
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLncRNA GAS5 facilitates nasopharyngeal carcinoma progression through epigenetically silencing PTEN via EZH2†
Dan
Zhao
,
Yujie
Li
 * and
Min
Yu
* and
Min
Yu
Department of Otorhinolaryngology Head and Neck Surgery, Zhengzhou Central Hospital Affiliated to Zhengzhou University, No. 195 Tongbai Road, Zhengzhou, 450000, China. E-mail: liyujiezzdxohns@163.com; Tel: +86-0371-67690915
First published on 7th October 2019
Abstract
Increasing evidence demonstrated that long non-coding RNA growth-arrest-specific transcript 5 (GAS5) serves as a critical regulator in cancer development and progression. However, its function and mechanism in nasopharyngeal carcinoma (NPC) is still not well elucidated. In this study, we investigate the functional role as well as the molecular mechanism of GAS5 in NPC progression. Our results indicated that GAS5 expression was elevated in NPC tissues and cells. High GAS5 expression was correlated with poor prognosis of NPC patients. GAS5 knockdown suppressed proliferation, migration and invasion, and induced apoptosis in NPC cells. Moreover, GAS5 could epigenetically suppress PTEN expression via recruiting enhancer of zeste homolog 2 (EZH2). PTEN knockdown could reverse the inhibitory effect of GAS5 inhibition on NPC progression. Furthermore, GAS5 knockdown suppressed the tumor growth in vivo. In summary, knockdown of GAS5 repressed proliferation, migration and invasion, and promoted apoptosis in NPC through epigenetically silencing PTEN via recruiting EZH2.
Introduction
Nasopharyngeal carcinoma (NPC) is a highly metastatic and invasive malignancy derived from the epithelial cell lining on the nasopharynx, and is prevalent in South Asia, North Africa and Southern China.1 The long-term survival was usually excellent in early stage NPC patients; however, the locally advanced and metastatic NPC patients were subjected to poor prognosis.2 Like many other cancers, multiple genetic and epigenetic aberrancies could lead to NPC tumorigenesis and progression.3 However, the precise molecular mechanisms responsible for NPC progression remain largely unknown. Therefore, it is still urgent to identify molecules involved in NPC progression, providing novel therapeutic targets for NPC patients.Long non-coding RNAs (lncRNAs) are a class of endogenous non-protein-coding RNAs greater than 200 nucleotides in length without obvious protein-coding functions.4 LncRNAs have recently been found to play vital regulatory roles in diverse biological processes, including tumorigenesis process.5 Increasing evidence suggests that lncRNAs are commonly deregulated in the progression of malignant tumors, including NPC.6–8 LncRNA growth-arrest-specific transcript 5 (GAS5) was originally identified in a subtraction complementary DNA (cDNA) library prepared for sequencing RNA expressed in growth arrested cells.9 Subsequently, accumulating documents have indicated that GAS5 is commonly downregulated in various malignancies, such as lung cancer, gastric cancer and ovarian cancer.10–12 Growing evidence suggests that GAS5 was linked to cell growth and metastasis, and played a crucial role in tumor progression.13,14 Moreover, GAS5 could overcome DDP-resistance in epithelial ovarian cancer through E2F4-PARP1-MAPK axis.15 Furthermore, low expression of GAS5 could predict poor prognosis in various cancers.16 However, limited knowledge is available concerning the functional role and underlying mechanism of GAS5 in NPC progression.
