
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffects of linker and liposome anchoring on lactose-functionalized glycomacromolecules as multivalent ligands for binding galectin-3†
Tanja Freichela,
Dominic Laafb,
Miriam Hoffmanna,
Patrick B. Konietznya,
Viktoria Heineb,
Robert Wawrzinekc,
Christoph Rademacher c,
Nicole L. Snyderd,
Lothar Ellingb and
Laura Hartmann*a
c,
Nicole L. Snyderd,
Lothar Ellingb and
Laura Hartmann*a
aDepartment of Organic and Macromolecular Chemistry, Heinrich-Heine-University Düsseldorf, Universitätsstraße 1, 40225 Düsseldorf, Germany. E-mail: laura.hartmann@hhu.de; Fax: +49-211-81-15840; Tel: +49-211-81-10360
bLaboratory for Biomaterials, Institute for Biotechnology, Helmholtz-Institute for Biomedical Engineering, RWTH Aachen University, Pauwelsstraße 20, 52074 Aachen, Germany
cMax Planck Institute of Colloids and Interfaces, Mühlenberg 1, 14424 Potsdam, Germany
dDepartment of Chemistry, Davidson College, North Carolina 28035, USA
First published on 29th July 2019
Abstract
In this work, we present a bottom-up approach for the synthesis of lactose-functionalized glycomacromolecules and glycofunctionalized liposomes and apply these compounds to investigate their effects of multivalent presentation on binding to galectin-3. Step-wise assembly of tailor-made building blocks on solid supports was used to synthesize a series of oligo(amidoamine) scaffolds that were further conjugated to lactose via copper catalyzed 1,3-dipolar cycloaddition. Binding studies with galectin-3 revealed affinities in the micromolar range that increased with increasing carbohydrate valency, and decreased with increasing size and linker flexibility. To further explore their multivalency, selected glycomacromolecules were conjugated to lipids and used in liposomal formulations. Binding studies show a further increase in binding in nanomolar ranges in dependence of both ligand structure and liposomal presentation, demonstrating the power of combining the two approaches.
Introduction
Carbohydrate–protein interactions play crucial roles in various biological binding processes such as cell–cell communication and tumor biology.1 An important family of carbohydrate-recognizing proteins are the galectins (Gal).2,3 Galectins have been shown to be involved in apoptosis, angiogenesis, cell–cell communication, immune maintenance and cell proliferation.4–7 There are currently fifteen members of the galectin family, which are further subclassified into three groups: prototype (e.g. Gal-1), tandem-repeat (e.g. Gal-9) and chimeric (Gal-3).8 Galectins bind to β-galactoside terminating saccharides, most notably LacNAc (Galβ1-4GlcNAc), through their conserved carbohydrate recognition domain (CRD).8 The galectin CRD is composed of five binding sites A–E, where C binds the galactose moiety and D the carbohydrate attached at the reducing end of the galactose residue.2,9 Gal-3, the only chimeric type galectin, is one of the best studied members4,5,10,11 and is an important target to develop synthetic ligands to better understand the role of Gal-3 in normal and diseased process as well as for the development of new diagnostics and therapeutics.5The natural multivalent ligands for Gal-3 are mostly glycans and glycoproteins such as laminin or fibronectin with O- and N-linked glycans that often terminate in repeating LacNAc units.12–15 Given the challenging synthesis of such polysaccharides or glycoproteins, the synthesis of more simplified multivalent glycomimetics as ligands of galectins is gaining attention.16–18 Multivalent glycomimetics often consist of a synthetic scaffold such as a peptide or polymer presenting multiple copies of a polysaccharide fragment.18–20 For example, successful implementation of glycomimetics targeting Gal-3 was shown by Becer and co-workers who synthesized a series of glycopolypeptides varying in the spacing and density of carbohydrate ligands.21 In another example, Kamerling and co-workers used solid phase synthesis to generate glycopeptide libraries to study Gal-3.22,23 Cloninger and co-workers18,24 used dendritic scaffolds for the multivalent presentation of carbohydrates and demonstrated their ability to induce Gal-3 aggregation and inhibit cancer cellular aggregation, while Percec and co-workers used glycodendrimers and dendrimersomes to present lactose to different galectins including Gal-1,25,26 Gal-3 and -4,27,28 and Gal-8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26,28,29 to explore their properties. Gabius and Roy evaluated different kinds of glycomimetics from di- to tetra-conjugated lactose-functionalized glycoclusters to nonacontavalent lactoside glycodendrimers and demonstrated their potential in solid phase as well as in cell assays.30–33 Bonduelle and co-workers used nanoparticles as a platform for the multivalent presentation of carbohydrates for galectin binding.34 Additionally, in the group of Elling, the enzymatic build-up of glycans and their subsequent conjugation to bovine serum albumin (BSA) to form so-called neo-glycoproteins was used to demonstrate the effect of multivalency in Gal-3 recognition.35,36
26,28,29 to explore their properties. Gabius and Roy evaluated different kinds of glycomimetics from di- to tetra-conjugated lactose-functionalized glycoclusters to nonacontavalent lactoside glycodendrimers and demonstrated their potential in solid phase as well as in cell assays.30–33 Bonduelle and co-workers used nanoparticles as a platform for the multivalent presentation of carbohydrates for galectin binding.34 Additionally, in the group of Elling, the enzymatic build-up of glycans and their subsequent conjugation to bovine serum albumin (BSA) to form so-called neo-glycoproteins was used to demonstrate the effect of multivalency in Gal-3 recognition.35,36
Another strategy for a multivalent presentation, which is, to the best of our knowledge unexplored for Gal-3, is the use of surface functionalized liposomes.37–42 The synthesis of carbohydrate–lipid conjugates and their incorporation into liposomes via self-assembly allows for the build-up of multivalent, supramolecular structures, which can be used as ligands, drug- or antigen-delivery systems.43–47 The use of natural membrane compounds like cholesterol and phosphatidylcholines (e.g. DSPC) for the formulation of liposomes can furthermore ensure a higher biocompatibility for biological applications. Thus, the presentation of glycomimetics on the surface of such supramolecular systems can be used for the targeting of proteins or cells for various fundamental and applied applications.37,48–51
In this work, we aimed to combine both approaches and show the impact on the binding of multivalent glycomimetics to Gal-3 (Fig. 1). First, we applied our previously introduced solid phase assembly of tailor-made building blocks to obtain monodisperse, sequence-controlled glycooligoamides, so-called precision glycomacromolecules, presenting carbohydrate fragments identified as ligands of Gal-3.52–56 To test for the influence of the scaffold structure on the lectin binding, glycomacromolecules were synthesized varying the overall valency and distance between individual carbohydrate ligands and in the length of the linker attaching ligands onto the scaffold (Fig. 2). Selected glycomacromolecules were then conjugated to lipids and the resulting ligand–lipid conjugates were used in liposome formulations. With this approach, we incorporate multivalency on two levels: the presentation of multiple lactose ligands along the macromolecular scaffold followed by the multiple presentation of the glycomacromolecules on the liposome (Fig. 1).
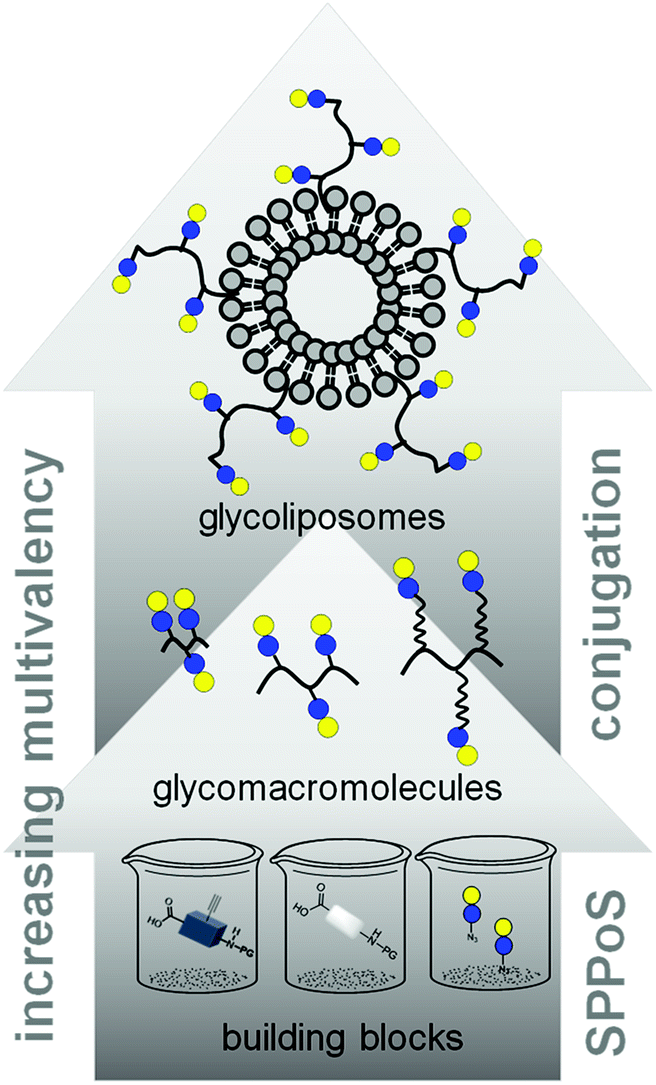 | ||
| Fig. 1 Schematic representation of assembling glycofunctionalized liposomes using solid phase synthesis of precision glycomacromolecules. | ||
