
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
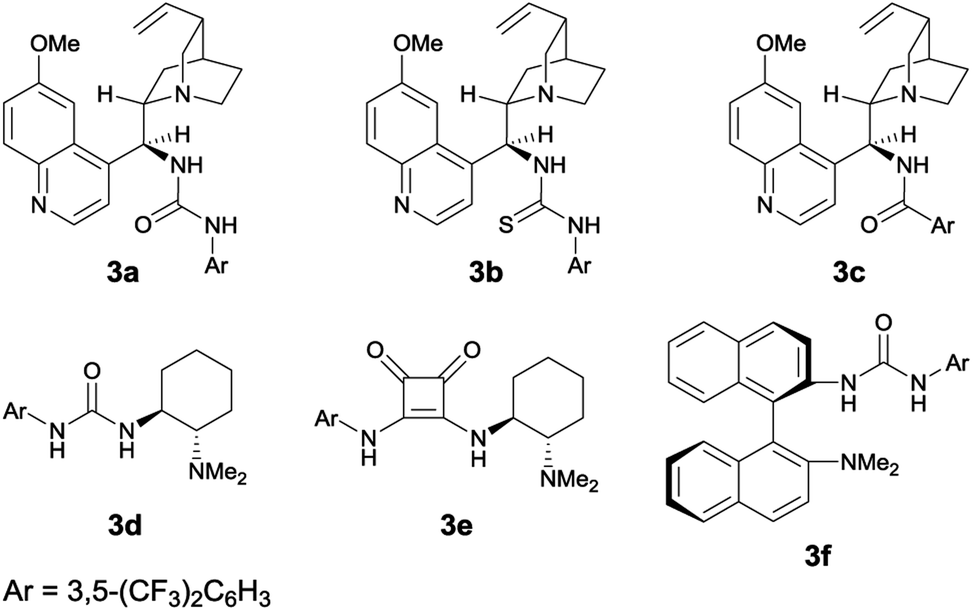
Enantioselective bromination of axially chiral cyanoarenes in the presence of bifunctional organocatalysts†
Yuuki
Wada
,
Akira
Matsumoto‡
 ,
Keisuke
Asano
* and
Seijiro
Matsubara
*
,
Keisuke
Asano
* and
Seijiro
Matsubara
*
Department of Material Chemistry, Graduate School of Engineering, Kyoto University, Kyotodaigaku-Katsura, Nishikyo, Kyoto 615-8510, Japan. E-mail: asano.keisuke.5w@kyoto-u.ac.jp; matsubara.seijiro.2e@kyoto-u.ac.jp; Fax: +81 75 383 2438; Tel: +81 75 383 7571 Tel: +81 75 383 7130
First published on 4th October 2019
Abstract
Enantioselective bromination of axially chiral cyanoarenes bearing high intrinsic rotational barriers via dynamic kinetic resolution using bifunctional organocatalysts is reported. Sequential addition of a brominating reagent in several portions at an optimized temperature was effective in accomplishing high enantioselectivities.
Axially chiral biaryls are privileged structures in pharmaceuticals,1 asymmetric catalysts,2 functional materials,3etc. Thus, the development of efficient methods for their synthesis is desirable to advance research in these scientific fields. Among recent accomplishments on catalytic atroposelective transformations4 toward the synthesis of densely substituted axially chiral biaryls, powerful strategies include organocatalytic dynamic kinetic resolution involving ortho-functionalization of existing biaryls via the introduction of additional rotational barriers.5 In this method, substrates should in principle have rotational barriers low enough to enable fast rotation about the biaryl axis, leading to their rapid racemization. Therefore, it is difficult to employ biaryl substrates bearing intrinsic rotational barriers, which impede their racemization. In 2015, the Miller group reported an elegant example of this type of dynamic kinetic resolution via bromination of 