In the present study, we found that GAS5 was up-regulated in NPC tissues and cells. Moreover, GAS5 knockdown repressed NPC cell proliferation, migration and invasion, and promoted apoptosis. Furthermore, GAS5 epigenetically suppressed PTEN expression through recruiting enhancer of zeste homolog 2 (EZH2), and then exerted its oncogenic function. Our study demonstrated a novel epigenetic regulatory mechanism between GAS5 and PTEN which could contribute to NPC progression.
Materials and methods
Tissue specimens and cell culture
Thirty-eight NPC patient tissue samples and thirty-eight normal nasopharyngeal tissue samples were obtained from Zhengzhou Central Hospital Affiliated to Zhengzhou University. This study was approved by the Ethic Review Committees of Zhengzhou Central Hospital Affiliated to Zhengzhou University. Informed consent was obtained from all subjects. To explore the expression level of GAS5 in NPC, we downloaded three sets of gene expression profiling data for NPC (GSE64634 and GSE12452) from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). All these data from databases were analyzed using R language.The human nasal epithelial cell (HNEpC) and nasopharyngeal carcinoma cell lines (5-8F, CNE1, CNE2 and HONE1) was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). All cells were cultured in RPMI-1640 (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS) (Invitrogen) at 37 °C with 5% CO2.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from NPC tissues and cells using the TRIzol reagent (Invitrogen) and reversely transcribed using a high capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA). GAS5 and PTEN expression levels were detected by A SYBR Premix Ex Taq™ kit (Takara Bio, Otsu, Japan) on the 7500 Real Time PCR System (Applied Biosystems). GAPDH acted as an internal control. The comparative Ct method (2−ΔΔCt) was used to analyze the data.Cell transfection
The empty vector pcDNA3.1 (Vector) or GAS5 overexpressing vector pcDNA3.1-HOXD-AS1 (pcDNA-GAS5) and small interfering RNAs against GAS5 (si-HOXD-AS1), EZH2 (si-EZH2) or PTEN (si-PTEN) or their scramble negative siRNA (si-con) were obtained from Genepharma (Shanghai, China). The Lipofectamine 2000 (Invitrogen) was used to perform cell transfections.Cell proliferation assay
CNE2 and HONE1 cells were seeded in 96-well plates and cultured for 24 h. After transfection for 24 h, 48 h, 72 h and 96 h, the cell viability was determined by MTT assay (Sigma, St. Louis, Missouri, USA). The absorbance at 490 nm was determined using a microtiter plate reader (Molecular Devices, Sunnyvale, CA, USA).Transwell migration and invasion assays
CNE2 and HONE1 cell migration and invasion were determined using transwell chambers (Millipore, Billerica, MA, USA). The detailed procedure is mentioned in the previous study.17Apoptosis assay
Transfected CNE2 and HONE1 cells were collected and stained according to the protocol of Annexin V-FITC/PI kit (BD Biosciences, San Jose, CA, USA). Then, the flow cytometry data were analyzed by BD FACSDiva software V6.1.3 (BD Biosciences).Subcellular fraction assays
The separation of nuclear and cytoplasmic RNA of CNE2 cells was performed using Cytoplasmic & Nuclear RNA Purification Kit (Norgen, Belmont, CA, USA) in accordance with the manufacturer's instructions.RNA pull-down assays
Biotinylated GAS5 and anti-sense-GAS5 RNAs were bound with Streptavidin Magnetic Beads (Life Technologies, USA) and incubated with CNE2 cell protein lysates. Then the bound proteins were purified for the further western blot analysis.RNA immunoprecipitation (RIP) assays
