
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA molecular electron density theory study of the mechanism, chemo- and stereoselectivity of the epoxidation reaction of R-carvone with peracetic acid†
Abdellah Zeroual *a,
Mar Ríos-Gutiérrezb,
Ouafa Amiric,
Mohammed El Idrissiad and
Luis R. Domingo*b
*a,
Mar Ríos-Gutiérrezb,
Ouafa Amiric,
Mohammed El Idrissiad and
Luis R. Domingo*b
aMolecular Modeling and Spectroscopy Research Team, Faculty of Science, ChouaïbDoukkali University, P.O. Box 20, 24000 El Jadida, Morocco
bDepartment of Organic Chemistry, University of Valencia, Dr. Moliner 50, 46100 Burjassot, Valencia, Spain. E-mail: domingo@utopia.uv.es
cLaboratory of Organic and Analytical Chemistry, Faculty of Sciences and Techniques, Sultan Moulay Slimane University, B. P. 523, Beni-Mellal, Morocco
dDepartment of Chemistry, Polydisciplinary Faculty, Sultan Moulay Slimane University, Beni-Mellal, Morocco
First published on 10th September 2019
Abstract
The epoxidation reaction of R-carvone 8 with peracetic acid 9 has been studied within the molecular electron density theory at the B3LYP/6-311(d,p) computational level. The chemo- and stereoisomeric reaction paths involving the two C–C double bonds of R-carvone 8 have been studied. DFT calculations account for the high chemoselectivity involving the C–C double bond of the isopropenyl group and the low diastereoselectivity, in complete agreement with the experimental outcomes. The Baeyer–Villiger reaction involving the carbonyl group of R-carvone 8 has also been analysed. A bonding evolution theory analysis of the epoxidation reaction shows the complexity of the bonding changes taking place along this reaction. Formation of the oxirane ring takes place asynchronously at the end of the reaction by attack of anionic oxygen on the two carbons of the isopropenyl C–C double bond.
1. Introduction
Oxiranes or epoxides are three-membered heterocyclic compounds having at least one oxygen in their structure, thus constituting an important motif in organic chemistry due to of their broad pharmacological and synthetic field of application.1 The introduction of an oxirane ring into organic compounds gives the latter an additional conformational restriction, a strategy widely used in the design of certain products to improve their pharmaceutical and chemical properties.2–4 These systems can be easily synthesized by the partial oxidation reactions of alkenes.5,6 Oxidizing agents, such us oxygen, ozone, peracids, are used for the oxidation of these chlorine and nitrogen oxides.7 However, the use of these agents generally leads to the formation of unwanted and potentially dangerous side products like acids (see Scheme 1).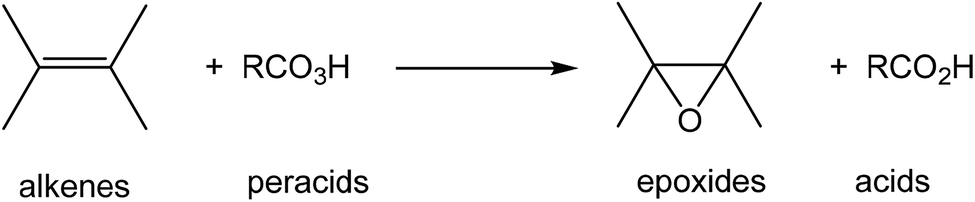 | ||
| Scheme 1 Epoxidation reactions of alkenes mediated by peracids. | ||
Faced with environmental concerns, the interest in the use of oxidizing agents which are safe for the environment is growing as the use of peracids leads to the formation of acids. Epoxidation of monoterpenes gives valuable fragrances, monomers, and food additives.8 Carvone is an oxygenated terpene, being a widely used monoterpene, isolated from more than 100 plants. It can be used in cosmetics, as fragrance in perfume and as a flavoring to mask the bitter taste of alkaloids. A variety of biological activities, including antimicrobial,9 anticancer,10 antifungal,11 antigerminative,12 and antimalarial activities as well as antioxidant activity13 have been ascribed to this monoterpene. Carvone is also a vastly useful starting material for numerous organic syntheses.14 In this context, epoxidation of carvone in heterogeneous and homogeneous catalysts15–20 provides isomeric mono- and diepoxides.21 Diepoxides can be converted into polymeric materials. Peroxides, hydrogen peroxide, m-chloroperbenzoic acid and peracetic acid are very convenient reagents to convert alkenes into their epoxides.22,23
Diverse theoretical models have been developed to explain the molecular mechanism, reactivity and regio-, chemo- and stereoselectivities in organic reactions. In this respect, quantum mechanics calculations have made it possible to understand the formation/breakage bonds and the ability to obtain changes in electron density along the reaction paths.24 For this reason, the conceptual density functional theory (CDFT),25 the electron localization function (ELF) method,26 and bonding evolution theory (BET)27 have been widely used to study reaction mechanisms28–30 within a recent model called Molecular Electron Density Theory (MEDT).31
Many theoretical studies devoted to the epoxidation of alkenes with peracids have been reported in the literature.32–38 In 2007, Rablen performed a B3LYP/6-31G(d) study of the stereoselective epoxidation of carene 1 with perfomic acid 2 (see Scheme 2). The transition state structure (TS) for the trans epoxidation, 9.2 kcal mol−1, was found below that for the cis one, 10.9 kcal mol−1.37 GIAO calculations were performed at the products and compared with the experimental spectra, finding that the proton shieldings calculated for the trans epoxide fitted much better the experimental values obtained for the major epoxidation product than those calculated for the cis epoxide.37
 | ||
| Scheme 2 Epoxidation of carene 1 with perfomic acid 2. | ||
The reversal stereoselectivity in the epoxidation of micheliolide 5 with meta-chloroperbenzoic acid (m-CPBA) 6 was theoretically studied by Zhang in 2012 at the B3LYP/6-31G(d) computational level (see Scheme 3). The inverse stereoselectivity was explained in base of the stability of α-epoxide 7 and the lower free energy of the TS for α-epoxidation.38
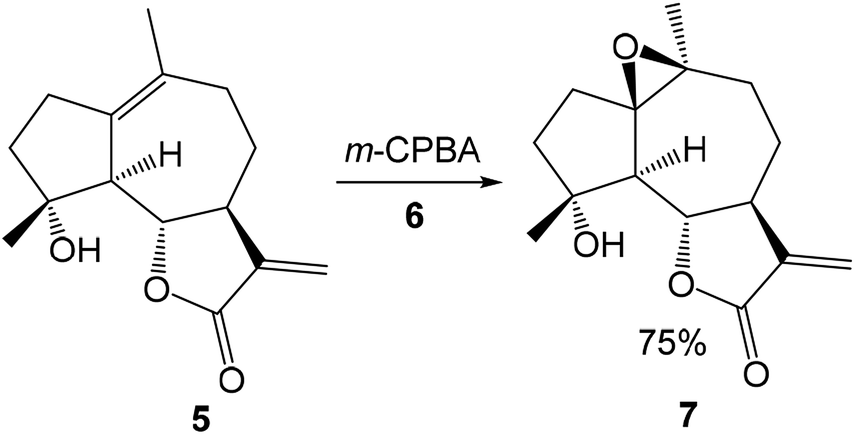 | ||
| Scheme 3 Epoxidation of micheliolide 5 with m-CPBA 6. | ||
Herein, an MEDT study of the epoxidation reaction of R-carvone 8 with peracetic acid 9, experimentally studied by Murphy,39 is carried out in order to understand the formation of epoxides 10 and 12 (Scheme 4). The molecular mechanism, as well as the chemo- and diastereofacial selectivities of these epoxidation reactions will be analysed. In addition, the Baeyer–Villiger (BV) reaction of R-carvone 8 with peracetic acid 9 is also studied.