3-arylquinazolin-4(3H)-ones, the rotational barrier of which is ∼19 kcal mol−1, by slow addition of a brominating agent.5e Here, we present enantioselective bromination of axially chiral cyanoarenes bearing intrinsic rotational barriers exceeding 18 kcal mol−1 (Scheme 1). To the best of our knowledge, there has been no report of a catalytic asymmetric reaction affording axially chiral cyanoarenes,6 despite their prevalence in bioactive agents7 and the rich chemistry of cyano compounds as synthetic intermediates.8
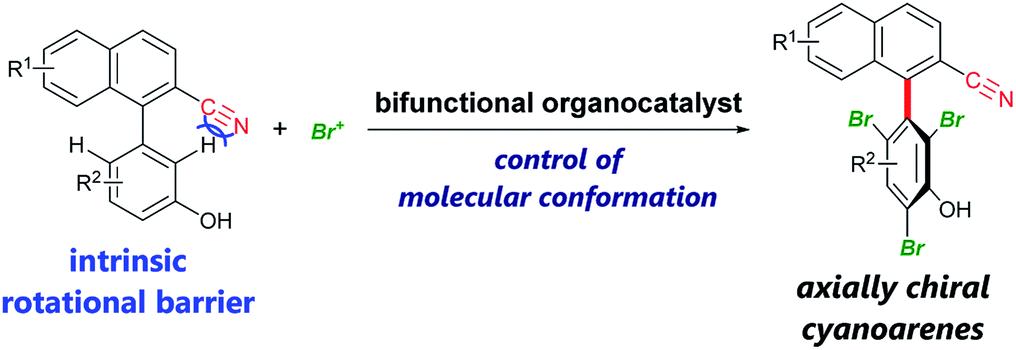 | ||
| Scheme 1 Enantioselective bromination of axially chiral cyanoarenes using bifunctional organocatalysts. | ||
Table 1 shows the optimization of reaction conditions. We started our investigation using 1-(3-hydroxyphenyl)-2-naphthonitrile (1a) and N-bromoacetamide (NBA, 4a) as the brominating reagent with 10 mol% quinine-derived bifunctional catalysts 3a–3c in CH2Cl2 at 25 °C. Urea and amide catalysts 3a and 3c, respectively, afforded 2a in higher enantioselectivities than the thiourea catalyst 3b (Table 1, entries 1–3).9 Other catalysts 3d and 3e, bearing a cyclohexanediamine framework, and 3f, bearing a binaphthyl framework, resulted in poor enantioselectivities (Table 1, entries 4–6).10 Using 3a and 3c, lower reaction temperatures were investigated (Table 1, entries 7–10); 3c gave higher enantioselectivity at −40 °C, although the reactions did not proceed at all at −60 °C. By screening different solvents, CH2Cl2 was identified as the most suitable solvent from the viewpoints of both yield and enantioselectivity (Table 1, entries 9 and 11–16). Other brominating reagents (Fig. 1) were also investigated; NBA (4a) still afforded the best enantioselectivities (Table 1, entries 9 and 17–19). The decrease in the loading of 3c to 5 mol% slightly improved the enantioselectivity, although a longer reaction time (48 h) was necessary (Table 1, entry 20). Next, using the thus-optimized