RIP experiments were performed using the Magna RIP RNA-binding protein immunoprecipitation kit (Millipore, Billerica, MA, USA) and EZH2 (#5246) and IgG antibodies (2729; Cell Signaling Technology, Danvers, MA, USA) following the manufacturer's instructions. The co-precipitated RNAs were purified and subjected to qRT-PCR analysis.Chromatin immunoprecipitation (ChIP) assays
ChIP assay was performed using EZ-ChIP kit (Millipore) with antibodies against EZH2 (#5246; Cell Signaling Technology), H3K27me3 (#17-622; Millipore) or IgG (#AP132P; Millipore). The immunoprecipitated chromatin was purified and analyzed by qRT-PCR analysis.Luciferase reporter assay
The PTEN promoter reporter vector was obtained from Genechem (Shanghai, China). CNE2 cells were co-transfected with (Vector or pcDNA-GAS5) or (si-con or si-GAS5) and PTEN promoter reporter plasmid using Lipofectamine 2000 reagent (Invitrogen). The luciferase activities were measured using the Dual-Glo luciferase reporter assay kit (Promega, Madison, WI, USA) 48 h post transfection.Western blot analysis
Western blotting was performed as described previously.18 The antibodies against EZH2 (#5246), PTEN (26H9) and GAPDH (#2118) were purchased from Cell Signaling Technology (Danvers, MA, USA).Animal experiments
The maintenance of the animals and the experimental procedures were in strict accordance with the guiding principles of the institutional animal ethics committee and was approved by the institutional research committee of Zhengzhou Central Hospital Affiliated to Zhengzhou University. CNE2 cells (1.0 × 107) stably infected with sh-GAS5 or sh-con were subcutaneously injected into the tail veins of BALB/c-nude mice (six weeks old) from Slac Laboratory (Shanghai, China). The tumor size was monitored every week. After 35 days, the mice were sacrificed, and the tumor weight was measured. GAS5 expression and PTEN protein levels were measured by qRT-PCR and western blot assays.Statistical analysis
All data are expressed as means ± SD. Statistical differences between groups were evaluated by Student's t-test and one-way ANOVA. P < 0.05 was considered significant.Results
GAS5 was up-regulated in NPC tissues and cells
To analyze the expression of lncRNA GAS5 in NPC tumor and normal tissues, we downloaded NPC gene expression data from GEO datasets (GSE64634 and GSE12452). GAS5 was highly expressed in NPC tumor tissues compared with normal tissues in both above-mentioned datasets (Fig. 1A and B). To confirm the result of the databases, qRT-PCR analysis was performed to measure the expression of GAS5 in NPC patient samples (n = 38) and normal tissue samples (n = 38). Compared with normal tissues, GAS5 was significantly elevated in NPC tumor tissues (Fig. 1C). Additionally, GAS5 expression was examined in NPC cell lines (5-8F, CNE1, CNE2 and HONE1) and HNEpC. qRT-PCR analysis revealed that GAS5 was evidently increased in all NPC cells compared with the normal HNEpC cells (Fig. 1D). Moreover, based on the median value of GAS5 expression, the patients were divided into GAS5 high expression (n = 19) and GAS5 low expression (n = 19) groups. Kaplan–Meier survival analysis revealed high GAS5 expression level was associated with a poor survival in NPC patients (P = 0.0083) (Fig. 1E). All these data suggested that aberrant GAS5 expression may play an important role in NPC tumorigenesis.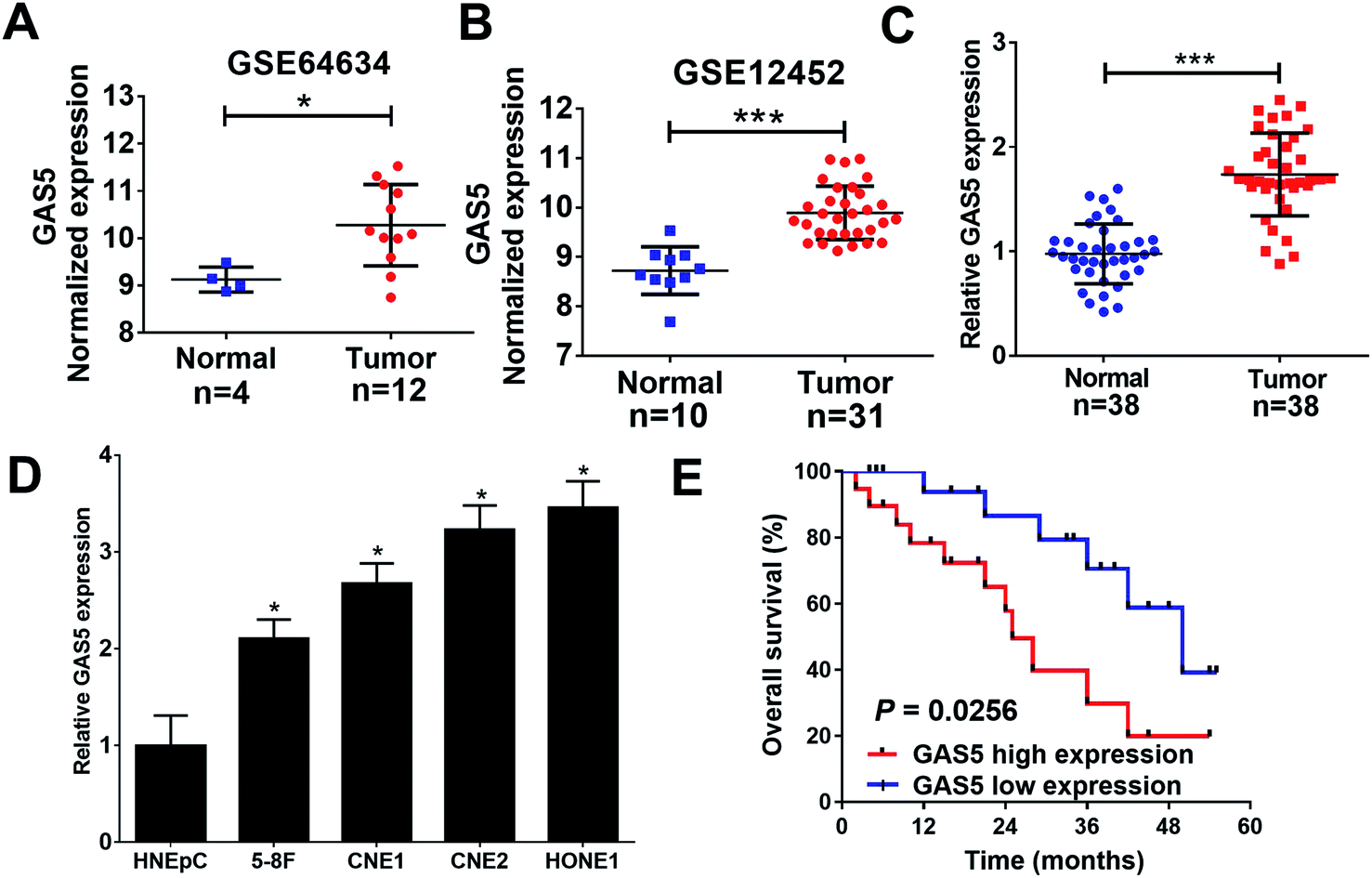 | ||