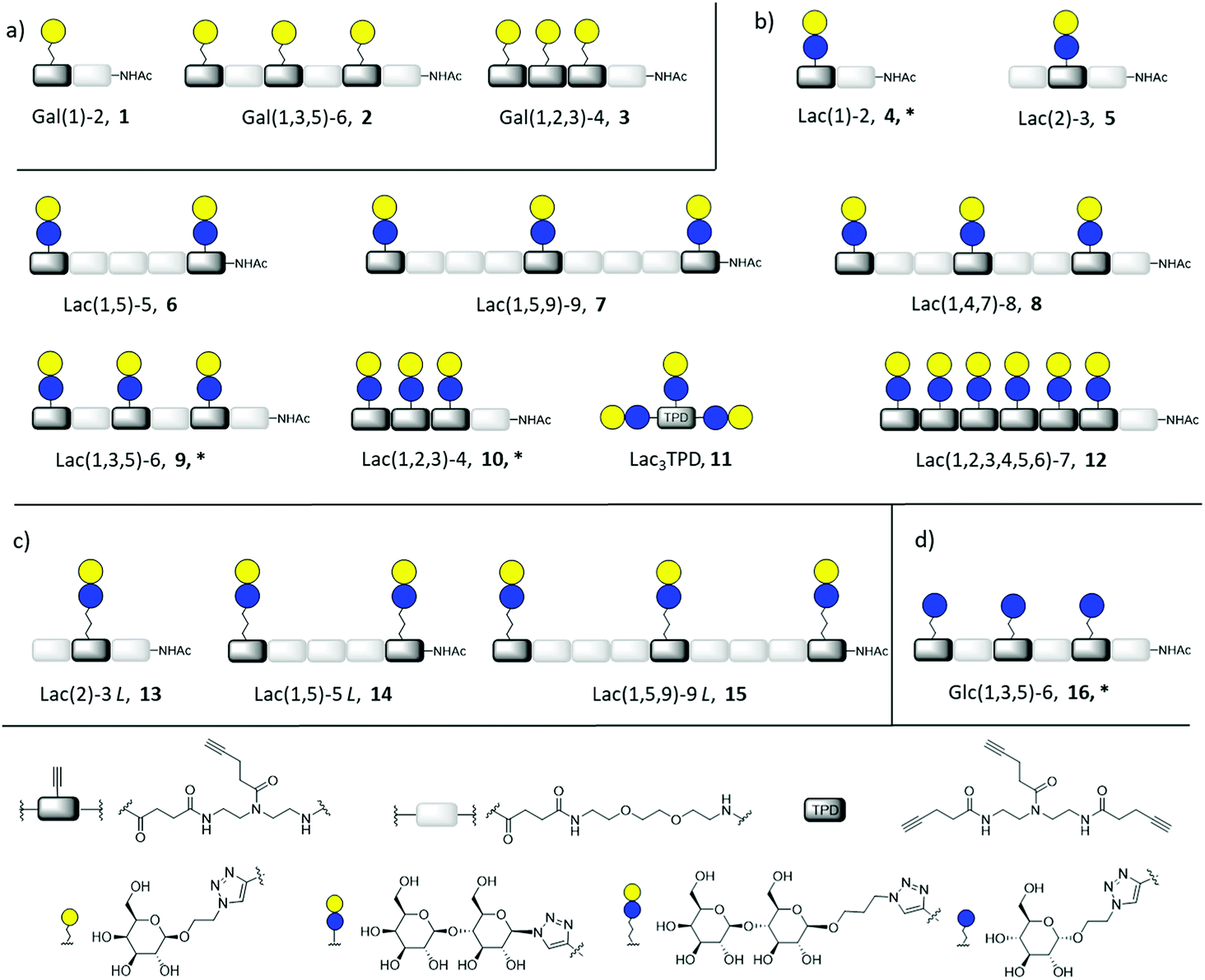 | ||
| Fig. 2 Overview of synthesized glycomacromolecules: (a) galactose-functionalized structures 1–3, (b) lactose-functionalized structures 4–12, (c) propyl lactose-functionalized structures 13–15, (d) glucose-functionalized structure 16. Structures denoted with * were synthesized as amine derivatives in addition to the acetyl-capped derivatives. | ||
Percec and co-workers used carbohydrate-functionalized dendrimers to incorporate multiple mono- or divalent constructs into liposomes and study the effects on clustered ligand presentation.25 Ratner and co-workers showed the incorporation of multivalent mannose- and galactose-functionalized polymers into liposomal formulations and the use of such glycopolymer-augmented liposomes to elucidate receptor-mediated uptake in macrophages.57 While they showed that the use of glycopolymers allowed for higher selectivity and specificity of cellular uptake of the glycoliposomes, they did not compare the effects of multivalent presentation of single ligands vs. multivalent glycopolymers on the liposomes. Such ‘multivalency of multivalency’ glycostructures are well-known in nature, e.g. the glycolipids or glycoproteins. Our synthetic platform allows for the systematic build-up of such structures starting from individual building blocks and building to multiple levels of multivalency (Fig. 1). This provides us with model compounds to study whether these two kinds of presentation in the glycomacromolecule–lipid conjugates are simply additive or benefit from additional factors. In this study, the binding to Gal-3 of both glycomacromolecules and glycomacromolecule-functionalized liposomes with variations in the number and spacing of carbohydrate ligands along the scaffold is investigated in inhibition-competition studies using an enzyme-linked immunosorbent inspired-assay (ELISA) and surface plasmon resonance (SPR).
Results and discussion
Synthesis of glycomacromolecules and glycomacromolecule–lipid conjugates
Synthesis of glycomacromolecules 1–10 and 12–16 was accomplished by applying a previously established solid phase polymer strategy (Fig. 2).52,56,58 In short, building blocks bearing a carboxylic acid and an Fmoc-protected amine group were coupled to an amine functionalized resin using PyBOP and DIPEA for activation. After successful coupling, Fmoc-deprotection with piperidine released the N-terminus of the first building block which could then be used for coupling of the next building block. This stepwise assembly allows for the synthesis of monodisperse, sequence-controlled oligomers. For the synthesis of glycomacromolecules in this study, TDS (triple bond diethylenetriamine succinyl, 1-(fluorenyl)-3,11-dioxo-7-(pent-4-ynoyl)-2-oxa-4,7,10-triazatetra-decan-14-oic acid)52 was used as an alkyne-functionalized building block for later conjugation with azide-derivatives of different carbohydrate ligands via copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC). EDS (ethylene glycol diamine succinyl, 1-(9H-fluoren-9-yl)-3,14-dioxo-2,7,10-trioxa-4,13-diazaheptadecan-17-oic acid)59 was chosen as a spacer building block introducing an ethylene glycol motif in the main chain of the glycomacromolecules. Through different combinations of these building blocks, glycomacromolecules with varying numbers of carbohydrate ligands and different spacing between ligands along the oligomeric scaffold were obtained (Fig. 2). β-Galactose (Gal) and β-lactose (Lac) were applied as known binding ligands of Gal-3. To investigate the effect of the linker between the carbohydrate ligand and the oligomeric scaffold, two different lactose-derivatives were conjugated, one with an anomeric azide, and one with a propyl linker terminating in an azide (Fig. 2). Finally, a non-binding α-glucose (Glc) residue was used to prepare glucose-functionalized glycomacromolecules as negative controls. All carbohydrate ligands were conjugated on solid support using previously reported conditions for CuAAC.60 De-O-acetylation of the carbohydrate residues under Zemplén conditions and cleavage from solid support gave the final glycomacromolecules.61 All structures were then purified by ion exchange and preparative HPLC to obtain final structures with high purities ≥ 95% (determined by UV 214 nm signal of RP-HPLC) (see ESI†).In contrast to glycomacromolecules 1–10 and 12–16, compound 11 was prepared in solution. Diethylenetriamine was treated with 4-pentynoyl chloride resulting in the precursor TPD 18 (tripentynoic acid diethylene triamine, N,N-bis(2-(pent-4-ynamido)ethyl)pent-4-ynamide) (Fig. 2). This was followed by conjugation of an azido-lactose analog via CuAAC (Scheme S1†). Global deprotection using Zemplén transesterification, followed by neutralization with Amberlite IR120 resin and preparative HPLC yielded Lac3TPD 11 with a purity ≥ 95%, determined by RP-HPLC. All final products were analyzed using 1H-NMR spectroscopy, analytical HPLC coupled with ESI-MS and HR-MS analysis (for more information see ESI†). Nomenclature of the final glycomacromolecules includes information on the type and position of the carbohydrate residue, as well as the overall valency of the oligomer. For example, Lac(1)-2, 4, represents a monovalent structure bearing a Lac ligand on the first position of a dimeric structure and Lac(1,5)-5 L, 14, is a divalent structure containing propyl linked (L) Lac in position 1 and 5 of a pentameric scaffold.
For the glycomacromolecule–lipid conjugation and later functionalization of the sensor surface for SPR measurements, amine functionalized glycoconjugates 4*, 9*, 10* and 16* were synthesized. For these derivatives, CuAAC was performed on scaffolds containing a terminal Fmoc group instead of the usual acetyl group. This was, followed by Fmoc-cleavage with piperidine and deprotection of the carbohydrates. After cleavage, the amine functionalized glycomacromolecules were purified via preparative chromatography resulting in purities ≥ 95%, as determined by RP-HPLC (see ESI†).
Lipid conjugation was conducted according to a previously published protocol.62 Commercially available DSPE-PEG-NHS ester was used as lipid (Fig. 3).
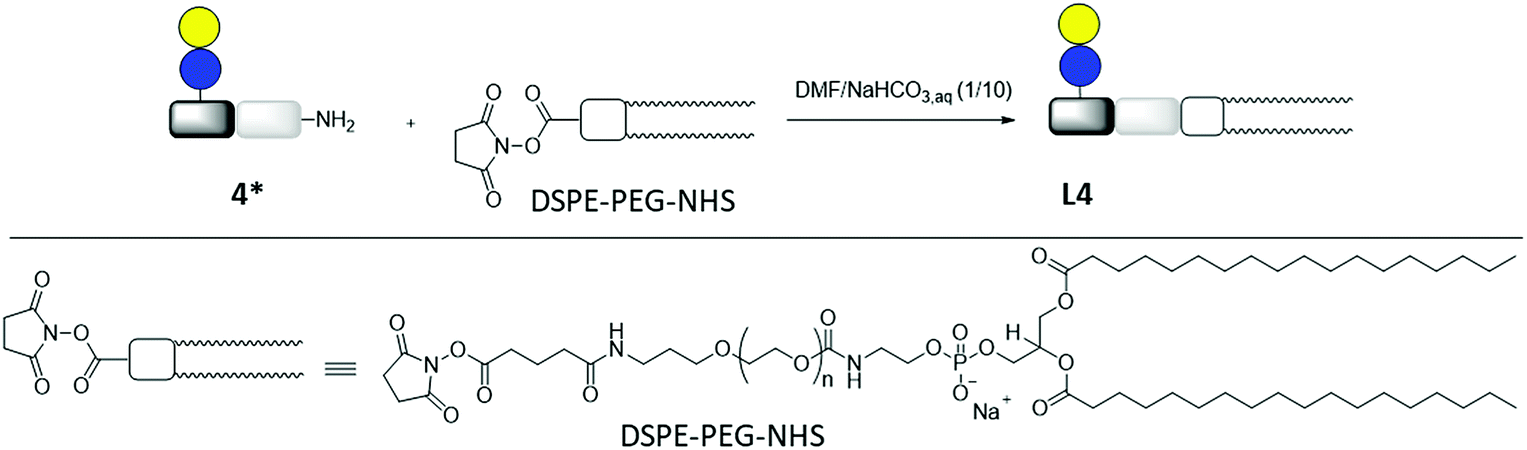 | ||
| Fig. 3 Synthesis of lipid-conjugate L4 through the reaction of DSPE-PEG-NHS and glycomacromolecule Lac(1)-2 (4*). | ||
| Glycomacromolecule–lipid-conjugate | MALDI-TOF-MSa | Conversionb [%]/yieldc [%] | |
|---|---|---|---|
| MW cal. for [M + Na]+ | m/z found | ||
| a MALDI-TOF-MS measurements were performed using positive ion mode. Molecular weights were calculated for the monitored maximum peak with a PEG repetition unit of 44.b Conversion ratio of conjugated lipid was determined by 1H-NMR via integration of the terminal methyl groups of the lipid chains signal normalized to the anomeric proton of the carbohydrate.c Determined by balance weight considering the conversion. | |||
| L4 | C173H330N11O73PNa 3786.5 | 3787.8 | 66/58 |
| L9 | C243H446N27O107PNa 5511.3 | 5511.6 | 56/44 |
| L10 | C223H410N23O99PNa 5051.8 | 5052.4 | 66/69 |
| L16 | C231H428N27O95PNa 5158.0 | 5158.7 | 62/35 |
Reaction conversions, equivalent to the ratio of conjugated lipid, were determined by 1H-NMR via integration of the terminal methyl groups of the lipid chains normalized to the anomeric proton of the carbohydrate moieties. They were found to be between 56–66%, which could be due to hydrolysis of the NHS-ester group of DSPE-PEG-NHS in aqueous solution. This hypothesis is strengthened by the MALDI-TOF-MS analysis, showing a corresponding MS-signal (see e.g. Fig. S69†) of the hydrolysis product of DSPE-PEG-NHS. For liposome formulation, the ratio of conjugated to unconjugated lipids was taken into account in order to obtain similar numbers of carbohydrate ligands per liposome.