conditions, reactions with shorter reaction times (24 h, 12 h, and 6 h) were carried out (Table 1, entries 21–23); the enantioselectivity was improved while the conversions decreased with decreasing reaction time. These results imply that the racemization of 1a at −40 °C is not rapid enough to make use of dynamic kinetic resolution.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Brominating reagent | Solvent | Temp. (°C) | Yieldb (%) | ee (%) |
| a Reactions were run using 1a (0.10 mmol), the brominating reagent (0.30 mmol), and the catalyst (0.010 mmol) in the solvent (10 mL). b Isolated yields. c Reactions were run using 3c (0.0050 mmol). d Reaction was run for 48 h. e Reaction was run for 24 h. f Reaction was run for 12 h. g Reaction was run for 6 h. | ||||||
| 1 | 3a | NBA (4a) | CH2Cl2 | 25 | 81 | 26 |
| 2 | 3b | NBA (4a) | CH2Cl2 | 25 | 87 | 6 |
| 3 | 3c | NBA (4a) | CH2Cl2 | 25 | 89 | 18 |
| 4 | 3d | NBA (4a) | CH2Cl2 | 25 | 83 | 6 |
| 5 | 3e | NBA (4a) | CH2Cl2 | 25 | 85 | 3 |
| 6 | 3f | NBA (4a) | CH2Cl2 | 25 | 79 | 3 |
| 7 | 3a | NBA (4a) | CH2Cl2 | −40 | 82 | 20 |
| 8 | 3a | NBA (4a) | CH2Cl2 | −60 | <1 | — |
| 9 | 3c | NBA (4a) | CH2Cl2 | −40 | 83 | 41 |
| 10 | 3c | NBA (4a) | CH2Cl2 | −60 | <1 | — |
| 11 | 3c | NBA (4a) | CHCl3 | −40 | 21 | 54 |
| 12 | 3c | NBA (4a) | Toluene | −40 | 18 | −10 |
| 13 | 3c | NBA (4a) | THF | −40 | 13 | −2 |
| 14 | 3c | NBA (4a) | Et2O | −40 | 46 | −20 |
| 15 | 3c | NBA (4a) | EtOAc | −40 | 68 | −4 |
| 16 | 3c | NBA (4a) | EtOH | −40 | <5 | — |
| 17 | 3c | DBH (4b) | CH2Cl2 | −40 | 79 | 1 |
| 18 | 3c | NBS (4c) | CH2Cl2 | −40 | 82 | 20 |
| 19 | 3c | NBP (4d) | CH2Cl2 | −40 | 79 | −5 |
| 20c,d | 3c | NBA (4a) | CH2Cl2 | −40 | 85 | 49 |
| 21c,e | 3c | NBA (4a) | CH2Cl2 | −40 | 51 | 59 |
| 22c,f | 3c | NBA (4a) | CH2Cl2 | −40 | 33 | 66 |
| 23c,g | 3c | NBA (4a) | CH2Cl2 | −40 | 17 | 71 |
|
||||||

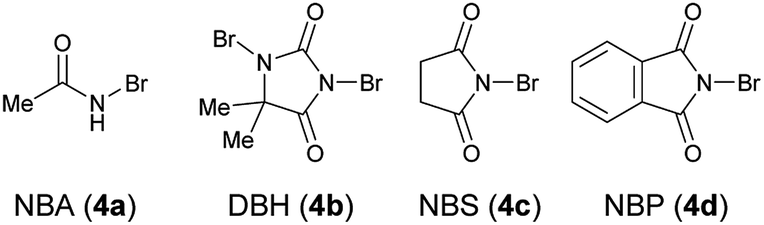 | ||
| Fig. 1 Brominating reagents. | ||