| Fig. 1 GAS5 was up-regulated in NPC tissues and cells. (A and B) The expression of GAS5 was analyzed in tumor or normal tissues from GEO dataset (GSE64634 and GSE12452). qRT-PCR analysis revealed the expression levels of lncRNA GAS5 in NPC patient tumor tissues (n = 35) and normal nasopharyngeal tissues (n = 35) (C), and NPC cell lines (5-8F, CNE1, CNE2 and HONE1) or HNEpC (D). (E) Kaplan–Meier curve was used to evaluate the overall survival between low and high GAS5 expression groups. The experiments were repeated at least three times. *P < 0.05. ***P < 0.001. | ||
GAS5 knockdown inhibited proliferation, migration and invasion, and promoted apoptosis in NPC cells
Considering CNE2 and HONE1 cells had higher GAS5 expression among NPC cell lines (shown in Fig. 1D), we chose CNE2 and HONE1 cells for further study. To investigate the functional role of GAS5 in NPC, the proliferation, migration and invasion, and apoptosis capacities of CNE2 and HONE1 cells were determined by MTT, transwell and flow cytometry assays after si-con or si-GAS5 transfection. Firstly, qRT-PCR analysis revealed that si-GAS5 transfection remarkably lowered GAS5 expression in CNE2 and HONE1 cells (Fig. 2A). MTT assay indicated that GAS5 knockdown evidently repressed the proliferation of CNE2 and HONE1 cells (Fig. 2B and C). Transwell assays showed that introduction of si-GAS5 pointedly suppressed migration and invasion of CNE2 and HONE1 cells (Fig. 2D and E). Flow cytometry analysis demonstrated that GAS5 inhibition dramatically improved apoptosis in CNE2 and HONE1 cells (Fig. 2F and G). Collectively, these results suggested that GAS5 knockdown suppressed proliferation, migration and invasion, and induced apoptosis in NPC cells.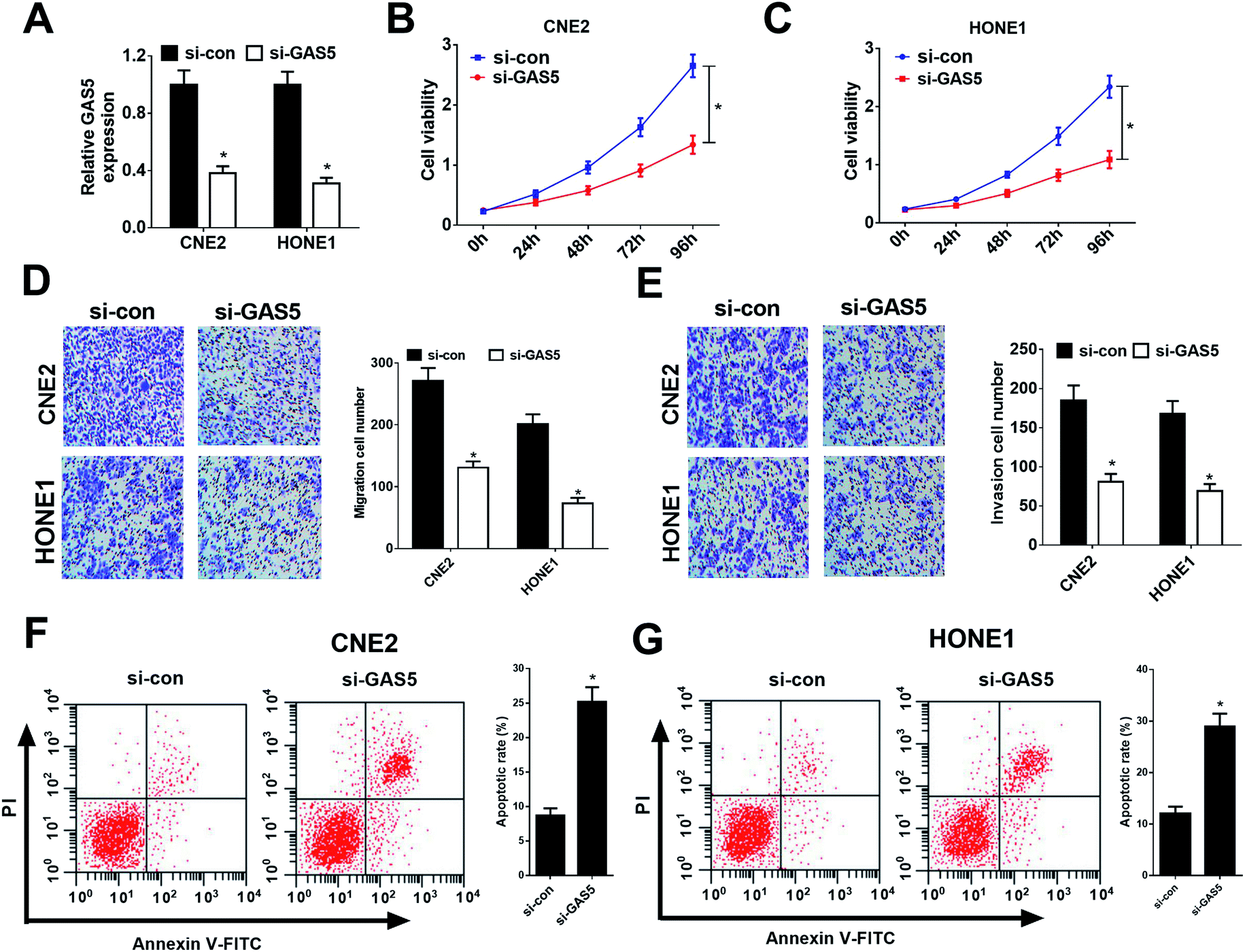 | ||
| Fig. 2 GAS5 knockdown suppressed proliferation, migration and invasion, and induced apoptosis in NPC cells. CNE2 and HONE1 cells were transfected with si-control or si-GAS5. (A) qRT-PCR analysis detected GAS5 expression in CNE2 and HONE1 cells. (B and C) The proliferation activity of CNE2 and HONE1 cells was evaluated by MTT assay. (D and E) Migration and invasion of CNE2 and HONE1 cells were determined by transwell assays. (F and G) Cell apoptosis was determined by flow cytometry analysis in CNE2 and HONE1 cells. The experiments were repeated at least three times. *P < 0.05. | ||
GAS5 epigenetically suppressed PTEN expression in NPC cells
GAS5 has been reported to regulate gene expression by recruiting EZH2, increasing the H3K27me3 level in the promoter regions of target genes.19 Moreover, PTEN could be suppressed by EZH2 through enrichment of H3K27me3 on its promoter region in various cancers.20,21 Hence, we further investigated whether GAS5 epigenetically suppressed PTEN expression in NPC cells by recruiting EZH2. Firstly, our results found that GAS5 was mainly located in the nucleus of CNE2 cells (Fig. 3A), suggesting that GAS5 may exert its regulatory role at the transcriptional level. Then, chipbase database (http://rna.sysu.edu.cn/chipbase/) was used to analyze the correlation between GAS5 and EZH2 in 502 HNSC tissue samples from TCGA databases. The results showed that GAS5 expression was positively associated with EZH2 level in HNSC tissue samples (Fig. 3B). Moreover, GAS5 silencing pointedly elevated PTEN expression in CNE2 cells (Fig. 3C). Additionally, introduction of si-EZH2 lowered EZH2 and increased PTEN expression in CNE2 cells (Fig. 3D). To confirm whether GAS5 directly bound EZH2 in CNE2 cells, RNA pull-down and RIP assays were performed in CNE2 cells. As expected, GAS5 could pull down EZH2 protein (Fig. 3E) and GAS5 was markedly enriched by EZH2 antibody (Fig. 3F). All these results revealed that GAS5 could directly bind EZH2 in NPC cells. To further confirm whether GAS5 transcriptionally suppressed PTEN expression through the enrichment of EZH2 to the PTEN promoter, ChIP assays were performed in CNE2 and HONE1 cells. The results revealed that GAS5 silencing remarkably reduced EZH2 binding and H3K27me3 occupancy on the promoter of PTEN in CNE2 and HONE1 cells (Fig. 3G and ESI Fig. 1A†). Also, luciferase reporter assay revealed that GAS5 inhibition improved the activity of PTEN promoter, oppositely, overexpression of GAS5 suppressed the promoter activity in CNE2 and HONE1 cells (Fig. 3H and ESI Fig. 1B†). Moreover, correlation analysis revealed that PTEN expression was negatively correlated with GAS5 expression in NPC tumor tissues (Fig. 3I). All these data demonstrated that GAS5 epigenetically silenced PTEN through recruiting EZH2 and increasing H3K27me3 level on the promoter region of PTEN in NPC cells.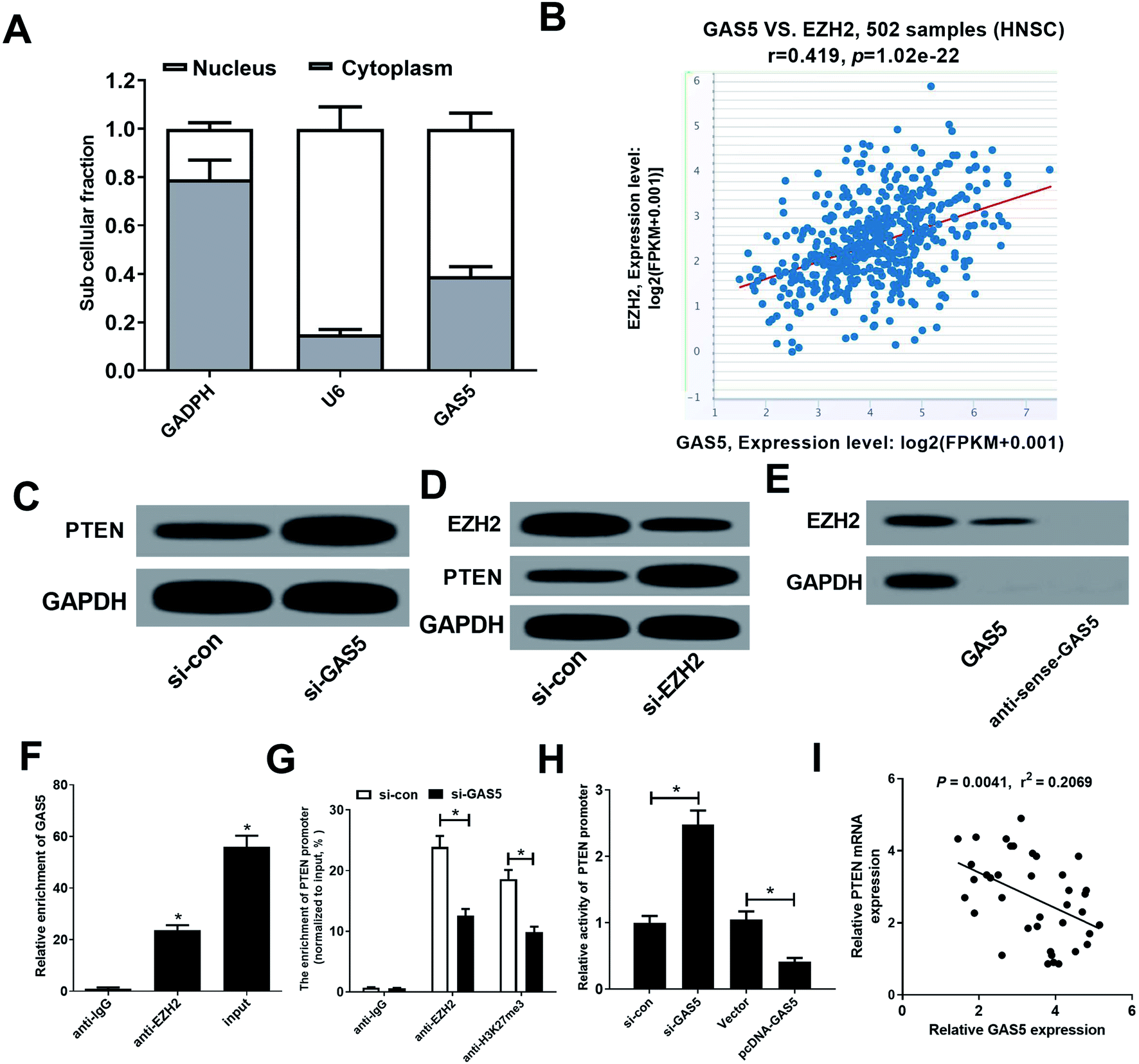 | ||
| Fig. 3 GAS5 epigenetically suppressed PTEN expression in NPC cells. (A) Pearson correlation analysis between GAS5 and EZH2 in 502 tumor tissue samples of HNSC from TCGA. (B) GAS5 subcellular location was explored in CNE2 cells. GAPDH acted as cytoplasm control and U6 used as a nucleus control. (C) PTEN protein levels were detected in CNE2 cells transfected with si-con or si-GAS5. (D) EZH2 and PTEN protein levels were measured in CNE2 cells transfected with si-con or si-EZH2. RNA pull-down assay (E) and RIP assay (F) were performed to identify the direct binding between GAS5 and EZH2. (G) qRT-PCR analysis following ChIP was performed to evaluate enrichment of EZH2 and H3K27me3 on the PTEN promoter in CNE2 cells. (H) Luciferase reporter assay was performed to evaluate the activity of the PTEN promoter in CNE2 cells transfected with (si-con or si-GAS5) or (Vector or pcDNA-GAS5). (I) Correlation analysis of GAS5 and PTEN expression in NPC tissues. The experiments were repeated at least three times.*P < 0.05. | ||
PTEN inhibition reversed the effect of GAS5 knockdown on proliferation, migration and invasion and apoptosis in NPC cells
Considering that GAS5 could repress the expression of PTEN, we further confirmed whether GAS5 exerted its function in NPC by regulating PTEN expression. First, CNE2 and HONE1 cells were transfected with si-con, si-GAS5 or si-GAS5 + si-PTEN. Then, qRT-PCR analysis, MTT assay, transwell assay, and flow cytometry analysis were performed. qRT-PCR analysis revealed that introduction of si-PTEN weakened si-GAS5-mediated enhancement in PTEN expression (Fig. 4A). Functional analysis revealed that the inhibitory effects of GAS5 knockdown on cell proliferation, migration and invasion and the inductive effect of si-GAS5 on apoptosis in CNE2 and HONE1 cells were eliminated by PTEN silencing (Fig. 4B–G). All these data demonstrated that GAS5 promoted NPC progression through epigenetically suppressed PTEN expression.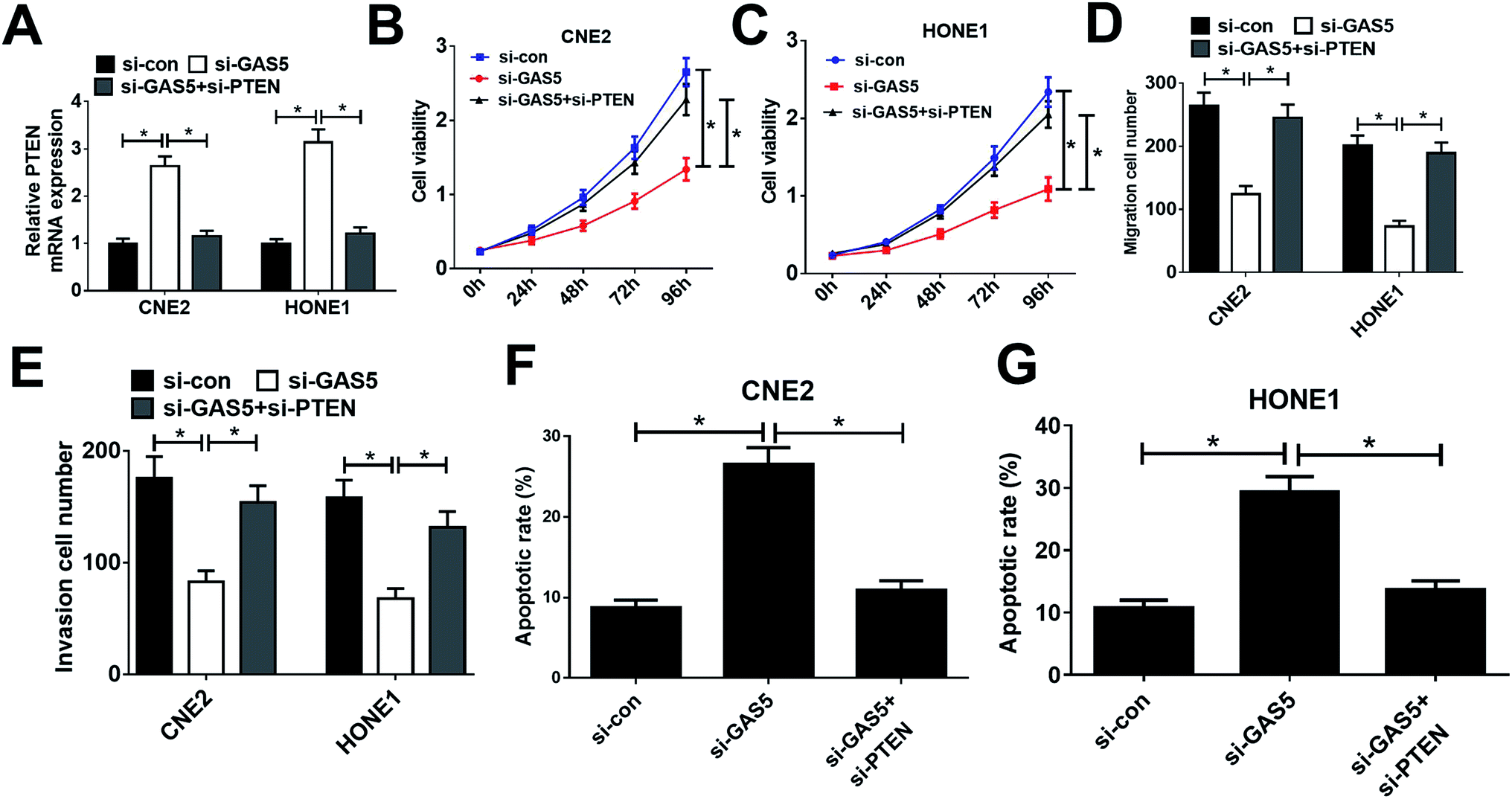 | ||
