
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNeuroprotective effects of myristargenol A against glutamate-induced apoptotic HT22 cell death
Jung-Soo
Park
,
Jong-Hwa
Park
and
Ki-Young
Kim
 *
*
Department of Genetic Engineering, College of Life Science, Graduate School of Biotechnology, Kyung Hee University, 1732, Deogyeong-daero, Giheung-gu, Yongin-si, Gyeonggi-do, Republic of Korea. E-mail: kiyoung@khu.ac.kr; Tel: +82-31-201-2633
First published on 2nd October 2019
Abstract
Glutamate is an important neurotransmitter in the central nervous system; however, at high concentrations, it causes excitotoxicity and many neurological disorders. Excitotoxicity induces cell death by apoptosis. Thus, factors that can inhibit the apoptotic pathways are a target of drug development for the treatment and prevention of neurodegenerative diseases. Herein, the antioxidative and neuroprotective effects of myristargenol A were examined in glutamate-induced mouse hippocampal neuronal HT22 cells. When the HT22 cells were stressed with glutamate, cell viability decreased to 44.4 ± 5.6% when compared with the case of the control cells (100 ± 4.8%); however, when these cells were treated with myristargenol A (10 μM), the cell viability was increased by 113.6 ± 2.3%. The protective effect of myristargenol A against the apoptosis of glutamate-induced HT22 cells was also confirmed using FITC-annexin V/propidium iodide double staining. In addition, myristargenol A protected the mitochondrial membrane potential (ΔΨm). Subsequently, the expression levels of proteins in the caspase pathway related with the induction of apoptosis were decreased. Moreover, the expression levels of mitochondrial-related proteins, such as Bcl-2 and Bax, were examined, and it was found that the expression ratio of Bax/Bcl-2 decreased. In addition, myristargenol A inhibited the activity of mitogen-activated protein kinases, including p38 and c-Jun N-terminal kinase, for an oxidative stress protection effect but increased the activity of the extracellular signal-regulated kinases 1 and 2 for cell proliferation. These results reveal that myristargenol A possesses a neuroprotective effect against the neuronal cell damage caused by glutamate.
Introduction
Glutamate is an important excitatory neurotransmitter in the mammalian central nervous system and plays an important role in neural development, learning, and memory.1 However, it has been reported that high concentrations of glutamate can cause excitotoxicity and cell death, resulting in ischemia and neurodegenerative diseases.2,3 Neurodegenerative diseases occur due to damage to brain cells caused by oxidative stress and excessive inflammation and include typical diseases such as Alzheimer's disease, Parkinson's disease, and Huntington's disease;4 these diseases are caused by damage to cranial nerve cells. Damage to the cranial nerve cells occurs due to the excessive activation of calcium (Ca2+) through the activation of glutamate receptors, the activation of caspase to induce apoptosis, and the generation of free radical oxidative stress, resulting in cell death.5 During oxidative stress, cells or tissues are damaged by toxic free radicals, especially neuronal cells, which are especially sensitive to oxidative stress.6The HT22 cells derived from mouse hippocampal cells have been widely used to study glutamate-induced neurons. This is due to the absence of ionic glutamate receptors that transduce the signal for cell death caused by glutamate-induced apoptosis; therefore, the HT22 cells exclude receptor-mediated stress.7,8
In the case of the death of neurons, glutamate at high concentrations can cause a malfunction of mitochondria due to changes in the homeostasis of Ca2+.9 In addition, glutamate toxicity induces altered expressions of pro-/anti apoptotic regulators, such as Bcl-2 and Bax, and causes the disruption of the mitochondrial membrane potential (ΔΨm), which leads to the release of apoptosis-inducing factors and cytochrome c.10,11 When cytochrome c is released from the mitochondria into the cytosol, it binds to adenosine triphosphate (ATP) and forms an apoptosome. This apoptosome binds to and cleaves the procaspase-9 protein to release activated caspase-9, which subsequently activates and cleaves caspase-3 for a subsequent caspase cascade that causes cell apoptosis.12,13
The production of reactive oxygen species (ROS) by intracellular oxidative stress is also triggered by the mitogen-activated protein kinase (MAPK) signaling pathway.14 The MAPK protein belongs to the serine/threonine protein kinase family and plays an important role in the expression of various proteins involved in cell differentiation, inflammation, and apoptosis.14 The extracellular signal-regulated kinases (ERKs) 1 and 2 (1/2) promote cell growth and differentiation; moreover, oxidative stress induces the sustained phosphorylation of ERKs 1/2.15 Furthermore, c-Jun N terminal kinase (JNK) and p38 have been shown to modulate cell differentiation and death by cytokines, ultraviolet light, heat shock, and a variety of other factors.16 Therefore, the identification of antioxidants that can weaken the level of glutamate-induced apoptosis is a good strategy for the prevention and treatment of neurodegenerative diseases.
Myristargenol A (MG), a lignin, has been isolated from Myristica fragrans and has tetrahydrofuran in its structure (Fig. 1). Schiglaucin A and B, which have a similar molecular structure, are known to have a neuroprotective effect.17 In addition, macelignan and myristargenol B are known to have antioxidant and anti-inflammatory activities.18,19 However, MG compounds are not well known for their protective effects on glutamate-induced hippocampal neuronal cell death. Therefore, in this study, we investigated the antioxidant mechanisms and mitochondrial-mediated proteins involved in the neuroprotective effects of MG on glutamate-induced HT22 cells; based on these studies, a new function, i.e. the prevention of cell death, of MG in neurons was discovered, and the effects of this compound were found to be more diverse.