Liposome preparation
For the liposome formulation, DSPC was used as the main lipid component with cholesterol as an additive for membrane stabilization through reduction of lipid ordering and increased melting temperatures (Fig. S1†).63–65 This liposomal formulation is approved by the FDA and therefore regularly used as standard in pharmaceutical research.66 The glycomacromolecule–lipid conjugates were used in a total quantity of 4.75 mol% in the whole formulation.Lipid film hydration and extrusion were used for the preparation of the liposomes. Extrusion allowed for size adjustment and homogenization. The liposomes were analyzed with DLS showing diameters (d) of approximately 150 nm and polydispersity indices (PDI) between 0.010–0.039. Vesicles in this PDI-range are termed monodisperse.67,68 Zeta potentials were measured to verify the negative charge of the surface, which correlates with the successful incorporation of the glycomacromolecule–lipids, and the overall stability of the liposomes. The results of the DLS and zeta potential measurements are summarized in Table 2 and correspond well to comparable systems in literature.50,62 Measurements were repeated after three months and showed comparable results indicating the stability of the liposomes over time (data not shown).
| Glycomacromolecule–lipid-conjugate used | Diameter (d)a [nm] | PDIb | Zeta potentialc [mV] |
|---|---|---|---|
| a Diameters (d) were determined by DLS analysis of the liposome solution.b PDI were determined via Gaussian fit of the DLS curve giving the standard deviation σ and applying PDI = (σ/d)2.c Zeta potentials were measured with a Zetasizer Nano-Z. | |||
| L4 | 156 ± 15 | 0.010 | −23.1 ± 7.6 |
| L9 | 150 ± 16 | 0.011 | −16.9 ± 8.5 |
| L10 | 154 ± 29 | 0.035 | −19.6 ± 8.2 |
| L11 | 146 ± 29 | 0.039 | −16.9 ± 8.5 |
| Glyco-conjugate | No. of carbohydrate | IC50 ± SDa [μM] | RIPb | RIP/Lac | IPc [%] |
|---|---|---|---|---|---|
| a Determined by ELISA-inspired inhibition studies on ASF coated plates. Measurements were performed two times in triplicates.b Relative inhibitory potency normalized to IC50 value of Lac (159 μM).c Reduced IC50 experiment determined by SPR inhibition studies with 100 μg mL−1 Gal-3 in PBS and 50 μM ligand. Binding signal of blank Gal-3 was set to 100% binding and 0% inhibition, inhibition values reported are referred to Gal-3. *RIP in brackets are the result of a direct comparison of the IC50 value of compound 5 to 13, 6 to 14 and 7 to 15. | |||||
| Lactose | 1 | 159 ± 13 | 1.0 | 1.0 | — |
| Lac(1)-2, 4 | 1 | 123 ± 3 | 1.3 | 1.3 | 31 |
| Lac(2)-3, 5 | 1 | 111 ± 4 | 1.4 | 1.4 | 46 |
| Lac(1,5)-5, 6 | 2 | 55 ± 5 | 2.9 | 1.5 | 61 |
| Lac(1,5,9)-9, 7 | 3 | 36 ± 3 | 4.4 | 1.5 | 66 |
| Lac(1,4,7)-8, 8 | 3 | 42 ± 3 | 3.8 | 1.3 | 65 |
| Lac(1,3,5)-6, 9 | 3 | 38 ± 2 | 4.2 | 1.4 | 68 |
| Lac(1,2,3)-4, 10 | 3 | 37 ± 1 | 4.3 | 1.4 | 72 |
| Lac3-TPD, 11 | 3 | 29 ± 1 | 5.5 | 1.8 | 77 |
| Lac(1,2,3,4,5,6)-7, 12 | 6 | 16 ± 4 | 9.8 | 1.6 | 78 |
| Lac(2)-3 L, 13 | 1 | 133 ± 8 | 1.2 (0.8*) | 1.2 | 36 |
| Lac(1,5)-5 L, 14 | 2 | 87 ± 3 | 1.8 (0.6*) | 0.9 | 50 |
| Lac(1,5,9)-9 L, 15 | 3 | 50 ± 2 | 3.2 (0.7*) | 1.1 | 55 |
Considering the incorporated glycomacromolecules could theoretically be presented on the inner or outer surface of the liposomes, the concentration of lactose, which can interact with Gal-3 in binding studies, was determined with a lactose-assay kit from BioAssay Systems.69,70 Here, the use of sterically demanding enzymes allows for the discrimination of the carbohydrates presented on the outer vs. the inner surface of the liposomes. Lactase is used to degrade Lac into Glc and Gal and the Gal concentration is determined indicating the concentration of accessible Lac. In contrast to the standard protocol, Lac was not a suitable standard because of the time-dependent behavior of free Lac hydrolysis compared to the conjugated Lac on the liposomes (see Fig. S2†). One reason for the observed discrepancy could be that the lactase converts the free Lac in solution more slowly than the Lac on the liposome surface, which also has been observed for other enzyme–substrate systems.71 Therefore, Gal was used instead of Lac as an alternative standard. An additional deviation from the protocol involved incubation of the samples with lactase only in assay buffer for 24 hours at 37 °C prior to dye-incubation instead of mixing the sample directly with lactase and dye-reagent simultaneously to achieve full degradation of lactose to its monosaccharides galactose and glucose. To verify the stability of liposomes during the measurements, 20 μL of the liposome–enzyme mixtures were diluted after the assay to a total volume of 1 mL with ultrapure water and measured with DLS. PDI and liposome diameter were in the pre-assay range indicating that the liposomes were stable during the assay (data not shown). Results from this protocol gave the surface concentration of Lac as shown in Table 4. Considering the amount of glycoligand–lipid-conjugate, which was used for the liposome formulation, the percentage of ligand on the liposome surface was calculated to be 94% for both L4 and L9 and 66% for L10.
| Glycoconjugate | No. of carbohydrate | Cmeas. ± SDa [μM] (Cmeas./C100% [%]) | IC50 ± SDb [μM] | RIPc | RIPligand/liposomed |
|---|---|---|---|---|---|
| a Cmeas. [μM]: concentration of the glycomacromolecules on the liposome surface determined with the lactose assay (percentage of glycooligomer relative to the theoretical concentration (Coligo/C100% [%])).b Determined by ELISA-inspired inhibition studies on ASF coated plates. Measurements were performed two times in triplicates.c Relative inhibitory potency (RIP) normalized to IC50 value of lactose (159 μM).d RIP of the liposomes compared to the corresponding single ligand in solution. | |||||
| Lac(1)-2, 4 | 1 | — | 123 ± 3 | 1.3 | — |
| Lac(1,3,5)-6, 9 | 3 | — | 38 ± 2 | 4.2 | — |
| Lac(1,2,3)-4, 10 | 3 | — | 37 ± 1 | 4.3 | — |
| Lac(1)-2, L4 | 1 | 143 ± 19 (94) | 12 ± 2 | 13 | 10 |
| Lac(1,3,5)-6, L9 | 3 | 127 ± 18 (94) | 1.0 ± 0.1 | 161 | 38 |
| Lac(1,2,3)-4, L10 | 3 | 107 ± 18 (66) | 0.3 ± 0.03 | 482 | 112 |
Galectin-3 binding studies
Binding of glycomacromolecules and liposomes to Gal-3 was evaluated by an inhibition competition study using an ELISA-inspired assay as previously introduced by Elling and co-workers72 and commonly used when evaluating the binding of glycomimetic structures to galectins.73–75 Asialofetuin, a natural multiantennary glycoprotein presenting nine terminal LacNAc residues, was coated onto microplates to enable the binding of Gal-3.76 Different concentrations of glycomacromolecules were added to the plates followed by Gal-3 carrying a His (histidine)-tag to achieve a competition event. Residual Gal-3/asialofetuin binding was then determined using a His-tag antibody carrying horseradish peroxidase (HRP) for the conversion of tetramethylbenzidine (TMB). After stopping the reaction by addition of HCl, absorbance was quantified at 640 nm. Inhibition of Gal-3 was determined by observing a decreasing signal in dependence of the ligand concentration.Plotting of the binding signal of Gal-3 against the ligand concentration gives inhibition curves as shown in Fig. 4a (for more information see ESI†). From these curves, the half maximal inhibition concentrations (IC50 values) could be determined. For the relative inhibitory potency (RIP), the obtained IC50 values were normalized to the IC50 value of non-conjugated Lac. Thus, stronger binding to Gal-3 results in a lower IC50 value and a higher RIP. To evaluate effects of valency further, the RIP was normalized to the number of carbohydrates per glycomacromolecule giving the RIP/Lac.