| Fig. 4 PTEN inhibition reversed the effects of GAS5 knockdown on proliferation, migration and invasion and apoptosis in NPC cells. CNE2 and HONE1 cells were with transfected with si-con, si-GAS5 or si-GAS5 + si-PTEN, followed by determination of PTEN expression by qRT-PCR analysis (A), cell viability by MTT assay (B and C), migration and invasion by transwell migration and invasion assays (D and E), and cell apoptosis by flow cytometry analysis (F and G). The experiments were repeated at least three times. *P < 0.05. | ||
GAS5 knockdown suppressed tumor growth in NPC in vivo
To further investigate the functional role of GAS5 in NPC in vivo, a xenograft nude mice model was established. GAS5 silencing extremely hindered tumor growth, evidenced by the diminished tumor volume (Fig. 5A) and tumor weight (Fig. 5B). Introduction of sh-GAS5 obviously decreased GAS5 levels but increased PTEN mRNA level in the excised tumor tissues (Fig. 5C). Moreover, knockdown of GAS5 noticeably lowered PTEN protein levels in the excised tumor tissues (Fig. 5D). Collectively, these results demonstrated that GAS5 depletion blocked tumor growth in NPC in vivo. Moreover, the schematic diagram of mechanism of GAS5 epigenetically regulating PTEN was shown in Fig. 6. It is possible that GAS5 interacts with EZH2 and recruits it on PTEN promoter to increase H3K27me3 level, reducing PTEN expression and subsequently leading to enhancement of proliferation, migration and invasion and suppression of apoptosis in NPC. The results indicate that lncRNA GAS5 contributed to NPC progression through epigenetically silencing PTEN. | ||
| Fig. 5 GAS5 knockdown suppressed tumor growth in NPC in vivo. CNE2 cells stably transfected with sh-con or sh-GAS5 were subcutaneously injected into the posterior flank of the nude mice. Mice were sacrificed to remove tumor masses after 35 days. (A) Tumor volumes were measured every 7 day using calipers. (B) The tumor tissues were excised and weighted. (C and D) The expressions of GAS5 and PTEN in the resected tumor tissues were determined by qRT-PCR and western blot analyses. The experiments were repeated at least three times. *P < 0.05. | ||
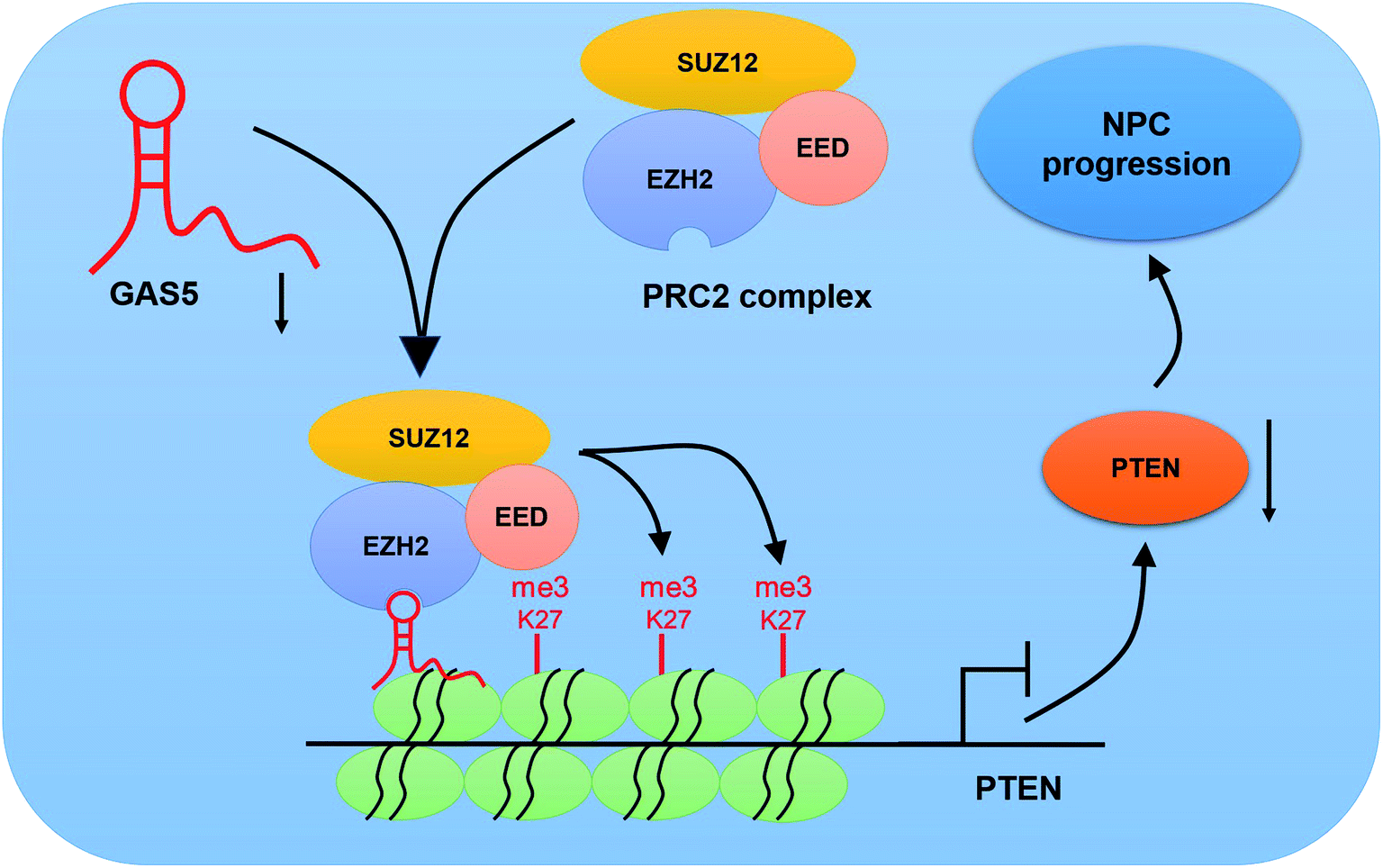 | ||
| Fig. 6 Schematic diagram of the mechanism of GAS5 epigenetically silencing PTEN in NPC. | ||
Discussion
NPC is a highly metastatic and invasive malignancy. Despite great advances achieved in NPC treatment, the prospect for NPC patients remains not optimistic. Therefore, to identify effective therapeutic targets for NPC is imperative. Increasing evidence has suggested that dysregulated lncRNAs play critical regulatory roles in carcinogenesis.22 Nevertheless, the function of lncRNAs as well as their underlying molecular mechanism in NPC remains largely unclear. In this work, GAS5 was found to be up-regulated in NPC tissues and cells. Moreover, GAS5 promoted NPC cell proliferation, migration and invasion, and suppressed apoptosis through epigenetically silencing PTEN via recruiting EZH2.Emerging evidence suggests that lncRNA GAS5 was abnormally down-regulated and exerted its tumor suppressor function in various malignancies. For example, GAS5 overexpression could obviously suppress migration and invasion of hepatocellular carcinoma cells in vitro, and suppress tumor metastasis in vivo through acting as a competing endogenous RNA (ceRNA) for miR-182 to upregulate the expression of anti-metastasis protein ANGPTL1.13 Moreover, elevated GAS5 expression repressed colorectal cancer cell invasion and migration, tumor growth, and liver metastases through suppressing the activation of the Wnt/β-catenin signaling pathway.23 In this study, we found that GAS5 expression was elevated in NPC tissues from GEO