Subsequently, to improve the efficiency of dynamic kinetic resolution by retarding the enantiodetermining bromination,5e4a was added sequentially in five portions (Fig. 2).11 Although the procedure hardly affected the results at −40 °C, the enantioselectivity was greatly improved for reactions carried out at −20 °C and −30 °C. Such effects were smaller at temperatures above −10 °C.
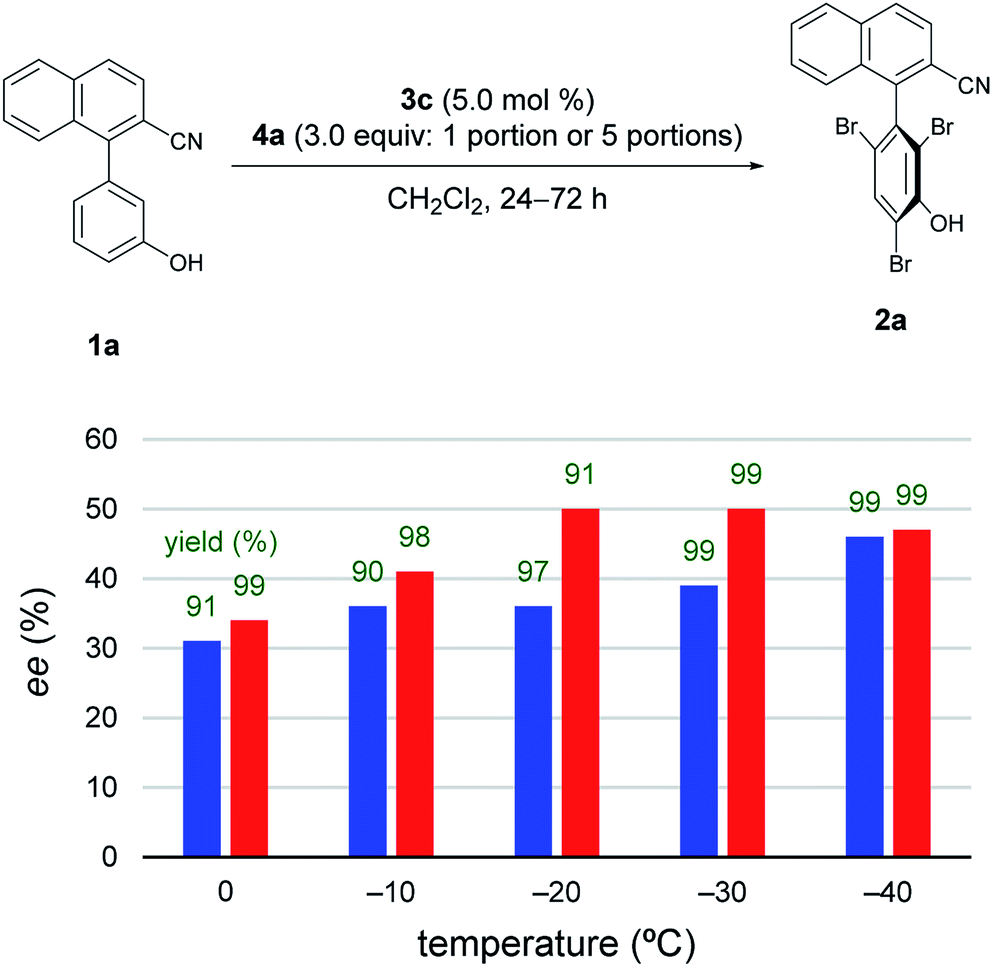 | ||
| Fig. 2 Investigations of temperatures and procedures. Blue bar: reactions were run with 4a added in 1 portion. Red bar: reactions were run with 4a added in 5 portions. Green values represent yields of 2a isolated after silica gel column chromatography. At 0, −10, −20, and −30 °C, reactions were run for 24 h; at −40 °C, reactions were run for 72 h. | ||
Next, at −30 °C and −40 °C, respectively, the relationships between enantioselectivity and yield were investigated (Fig. 3). Reactions were carried out using various amounts of 4a. At both temperatures, the enantioselectivity decreased as the yield increased; however, the quantitative reactions also exhibited some enantioselectivity (−30 °C: 99% yield, 50% ee; −40 °C: 99% yield, 47% ee), implying the presence of the characteristics of dynamic kinetic resolution. In addition, although the enantioselectivity was better at −40 °C than −30 °C when the yield was low, the relationship became reversed as the yield increased; hence, the efficiency of dynamic kinetic resolution was revealed to be better at −30 °C than at −40 °C. Furthermore, when 1.5 equiv. of 4a were used at −30 °C affording 2a in 22% yield with 65% ee, the ortho-monobrominated product 1a-Br (Fig. 4) was also obtained with 75% ee (see Scheme S1 in the ESI† for details). It shows that the bromination at one of the ortho-positions introduces a rotational barrier high enough to set the chiral axis, which is consistent with the rotational barriers calculated at the M06-2X/6-311++G(2d,3p)//B3LYP/6-31+G(d,p) level of theory (Fig. 4).