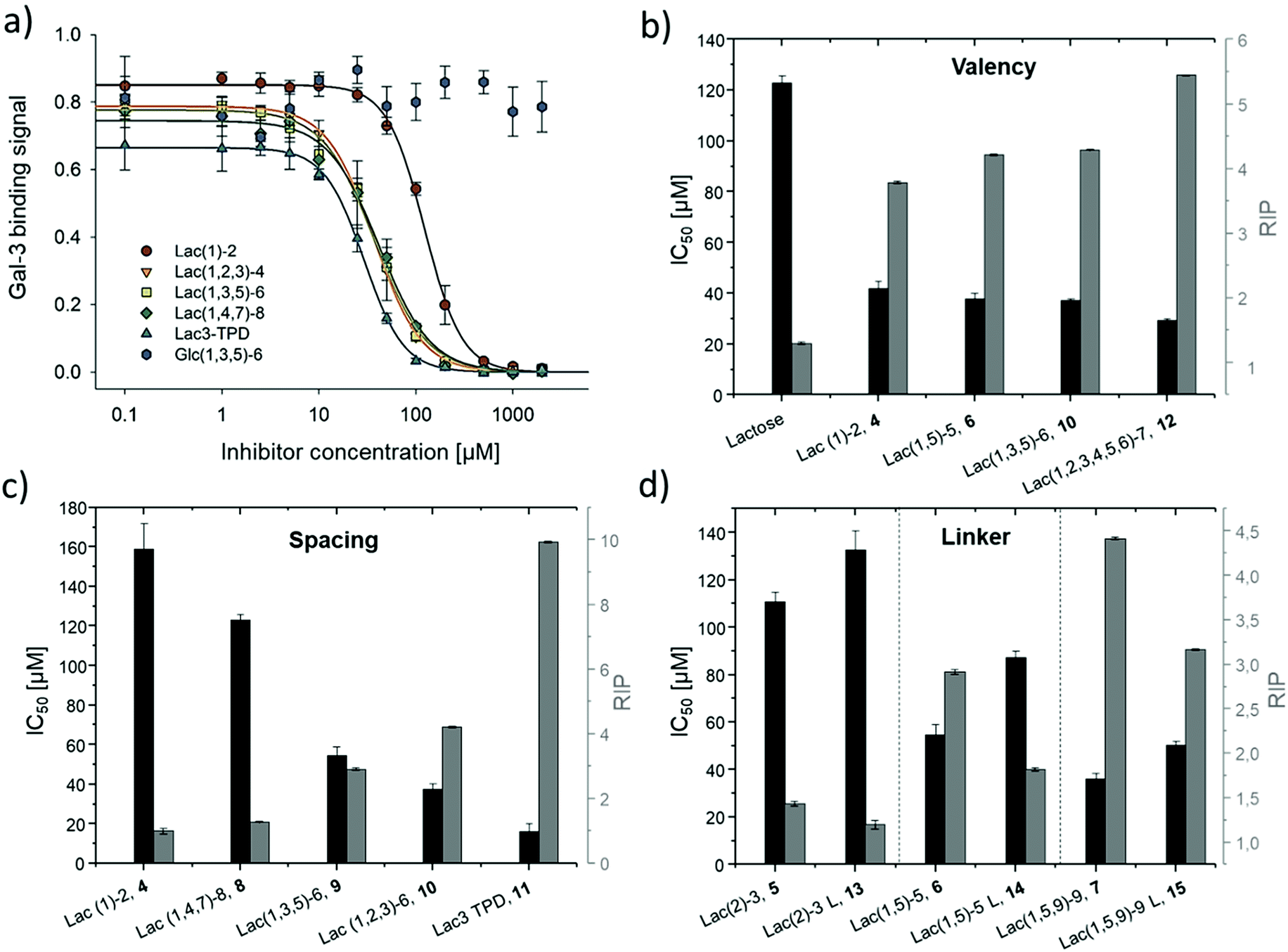 | ||
| Fig. 4 Results from the inhibition of Gal-3 in an ELISA-inspired assay. (a) Exemplary inhibition curves of the inhibition with structures 4, 8, 9, 10, 11 and negative control 16. Values are normalized to the signal of pure Gal-3. (b–d) IC50 values [μM] (black) and RIP (grey) for: (b) Lac and glycomacromolecules 4, 6, 10 and 12 with increasing valency; (c) glycomacromolecules 4, 8–11 with decreasing spacing and (d) glycomacromolecules 5–7 and their propyl-Lac counterparts 13–15. RIP are referenced to the IC50 value of lactose. | ||
The IC50 values and relative inhibitory potencies (RIP) as well as the RIP normalized to carbohydrate moieties for ligands 4–15 are listed in Table 3. Binding studies with galactose structures 1–3 (Fig. S4†) and the negative control 16 (Fig. 3) did not show any significant inhibition. Gal is known to be a poor binder for Gal-3 with Kd values around 10 mM,77 50-fold lower compared to the disaccharide Lac with a Kd value of 0.2 mM.78 Thus, the multivalent presentation of Gal on the macromolecular scaffold did not lead to a sufficient increase in binding to efficiently inhibit Gal-3 in this assay.
In general, a decrease in IC50 values and a corresponding increase in RIP values indicates an increase in inhibitory potency and thereby presumably binding affinity. For Lac structures 4–15, slightly increased inhibitory potencies are observed for glycomacromolecules with increasing valency (number of Lac residues) and decreasing spacing (number of EDS building blocks in between Lac-functionalized building blocks) (Fig. 4b and c). For the trivalent ligands showing the same valency and linker length, the lowest IC50 value was observed for the smallest ligand of this series, glycomacromolecule 11, with 29 ± 1 μM, and the highest IC50 value was observed for one of the largest structures, glycomacromolecule 8, with 42 ± 3 μM. An explanation could be in the sterical and geometrical effects related to the distances between the carbohydrate-epitopes which can have an impact on protein-clustering. Considering the distances of the N-atoms of the three triazoles in the stretched trivalent structures, the smallest structure Lac3TPD 11 was found to have theoretical distances between 8–17 Å, whereas the glycomacromolecule 8 was found to be in a range of 44–87 Å (see ESI Fig. S9 and S10†).
To the best of our knowledge, the distances between the CRDs in oligomeric Gal-3 lattices have not yet been reported, and are likely to vary significantly based on their complexity and flexible geometry.79 The results of the spacing are strengthened by studying the influence of the linker length and the distance of the triazole moiety from the carbohydrate on the inhibition of Gal-3 as shown in Fig. 4d. When comparing compounds 5–7 and 13–15, we observe that the introduction of a longer linker between the lactose ligand and oligomer backbone leads to a decrease in binding showing just 0.6–0.8 relative potency when compared to their shorter linker counterparts (Table 3, marked with *). This effect is even more pronounced with the higher valent glycomacromolecules.
We hypothesize the differences in avidity could also be due in part to the triazole motif in the linker participating in hydrophobic interactions in the binding groove of Gal-3. Similar results were found by Nilsson and co-workers where replacing ester or amide bonds through triazoles had an impact on the affinity and specificity towards Gal-3.80 In addition, the influence of hydrophobic, aromatic residues such as triazole, substituted phenyl and coumaryl methyl on the binding of Gal-3 is well-known from literature and might also effect binding of the glycomacromolecules.16,33,81–83
Besides geometrical effects and hydrophobic interactions, entropy can also play an important role where the loss of flexibility due to increased rigidity can have a positive impact on the entropy of the system.84,85 In this study, Lac3TPD 11 is assumed to be the most rigid structure showing the highest avidity towards Gal-3 in comparison to the other trivalent systems. Pieters and co-workers performed solid phase inhibition and fluorescence polarization studies on rigidified multivalent lactose ligands where they could see a twofold higher binding of rigidified structures compared to their more flexible counterparts.19 Furthermore, they could observe a high multivalent effect with increasing valency showing a 300 times higher binding for a tetravalent structure with a IC50 value of 0.07 μM compared to the monovalent derivative with an IC50 value of 21 μM and free lactose with 300 μM.19 In our case, the monovalent glycomacromolecules 4 and 5 show only slightly increased binding by a factor of 1.3–1.4 in comparison to free Lac, whereas the highest increase was observed for hexavalent ligand 12 by a factor of about 9–10. Comparing the characteristics of the herein reported glycooligo(amidoamines) and the discussed rigidified lactosidic structures, the more aromatic and thus hydrophobic nature of the structures by Pieters could be the reason for the more distinguished enhancement in binding, caused by participation of the aromatic residues on the binding event as mentioned before.
Overall, looking at all examined structures, the relative increase in inhibition for the glycomacromolecules is comparable to other multivalent constructs of similar valencies from literature, though not as potent as the previously discussed study by Pieters.19 In another example, Cagnoni and co-workers observed relative potencies of 10 and 6 by presenting dithiogalactose as di- and tetrasaccharides in comparison to the monovalent analogues.86 The aforementioned glycoclusters synthesized by Gabius and Roy evaluated in a solid-phase assay resulted in IC50 values of 165 μM for the divalent structure compared to 62–125 μM for the tetravalent structures. Compared to free lactose with 700 μM, the divalent structure showed an RIP of 4.3 and the tetravalent 5.6–11.2, which is again in the same range or even lower than those reported here from glycooligo(amido amines).30
In general, the obtained IC50 values of the glycomacromolecules are within the concentration range expected for this type of ELISA-inspired inhibition study on Gal-3. For example, Elling and co-workers reported IC50 values between 6 and 42 μM for LacNAc-based di- to heptasaccharides, respectively.73 Notably, LacNAc is an even better binder for Gal-3 with a Kd of 70 μM.72
SPR was used to perform a reduced IC50 experiment as a comparable method to support the aforementioned ELISA-inspired assay. Trivalent ligand Lac(1,3,5)-6 (9*) with a terminal amine group was used for sensor surface functionalization to provide high loading. The experimental conditions were based on the results from the ELISA study. In this case a fixed concentration of Gal-3 (100 μg mL−1) and a fixed concentration of ligands (50 μmol L−1) were used. Comparing Gal-3 binding in presence of the different ligands gives the inhibition potency (IP) at fixed ligand and receptor concentrations. In this context, higher affinity ligands result in a lower Gal-3 binding signal (Fig. 5 and ESI†) and higher IP values (Table 3).
SPR measurements (Fig. 5) support the results and trends observed in the ELISA-inspired assay (Fig. 4). Decreased spacing, e.g. going from structure 7 to 11, led to a slight increase in inhibitory potency from 65% to 77% (Table 3). Structures with the longer propyl-based linker 13–15 again showed lower inhibitory potency compared to derivatives 5–7 with the shorter linker.