datasets and our NPC tumor tissue samples and cells. Additionally, high GAS5 expression predicted a poor prognosis in NPC patients. GAS5 up-regulation promoted proliferation, migration and invasion, and inhibited apoptosis in NPC cells. Similarly, a recent study indicated that GAS5 promotes proliferation, migration and invasion, and inhibited apoptosis in esophageal cancer cells by sponging miR-301a. All these studies demonstrated that GAS5 could act as a critical regulator involved in cancer progression, which may be a promising therapeutic target for cancer patients.
The precise mechanism by which elevated GAS5 contributes to NPC progression is undecided. Therefore, the functional mechanism of GAS5 was further explored in this study. Previous studies found that about 20% of lncRNAs can bind to polycomb repressive complex 2 (PRC2), which subsequently induced the silence of targeted genes through harboring methyltransferase activity.24 Moreover, GAS5 could epigenetically suppress genes expression through recruiting EZH2.19 EZH2, a vital catalytic subunit of polycomb repressive complex 2 (PRC2), is a histone methyltransferase that epigenetically represses gene expression by promoting histone H3 lysine 27 trimethylation (H3K27me3).25,26 PTEN, a tumor suppressor, was recently demonstrated to be negatively regulated by lncRNAs, via recruiting EZH2 and subsequently changing H3K27me3 level in various cancers.27–29 Therefore, we further studied whether GAS5 regulated PTEN expression by recruiting EZH2. Our study found that GAS5 or EZH2 silencing increased PTEN protein levels. Moreover, RNA pull-down and RIP assays further validated that GAS5 could bind to EZH2. ChIP and luciferase reporter assays further proved that GAS5 inhibition improved the promoter activity of PTEN through reducing the recruiting of EZH2 on PTEN promoter region. These data demonstrated that GAS5 epigenetically suppressed PTEN expression through recruiting EZH2 in NPC cells. PTEN was revealed to act as a tumor suppressor in many malignances.30,31 Moreover, PTEN could inhibit proliferation, migration and invasion, and induced apoptosis in cancers, including NPC.32–34 Consistently, our data also revealed that PTEN inhibition reversed the suppressive effect of GAS5 knockdown on the proliferation, migration and invasion and the inductive effect on apoptosis in CNE2 and HONE1 cells. All these data demonstrated that GAS5 contributed to the cancer progression through epigenetically silencing PTEN in NPC. However, whether GAS5 could mediate tumor progression through EZH2-mediated epigenetic suppression of target genes in other cancers needs further researches to confirm.
In summary, our findings suggested that GAS5 was up-regulated and implicated with poor prognosis in NPC. Moreover, knockdown of GAS5 inhibited proliferation, migration and invasion, and promoted apoptosis in NPC via epigenetically silencing PTEN, which may be a novel therapeutic target for NPC patients.
Conflicts of interest
The authors have no conflict of interest to declare.References
- I. Nawaz, K. Moumad, D. Martorelli, M. M. Ennaji, X. Zhou, Z. Zhang, R. Dolcetti, M. Khyatti, I. Ernberg and L.-F. Hu, Clin. Epigenet., 2015, 7, 89 CrossRef.
- K. C. Chan, L. S. Chan, J. C. Y. Ip, C. Lo, T. T. C. Yip, R. K. C. Ngan, R. N. S. Wong, K. W. Lo, W. T. Ng and A. W. M. Lee, Sci. Rep., 2015, 5, 9979 CrossRef CAS.
- J. Chou, Y. C. Lin, J. Kim, L. You, Z. Xu, B. He and D. M. Jablons, Head Neck, 2008, 30, 946–963 CrossRef.