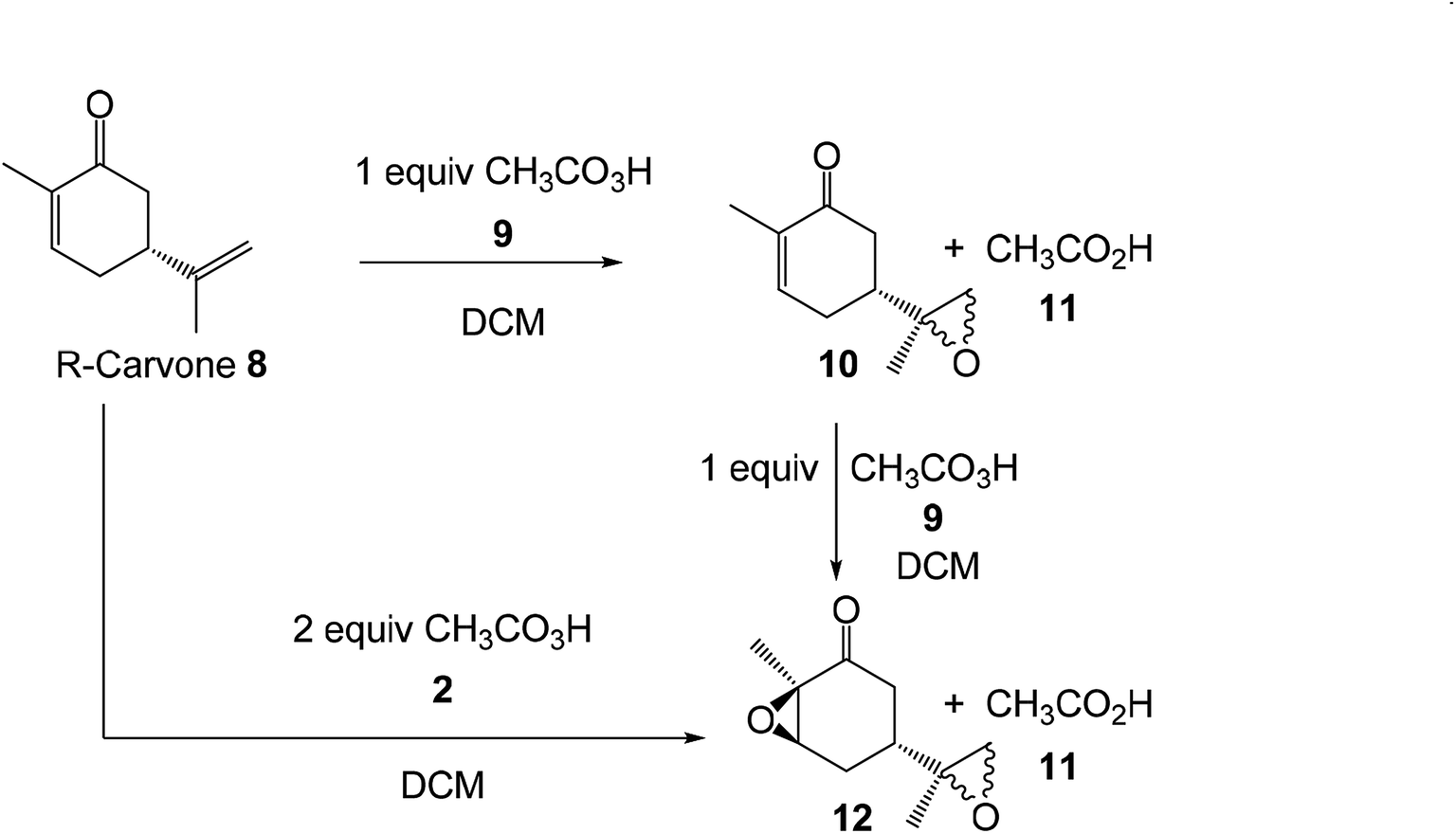 | ||
| Scheme 4 Reactions of R-carvone 8 with peracetic acid 9 studied by Murphy. | ||
2. Computational methods
DFT calculations were performed using the B3LYP functional40,41 together with the 6-311G(d,p) basis set.42 This functional has been widely used in previous studied of epoxidations reactions.32,34,36–38 Optimisations were carried out using the Berny analytical gradient optimisation method.43,44 The stationary points were characterised by frequency computations in order to verify that TSs have one and only one imaginary frequency. The intrinsic reaction coordinate45 (IRC) paths were traced in order to check the energy profiles connecting each TS to the two associated minima of the proposed mechanism using the second order González–Schlegel integration method.46,47 Solvent effects of dichloromethane (DCM) were taken into account by full optimization of the gas phase structures using the polarisable continuum model (PCM) developed by Tomasi's group48 in the framework of the self-consistent reaction field (SCRF).49–51 Values of enthalpies, entropies and Gibbs free energies in DCM were calculated with standard statistical thermodynamics at 25 °C and 1 atm.Conceptual DFT (CDFT) global reactivity indices25,52 and Parr functions were computed using the equations given in ref. 53. All computations were carried out with the Gaussian 09 suite of programs.54
Topological analyses of the ELF26 were performed with the TopMod55 package using the corresponding monodeterminantal wave functions. For the BET study,27 the most favourable reaction path of the monoepoxidation of R-carvone 8 was followed by performing the topological analysis of the ELF for about 195 and 196 nuclear configurations along the IRC paths.