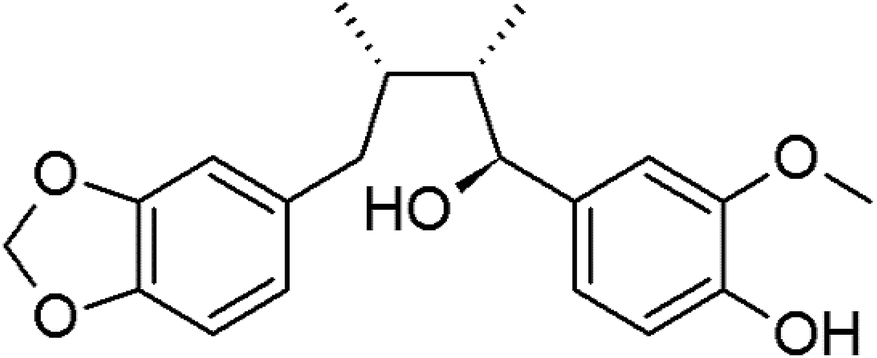 | ||
| Fig. 1 The chemical structure of myristargenol A. | ||
Methods
Cell culture and treatment
The HT22 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) containing 10% fetal bovine serum at 37 °C under a 5% CO2 humidified atmosphere. The cells were placed on plates or dishes, incubated for 24 h to adhere before compound treatment, and then exposed to 10 mM of glutamate (Sigma-Aldrich, St. Louis, MO, USA) in the presence or absence of MG for 24 h.Cell viability assay
The neuroprotective effect of MG against glutamate-induced HT22 cell death was measured using the MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide; Sigma-Aldrich, St. Louis, MO, USA) assay. The HT22 cells were seeded onto 96-well plates at the density of 4 × 103 cells per well and incubated for 24 h. The cells were treated with various concentrations of MG. After 24 h, 5 mg mL−1 of MTT in a phosphate-buffered saline (PBS) solution was added to the cells, followed by reaction for 2 h. The activity was measured at an absorbance of 540 nm using a microplate reader (BioTek Instruments, Inc. Winooski, VT, USA). The cell viability was established as a percentage of the control cells (100%). Since quercetin (QC) is known to have neuroprotective effects, it has been used as a positive control.20DPPH assay
The oxidation of 2,2-diphenyl-1-picrylhydrazyl (DPPH) radicals was measured to determine the antioxidative activity of MG. The DPPH solution (final concentration: 0.4 mM) and MG (0.5–64 μM) in a total volume of 100 μL were placed on a 96-well plate. After 30 min in the dark, the optical density (OD) was measured at 517 nm. The DPPH scavenging activity was calculated as follows:| DPPH radical scavenging activity (%) = [(ODcontrol − ODsample)/ODcontrol] × 100 |
Quantitative analysis of apoptotic cells (flow cytometry)
The HT22 cells were seeded at a density of 5 × 105 cells per dish in dishes and incubated for 24 h. After the cells were treated with various concentrations of MG for 24 h, they were harvested with scrapers, washed with PBS and then suspended in a binding buffer. After adding the fluorochrome-conjugated annexin V to the cells and staining for 15 min, the cells were washed with a binding buffer containing the propidium iodide staining solution for 20 min in the dark. The cells were then analyzed by flow cytometry (ACEA Biosciences Inc.).Measurement of mitochondrial membrane potential (ΔΨm)
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 15 min at 4 °C, protein extracts were obtained, and the protein concentrations were determined. Protein samples were separated via the sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to the polyvinylidene fluoride membrane (PALL Corp., Port Washington, NY, USA). Membranes were incubated with a blocking solution [5% skim milk in Tris-buffered saline containing 0.1% Tween-20 (TBS-T)] for 1 h and then incubated with primary antibodies such as anti-β-actin, anti-Bcl-2, anti-caspase-3, anti-poly (ADP-ribose) polymerase (PARP), anti-p38 MAPK, anti-p38 MAPK, anti-SAPK/JNK, and anti-p42/44 MAPK (Cell Signaling Technology Inc.), anti-Bax, anti-caspase-9, anti-pJNK, anti-ERK, and anti-GAPDH (Santa Cruz Biotechnology Inc.), and anti-p53 (BD PharmingenTM) at 4 °C overnight. All primary antibodies were diluted 1:2000 in the blocking solution. The membranes were washed three times with TBS-T and then probed with horseradish peroxidase-conjugated anti-mouse immunoglobulin (Ig)G or anti-rabbit IgG (1:4000 diluted in the blocking solution; Sigma-Aldrich) antibody at room temperature for 2 h. Proteins were detected using the SuperSignal™ West Pico or Femto Chemiluminescence System (Thermo Fisher Scientific Inc.), and the optical densities of the immunoreactive bands were quantified using the ImageJ software and normalized with those of the control group.
000 rpm for 15 min at 4 °C, protein extracts were obtained, and the protein concentrations were determined. Protein samples were separated via the sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to the polyvinylidene fluoride membrane (PALL Corp., Port Washington, NY, USA). Membranes were incubated with a blocking solution [5% skim milk in Tris-buffered saline containing 0.1% Tween-20 (TBS-T)] for 1 h and then incubated with primary antibodies such as anti-β-actin, anti-Bcl-2, anti-caspase-3, anti-poly (ADP-ribose) polymerase (PARP), anti-p38 MAPK, anti-p38 MAPK, anti-SAPK/JNK, and anti-p42/44 MAPK (Cell Signaling Technology Inc.), anti-Bax, anti-caspase-9, anti-pJNK, anti-ERK, and anti-GAPDH (Santa Cruz Biotechnology Inc.), and anti-p53 (BD PharmingenTM) at 4 °C overnight. All primary antibodies were diluted 1:2000 in the blocking solution. The membranes were washed three times with TBS-T and then probed with horseradish peroxidase-conjugated anti-mouse immunoglobulin (Ig)G or anti-rabbit IgG (1:4000 diluted in the blocking solution; Sigma-Aldrich) antibody at room temperature for 2 h. Proteins were detected using the SuperSignal™ West Pico or Femto Chemiluminescence System (Thermo Fisher Scientific Inc.), and the optical densities of the immunoreactive bands were quantified using the ImageJ software and normalized with those of the control group.