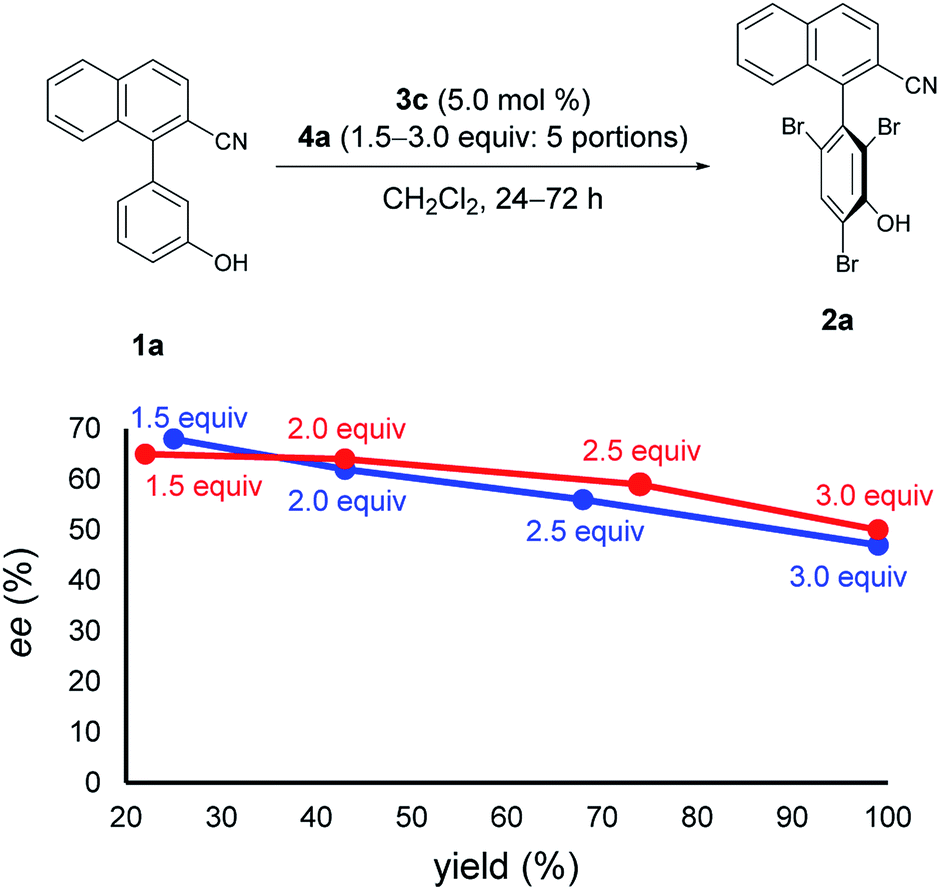 | ||
| Fig. 3 Relationships between ee and yield. Red line: reactions were run at −30 °C for 24 h with 4a added in 5 portions. Blue line: reactions were run at −40 °C for 72 h with 4a added in 5 portions. Red and blue values represent amounts of 4a used for each reaction. | ||
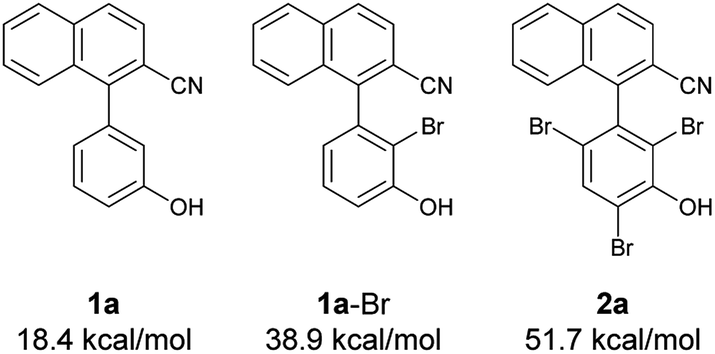 | ||
| Fig. 4 Rotational barriers of substrate, intermediate, and product calculated at the M06-2X/6-311++G(2d,3p)//B3LYP/6-31+G(d,p) level of theory. | ||
Under the conditions of using 3c as the catalyst at −30 °C with 3 equiv. of 4a added sequentially in five portions, other substrates bearing substituted phenols were also investigated (Scheme 2).12 First, substrates 1b–1e bearing a substituent at the meta-position were investigated. While the electron-deficient substrate 1b resulted in poor enantioselectivity, 1c and 1d bearing aliphatic substituents gave improved enantioselectivities; however, substrate 1e with a methoxy group resulted in low enantioselectivity. In addition, substrates 1f–1i bearing substituents at the para-positions of the biaryl axis were then examined; phenol 2h bearing a methyl group resulted in higher enantioselectivities than phenols 2f and 2g bearing electron-withdrawing groups and 2i bearing a methoxy group. These results suggest that aliphatic substituents might efficiently facilitate the racemization of 1 during bromination, leading to dynamic kinetic resolution with greater enantioselectivity. Utilizing this methodology with the characteristics of dynamic kinetic resolution, the reactions of 1c and 1g were also carried out using a sub-stoichiometric amount of 4a (Scheme 3); higher enantioselectivities were accomplished albeit with lower yields.13 The absolute configuration of 2c was determined by X-ray crystallography (see the ESI† for details), and the configurations of all other products were assigned analogously.