3. Results and discussion
The present MEDT study has been divided in three sections: (i) first, the CDFT reactivity indices of the reagents are analysed; (ii) in the second part, the competitive reaction paths associated with the epoxidation reactions given in Scheme 5, and the BV reaction of R-carvone 8 with peracetic acid 9 are studied; and finally, (iii) in the third part, the formation of the two new C–O single bonds along the most favourable reaction path is characterised through the topological analysis of the ELF.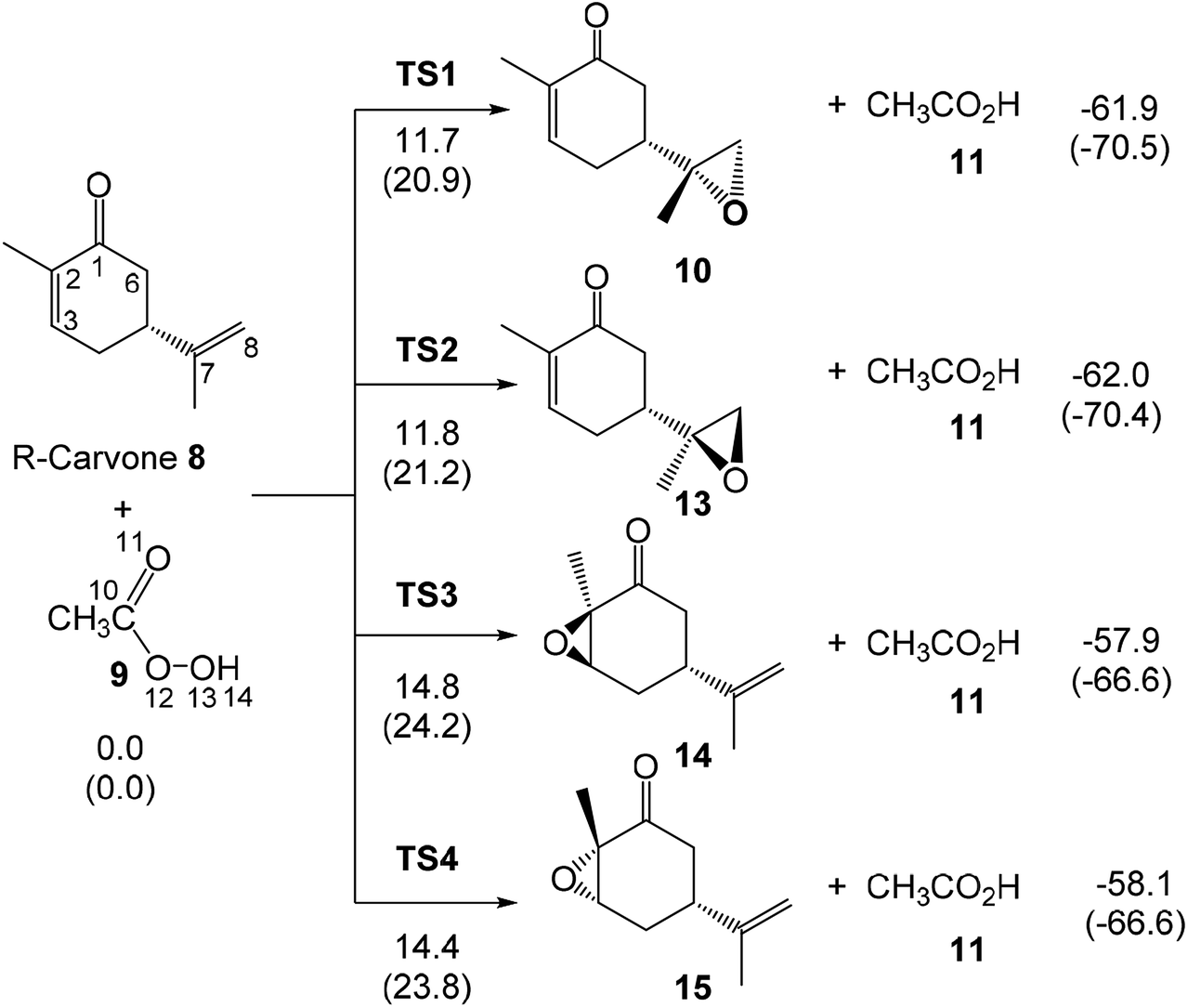 | ||
| Scheme 5 Competitive reaction paths associated to the epoxidation reaction of R-carvone 8 with peracetic acid 9. B3LYP/6-311G(d,p) relative enthalpies and Gibbs free energies, in parentheses, at 25 °C in DCM, are given in kcal mol−1. | ||
3.1 Analysis of the CDFT indices of the reagents
Numerous studies devoted to organic reactions have shown that the examination of the reactivity indices defined within CDFT is a powerful tool to understand organic chemical reactivity. Thus, in order to predict the reactivity of R-carvone 8 and peracetic acid 9 in epoxidation reaction, the global indices gathered in Table 1, i.e. the electronic chemical potential, μ, chemical hardness, η, electrophilicity, ω, and nucleophilicity, N, are analysed.| μ | η | ω | N | |
|---|---|---|---|---|
| R-Carvone 8 | −3.85 | 5.18 | 1.43 | 2.68 |
| Peracetic acid 9 | −4.00 | 7.39 | 1.08 | 1.42 |
The electronic chemical potential μ of R-carvone 8, −3.85 eV, is only slightly higher than that of peracetic acid 9, μ = −4.00 eV. Consequently, the reaction of R-carvone 8 with peracetic acid 9 is expected to have a low polar character.
The electrophilicity ω and the nucleophilicity N indices of R-carvone 8 are 1.43 and 2.68 eV, respectively. These values allow classifying R-carvone 8 as a strong electrophile and as a moderate nucleophile on the electrophilicity and the nucleophilicity scales.52 The electrophilicity ω and the nucleophilicity N indices of peracetic acid 9 are 1.08 and 1.42 eV, respectively. These values allow classifying peracetic acid 2 as being on the borderline of a strong electrophile and of a marginal nucleophile. These low electrophilicity ω and nucleophilicity N values indicate that the epoxidation reaction will have a low polar character, in agreement with low difference between their electronic chemical potentials μ.
In recent years, the electrophilic Pk+ and nucleophilic Pk− Parr functions have been proposed to examine the local reactivity involving reactions between a nucleophile/electrophile pair.53 Therefore, the nucleophilic Pk− Parr functions for R-carvone 8 and the electrophilic Pk+ Parr functions for peracetic acid 9 are analysed (see Fig. 1).
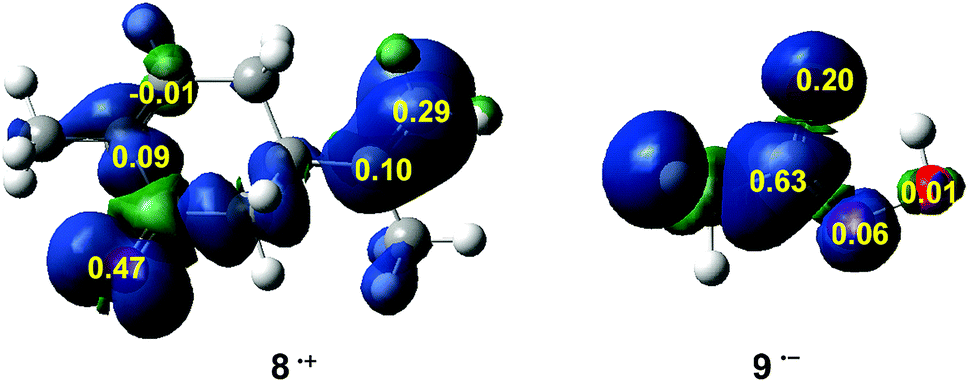 | ||
| Fig. 1 Three-dimensional (3D) representations of the Mulliken atomic spin densities of the radical cation of R-carvone 8 and the radical anion of peracetic acid 9 together with the nucleophilic Pk− Parr functions for 8, and the electrophilic Pk+ Parr functions for 9. | ||
Analysis of the nucleophilic Pk− Parr functions for R-carvone 8 indicates that the two carbons of the isopropenyl C–C double bond are more nucleophilically activated, Pk− = 0.10 and 0.29, than those of the conjugated C–C double bond, Pk− = 0.09 and −0.01. Consequently, as expected, the isopropenyl C–C double bond is the more reactive one of the two C–C double bonds of R-carvone 8. The asymmetric nucleophilic activation of the exocyclic double bond accounts for the asynchronicity found in the formation of the two C–O single bonds in these epoxidation reactions. Therefore, it is expected that the attack of peracetic acid 9 takes place on the isopropenyl C–C double bond of R-carvone 8, in excellent agreement with experimental outcomes (see Scheme 5). Note that the most nucleophilic center of R-carvone 8 is the carbonyl oxygen, Pk− = 0.47, but it does not participate in epoxidation reactions. Analysis of the electrophilic Pk+ Parr functions for peracetic acid 9 indicates that the carboxyl carbon is the most electrophilic center of this molecule, Pk+ = 0.63, followed by the carboxyl oxygen, Pk+ = 0.20. Thus, the electrophilic Pk+ Parr functions of peracetic acid 9 do not account for the local reactivity of this species acting as oxidant.