Statistical analysis
All results were represented as means ± standard deviations (SDs) or standard errors (SEs).Results and discussion
Neuroprotective effects of MG against glutamate-induced HT22 cell death and antioxidative effect of MG
The protective activity of MG against glutamate-induced cell death was investigated by the MTT assay (Fig. 2B). QC has been used as a positive control because it has been shown to have neuroprotective activity against glutamate-induced apoptosis. The viability of the cells exposed to 10 mM of glutamate decreased to 44.4 ± 5.6% when compared with the case of the untreated control cells. MG prevented glutamate-induced HT22 cell death in a dose-dependent manner, and the highest protective effect was achieved with 10 μM of MG (113.6 ± 2.3%) (Fig. 2B). Upon comparing the cell viabilities in the cases of siglausin A, siglausin B, macelignan and oleiferin F, which have a structure similar to that of MG, it has been found that the neuroprotective effect of MG is best. The HT22 cells treated with 10 μM of MG shared a similar morphology with the normal control cells when compared with the glutamate-treated cells (Fig. 2A). These results suggest that MG may be a good compound with neuroprotective effects.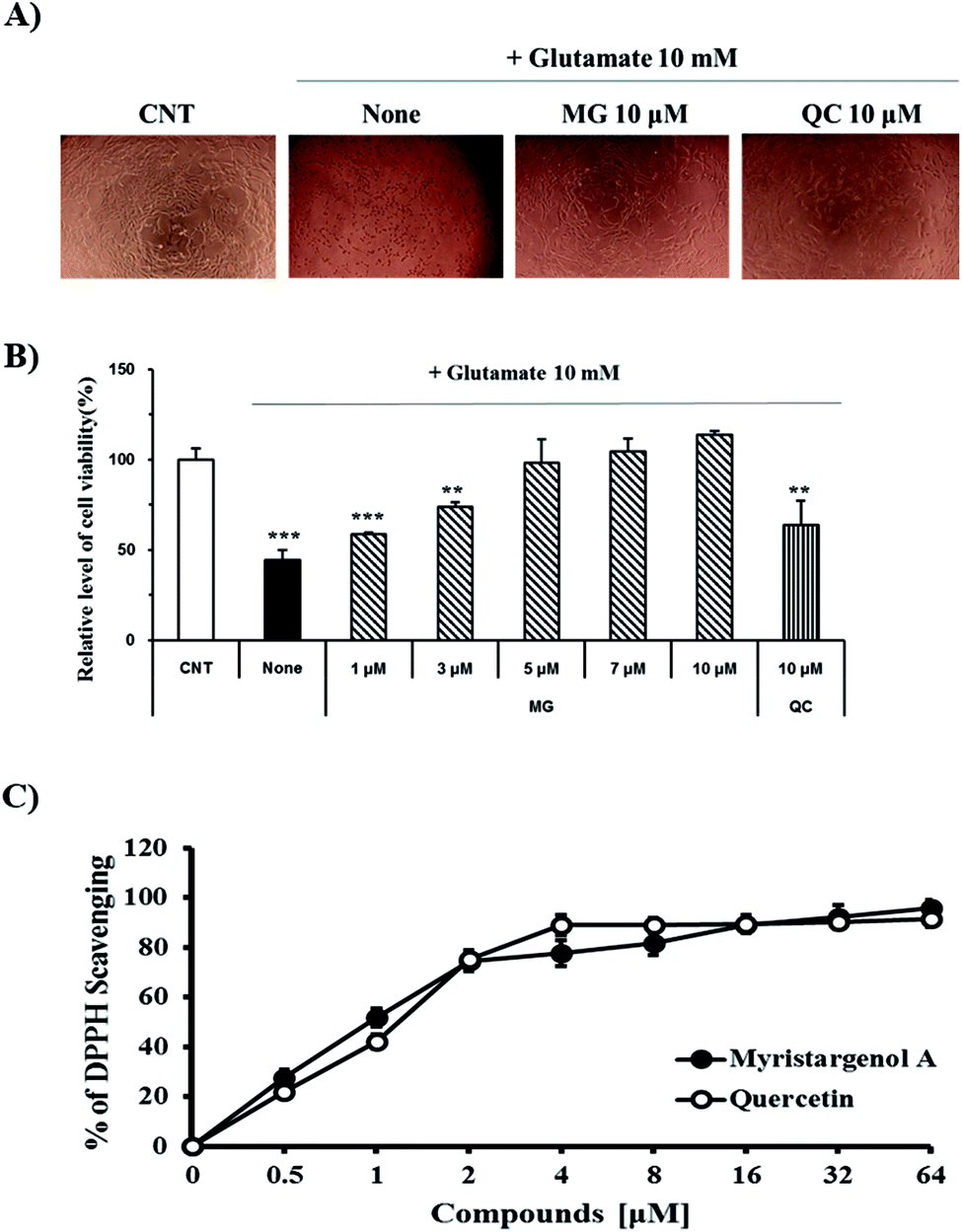 | ||
| Fig. 2 The neuroprotective effects of MG on glutamate-induced HT22 cells. (A) Morphology of the HT22 cells. (B) Cell viability of the HT22 cells was measured by the MTT assay. (C) DPPH radical scavenging assay for the anti-oxidant effect of compounds. ***p < 0.001 and **p < 0.01 compared to control cells. QC was used as a positive control. | ||
To assess the inhibitory effect of glutamate-induced oxidative stress, the antioxidative effect of MG was measured by the DPPH assay (Fig. 2C). MG (64 μM) had an antioxidant effect with the DPPH radical scavenging activity of 95.63 ± 3.09% (Fig. 2C). This finding suggests that oxidative stress can be suppressed and neuronal cells can be protected by MG.
Protective effects of MG against the glutamate-induced apoptosis in HT22 cells
Glutamate induces neuronal cell death through necrosis and apoptotic pathways. To observe the protective effects of MG on glutamate-induced apoptosis, annexin V/propidium iodide double staining was performed and analyzed via flow cytometry (Fig. 3A). The number of alive cells in the glutamate-treated sample decreased (56.2%) when compared with the case of the control group (91.8%); however, when the glutamate-treated cells were treated with MG, cell death was inhibited by MG in a dose-dependent manner (Fig. 3B). In addition, the early apoptotic cell rate (7.1%) increased in glutamate-induced cells (18.1%) as compared to that in the case of the control cells; however, MG decreased the early apoptotic cell rate in a dose-dependent manner (Fig. 3C). These results suggested that MG prevented glutamate-induced HT22 apoptosis.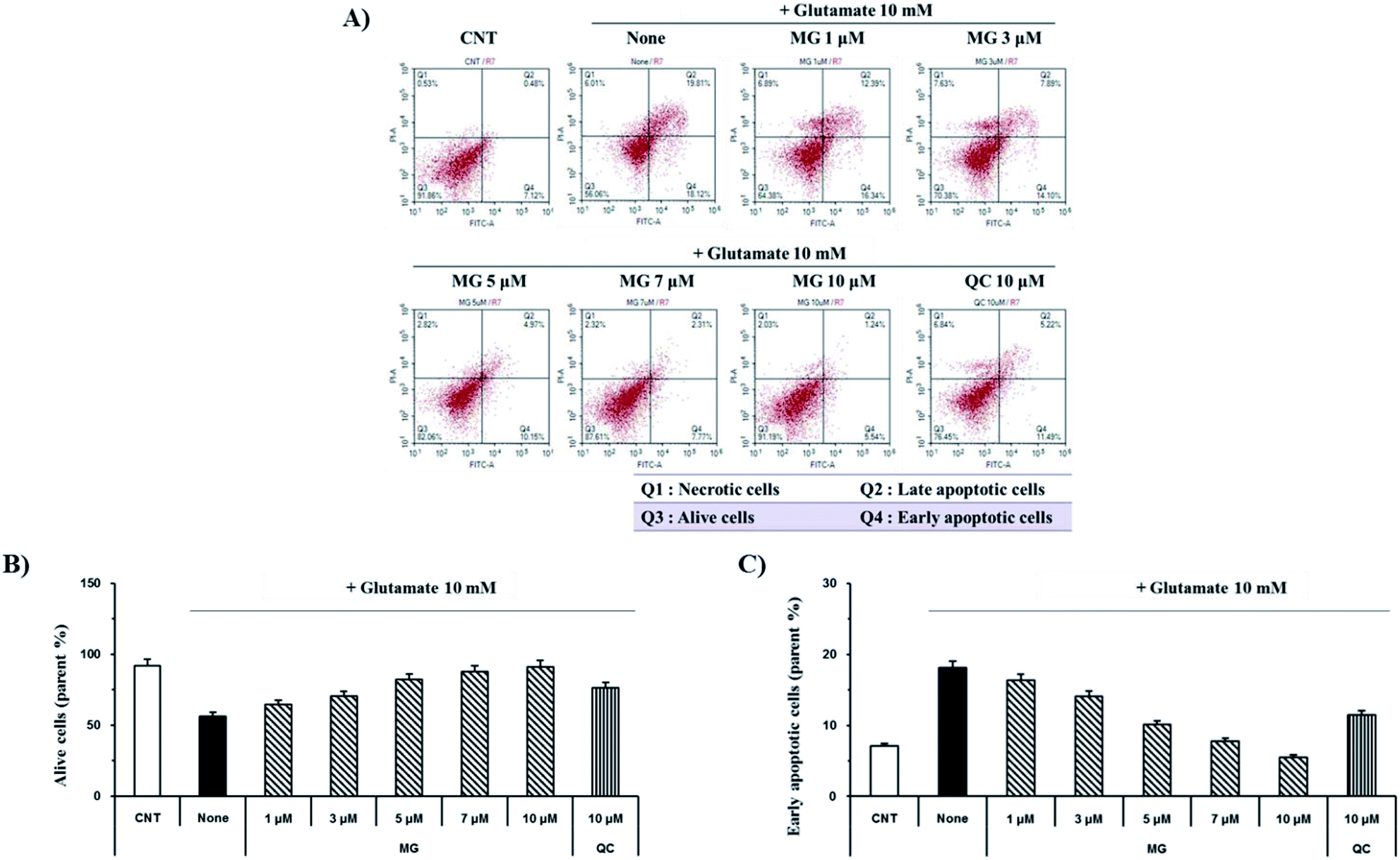 | ||