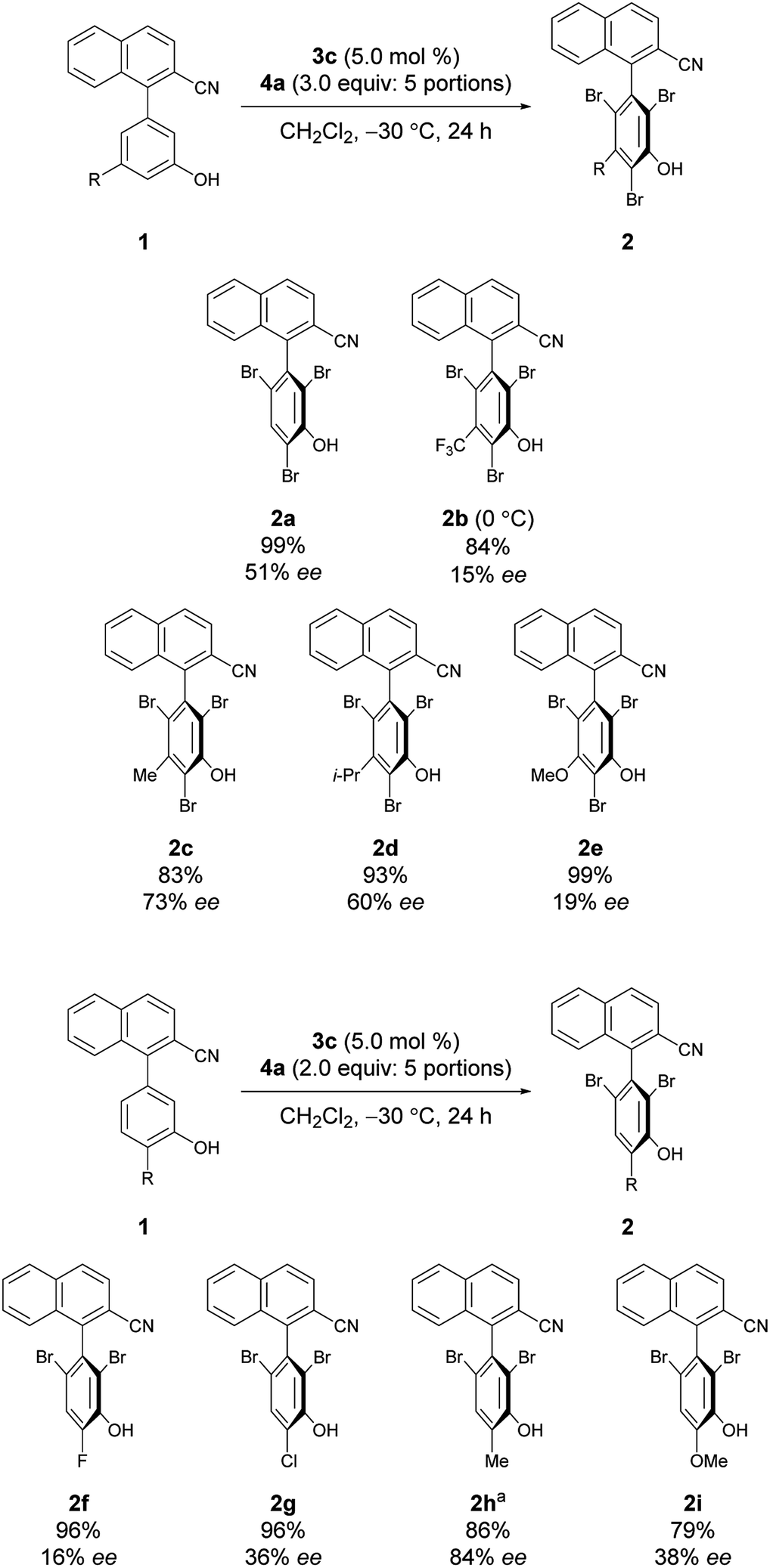 | ||
| Scheme 2 Reactions of substrates with substituted phenols. aReaction was run for 72 h. | ||
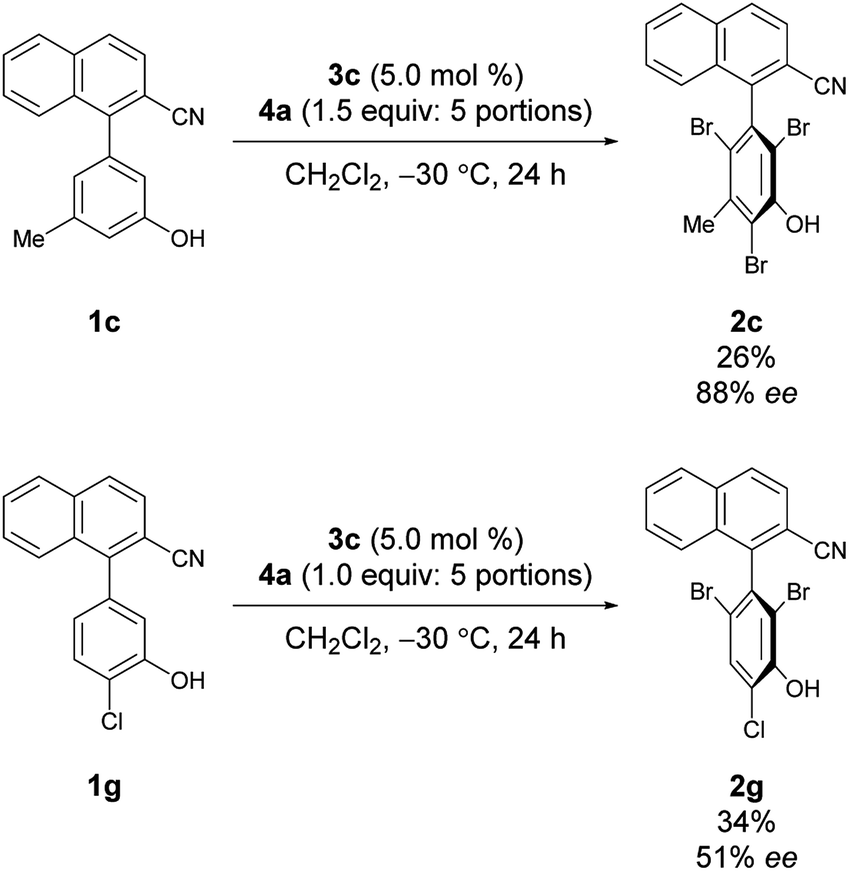 | ||
| Scheme 3 Reactions with a substoichiometric amount of 4a. | ||
In summary, we present enantioselective bromination of axially chiral cyanoarenes bearing high intrinsic rotational barriers via dynamic kinetic resolution using bifunctional organocatalysts. The sequential addition of 4a in several portions at the optimized temperature was effective in improving the enantioselectivity. Although the enantioselectivities are still moderate using the current catalytic system, the guidelines for designing catalytic asymmetric syntheses of axially chiral cyanoarenes were established. Further studies on the additional optimization and application of this methodology to the construction of densely substituted axially chiral biaryls are currently underway.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by the Japanese Ministry of Education, Culture, Sports, Science and Technology (JP15H05845, JP16K13994, JP17K19120, JP18K14214, and JP18H04258). K. A. also acknowledges the Naito Foundation, Research Institute for Production Development, the Tokyo Biochemical