3.2 Study of the reaction of R-carvone 8 with peracetic acid 9
The present section has been divided in four parts: (i) epoxidation reaction of R-carvone 8 with peracetic acid 9; (ii) BV reaction of R-carvone 8 with peracetic acid 9; (iii) epoxidation reaction of monoepoxide 10 with peracetic acid 9; and (iv) comparative analysis of the Gibbs free energies involved in the chemo- and stereoisomeric reaction paths.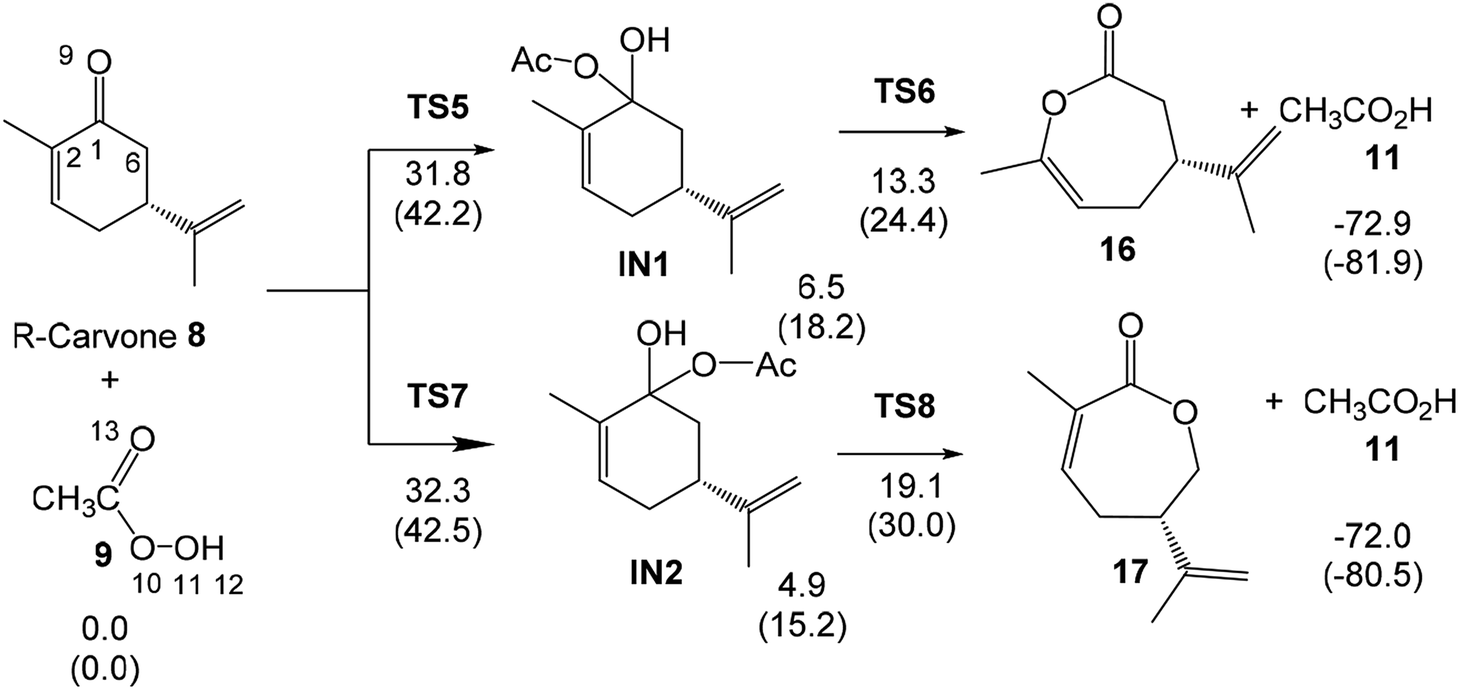 | ||
| Scheme 6 BV reaction of R-carvone 8 with peracetic acid 9. B3LYP/6-311G(d,p) relative enthalpies and Gibbs free energies, in parentheses, at 25 °C in DCM, are given in kcal mol−1. | ||
The activation enthalpies associated to the four competitive reaction paths of the epoxidation reaction of R-carvone 8 with peracetic acid 9 range from 11.7 (TS1) to 14.8 (TS3), the epoxidation reaction being strongly exothermic from −57.9 (14) to −62.0 (13) kcal mol−1 (see Scheme 5). Some appealing conclusions can be drawn from these relative enthalpies: (i) formation of the experimental epoxide 10 via TS1 presents an activation enthalpy of 11.7 kcal mol−1, a closer value to the activation energy computed by Rablen for the epoxidation of carene 1 with perfomic acid 2, 9.2 kcal mol−1 (see Scheme 2);37 (ii) this epoxidation reaction is non-diastereoselective as TS2 is only 0.1 kcal mol−1 higher in energy than TS1; (iii) this epoxidation reaction is highly chemoselective as TS4 is 2.7 kcal mol−1 higher in energy than TS1; (v) formation of epoxide 10 is strongly exothermic by 61.9 kcal mol−1. Consequently, this epoxidation reaction can be considered irreversible.
The optimised geometries in DCM of the TSs involved in the epoxidation reaction of R-carvone 8 with peracetic acid 9 are given in Fig. 2. At the two pairs of chemoisomeric TSs, the distances between the O11 oxygen and the C7 and C8 carbons are 2.315 and 2.058 Å at TS1 and 2.302 and 2.064 Å at TS2, while the distances between the O11 oxygen and the C2 and C3 carbons are 2.208 and 2.024 Å at TS3 and 2.143 and 2.060 Å at TS4. These distances indicate that the formation of the two C–O single bonds at the four TSs is somewhat asynchronous. At the diastereoisomeric TS1 and TS2 the formation of the C8–O11 single bond involving the most nucleophilic carbon of R-carvone 8 is more advanced than the formation of the C7–O11 single bond, while at the diastereoisomeric TS3 and TS4 the formation of the C3–O11 single bond is more advanced than the formation of the C2–O11 single bond. At the four TSs, the distances of the O10–O11 oxygens of peracetic acid 9 are in the short range of 1.82 to 1.86 Å. Considering that the formation of the C–O single bond begins at the distance of 1.8–1.7 Å, and that the O–O single bond is shorter than the C–O one. These geometrical parameters indicate that the O–O single bond has been broken at the four TSs, while the formation of the two C–O single bonds has not yet begun. At the four TSs, the distance between the H12 proton of peracetic acid 9 and carboxyl O13 oxygen, ca. 1.7 Å indicates the presence of a strong hydrogen bond.
 | ||
| Fig. 2 B3LYP/6-311G(d,p) geometries in DCM of the TSs involved in the epoxidation reaction of R-carvone 8 with peracetic acid 9. Distances are given in Angstroms. | ||
The two competitive reaction paths associated to the BV reaction of R-carvone 8 with peracetic acid 9 begin with the nucleophilic attack of the O11 oxygen of peracetic acid 9 on the C1 carbon of R-carvone 8, via TS6 and TS8, yielding a pair of tetrahedric intermediates, IN1 and IN2. Finally, a structural rearrangement in these intermediates yields lactones 16 and 17, respectively, plus acetic acid 11. The activation enthalpies associated with TS5 and TS7 are very high, 31.8 and 32.3 kcal mol−1; formation of the corresponding tetrahedric intermediate is endothermic by 6.5 (IN1) and 4.9 (IN2) kcal mol−1. Finally, the formation of lactones 16 and 17 via TS6 and TS8 presents an activation energy of 6.7 and 14.3 kcal mol−1, respectively; the overall BV reaction being strongly exothermic by 72.9 (16) and 72.0 (17) kcal mol−1. Some appealing conclusions can be drawn from these relative enthalpies: (i) the activation enthalpies associated with the nucleophilic attack of peracetic acid 9 on the C1 carbon of R-carvone 8 is very high, ca. 32 kcal mol−1; consequently, the BV reaction of R-carvone 8 is non-competitive with the epoxidation reactions; (ii) the TS6 and TS9, and INI and IN2 pairs present very similar relative enthalpies since they are two pairs of diastereomers species with similar structures (see later); (iii) while the first step is the rate-determining-step of the BV reaction, the second step is the regioisomer-determining-step; (iv) this reaction is completely regioselective as TS8 is 5.8 kcal mol−1 higher in energy than TS6; and finally, (v) the high exothermic character of the formation of lactone 16 makes the BV reaction irreversible.