| Fig. 3 MG prevented glutamate-induced apoptosis in the HT22 cells. (A) The HT22 cells were exposed to 10 mM of glutamate in the presence of 1–10 μM of MG for 24 h and double stained with annexin V/propidium iodide to evaluate the number of apoptotic and necrotic cells, respectively. (B and C) Images were quantitatively analyzed using the NovoExpress software (ACEA Biosciences, Inc.). (B) Bars denote the percentage of alive cells. (C) Bars denote the percentage of early apoptotic cells. | ||
Reduction of glutamate-induced disruption of mitochondrial membrane potential (ΔΨm) by MG
Mitochondrial membrane depolarization is an important step in the induction of apoptosis, which increases the production of free radicals. Therefore, mitochondrial membrane potential (ΔΨm) was measured by JC-1 staining to examine the protective effect of MG on glutamate-induced mitochondrial damage (Fig. 4). Fluorescence microscopy results showed that 10 mM of glutamate treatment caused a marked decrease in the JC-1 red and green fluorescence ratios (0.64 ± 0.04%), indicating the destruction of the mitochondrial membrane potential (ΔΨm) (Fig. 4A). The red and green fluorescence ratios were dose-dependently increased via treatment with MG, indicating that the mitochondrial membrane potential (ΔΨm) was restored (Fig. 4B).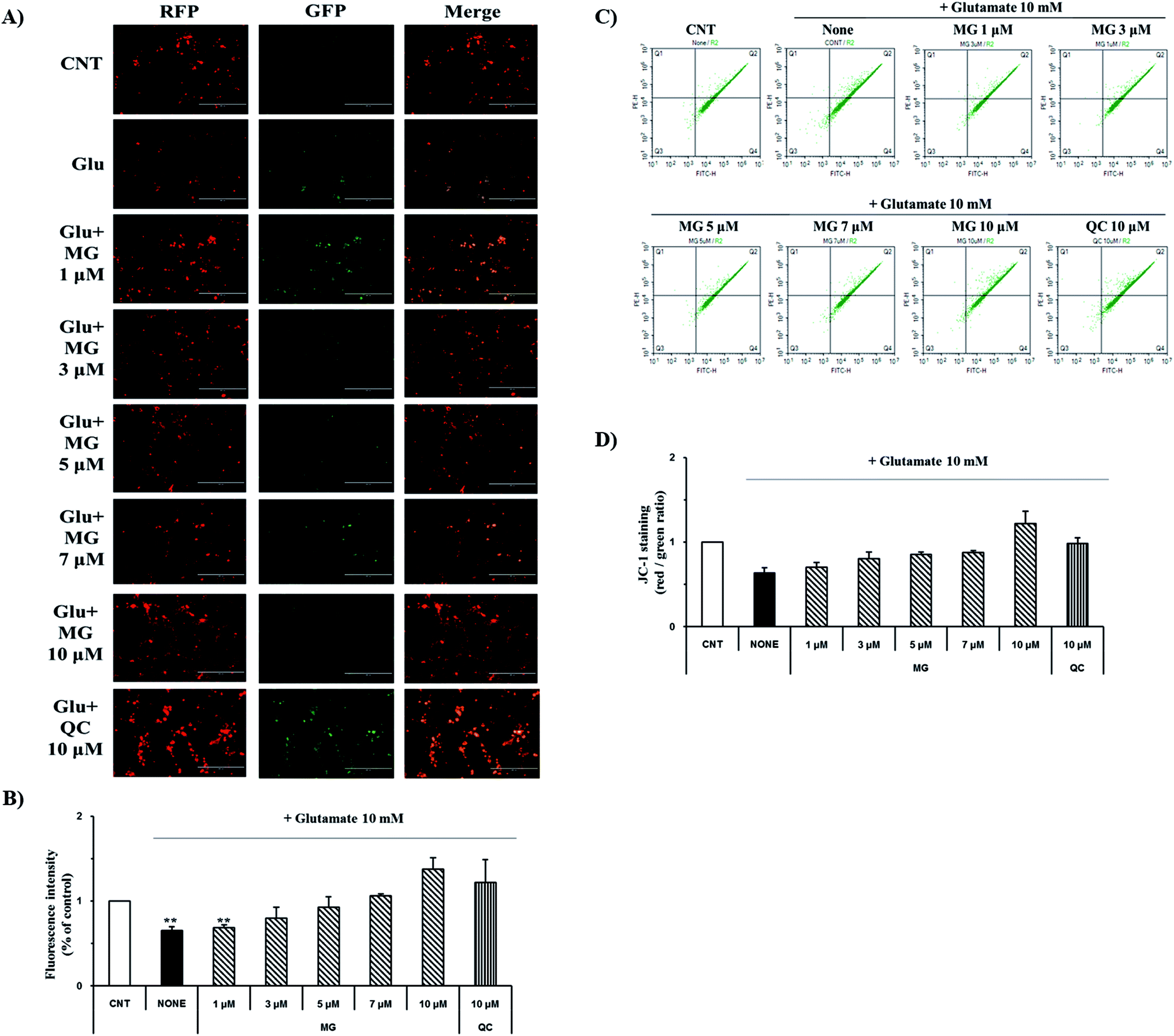 | ||
| Fig. 4 Inhibitory effects of MG on the disruption of mitochondrial membrane potential (ΔΨm). (A) JC-1 staining images were obtained by fluorescence microscopy. (B) The fluorescence intensity was measured using the ImageJ software. (C) JC-1 staining was performed and analyzed using a flow cytometer. (D) The red/green percentage was analyzed using the NovoExpress software. **p < 0.01 as compared to the control cells. | ||
For more detailed and accurate experiments, the mitochondrial membrane potentials were examined by flow cytometry analysis (Fig. 4C). Glutamate treatment decreased the ratio (0.63 ± 0.06%) of the red and green fluorescence in the experimental cells when compared with that in the control cells (1%), whereas the red and green fluorescence ratio increased after MG treatment in a dose-dependent manner (Fig. 4D). These results indicate that MG has effectively protected the mitochondria, which plays a crucial role in ATP production, by inhibiting the destruction of the glutamate-induced mitochondrial membrane potential (ΔΨm).