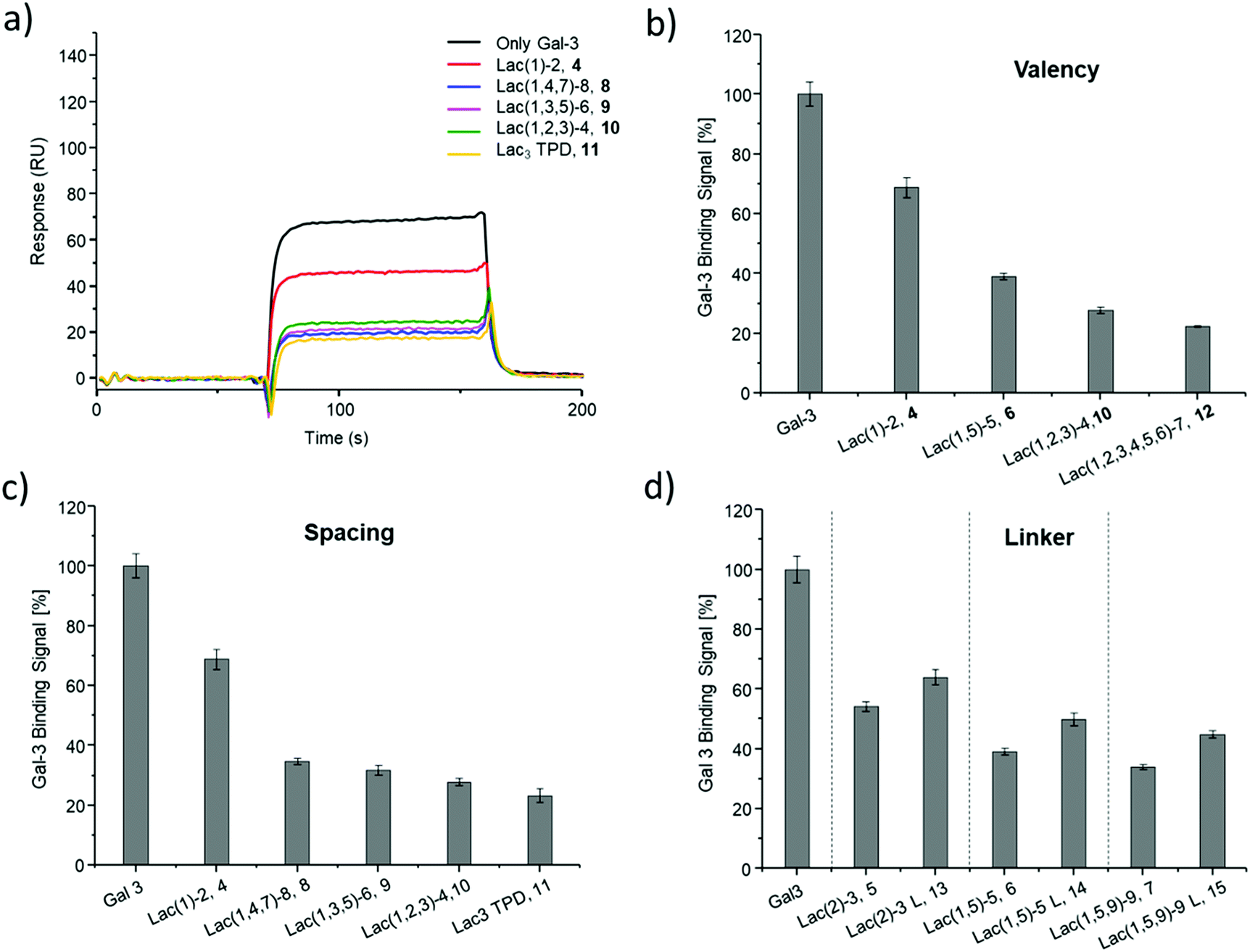 | ||
| Fig. 5 Results from the reduced IC50 SPR inhibition experiment of Gal-3 with samples 4–15. (a) Exemplary SPR-sensorgrams of only Gal-3 and Gal-3 incubated with macromolecule 4, 8–11. (b and c) Gal-3 binding signal ± SD [%] with reference of only Gal-3 signal as 100% binding for (b) glycomacromolecules 4, 6, 10 and F with increasing valency; (c) glycomacromolecules 4, 8–11 with decreasing spacing and (d) glycomacromolecules 5–7 and their propyl Lac counterparts 13–15. All measurements were performed in triplicates. | ||
Based on these findings, lipid-conjugation was performed with two trivalent ligands with different spacing and overall size (9* and 10*) as well as a monovalent glycomacromolecule (4*). As a negative control, a trivalent Glc-functionalized glycomacromolecule (16*) of the same sequence as Lac-functionalized ligand (9*) was included. The inhibitory potency of the liposomes (L4, L9 and L10) were studied by the same ELISA-inspired assay previously used for the free ligands (Fig. 4).
To compare results based on the number of Lac ligands available for binding to Gal-3 on the surface of the liposomes, IC50 values were normalized to the concentration of Lac as determined by the lactase-assay described above. To further compare the inhibitory potency of the ligands attached to the liposomes vs. the free ligands in solution, IC50 values were normalized to the IC50 value of Lac giving the RIP of the liposomes (Fig. 6).
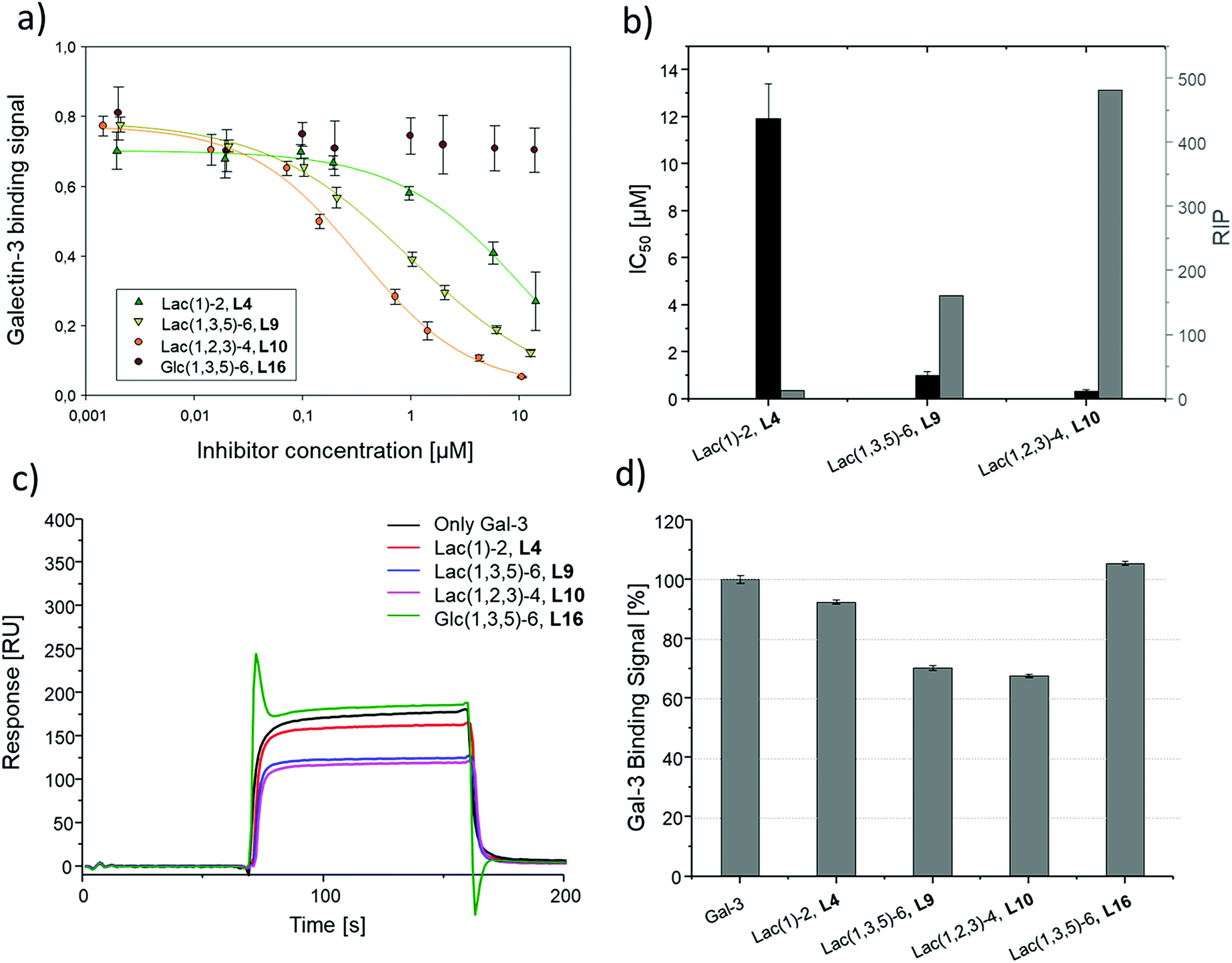 | ||
| Fig. 6 Results of the inhibition studies of the liposomes L4, L9, L10 and L16. (a) Inhibition curves of the inhibition of Gal-3 in ELISA-inspired assay. (b) Resulting IC50 values and RIP. Results from the reduced IC50 SPR inhibition experiment of Gal-3 (100 μg mL−1) and 10 μM of L4, L9, L10 and L16. (c) Exemplary SPR sensorgrams of only Gal-3 and incubated with L4, L9, L10 and L16. (d) Gal-3 binding signal ± SD [%] with reference of only Gal-3 signal as 100% binding. All measurements were performed in triplicates. | ||
To better demonstrate and compare the avidity enhancement enabled through the presentation of the glycomacromolecules on the liposomes, the RIPs of the glycomacromolecules were divided by the RIPs of the corresponding liposomes giving the RIPligand/liposome (Table 4). Results show the same trends for the different ligands presented on the liposomes in comparison to the free ligands, with the smaller trivalent structure (L10) showing a slightly higher inhibition potency than the longer trivalent ligand (L9), and trivalent ligands showing higher inhibition potency than the monovalent ligand (L4). Negative control L16 showed no inhibitory effect on Gal-3 binding (Fig. 6).