The optimised geometries in DCM of the TSs involved in the BV reaction of R-carvone 8 with peracetic acid 9 are given in Fig. 3. At TS5 and TS7 associated to the nucleophilic attack of the O11 oxygen on the carbonyl C1 carbon, the C1–O11 distances are 2.310 and 2.289 Å, respectively. The O9–H12 and O11–H12 distances at these TSs, 1.016 and 1.632 Å at TS5 and 1.020 and 1.617 Å at TS5, indicate that at these TSs the H12 proton of the peracetic acid 9 has already been transferred to the carbonyl O9 oxygen. At TS6 and TS8 associated with the structural reorganisation of intermediates IN1 and IN2, respectively, the distance between the O11 oxygen and the C2 or the C6 carbons is 1.911 and 2.099 Å, while the distance between the C1 carbon and the C6 or the C2 carbons is 1.595 and 1.812 Å, respectively. Consequently, the more favourable TS6 is earlier than TS8.
 | ||
| Fig. 3 B3LYP/6-311G(d,p) geometries in DCM of the TSs involved in the BV reaction of R-carvone 8 with peracetic acid 9. Distances are given in Angstroms. | ||
 | ||
| Scheme 7 Competitive reaction paths associated with the epoxidation reaction of α,β-conjugated ketone 10 with peracetic acid 9. B3LYP/6-311G(d,p) relative enthalpies and Gibbs free energies, in parentheses, in at 25 °C DCM, are given in kcal mol−1. | ||
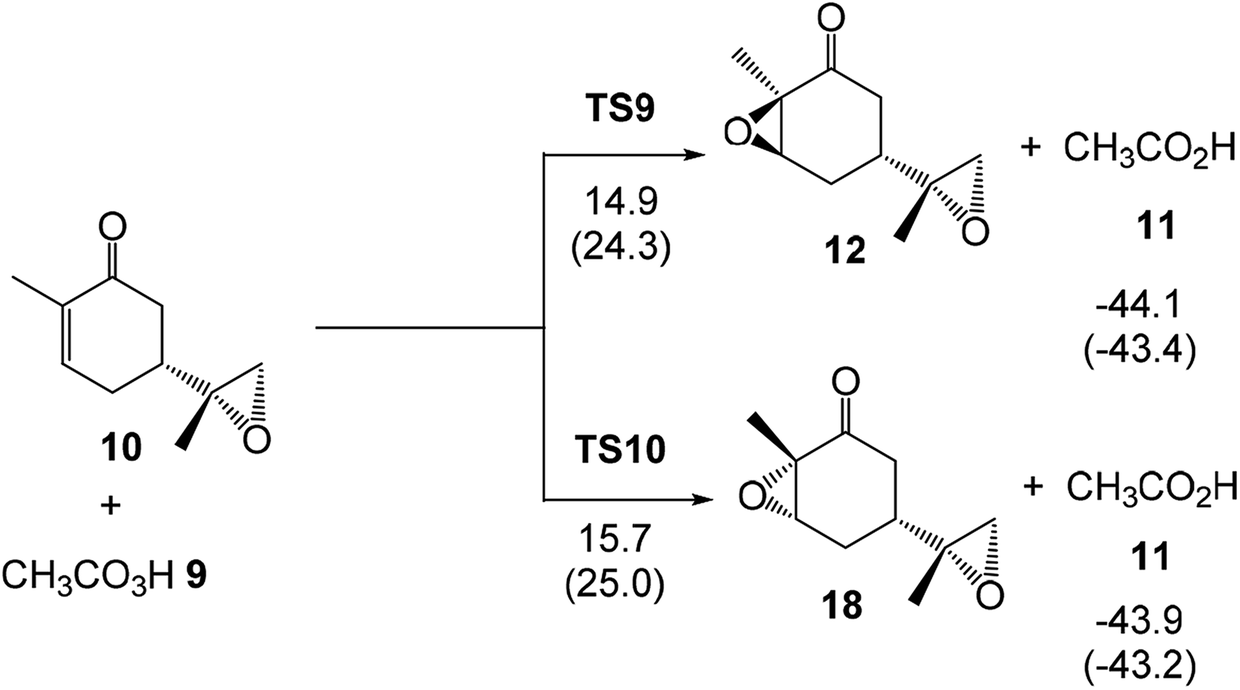
The activation enthalpies associated with the two diastereoselective reaction paths of the epoxidation reaction of α,β-conjugated ketone 10 are 14.9 (TS9) and 15.7 (TS10) kcal mol−1, the epoxidation reaction being strongly exothermic by 44.1 (12) and 43.9 (18) kcal mol−1 (see Scheme 7). Some appealing conclusions can be drawn from these relative enthalpies: (i) the activation enthalpy associated with the formation of the diepoxide 12 via TS9 is 3.2 kcal mol−1 higher in energy than formation of the monoepoxide 10 via TS1; consequently, in the presence of 1 equivalent of peracetic acid 9, only monoepoxidation products will be obtained, in clear agreement with the experimental outcomes (see Scheme 5); (ii) the diastereoselectivity of this reaction is very low as TS10 is only 0.8 kcal mol−1 higher in enthalpy than TS9; (iii) formation of epoxide 12 is strongly exothermic by 44.1 kcal mol−1. Consequently, this epoxidation reaction can be considered irreversible.
The optimised geometries in DCM of the two diastereoselective TSs involved in the epoxidation reaction of the α,β-conjugated ketone 10 with peracetic acid 9 are given in Fig. 4. At the two TSs, the distances between the O11 oxygen and the C2 and C3 carbons are 2.243 and 1.972 Å at TS9 and 2.158 and 2.037 Å at TS4. At the two TSs, the distance of the O10–O11 oxygens of peracetic acid 9 are ca. 18.5 Å, while the distance between the H12 proton of peracetic acid 9 and carboxyl O13 oxygen, ca. 1.7 Å, indicates the presence of a strong hydrogen bond. As expected, the geometries of TS9 and TS10 are very similar to those of TS2 and TS3 (see Fig. 2).