Effect of MG on the Bcl-2, Bax, p53 and caspase cascade levels
The caspase cascade pathway is important in the apoptotic pathway. This begins with the release of cytochrome c from the mitochondria, which is regulated by the Bcl-2 family. In the glutamate-induced neuronal cells, the amount of the antiapoptotic Bcl-2 protein decreases and the amount of the proapoptotic Bax protein increases. Moreover, Bax is activated by the p53 protein.21 This process promotes the release of cytochrome c. When cytochrome c is released, it binds to dATP and forms an apoptosome, which binds to and cleaves the procaspase-9 protein to produce an activated form. The activated caspase-9 activates and cleaves caspase-3, promoting a subsequent caspase cascade that kills cells by activating apoptosis. In addition, DNA cleavage occurs by the activation and cleavage of the transcription factor PARP.The Bcl-2, Bax, p53, caspase-3, caspase-9, and PARP protein levels were analyzed by western blotting to determine whether MG inhibited the activation of caspase pathways caused by the glutamate-induced disruption of mitochondrial membranes. In the HT22 cells treated with 10 mM of glutamate, the Bcl-2 levels decreased and the Bax and p53 levels increased. However, MG treatment prevented these changes in the Bcl-2, p53, and Bax expression levels (Fig. 5). The caspase cascade was activated and cleaved in response to apoptotic stimulation, but MG treatment prevented the induction of apoptosis by blocking the caspase-3 cleaved form formation in a dose-dependent manner (Fig. 6). These results suggested that MG might upregulate mitochondrial-mediated Bcl-2 family protein expression and inhibit caspase activity to prevent apoptosis induction.
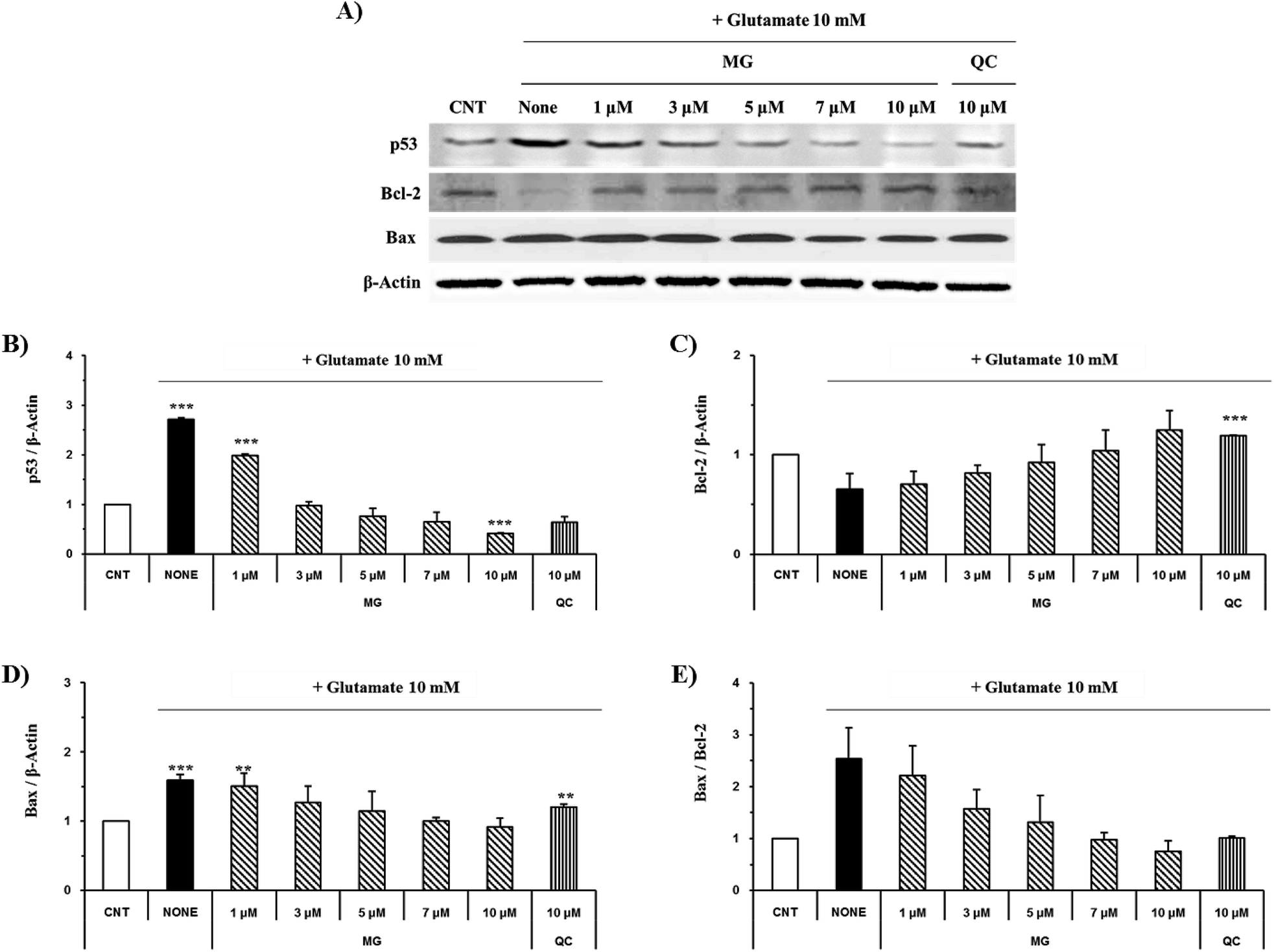 | ||