Research Foundation, the Uehara Memorial Foundation, the Kyoto University Foundation, the Institute for Synthetic Organic Chemistry, Toyo Gosei Memorial Foundation, the Sumitomo Foundation, and Fukuoka Naohiko Memorial Foundation. A. M. also acknowledges the Japan Society for the Promotion of Science for Young Scientists for the fellowship support.Notes and references
- (a) G. Bringmann and D. Menche, Acc. Chem. Res., 2001, 34, 615 CrossRef CAS; (b) M. C. Kozlowski, B. J. Morgan and E. C. Linton, Chem. Soc. Rev., 2009, 38, 3193 RSC; (c) G. Bringmann, T. Gulder, T. A. M. Gulder and M. Breuning, Chem. Rev., 2011, 111, 563 CrossRef CAS; (d) A. Zask, J. Murphy and G. A. Ellestad, Chirality, 2013, 25, 265 CrossRef CAS; (e) J. E. Smyth, N. M. Butler and P. A. Keller, Nat. Prod. Rep., 2015, 32, 1562 RSC; (f) C. L. Covington, F. M. S. Junior, J. H. S. Silva, R. M. Kuster, M. B. de Amorim and P. L. Polavarapu, J. Nat. Prod., 2016, 79, 2530 CrossRef CAS.
- (a) M. McCarthy and P. J. Guiry, Tetrahedron, 2001, 57, 3809 CrossRef CAS; (b) Y. Chen, S. Yekta and A. K. Yudin, Chem. Rev., 2003, 103, 3155 CrossRef CAS; (c) J. M. Brunel, Chem. Rev., 2005, 105, 857 CrossRef CAS; (d) T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691 CrossRef CAS PubMed.