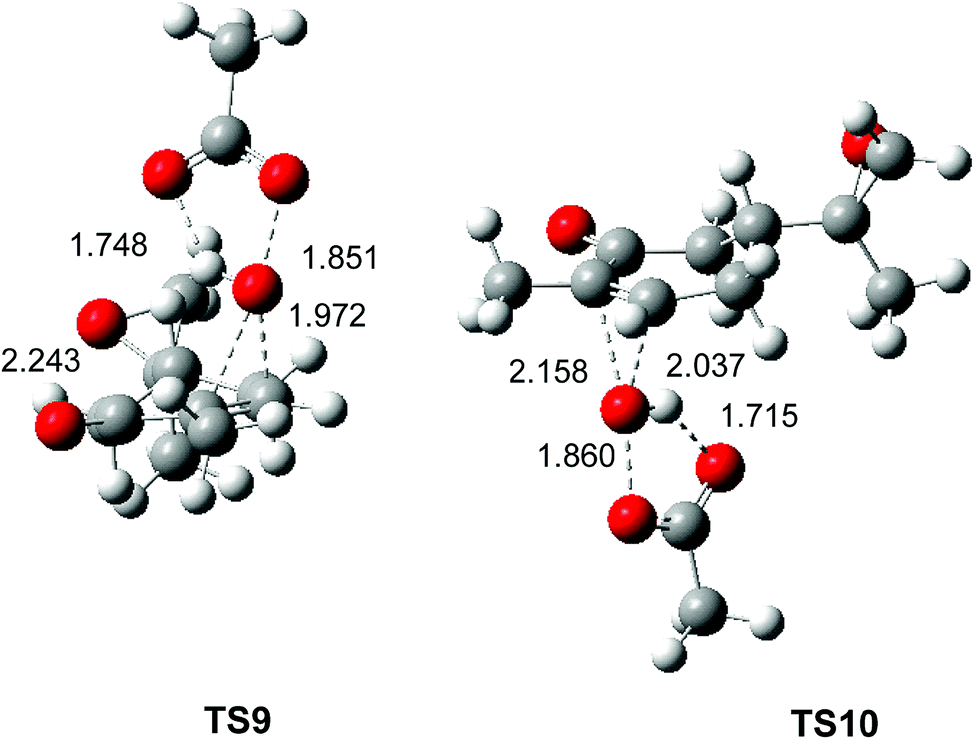 | ||
| Fig. 4 B3LYP/6-311G(d,p) geometries in DCM of the TSs involved in the epoxidation reaction of α,β-conjugated ketone 10 with peracetic acid 9. Distances are given in Angstroms. | ||
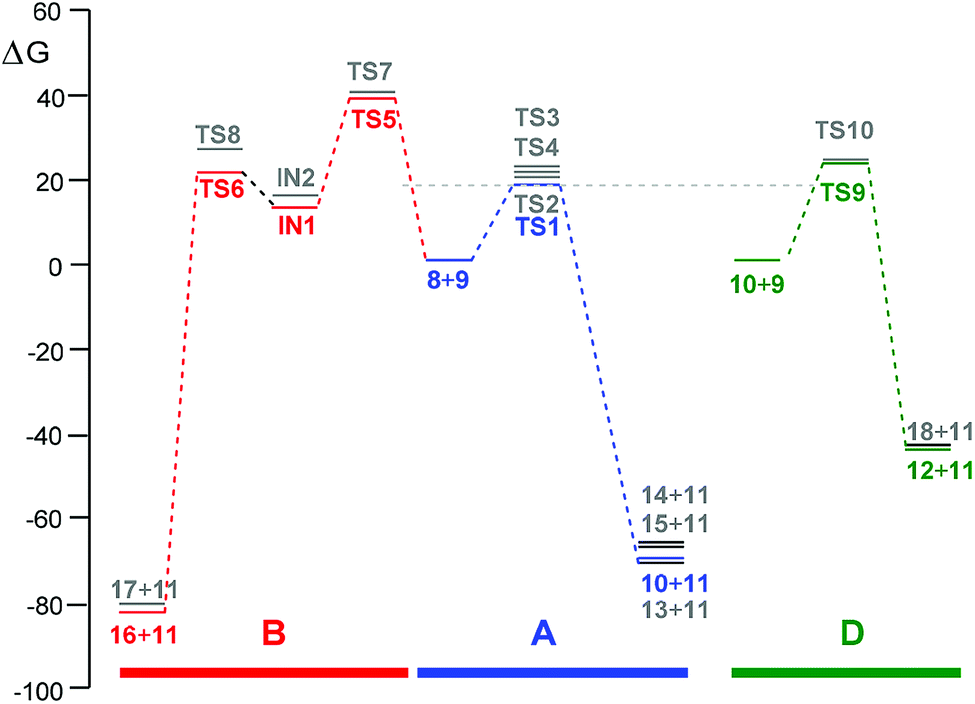 | ||
| Fig. 5 Gibbs free energy profiles, ΔG in kcal mol−1, for the competitive reaction paths associated with the reaction of R-carvone 8 with peracetic acid 9, computed at 25 °C in DCM. (A) Reaction of R-carvone 8 with 1 equivalent of peracetic acid 9 in blue; (B) BV reaction of R-carvone 8 with peracetic acid 9; and reaction of epoxide 10 with 1 equivalent of peracetic acid 9 in green. The stationary points associated with the chemo- regio- and stereoisomeric reaction paths are given in grey. | ||
The most favourable reaction paths of the reaction of R-carvone 8 with peracetic acid 9 are those associated with the attack of peracetic acid 9 on the terminal C–C double bond of the isopropenyl group. The activation Gibbs free energies associated to TS1 and TS2 are very similar, 20.9 and 21.2 kcal mol−1, respectively; consequently, this epoxidation reaction has a low diastereoselectivity. The high exergonic character of this epoxidation reaction, ca. −70 kcal mol−1, makes it irreversible. TS4 associated with the attack of peracetic acid 9 on the conjugated C–C double bond of R-carvone 8 is found 2.9 kcal mol−1 higher in Gibbs free energy than TS2; consequently, this epoxidation reaction presents a high chemoselectivity, in agreement with the analysis of the nucleophilic Pk− Parr functions of R-carvone 8. The nucleophilic attack of peracetic acid 9 on the carbonyl C1 carbon of R-carvone 8 presents a very high activation Gibbs free energy, ca. 42 kcal mol−1. In spite of the strong exergonic character of the formation of lactones 16 and 17, ca. −81 kcal mol−1, the corresponding BV reaction of R-carvone 8 is non-competitive with the epoxidation reaction as TS5 is 21.2 kcal mol−1 higher in Gibbs free energy than TS2.
Finally, the activation Gibbs free energy associated with the epoxidation of monoepoxide 10 via TS9 is slightly higher than that associated to TS4, 0.5 kcal mol−1, but it is 3.4 kcal mol−1 higher than TS1. This Gibbs free energy difference accounts for the formation of monoepoxide 10 in the presence of 1 equivalent of peracetic acid 9, while in the presence of 2 equivalents of peracetic acid 9, di-epoxide 12 is expected to be the major product of the epoxidation reaction (see Scheme 4).
Taking into account that the monoepoxidation reaction of R-carvone 8 with peracetic acid 9 takes place through favourable kinetic control, the Eyring–Polanyi equation56 was used to estimate the composition of the reaction mixture.