| Fig. 5 The effects of MG on the Bcl-2, Bax, and p53 levels in the glutamate-induced HT22 cells. (A) The HT22 cells were exposed to 10 mM of glutamate in the presence of 1–10 μM of MG for 24 h, and then, western blot analysis was performed. β-Actin was used as a loading control. (B and D) Bars indicate the increase in the level of p53 and Bax when compared with the case of the control cells. (C) Bars indicate the decrease in the level of Bcl-2 when compared with the case of the control cells. (E) Bars indicate the increase in the Bax/Bcl-2 ratio when compared with the case of the control cells. ***p < 0.001 and **p < 0.01 when compared with the control cells. | ||
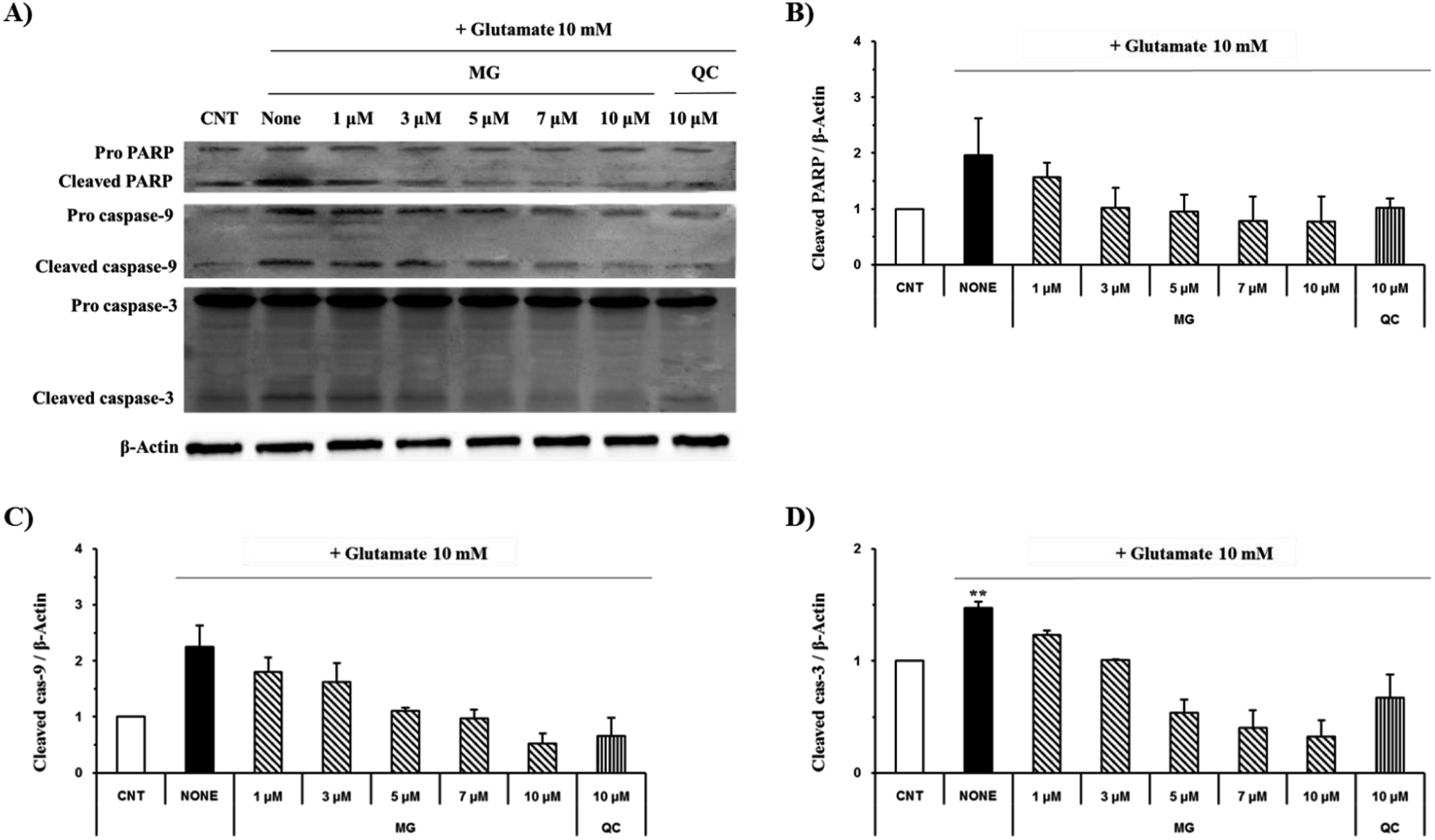 | ||
| Fig. 6 The effects of MG on the caspase-3, caspase-9 and PARP levels in glutamate-induced HT22 cells. (A) The HT22 cells were exposed to 10 mM of glutamate in the presence 1–10 μM MG for 24 h, and western blot analysis was performed. β-Actin was used as a loading control. (B–D) Bars indicate the increase in the level of cleaved caspase-3, cleaved caspase-9, and cleaved PARP when compared with the case of the control cells. **p < 0.01 as compared to the control cells. | ||
Regulation of MG on glutamate-induced MAPK activation
Mitogen-activated protein kinase plays a key role in cell survival and death. During neuronal cell death prompted by oxidative stress, MAPKs, including ERK 1/2, p38, and JNK, are overly phosphorylated and activated. Previous studies have suggested that glutamate-induced oxidative stress can trigger the activation of ERK 1/2, p38, and JNK, resulting in neuronal cell death.22 MG significantly reduced the phosphorylation of glutamate-induced JNK and p38 in a dose-dependent manner (Fig. 7).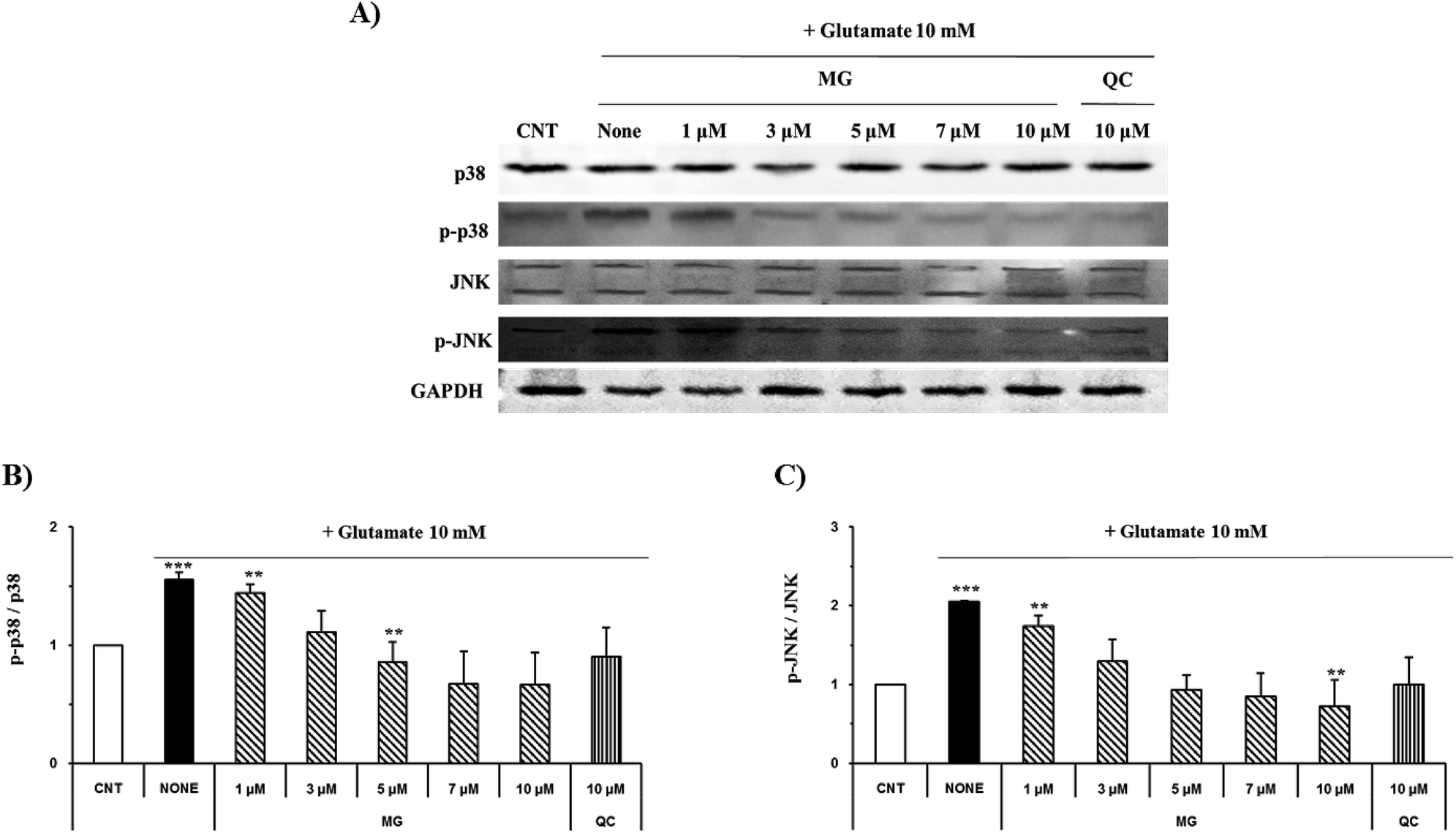 | ||