To support the results of the ELISA, the liposomes were tested in the reduced IC50 assay using SPR as previously described for the glycomacromolecules. However, using the same concentration of liposomes as the free glycomacromolecules led to complete inhibition of Gal-3 (data not shown). Reducing the concentration to 10 μM yielded detectable differences in Gal-3 binding (see sensorgrams shown in Fig. 6). Results of the ELISA were again supported with smaller and higher valent structures showing higher binding.
Comparing the IC50 values of the liposomes with the corresponding inhibition of the glycomacromolecules revealed that presentation on the liposome surface leads to an increase in inhibitory potencies. For the best binder of the liposomal formulations (L10), inhibition potency increases 112-fold from 37 to 0.3 μM in comparison to the free ligand 10. This is in the order of magnitude for a comparable ‘multivalency of multivalency’ system from the work of Laaf and co-workers who observed 180 to 350-fold higher inhibition for different saccharides presented in a multivalent fashion on BSA compared to the free saccharide in solution.35,36 In addition, the glycodendrimers evaluated by Gabius and Roy presenting 90 lactose residues showed IC50 values of 0.16 μM compared to 164 μM for the monovalent ligand, resulting in an inhibitory potency of 1025 and 11 per carbohydrate.31
Several studies suggest that Gal-3 oligomerizes upon glycan binding through its N-terminal domain,29,32,87 however, CRD mediated multimerization has also been described.88–90 With multivalent glycans on cell surfaces, Gal-3 can induce cell–cell interactions, crosslink receptors and even form lattice on cell surfaces.29,79 Due to the complexity and flexibility of galectin oligomers an increase in inhibitory potential can also point towards the formation of larger aggregates with Gal-3 based on the crosslinking property of galectin oligomers. This has been demonstrated for Gal-3 induced glycodendrimers24 and glycodendrisomes.25–29 Lactose was used as relative low affinity glycan and induced aggregation as multivalent ligand through Gal-3 binding. Interesting in our study is that the presentation of lower affinity monovalent ligand 4 and the trivalent ligand 9 on the liposomes (L4, L9), led to an increase of 10-fold or 38-fold, respectively. This shows that both the multivalency of the glycomacromolecules as, well as the multivalency of the presentation on the liposomes, contribute to the increased binding of Gal-3. Whether Gal-3 oligomerizes and forms aggregates with multivalent liposomes remains to be studied in future work.
Conclusions
Within this work, we investigated effects of the multivalent presentation of Lac using precision glycomacromolecules in binding to Gal-3. The use of solid phase synthesis allowed for the controlled variation of carbohydrate valency, spacing and linkage on an oligo(amidoamine) scaffold. ELISA-inspired and SPR assays revealed an influence of all three parameters on Gal-3 inhibition giving inhibition constants in the lower μM range. As expected, higher valency leads to higher binding. Surprisingly, decreasing the linker length and overall size of the scaffold also leads to an increase in binding. We partially attribute this to secondary binding interactions of the hydrophobic triazole linkages which are in closer proximity to the lectin for glycomacromolecules with shorter linkers. Further conjugation of selected glycomacromolecules to lipids allowed for additional multivalent presentation on the surface of liposomes, which increased binding and resulted in nM inhibition. However, the presentation of monovalent ligands in the liposomal formulations resulted in a much less pronounced increase in inhibitory potency, showing the importance of multivalency on both length scale, the macromolecular scaffold and liposome decoration, to effectively yield high avidity ligands. Indeed, this is a key feature of many glycoconjugates in nature such as glycolipids or glycoproteins. Our synthetic platform and the approach presented here give straightforward access to the design and synthesis of ligands using ‘multivalency of multivalency’ effects to achieve high avidity biomimetic ligands and to further study the underlying mechanisms involved in receptor binding and clustering.Experimental
Materials
All reagents and solvents were used without further purification. Acetic anhydride and sulfonic acid were purchased from VWR chemicals. Piperidine, trifluoro acetic acid, sodium methoxide, pentynoic acid and sodium diethyldithiocarbamate were purchased from Acros Organics. Dimethylformamide (for peptide synthesis) was purchased from Biosolve. Triisopropylsilane (TIPS) was purchased from Sigma Aldrich. Oxalyl chloride was purchased from Alfa Aesar. HOBt was purchased from Iris Biotech. N,N-Diisopropylethylamine (DIPEA) and diethylenetriamine were purchased from Carl Roth. Sodium ascorbate, phenol and potassium carbonate were purchased from PanReac AppliChem. Dichloromethane and triethylamine were purchased from Merck. PyBOP was purchased from Fluorochem and CuSO4 anhydrous from Fluka Chemika. Phosphate-buffered saline (PBS) tablets were purchased from Sigma Aldrich and ready-made PBS buffer from Gibco.Solid phase synthesis was performed on TentaGel® SRAM resin purchased from Rapp Polymere using polypropylene reactors with polyethylene frits closed with Luer-stoppers from MultiSyntech GmbH. Ion exchange resin AG1-X8, quat. ammonium, 100–200 mesh, acetate form was purchased from BioRad and Amberlite IR120 (hydrogen form) from Sigma Aldrich. For the liposomes, 1,2-distearoyl-sn-glycero-3-phosphocholine = COATSOME MC-0808® (DSPC), N-(methylpolyoxyethylene oxycarbonyl)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine sodium salt = SUNBRIGHT DSPE-020CN (DSPE-PEG) and N-[N′-(succinimidyloxy glutaryl)aminopropylpolyoxyethylene oxycarbonyl]-1,2-distearoyl-sn-glycero-3-phosphoethanolamine sodium salt = SUNBRIGHT DSPE-020GS (DSPE-PEG-NHS) were purchased from NOF Europe. Cholesterol was purchased from Sigma Aldrich. Filter supports and a Mini-Extruder Kit were purchased from Avanti Polar Lipids and used with Hamilton-syringes (1000 μL) with polycarbonate membranes, pore sizes 0.1 and 0.2 μm. Slide-A-Lyzer™ Dialysis Cassettes were purchased from Thermo Scientific, ultrapure water was supplied from Invitrogen™ UltraPure™ from FisherScientific. EnzyChrom TM Lactose-Assay Kit was purchased from BioAssay Systems and used together with a Multiskan Go Microplate Spectrophotometer from Thermo Scientific and clear flat-bottom 96-well microplates from Greiner Bio-One.
(2-Azidoethyl)-2,3,4,6-tetra-O-acetyl-β-D-galactopyranoside, (2-azidoethyl)-2,3,4,6-tetra-O-acetyl-α-D-glucopyranoside and hepta-O-acetyl-β-lactosylazide were synthesized following established protocols.91 Reactions were observed via analytical thin layer chromatography, performed on Merck silica gel 60 F254 plates and were visualized with ninhydrin and anisaldehyde staining. 1H-NMR and 13C-NMR spectra were measured on Bruker Avance III 300 or Bruker Avance III 600. Analytical reversed phase HPLC (RP-HPLC) measurements were performed on Agilent Technologies 6120 series coupled with an Agilent quadrupole mass spectrometer. All spectra were measured with solvent A: 95% H2O, 5% ACN, +0.1% formic acid, and solvent B: 5% H2O, 95% ACN, +0.1% formic acid with a gradient of 5 to 50% B over 30 min. Purities of the compounds were determined by the integration of the signals absorbing at 214 nm. Preparative RP-HPLC was performed on an Agilent 1200 series. High resolution ESI (HR-ESI) spectra were measured on UHR-QTOF maXis 4G (Bruker Daltonics).
Methods
Diethylenetriamine (3 mmol, 0.3 mL, 1 eq.) was dissolved in 300 mL DCM and 4-pentynoic acid chloride (3 eq.) was added carefully over 30 min. The reaction was stirred for 1 h at room temperature. After adding 100 mL of a saturated NaHCO3 solution, the organic layer was separated, washed two times with 50 mL of saturated NaHCO3 solution and dried over Na2SO4. The solvent was removed under reduced pressure and the crude product was recrystallized from ethyl acetate resulting in the desired product 18 as a white solid in yield of 31% (300 mg, 0.87 mmol).
TPD-precursor 18 (0.1 mmol 34 mg, 1 eq.) and azido-lactose (0.45 mmol, 300 mg, 4.5 eq.) were dissolved in 2 mL DMF. Sodium ascorbate (30 mg, 50 mol%/alkyne) and CuSO4 (38 mg, 50 mol%/alkyne) were each dissolved in 0.2 mL H2O and added to the TPD-lactose solution. The mixture was stirred for 3 d at rt. The reaction mixture was added to 40 mL H2O to precipitate the product. After centrifugation, the precipitate was redissolved in 1 mL DMF and precipitated in 40 mL H2O for a second time. The product was deprotected by treating the crude precipitate with 6 mL of 0.2 M NaOMe in MeOH for 1 h. After adding 4 mL H2O, the solution was neutralized using Amberlite IR120. After filtration and removal of the solvent, the crude product was purified using preparative RP-HPLC. The product was obtained as a white solid with a yield of 50% (73 mg, 0.05 mmol).
Stock solutions of each 8 mg mL−1 DSPE-PEG and DSPE-PEG-ligand in DMSO-d6, and 20 mg mL−1 DSPC and 10 mg mL−1 cholesterol in CDCl3 were prepared. The batch for the formulation was calculated for a final total lipid concentration of 3 μmol in 624 μL PBS-buffer (4.81 mM).
All DMSO-dissolved compounds were added to a test tube and the sample was freeze-dried. Then, DSPC and cholesterol were added and the mixture was further dried in a stream of nitrogen and subsequently under high vacuum for 1 h. 624 μL PBS-buffer were added and the test tube was sonicated for four times at 50 °C for 3–4 s, then vortexed and allowed to rest for 30 s. This procedure was repeated until all of the precipitate was suspended.
In the following extrusion step, the extruder was build-up and prepared as described by the supplier. The liposome suspension was taken up with the Hamilton syringe of the mini-extruder kit and was first extruded 30 times through a 0.2 μm filter and then through a 0.1 μm filter. The suspension was allowed to stand for 30 min at room temperature and then stored at 4 °C.
Surface plasmon resonance. The SPR inhibition studies were performed on a CM5 sensor chip on a Biacore X100 from GE Healthcare Life Science. For immobilization, the “Surface Preparation Wizard” for the sensor chip CM5 was used. The functionalization of the two flow cells was performed through an amine-coupling procedure with NHS/EDC (contact time 420 s, flow rate 10 μL min−1). Flow cell 2 (mess cell) was immobilized with 1 mM Lac(1,3,5)-6, 9*, in HBS-P buffer from GE Healthcare with a contact time of 600 s. For flow cell 1 (reference cell) a blank immobilization with ethanolamine was performed according to the software. As running buffer, HBS-P buffer from GE Healthcare was used. The immobilization levels reached 411 RU for flow cell 2 and 186 RU for flow cell 1.