- X. Yu and Z. Li, Oncol. Lett., 2015, 10, 1953–1958 CrossRef CAS.
- F. Zhang, L. Zhang and C. Zhang, Tumour Biol., 2015, 37, 163–175 CrossRef.
- A. M. Schmitt and H. Y. Chang, Nature, 2013, 500, 536–537 CrossRef CAS.
- Q. Q. Yang and Y. F. Deng, Histopathology, 2015, 66, 1022–1030 CrossRef.
- Y. Nie, X. Liu, S. Qu, E. Song, H. Zou and C. Gong, Cancer Sci., 2013, 104, 458–464 CrossRef CAS.
- C. Schneider, R. M. King and L. Philipson, Cell, 1988, 54, 787–793 CrossRef CAS.
- Q. Tan, J. Zuo, S. Qiu, Y. Yu, H. Zhou, N. Li, H. Wang, C. Liang, M. Yu and J. Tu, Int. J. Oncol., 2017, 50, 1729 CrossRef CAS.
- N. Zhang, A. Y. Wang, X. K. Wang, X. M. Sun and H. Z. Xue, Eur. Rev. Med. Pharmacol. Sci., 2016, 20, 3199–3205 CAS.
- H. M. Zhao, H. L. Yu, J. H. Zheng, N. Ning, F. L. Tang, Y. Yang and Y. Wang, Gynecol. Oncol., 2018, 151, 345–355 CrossRef CAS.
- F. Chen, Y. H. Li, M. J. Li and L. Wang, Am. J. Cancer Res., 2019, 9, 108–121 Search PubMed.
- B. Y. Liu, S. Q. Wu, J. Ma, S. Yan, Z. G. Xiao, L. H. Wan, F. Zhang, M. Y. Shang and A. W. Mao, Mol. Ther.--Nucleic Acids, 2018, 13, 472–482 CrossRef CAS.
- X. R. Long, K. Q. Song, H. Hu, Q. Tian, W. J. Wang, Q. Dong, X. Yin and W. Di, J. Exp. Clin. Cancer Res., 2019, 38, 345 CrossRef.
- Q. J. Gao, H. B. Xie, H. J. Zhan, J. F. Li, Y. C. Liu and W. R. Huang, Front. Physiol., 2017, 8, 814 CrossRef.
- L. Niu, A. Liu, W. Xu, L. Yang, W. Zhu and Y. Gu, Oncol. Lett., 2018, 16, 4551–4560 Search PubMed.
- H. C. Sun, Q. H. Ye, N. Ren, H. L. Jia, B. S. Sun, P. C. Lu, X. Q. Zhu, Y. F. Wang, K. F. Lei and L. Wang, BMC Cancer, 2007, 7, 172 CrossRef.
- D. Sun, Z. Yu, X. Fang, M. Liu, Y. Pu, Q. Shao, D. Wang, X. Zhao, A. Huang and Z. Xiang, EMBO Rep., 2017, 18, 1801 CrossRef CAS.
- L. Gan, M. D. Xu, R. X. Hua, C. Tan, J. Y. Zhang, Y. W. Gong, Z. H. Wu, W. W. Weng, W. Q. Sheng and W. J. Guo, J. Hematol. Oncol., 2018, 11, 9 CrossRef.
- J. F. Zhou, D. N. Nie, J. Li, X. Du, Y. H. Lu, Y. Q. Li, C. Liu, W. Dai, Y. Wang, Y. L. Jin and J. X. Pan, Clin. Cancer Res., 2018, 24, 145–157 CrossRef CAS.
- C. P. Ponting, P. L. Oliver and W. Reik, Cell, 2009, 136, 629–641 CrossRef CAS.
- J. Song, H. Shu, L. Zhang and J. Xiong, J. Cell. Biochem., 2019, 120, 6937–6951 CrossRef CAS.
- C. C. Sun, S. J. Li, G. Li, R. X. Hua, X. H. Zhou and D. J. Li, Mol. Ther.--Nucleic Acids, 2016, 5, e385 CrossRef CAS.
- R. Cao, L. Wang, H. Wang, L. Xia, H. Erdjumentbromage, P. Tempst, R. S. Jones and Y. Zhang, Science, 2002, 300, 131 Search PubMed.
- R. Cao and Y. Zhang, Curr. Opin. Genet. Dev., 2004, 14, 155–164 CrossRef CAS.
- X. K. Ma, J. Y. Zhou, J. Y. Liu, G. P. Wu, Y. Yu, H. Y. Zhu and J. S. Liu, OncoTargets Ther., 2018, 11, 8399–8408 CrossRef.
- L. Yang, D. W. Ge, X. Chen, J. J. Qiu, Z. W. Yin, S. N. Zheng and C. Z. Jiang, Biochem. Biophys. Res. Commun., 2018, 502, 493–500 CrossRef CAS.
- Y. D. Han, M. Z. Chen, A. L. Wang and X. P. Fan, Biochem. Biophys. Res. Commun., 2019, 508, 472–479 CrossRef CAS.
- J. Yuan, Z. Y. Su, W. C. Gu, X. J. Shen, Q. M. Zhao, L. Y. Shi, C. J. Jin, X. D. Wang, H. Cong and S. Q. Ju, Cancer Biomarkers, 2019, 24, 279–289 CAS.
- Y. N. Wang, F. Xu, P. Zhang, P. Wang, Y. N. Wei, C. Wu and S. J. Cheng, Biomed. Pharmacother., 2019, 113, 108716 CrossRef CAS.
- Q. Z. Gao, L. H. Tang, L. Wu, K. T. Li, H. Wang, W. D. Li, J. Wu, M. Y. Li, S. Wang and L. Zhao, Cell Death Dis., 2018, 9, 393 CrossRef.
- Q. B. Wu, T. T. Han, X. Sheng, N. Zhang and P. Wang, Biomed. Pharmacother., 2018, 108, 741–751 CrossRef CAS.
- L. Y. Zhang, L. V. Ho-Fun, A. M. Wong, D. L. Kwong, Y. H. Zhu, S. S. Dong, K. K. Kong, J. Chen, S. W. Tsao, X. Y. Guan and L. Fu, Carcinogenesis, 2013, 34, 454–463 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05405g |
| This journal is © The Royal Society of Chemistry 2019 |