| Fig. 7 MG inhibits glutamate-induced MAPK activation in the HT22 cells. (A) The HT22 cells were exposed to 10 mM of glutamate in the presence of 1–10 μM MG for 24 h, and western blot analysis was performed. JNK, p38, and GAPDH were used as loading controls. (B and C) Bars indicate the increase in the levels of p38, JNK, and phosphorylation when compared with the case of the control cells. ***p < 0.001 and **p < 0.01 as compared to the control cells. | ||
ERK 1/2 was found to exist in two forms depending on the concentration and time of glutamate treatment. This is related to the sustained phosphorylation of ERK, which is known to cause apoptosis due to the persistent phosphorylation of ERK 1/2 in the glutamate-induced neuronal cells. To understand how MG influences the ERK 1/2 activity, the HT22 cells were treated with 10 mM of glutamate in the presence or absence of MG for 24 h, and western blotting analysis was performed (Fig. 8B). The phosphorylation of ERK 1/2 differed depending on the concentration (5 or 10 mM) and time (6, 12, or 24 h) of the glutamate treatment (Fig. 8A). The HT22 cells were continuously phosphorylated for 6 and 12 h after glutamate (5 and 10 mM) treatment and decreased at 24 h.
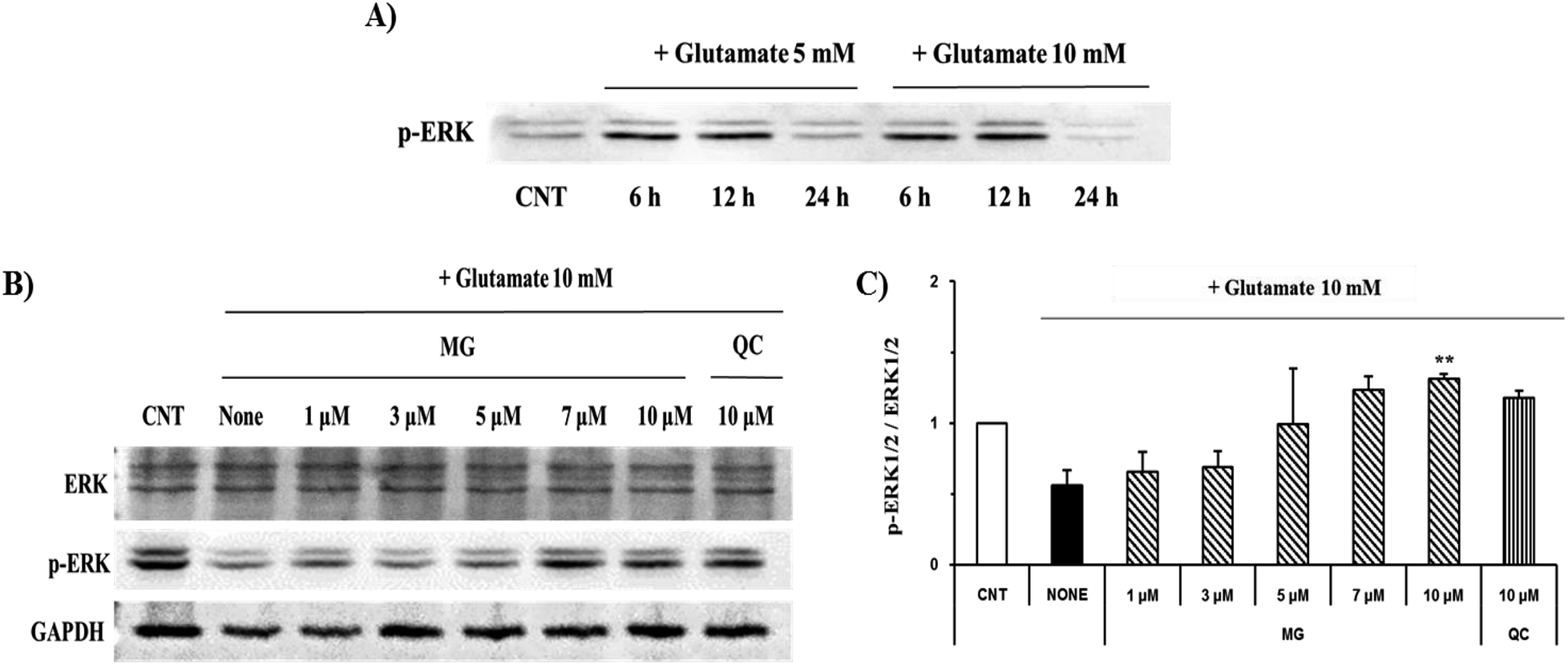 | ||
| Fig. 8 The effects of MG on the glutamate-induced ERK 1/2 phosphorylation. (A) The HT22 cells were exposed to glutamate at 5 and 10 mM, and phosphorylated ERK 1/2 was measured during the time course. (B) The HT22 cells were exposed to 10 mM of glutamate in the presence of 1–10 μM MG for 24 h, and western blot analysis was performed. ERK 1/2 GAPDH were used as loading controls. (C) Bars indicate the increase in the levels of ERK 1/2 phosphorylation as compared to the case of the control cells. **p < 0.01 as compared to the control cells. | ||
Glutamate (10 mM) and MG were applied for 24 h to determine the effect of MG on the ERK 1/2 phosphorylation. MG dose-dependently activated ERK 1/2 phosphorylation associated with cell growth and proliferation and therefore appeared helpful for cell growth (Fig. 8B and C).