The inhibition assay was performed in a “Custom Assay Wizard-Binding Analysis” in a multi cycle measurement. For the inhibition studies, stock solutions of 200 μg mL−1 of Gal-3 and 100 μM for each ligand in PBS buffer (150 mM NaCl, 50 mM NaH2PO4, pH 7.5) were prepared. Gal-3 was incubated with each ligand by mixing the solutions of the protein and ligands in a 1:1 ratio, resulting in final concentrations of 100 μg mL−1 for Gal-3 and 50 μM for the ligands. The assay was performed with PBS as running buffer using 90 s for association time and 60 s for dissociation time with a flow rate of 10 μL min−1 over both flow cells. The cell surface was regenerated by injecting 3 M MgCl2 in MilliQ water for 60 s with a flow rate of 10 μL min−1 over the surface after each cycle. Liposomes were measured at concentrations of 10 μM using a stock solution of 20 μM in PBS buffer due to their higher binding affinities to Gal-3.
The report points for the binding event of Gal-3 without and with ligand were taken after 155 s after sensorgram adjustment to baseline. The response unit of only Gal-3 was set as a reference point to 100% binding and 0% inhibition. Inhibition of the glycomacromolecules were referred to the response unit of only Gal-3. All measurements were performed in triplicates.
Enzyme-linked immunosorbent assay (ELISA). Inhibition studies were performed according to an established protocol by Elling and co-workers.35 Glycoligands 1–16 were measured with final concentrations between 0.1 and 2000 μM and liposomes L4, L9, L10 and L16 with final concentrations calculated according to the results of the Lactose-Assay Kit between 0.002 and 14 μM of the glycoligands.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Jan Dirks for his support in synthesizing the H2SO4-cat. We acknowledge support by the Heinrich Heine University Duesseldorf. We thank the Boehringer Ingelheim Foundation for financial support through the Plus 3 program and the DFG for support through the large equipment grant INST 208/735-1. L. E. and D. L. gratefully acknowledge financial support by the DFG (project EL 135/12-1).References
- D. H. Dube and C. R. Bertozzi, Nat. Rev. Drug Discovery, 2005, 4, 477 CrossRef CAS PubMed.
- H. Leffler, S. Carlsson, M. Hedlund, Y. Qian and F. Poirier, Glycoconjugate J., 2002, 19, 433–440 CrossRef CAS PubMed.
- P. Nangia-Makker, J. Conklin, V. Hogan and A. Raz, Trends Mol. Med., 2002, 8, 187–192 CrossRef CAS PubMed.
- A. C. F. Cardoso, L. N. d. S. Andrade, S. O. Bustos and R. Chammas, Front. Oncol., 2016, 6, 1–12 Search PubMed.
- H. Blanchard, X. Yu, P. M. Collins and K. Bum-Erdene, Expert Opin. Ther. Pat., 2014, 24, 1053–1065 CrossRef CAS PubMed.
- M. L. Bacigalupo, M. Manzi, G. A. Rabinovich and M. F. Troncoso, World J. Gastroenterol., 2013, 19, 8831–8849 CrossRef CAS PubMed.
- H. Blanchard, K. Bum-Erdene, M. H. Bohari and X. Yu, Expert Opin. Ther. Pat., 2016, 26, 537–554 CrossRef CAS PubMed.
- Y.-C. Chan, H.-Y. Lin, Z. Tu, Y.-H. Kuo, S.-T. Hsu and C.-H. Lin, Int. J. Mol. Sci., 2018, 19, 392 CrossRef PubMed.
- C. T. Öberg, H. Leffler and U. J. Nilsson, Chimia, 2011, 65, 18–23 CrossRef.
- T. Funasaka, A. Raz and P. Nangia-Makker, Semin. Cancer Biol., 2014, 30–38 CrossRef CAS PubMed.
- K. C. Haudek, K. J. Spronk, P. G. Voss, R. J. Patterson, J. L. Wang and E. J. Arnoys, Biochim. Biophys. Acta, 2010, 1800, 181 CrossRef CAS PubMed.
- K. H. Mayo, in Galectins and Disease Implications for Targeted Therapeutics, Am. Chem. Soc., 2012, vol. 1115, pp. 61–77 Search PubMed.
- L. Díaz-Alvarez and E. Ortega, Mediators Inflammation, 2017, 9247574 Search PubMed.
- A. Lagana, J. G. Goetz, P. Cheung, A. Raz, J. W. Dennis and I. R. Nabi, Mol. Cell. Biol., 2006, 26, 3181–3193 CrossRef CAS PubMed.
- J. Ochieng, M. L. Leite-Browning and P. Warfield, Biochem. Biophys. Res. Commun., 1998, 246, 788–791 CrossRef CAS PubMed.
- V. L. Campo, M. F. Marchiori, L. C. Rodrigues and M. Dias-Baruffi, Glycoconjugate J., 2016, 33, 853–876 CrossRef CAS PubMed.
- J. M. Cousin and M. J. Cloninger, Int. J. Mol. Sci., 2016, 17, 1566 CrossRef PubMed.
- A. K. Michel, P. Nangia-Makker, A. Raz and M. J. Cloninger, ChemBioChem, 2014, 15, 2106–2112 CrossRef CAS PubMed.
- I. Vrasidas, S. André, P. Valentini, C. Böck, M. Lensch, H. Kaltner, R. M. J. Liskamp, H.-J. Gabius and R. J. Pieters, Org. Biomol. Chem., 2003, 1, 803–810 RSC.
- Y. Hou, S. Cao, X. Li, B. Wang, Y. Pei, L. Wang and Z. Pei, ACS Appl. Mater. Interfaces, 2014, 6, 16909–16917 CrossRef CAS PubMed.
- C. Lavilla, G. Yilmaz, V. Uzunova, R. Napier, C. R. Becer and A. Heise, Biomacromolecules, 2017, 18, 1928–1936 CrossRef CAS PubMed.
- S. André, C. E. P. Maljaars, K. M. Halkes, H.-J. Gabius and J. P. Kamerling, Bioorg. Med. Chem. Lett., 2007, 17, 793–798 CrossRef PubMed.
- C. E. P. Maljaars, S. André, K. M. Halkes, H.-J. Gabius and J. Kamerling, Anal. Biochem., 2008, 378, 190 CrossRef CAS PubMed.
- C. K. Goodman, M. L. Wolfenden, P. Nangia-Makker, A. K. Michel, A. Raz and M. J. Cloninger, Beilstein J. Org. Chem., 2014, 10, 1570–1577 CrossRef PubMed.
- S. Zhang, R.-O. Moussodia, C. Murzeau, H.-J. Sun, M. L. Klein, S. Vértesy, S. André, R. Roy, H.-J. Gabius and V. Percec, Angew. Chem., Int. Ed., 2015, 54, 4036–4040 CrossRef CAS PubMed.
- C. Rodriguez-Emmenegger, Q. Xiao, N. Y. Kostina, S. E. Sherman, K. Rahimi, B. E. Partridge, S. Li, D. Sahoo, A. M. Reveron Perez, I. Buzzaccera, H. Han, M. Kerzner, I. Malhotra, M. Möller, C. J. Wilson, M. C. Good, M. Goulian, T. Baumgart, M. L. Klein and V. Percec, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 5376–5382 CrossRef CAS PubMed.
- V. Percec, P. Leowanawat, H.-J. Sun, O. Kulikov, C. D. Nusbaum, T. M. Tran, A. Bertin, D. A. Wilson, M. Peterca, S. Zhang, N. P. Kemat, K. Vargo, D. Moock, E. D. Johnston, D. A. Hammer, D. J. Pochan, Y. Chen, Y. M. Chabre, T. C. Shiao, M. Bergeron-Brleck, S. André, R. Roy, H.-J. Gabius and P. A. Henry, J. Am. Chem. Soc., 2013, 135, 9055–9077 CrossRef CAS PubMed.
- S. Zhang, Q. Xio, S. E. Sherman, A. Muncan, A. D. M. Rmaos Vincente, Z. Wang, D. A. Hammer, D. Willams, Y. Chen, D. J. Pouchan, S. André, M. L. Klein, H.-J. Gabius and V. Percec, J. Am. Chem. Soc., 2015, 137, 13334–13344 CrossRef CAS PubMed.
- Q. Xiao, A.-K. Ludwig, C. Romanò, I. Buzzacchera, S. E. Sherman, M. Vetro, S. Vértesy, H. Kaltner, E. H. Reed, M. Möller, C. J. Wilson, D. A. Hammer, S. Oscarson, M. L. Klein, H.-J. Gabius and V. Percec, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E2509–E2518 CrossRef CAS PubMed.
- S. André, B. Liu, H.-J. Gabius and R. Roy, Org. Biomol. Chem., 2003, 1, 3909–3916 RSC.
- L. Abbassi, Y. M. Chabre, N. Kottari, A. A. Arnold, S. André, J. Josserand, H.-J. Gabius and R. Roy, Polym. Chem., 2015, 6, 7666–7683 RSC.
- N. Ahmad, H.-J. Gabius, S. André, H. Kaltner, S. Sabesan, R. Roy, B. Liu, F. Macaluso and C. F. Brewer, J. Biol. Chem., 2004, 279, 10841–10847 CrossRef CAS PubMed.
- S. R. Rauthu, T. C. Shiao, S. André, M. C. Miller, É. Madej, K. H. Mayo, H.-J. Gabius and R. Roy, ChemBioChem, 2015, 16, 126–139 CrossRef CAS PubMed.
- C. Bonduelle, H. Oliveira, C. Gauche, J. Huang, A. Heise and S. Lecommandoux, Chem. Commun., 2016, 52, 11251–11254 RSC.
- D. Laaf, P. Bojarová, H. Pelantová, V. Křen and L. Elling, Bioconjugate Chem., 2017, 28, 2832–2840 CrossRef CAS PubMed.
- S. Böcker, D. Laaf and L. Elling, Biomolecules, 2015, 5, 1671 CrossRef PubMed.
- G.-J. Boons, Expert Rev. Vaccines, 2010, 9, 1251–1256 CrossRef CAS PubMed.
- V. P. Torchilin, Nat. Rev. Drug Discovery, 2005, 4, 145 CrossRef CAS PubMed.
- B. S. Pattni, V. V. Chupin and V. P. Torchilin, Chem. Rev., 2015, 115, 10938–10966 CrossRef CAS PubMed.