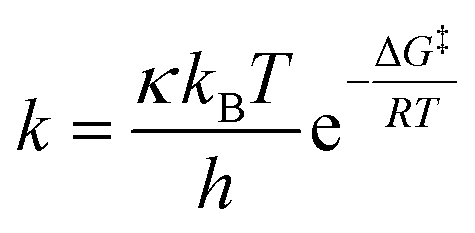
From this equation, the relative reaction rate constants krel can be obtained as:
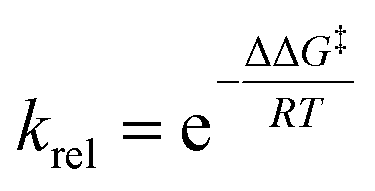
Considering the Gibbs free energies associated to TS1, TS2, TS3 and TS4, given in Scheme 5, and the reaction temperature, 25 °C, the following relationship between the four monoepoxide isomers can be obtained: 62.2 (10): 37.1 (13): 0.3 (14): 0.5 (15). This analysis indicates that the stereoisomeric monoepoxides 10 and 13, obtained in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, are the majority reaction products, ca. 99%, while formation of the monoepoxides 14 and 15 is experimentally negligible. These results account for the high chemoselectivity involving the C–C double bond of the isopropenyl group and the low diastereoselectivity, in complete agreement with the experimental outcomes.39
:1 ratio, are the majority reaction products, ca. 99%, while formation of the monoepoxides 14 and 15 is experimentally negligible. These results account for the high chemoselectivity involving the C–C double bond of the isopropenyl group and the low diastereoselectivity, in complete agreement with the experimental outcomes.39
3.3 BET analysis of the bonding changes along the most favourable reaction path associated with the monoepoxidation of R-carvone 8 with peracetic acid 9
In order to understand the bonding changes taking place along the epoxidation reaction of R-carvone 8 with peracetic acid 9, and thus, the formation of the two new C–O single bonds of the oxirane ring, a BET study27 along the IRC associated with the most favourable reaction path leading to the formation epoxide 10 was performed. The detailed description of the BET analysis is given in the ESI,† while the most relevant conclusions arising from this analysis are summarised below.Some appealing conclusions can be drawn from this BET study: (i) the IRC of the epoxidation reaction is divided in sixteen differentiated phases associated with the rupture and formation of at least five single bonds; (ii) the reaction begins with the rupture of the O12–O13 single bond of peracetic acid 9 in the early Phase II, at an O12–O13 distance of 1.48 Å, by sharing 0.64 e of the O–O single bond between the two O12 and O13 oxygen nuclei in an approximate 20:80% relationship, respectively; (iii) the rupture of the O12–O13 single bond, which demands a relatively low energy cost of 7 kcal mol−1 and accounts for the labile character of this O–O single bond, releases one hydroxyl and one acyloxy pseudoradical framework; (iv) the high activation energy associated to the epoxidation reaction of R-carvone 8 with peracetic acid 9, 16.4 kcal mol−1, can mainly be related to the ruptures of the O12–O14 single bond of peracetic acid 9, and the depopulation of C7–C8 double bond of R-carvone 8, as well as to the electronic reorganisation at both O12 and O13 oxygens; (v) the rupture of the O13–H14 single bond at the hydroxyl framework takes place in Phase XI, at an O–H distance of 1.08 Å, by sharing its 2.01 e between the O13 oxygen and H14 hydrogen nuclei in an approximate 75:25% relationship, respectively, thus releasing an anionic O13 oxygen integrating 6.65 e, and a free pseudoradical H14 hydrogen integrating 0.56 e; (vi) formation of the two new C7–O13 and C8–O13 single bonds takes place at a C–O distance of 1.52 and 1.55 Å in Phases XII and XVI, respectively, and with an initial population of 0.82 e and 0.74 e, entirely by donation of non-bonding electron density of the anionic O13 oxygen to the C7 and C8 carbons (see structures S13 and S16 in Fig. 6); (vii) formation of both oxiranic O–C single bonds is highly asynchronous. Although formation of both O–C single bonds occurs at the end of the reaction path, formation of the second C7–O13 single bond takes place when the first C8–O13 single bond is completely formed; (viii) formation of the new H14–O6 single bond present in acetic acid 11 takes place in Phase XIV, after formation of the first C8–O13 single bonds and before formation of the C7–O13 one, at an H14–O11 distance of 1.14 Å, and with an initial population of 1.73 e, by sharing the electron density of the H14 pseudoradical center and some non-bonding electron density of the O11 oxygen in an approximate 25:75% relationship, respectively; and, finally, (ix) the present BET analysis clearly shows that the molecular mechanism of this epoxidation reaction involving at least the rupture and formation of five single bonds is non-concerted; any formation of a new single bond demands the previous rupture of old single bonds.
 | ||
| Fig. 6 B3LYP/6-311G(d,p) ELF localisation domains of the structures S12, S13, S15 and S14 involved in the formation of the two oxiranic C–O single bonds along the most favourable reaction path associated with the monoepoxidation of R-carvone 8 with peracetic acid 9, represented at an isosurface value of ELF = 0.75. | ||
4. Conclusions
The epoxidation reaction of R-carvone 8 with peracetic acid 9 has been studied within the MEDT through DFT calculations at the B3LYP/6-311G(d,p) computational level. The chemo- and stereoisomeric reaction paths associated with the mono- and diepoxidation of R-carvone 8, and the regioisomeric reaction paths associated with the BV reaction have been explored and characterized.Analysis of the CDFT indices accounts for the reactivity of R-carvone 8, but they fail when predicting the reactivity of peracetic acid 9 acting as oxidant. R-carvone 8 is classified as a strong electrophile and a moderate nucleophile. Analysis of the nucleophilic Pk− Parr functions indicates that the non-substituted carbon of the isopropenyl C–C double bond is the most nucleophilic carbon of R-carvone 8. This feature accounts for the chemoselectivity and asynchronicity found in the C–O bond formation at the most favourable TSs associated with the epoxidation reaction.
The epoxidation reaction takes place through a one-step mechanism, in which the formation of the two new C–O single bonds is somewhat asynchronous. Analysis of the Gibbs free energy profiles associated with the epoxidation reaction indicates that this reaction presents a poor diastereoselectivity and a high chemoselectivity. The most favourable reaction paths are those associated with the attack of peracetic acid 9 on the isopropenyl C–C double bond of R-carvone 8.
Although the attack to the conjugated C–C double bond of R-carvone 8 is only 2.8 kcal mol−1 higher in Gibbs free energy than the attack to the isopropenyl C–C double bond, this attack on the monoepoxide obtained along the first epoxidation reaction is 5.4 kcal mol−1 higher in Gibbs free energy than the attack on R-carvone 8. These energy results allow explaining the complete monoepoxidation of R-carvone 8 at −20 °C by using only one equivalent of peracetic acid 9.
Analysis of the energetic results associated with the BV reaction indicates that it is completely regioselective, but it is not competitive with the epoxidation reaction.
Finally, the BET analysis of the bonding changes along the most favourable reaction path associated with the monoepoxidation of R-carvone 8 shows the complexity of the mechanism of this epoxidation reaction involving a non-symmetric ethylene. The epoxidation reaction begins with the rupture of the labile O12–O13 single bond of peracetic acid 9 and with the depopulation of the isopropenyl C7–C8 double bond of R-carvone 8. The subsequent rupture of the O13–H14 single bond at the released hydroxyl framework generates anionic O13 oxygen which attacks asynchronously the C7 and C8 carbons of the double bond of R-carvone 8, yielding the formation of the oxirane ring at the end of the reaction path.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the Ministerio of Investigación, Innovación y Universidades of the Spanish Government, project CTQ2016-78669-P. This project has also received funding from the European Union's Horizon 2020 research and Innovation Programme under the Marie Skłodowska-Curie grant agreement no. 846181 (MRG).References
- M. Tarleton, J. Gilbert, J. A. Sakoff and A. Mc Cluskey, Eur. J. Med. Chem., 2012, 54, 573–581 CrossRef CAS PubMed.