Conclusions
Neuronal cell death due to high concentrations of glutamate is a cause of Alzheimer's disease and neurodegenerative diseases. Therefore, finding a substance that inhibits the mechanism of neuronal cell death caused by glutamate can be a good way to prevent neurological diseases. This study demonstrated that MG had a potent neuroprotective effect on glutamate-induced cell death in the HT22 cells. MG inhibited glutamate-induced mitochondrial membrane disruption and thereby contributed to energy synthesis. MG also inhibited apoptosis by controlling the expression levels of pro- and anti-apoptotic proteins (Bax and Bcl-2). MG suppressed the activity of the caspase pathway in the glutamate-induced HT22 cells and inhibited apoptosis induction and DNA damage. High concentrations of glutamate-induced oxidative stress could be inhibited by the antioxidant effect of MG. In addition, MG inhibited the activation of MAPKs, including p38 and JNK, caused by oxidative stress and thereby prevented apoptosis. MG increased the phosphorylation of ERK 1/2, which was helpful for cell proliferation and growth. Our experimental results suggest that natural raw materials containing MG are good tools for the prevention and treatment of neurological disorders.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2015R1D1A1A01061225).References
- M. Suzuki, A. D. Nelson, J. B. Eickstaedt, K. Wallace, L. S. Wright and C. N. Svendsen, Eur. J. Neurosci., 2006, 24, 645–653 CrossRef
.
- S. M. Greenwood and C. N. Connolly, Neuropharmacology, 2007, 53, 891–898 CrossRef CAS
- L. I. Bruijn, T. M. Miller and D. W. Cleveland, Annu. Rev. Neurosci., 2004, 27, 723–749 CrossRef CAS PubMed
- P. J. Houghton and M. J. Howes, Neurosignals, 2005, 14, 6–22 CrossRef CAS
- P. Maher and D. Schubert, Cell. Mol. Life Sci., 2000, 57, 1287–1305 CrossRef CAS
- C. T. Chu, D. J. Levinthal, S. M. Kulich, E. M. Chalovich and D. B. Defranco, Eur. J. Biochem., 2004, 271, 2060–2066 CrossRef CAS
- M. Fukui, J. H. Song, J. Choi, H. J. Choi and B. T. Zhu, Eur. J. Pharmacol., 2009, 617, 1–11 CrossRef CAS
- M. P. Mattson, Nat. Rev. Mol. Cell Biol., 2000, 1, 120–130 CrossRef CAS PubMed
- M. R. Duchen, J. Physiol., 2000, 529, 57–68 CrossRef CAS
- S. Tobaben, J. Grohm, A. Seiler, M. Conrad, N. Plesnila and C. Culmsee, Cell Death Differ., 2011, 18, 282–292 CrossRef CAS
- E. J. Yang, M. S. Min, H. Y. Ku, H. S. Choi, M. K. Park, M. K. Kim, K. S. Song and D. S. Lee, Biochem. Biophys. Res. Commun., 2012, 421, 658–664 CrossRef CAS
- F. P. Pablo and G. S. Salvesen, Biochem. J., 2004, 384, 201–232 CrossRef
- K. M. Boatright and G. S. Salvesen, Curr. Opin. Cell Biol., 2003, 15, 725–731 CrossRef CAS
- G. Pearson, F. Robinson, T. B. Gibson, B. Xu, M. Karandikar, K. Berman and M. H. Cobb, Endocr. Rev., 2001, 22, 153–158 CAS
- K. Sato, Y. Yamanaka, Y. Asakura and T. Nedachi, Biosci., Biotechnol., Biochem., 2016, 80, 712–718 CrossRef CAS
- L. Chang and M. Karin, Nature, 2001, 410, 37–40 CrossRef CAS
- H. Y. Yu, C. Hao, F. Y. Meng, X. Li, Z. Y. Chen, X. Liang and H. L. Ruan, Planta Med., 2012, 78, 1962–1966 CrossRef CAS
- D. Q. Jin, C. S. Lim, J. K. Hwang, I. Ha and J. S. Han, Biochem. Biophys. Res. Commun., 2005, 331, 1264–1269 CrossRef CAS
- S. K. Sadhu, E. Okuyama, H. Fujimoto and M. Ishibashi, Chem. Pharm. Bull., 2003, 51, 595–598 CrossRef CAS
- E. J. Yang, G. S. Kim, J. A. Kim and K. S. Song, Pharmacogn. Mag., 2013, 9, 302–308 CrossRef CAS
- X. Hong, K. Yoshito, K. C. Michael, J. K. Stanley, A. S. Philip and S. M. Richard, J. Neurosci., 1998, 18, 1363–1373 CrossRef
- D. Ortuno-Sahagun, R. M. Gonzalez, E. Verdaguer, V. C. Huerta, B. M. Torres-Mendoza, L. Lemus, M. C. Rivera-Cervantes, A. Camins and C. B. Zarate, J. Mol. Neurosci., 2014, 52, 366–377 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2019 |