- R. Stahn, H. Schäfer, J. Schreiber and M. Brudel, J. Liposome Res., 1995, 5, 61–73 CrossRef CAS.
- C. Sandoval-Altamirano, S. A. Sanchez, N. F. Ferreyra and G. Gunther, Colloids Surf., B, 2017, 158, 539–546 CrossRef CAS PubMed.
- R. Zeisig, R. Stahn, K. Wenzel, D. Behrens and I. Fichtner, Biochim. Biophys. Acta, Biomembr., 2004, 1660, 31–40 CrossRef CAS PubMed.
- C. Kelly, C. Jefferies and S.-A. Cryan, J. Drug Delivery, 2011, 2011, 727241 Search PubMed.
- M. Srinivasarao and P. S. Low, Chem. Rev., 2017, 117, 12133–12164 CrossRef CAS PubMed.
- N. Jayaraman, K. Maiti and K. Naresh, Chem. Soc. Rev., 2013, 42, 4640–4656 RSC.
- J. J. Weingart, P. Vabbilisetty and X. L. Sun, Carbohydrate Nanotechnology, ed. K. J. Stine, John Wiley & Sons, Inc., 1st edn, 2016 Search PubMed.
- L. Cai, Z. Gu, J. Zhong, D. Wen, G. Chen, L. He, J. Wu and Z. Gu, Drug Discovery Today, 2018, 23, 1126–1138 CrossRef CAS PubMed.
- W. C. Chen, G. C. Completo, D. S. Sigal, P. R. Crocker, A. Saven and J. C. Paulson, Blood, 2010, 115, 4778–4786 CrossRef CAS PubMed.
- N. Kawasaki, J. L. Vela, C. M. Nycholat, C. Rademacher, A. Khurana, N. van Rooijen, P. R. Crocker, M. Kronenberg and J. C. Paulson, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 7826–7831 CrossRef CAS PubMed.
- M. S. Macauley, F. Pfrengle, C. Rademacher, C. M. Nycholat, A. J. Gale, A. von Drygalski and J. C. Paulson, J. Clin. Invest., 2013, 123, 3074–3083 CrossRef CAS PubMed.
- J. Zhu, J. Xue, Z. Guo, L. Zhang and R. E. Marchant, Bioconjugate Chem., 2007, 18, 1366–1369 CrossRef CAS PubMed.
- D. Ponader, F. Wojcik, F. Beceren-Braun, J. Dernedde and L. Hartmann, Biomacromolecules, 2012, 13, 1845–1852 CrossRef CAS PubMed.
- D. Ponader, P. Maffre, J. Aretz, D. Pussak, N. M. Ninnemann, S. Schmidt, P. H. Seeberger, C. Rademacher, G. U. Nienhaus and L. Hartmann, J. Am. Chem. Soc., 2014, 136, 2008–2016 CrossRef CAS PubMed.
- C. Gerke, M. F. Ebbesen, D. Jansen, S. Boden, T. Freichel and L. Hartmann, Biomacromolecules, 2017, 18, 787–796 CrossRef CAS PubMed.
- T. Freichel, S. Eierhoff, N. L. Snyder and L. Hartmann, J. Org. Chem., 2017, 82, 9400–9409 CrossRef CAS PubMed.
- M. Baier, M. Giesler and L. Hartmann, Chem.–Eur. J., 2018, 24, 1619–1630 CrossRef CAS PubMed.
- J. Chen, H.-N. Son, J. J. Hill, S. Srinivasan, F.-Y. Su, P. S. Stayton, A. J. Convertine and D. M. Ratner, Nanomedicine, 2016, 12, 2031–2041 CrossRef CAS PubMed.
- S. Boden, K. Wagner, M. Karg and L. Hartmann, Polymers, 2017, 9, 716 CrossRef PubMed.
- M. F. Ebbesen, C. Gerke, P. Hartwig and L. Hartmann, Polym. Chem., 2016, 7, 7086–7093 RSC.
- K. S. Bücher, H. Yan, R. Creutznacher, K. Ruoff, A. Mallagaray, A. Grafmüller, J. S. Dirks, T. Kilic, S. Weickert, A. Rubailo, M. Drescher, S. Schmidt, G. Hansman, T. Peters, C. Uetrecht and L. Hartmann, Biomacromolecules, 2018, 19, 3714–3724 CrossRef PubMed.
- G. Zemplén and E. Pacsu, Ber. Dtsch. Chem. Ges., 1929, 62, 1613–1614 CrossRef.
- E.-C. Wamhoff, J. Schulze, L. Bellmann, G. Bachem, F. F. Fuchsberger, J. Rademacher, M. Hermann, B. Del Frari, R. van Dalen, D. Hartmann, N. van Sorge, O. Seitz, P. Stoitzner and C. Rademacher, ACS Cent. Sci., 2019, 5, 808–820 CAS.
- P. L. Yeagle, Biochim. Biophys. Acta, Rev. Biomembr., 1985, 822, 267–287 CrossRef CAS.
- S. Raffy and J. Teissié, Biophys. J., 1999, 76, 2072–2080 CrossRef CAS PubMed.
- J. Senior and G. Gregoriadis, Life Sci., 1982, 30, 2123–2136 CrossRef CAS PubMed.
- Y. Barenholz, J. Controlled Release, 2012, 160, 117–134 CrossRef CAS PubMed.
- M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard, A. Dokhani, S. Khorasani and M. R. Mozafari, Pharmaceutics, 2018, 10, 57 CrossRef PubMed.
- K. N. Clayton, J. W. Salameh, S. T. Wereley and T. L. Kinzer-Ursem, Biomicrofluidics, 2016, 10, 054107 CrossRef PubMed.
- R. I. Jolck, L. N. Feldborg, S. Andersen, S. M. Moghimi and T. L. Andresen, Adv. Biochem. Eng./Biotechnol., 2011, 125, 251–280 CrossRef PubMed.
- P. Vabbilisetty and X.-L. Sun, Org. Biomol. Chem., 2014, 12, 1237–1244 RSC.
- I. Taniguchi, K. Akiyoshi and J. Sunamoto, Macromol. Chem. Phys., 1999, 200, 1386–1392 CrossRef CAS.
- H. Zhang, D. Laaf, L. Elling and R. J. Pieters, Bioconjugate Chem., 2018, 29, 1266–1275 CrossRef CAS PubMed.
- L. Bumba, D. Laaf, V. Spiwok, L. Elling, V. Křen and P. Bojarová, Int. J. Mol. Sci., 2018, 19, 372 CrossRef PubMed.
- M. Wolfenden, J. Cousin, P. Nangia-Makker, A. Raz and M. Cloninger, Molecules, 2015, 20, 7059–7096 CrossRef CAS PubMed.
- P. Sörme, B. Kahl-Knutsson, U. Wellmar, B.-G. Magnusson, H. Leffler and U. J. Nilsson, in Methods in Enzymology, Academic Press, 2003, vol. 363, pp. 157–169 Search PubMed.
- T. K. Dam, H.-J. Gabius, S. André, H. Kaltner, M. Lensch and C. F. Brewer, Biochemistry, 2005, 44, 12564–12571 CrossRef CAS PubMed.
- J. Stegmayr, A. Lepur, B. Kahl-Knutson, M. Aguilar-Moncayo, A. A. Klyosov, R. A. Field, S. Oredsson, U. J. Nilsson and H. Leffler, J. Biol. Chem., 2016, 291, 13318–13334 CrossRef CAS PubMed.
- K. Saraboji, M. Håkansson, S. Genheden, C. Diehl, J. Qvist, U. Weininger, U. J. Nilsson, H. Leffler, U. Ryde, M. Akke and D. T. Logan, Biochemistry, 2012, 51, 296–306 CrossRef CAS PubMed.
- I. R. Nabi, J. Shankar and J. W. Dennis, J. Cell Sci., 2015, 128, 2213–2219 CrossRef CAS PubMed.
- M. van Scherpenzeel, E. E. Moret, L. Ballell, R. M. J. Liskamp, U. J. Nilsson, H. Leffler and R. J. Pieters, ChemBioChem, 2009, 10, 1724–1733 CrossRef CAS PubMed.
- M. F. Marchiori, D. E. Pires Souto, L. Oliveira Bortot, J. Francisco Pereira, L. T. Kubota, R. D. Cummings, M. Dias-Baruffi, I. Carvalho and V. L. Campo, Bioorg. Med. Chem., 2015, 23, 3414–3425 CrossRef CAS PubMed.
- J. Tejler, H. Leffler and U. J. Nilsson, Bioorg. Med. Chem. Lett., 2005, 15, 2343–2345 CrossRef CAS PubMed.
- V. Denavit, D. Lainé, T. Tremblay, J. St-Gelais and D. Giguère, Trends Glycosci. Glycotechnol., 2018, 30, SE21–SE40 CrossRef.
- J. M. Fox, M. Zhao, M. J. Fink, K. Kang and G. M. Whitesides, Annu. Rev. Biophys., 2018, 47, 223–250 CrossRef CAS PubMed.
- C.-e. A. Chang, W. Chen and M. K. Gilson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1534–1539 CrossRef CAS PubMed.
- A. J. Cagnoni, J. Kovensky and M. L. Uhrig, J. Org. Chem., 2014, 79, 6456–6467 CrossRef CAS PubMed.
- H. Halimi, A. Rigato, D. Byrne, G. Ferracci, C. Sebban-Kreuzer, L. El Antak and F. Guerlesquin, PLoS One, 2014, 9, e111836 CrossRef PubMed.
- M. Sundqvist, A. Welin, J. Elmwall, V. Osla, U. J. Nilsson, H. Leffler, J. Bylund and A. Karlsson, J. Leukocyte Biol., 2018, 103, 341–353 CrossRef CAS PubMed.
- R.-Y. Yang, P. N. Hill, D. K. Hsu and F.-T. Liu, Biochemistry, 1998, 37, 4086–4092 CrossRef CAS PubMed.
- A. Lepur, E. Salomonsson, U. J. Nilsson and H. Leffler, J. Biol. Chem., 2012, 287, 21751–21756 CrossRef CAS PubMed.
- L. Wu and N. S. Sampson, ACS Chem. Biol., 2014, 9, 468–475 CrossRef CAS PubMed.
- S. Chen, L. Li, C. Zhao and J. Zheng, Polymer, 2010, 51, 5283–5293 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR, HR-ESI, LCMS, MALDI-TOF. See DOI: 10.1039/c9ra05497a |
| This journal is © The Royal Society of Chemistry 2019 |