. Smith, S. Gerecht and G. Drazer, Biomed. Microdevices, 2012, 14, 145–152 CrossRef CAS
- S. C. Oppegard, A. J. Blake, J. C. Williams and D. T. Eddington, Lab Chip, 2010, 10, 2366–2373 RSC
- J. J. Lemasters, J. Biol. Chem., 1984, 259, 13123–13130 CAS
- P. C. Hinkle, Biochim. Biophys. Acta Bioenerg., 2005, 1706, 1–11 CrossRef CAS
- M. Opreanu, M. Tikhonenko, S. Bozack, T. A. Lydic, G. E. Reid, K. M. McSorley, A. Sochacki, G. I. Perez, W. J. Esselman, T. Kern, R. Kolesnick, M. B. Grant and J. V. Busik, Diabetes, 2011, 60, 2370–2378 CrossRef CAS
- D. J. Hausenloy, M. R. Duchen and D. M. Yellon, Cardiovasc. Res., 2003, 60, 617–625 CrossRef CAS
- X. Zhou, F. Su, Y. Tian, R. H. Johnson and D. R. Meldrum, Sens. Actuators, B, 2011, 159, 135–141 CrossRef CAS
- L. Chau, M. Doran and J. Cooper-White, Lab Chip, 2009, 9, 1897–1902 RSC
- J. Shao, L. Wu, J. Wu, Y. Zheng, H. Zhao, Q. Jin and J. Zhao, Lab Chip, 2009, 9, 3118–3125 RSC
- S. Oyre, E. M. Pedersen, S. Ringgaard, P. Boesiger and W. P. Paaske, Eur. J. Vasc. Endovasc. Surg., 1997, 13, 263–271 CrossRef CAS
- E. Fröhlich, G. Bonstingl, A. Höfler, C. Meindl, G. Leitinger, T. R. Pieber and E. Roblegg, Toxicol. in Vitro, 2013, 27, 409–417 CrossRef
- R. P. Rimington, A. J. Capel, D. J. Player, R. J. Bibb, S. D. R. Christie and M. P. Lewis, Macromol. Biosci., 2018, 18, 1–12 CrossRef
- F. Chen, Y. Luo, N. G. Tsoutsos, M. Maniatakos, K. Shahin and N. Gupta, Adv. Eng. Mater., 2018, 1800495, 1–8 Search PubMed
- M. Robins, J. B. Solomon and E. Samei, Medical Imaging 2018: Physics of Medical Imaging, 2018, 10573, p. 148 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05289e |
| This journal is © The Royal Society of Chemistry 2019 |