- Z. Liu, L. Ma and G. B. Zhou, Molecules, 2011, 16, 5283–5297 CrossRef CAS PubMed.
- A. Lewinska, P. Chochrek, K. Smolag, E. Rawska and M. Wnuk, Redox Rep., 2015, 20, 116–125 CrossRef CAS PubMed.
- B. A. Adeniyi, M. F. Robert, H. Chai and H. H. S. Fong, Phytother. Res., 2003, 17, 282–284 CrossRef CAS PubMed.
- K. Yamada, Y. Igarashi, T. Betuyaku, M. Kitamura, K. Hirata, K. Hioki and M. Kunishima, Org. Lett., 2018, 20, 2015–2019 CrossRef CAS PubMed.
- R. A. Moretti, J. Du Bois and T. D. P. Stack, Org. Lett., 2016, 18, 2528–2531 CrossRef CAS PubMed.
- V. Hulea, E. Dumitriu, F. Patcas, R. Ropot, P. Graffin and P. Moreau, Appl. Catal., A, 1998, 170, 169–175 CrossRef CAS.
- J. L. Bicas, A. P. Dionísio and G. M. Pastore, Chem. Rev., 2009, 109, 4518–4531 CrossRef CAS PubMed.
- K. K. Aggarwal, S. P. S. Khanuja, A. Ahmad, T. R. Santha Kumar, V. K. Gupta and S. Kumar, Flavour Fragrance J., 2002, 17, 59–63 CrossRef CAS.
- R. Paduch, M. Kandefer-Szerszeń, M. Trytek and J. Fiedurek, Arch. Immunol. Ther. Exp., 2007, 55, 315–327 CrossRef CAS.
- G. K. Marei, M. A. Abdel Rasoul and S. A. M. Abdelgaleil, Pestic. Biochem. Physiol., 2012, 103, 56–61 CrossRef CAS.
- L. D. Martino, E. Mancini, L. F. Rolim de Almeida and V. De Feo, Molecules, 2010, 15, 6630–6637 CrossRef PubMed.
- M. Elmastas, I. Dermirtas and O. Isildak, J. Liq. Chromatogr. Relat. Technol., 2006, 29, 1465–1475 CrossRef CAS.
- M. Tommi, A. Meulemans Gerrit, Z. Stork Fliur, J. M. Macaev Ben and J. Aede de Groot, J. Org. Chem., 1999, 64, 9178–9188 CrossRef.
- B. Song, Y. Zhou, H.-M. Yang, J.-H. Liao, L.-. Yang, X.-B. Yang and E. Ganz, J. Am. Chem. Soc., 2019, 418, 3630–3640 CrossRef PubMed.
- L.-M. Yang, V. Bačić, I. A. Popov, A. I. Boldyrev, T. Heine, T. Frauenheim and E. Ganz, J. Am. Chem. Soc., 2015, 137, 2757–2762 CrossRef CAS PubMed.
- J.-H. Liu, L.-M. Yang and E. Ganz, ACS Sustainable Chem. Eng., 2018, 6, 15494–15502 CrossRef CAS.
- J.-H. Liu, L.-M. Yang and E. Ganz, J. Mater. Chem. A, 2019, 7, 3805–3814 RSC.
- J.-H. Liu, L.-M. Yang and E. Ganz, J. Mater. Chem. A, 2019, 7, 11944–11952 RSC.
- L. Xu, L.-M. Yang and E. Ganz, Theor. Chem. Acc., 2018, 137, 98 Search PubMed.
- I. Garcia-Bosch, X. Ribas and M. Costas, Adv. Synth. Catal., 2009, 351, 348–352 CrossRef CAS.
- D. Sureshkumar, S. Maity and S. Chandrasekaran, J. Org. Chem., 2006, 71, 1653–1657 CrossRef CAS PubMed.
- G. Majetich, R. Hicks, G. Sun and P. McGill, J. Org. Chem., 1998, 63, 2564–2573 CrossRef CAS PubMed.
- L. R. Domingo, M. Ríos-Gutiérrez and N. Acharje, Molecules, 2019, 24, 832 CrossRef PubMed.
- P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793–1873 CrossRef CAS PubMed.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- X. Krokidis, S. Noury and B. Silvi, J. Phys. Chem. A, 1997, 101, 7277–7282 CrossRef CAS.
- S. J. Hosseinia, S. Emamian and L. R. Domingo, Struct. Chem., 2019, 30, 317–326 CrossRef.
- L. R. Domingo, M. Ríos-Gutiérrez and S. Emamian, RSC Adv., 2017, 7, 15586–15595 RSC.
- A. Zeroual, M. Ríos-Gutiérrez, M. El Idrissi, H. El Alaoui El Abdallaoui and L. R. Domingo, Int. J. Quantum Chem., 2019, e25980 Search PubMed.
- L. R. Domingo, Molecules, 2016, 21, 1319 CrossRef PubMed.
- M. Freccero, R. Gandolfi, M. Sarzi-Amade and A. Rastelli, J. Org. Chem., 2000, 65, 8948–8959 CrossRef CAS PubMed.
- S. I. Okovityi, R. G. Gaponova, Y. A. Seredyuk and L. I. Kas'yan, Russ. J. Org. Chem., 2002, 38, 160–164 CrossRef CAS.
- R. D. Bach and O. Dmitrenko, J. Phys. Chem. A, 2003, 107, 4300–4306 CrossRef CAS.
- R. D. Bach, O. Dmitrenko, W. Adam and S. Schambony, J. Am. Chem. Soc., 2003, 125, 924–934 CrossRef CAS PubMed.
- H. C. Shi, Z. G. Zhang and Y. L. Wang, J. Mol. Catal. A: Chem., 2005, 238, 13–25 CrossRef CAS.
- S. M. Koskowich, W. C. Johnson, R. S. Paley and P. R. Rablen, J. Org. Chem., 2008, 73, 3492–3496 CrossRef CAS PubMed.
- J. D. Zhai, D. Li, J. Long, H.-L. Zhang, J.-P. Lin, C.-J. Qiu, Q. Zhang and Y. Chen, J. Org. Chem., 2012, 77, 7103–7107 CrossRef CAS PubMed.
- A. Murphy, G. Dubois and T. D. P. Stack, J. Am. Chem. Soc., 2003, 125, 5250–5251 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- H. B. Schlegel, J. Comput. Chem., 1982, 2, 214–218 CrossRef.
- H. B. Schlegel, in Modern Electronic Structure Theory, ed. D. R. Yarkony, World Scientific Publishing, Singapore, 1994 Search PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161–4163 CrossRef CAS.
- C. González and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef.
- C. González and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853–5860 CrossRef.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327–335 CrossRef CAS.
- E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032–3041 CrossRef CAS.
- V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404–417 CrossRef CAS.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, Molecules, 2016, 21, 748 CrossRef PubMed.
- L. R. Domingo, P. Pérez and J. A. Sáez, RSC Adv., 2013, 3, 1486–1494 RSC.
- M. J. Frisch, et al., Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- S. Noury, X. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597–604 CrossRef CAS.
- M. G. Evans and M. Polanyi, Trans. Faraday Soc., 1935, 31, 875–894 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05309c |
| This journal is © The Royal Society of Chemistry 2019 |