- (a) Q. Li, L. Green, N. Venkataraman, I. Shiyanovskaya, A. Khan, A. Urbas and J. W. Doane, J. Am. Chem. Soc., 2007, 129, 12908 CrossRef CAS; (b) H. Hayasaka, T. Miyashita, M. Nakayama, K. Kuwada and K. Akagi, J. Am. Chem. Soc., 2012, 134, 3758 CrossRef CAS; (c) Y.-L. Wu, F. Ferroni, S. Pieraccini, W. B. Schweizer, B. B. Frank, G. P. Spada and F. Diederich, Org. Biomol. Chem., 2012, 10, 8016 RSC; (d) L. Pu, Acc. Chem. Res., 2012, 45, 150 CrossRef CAS PubMed; (e) G. Wei, S. Zhang, C. Dai, Y. Quan, Y. Cheng and C. Zhu, Chem.–Eur. J., 2013, 19, 16066 CrossRef CAS.
- For selected recent reviews on enantioselective syntheses of axially chiral compounds, see ref. 1 and the following: (a) P. G. Cozzi, E. Emer and A. Gualandi, Angew. Chem., Int. Ed., 2011, 50, 3847 CrossRef CAS; (b) O. Quinonero, C. Bressy and X. Bugaut, Angew. Chem., Int. Ed., 2014, 53, 10861 CrossRef CAS; (c) J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418 RSC; (d) G. Ma and M. P. Sibi, Chem.–Eur. J., 2015, 21, 11644 CrossRef CAS; (e) G. Bencivenni, Synlett, 2015, 26, 1915 CrossRef CAS; (f) E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Chem. Rev., 2015, 115, 11239 CrossRef CAS PubMed; (g) S. Shirakawa, S. Liu and S. Kaneko, Chem.–Asian J., 2016, 11, 330 CrossRef CAS; (h) P. Renzi, Org. Biomol. Chem., 2017, 15, 4506 RSC; (i) B. Zilate, A. Castrogiovanni and C. Sparr, ACS Catal., 2018, 8, 2981 CrossRef CAS; (j) O. Baudoin, Eur. J. Org. Chem., 2005, 4223 CrossRef CAS; (k) G. Bringmann, A. J. P. Mortimer, P. A. Keller, M. J. Gresser, J. Garner and M. Breuning, Angew. Chem., Int. Ed., 2005, 44, 5384 CrossRef CAS; (l) D. Zhang and Q. Wang, Coord. Chem. Rev., 2015, 286, 1 CrossRef CAS; (m) A. H. Cherney, N. T. Kadunce and S. E. Reisman, Chem. Rev., 2015, 115, 9587 CrossRef CAS; (n) P. Loxq, E. Manoury, R. Poli, E. Deydier and A. Labande, Coord. Chem. Rev., 2016, 308, 131 CrossRef CAS; (o) K. Tanaka, Chem.–Asian J., 2009, 4, 508 CrossRef CAS.
- (a) J. L. Gustafson, D. Lim and S. J. Miller, Science, 2010, 328, 1251 CrossRef CAS; (b) T. P. Pathak and S. J. Miller, J. Am. Chem. Soc., 2012, 134, 6120 CrossRef CAS; (c) K. T. Barrett and S. J. Miller, J. Am. Chem. Soc., 2013, 135, 2963 CrossRef CAS; (d) K. T. Barrett, A. J. Metrano, P. R. Rablen and S. J. Miller, Nature, 2014, 509, 71 CrossRef CAS; (e) M. E. Diener, A. J. Metrano, S. Kusano and S. J. Miller, J. Am. Chem. Soc., 2015, 137, 12369 CrossRef CAS PubMed; (f) R. Miyaji, K. Asano and S. Matsubara, J. Am. Chem. Soc., 2015, 137, 6766 CrossRef CAS; (g) R. Miyaji, K. Asano and S. Matsubara, Chem.–Eur. J., 2017, 23, 9996 CrossRef; (h) R. Miyaji, Y. Wada, A. Matsumoto, K. Asano and S. Matsubara, Beilstein J. Org. Chem., 2017, 13, 1518 CrossRef CAS PubMed.
- For the synthesis of axially chiral cyanoarenes using optically active starting materials, see: J. Liu, H.-X. Zheng, C.-Z. Yao, B.-F. Sun and Y.-B. Kang, J. Am. Chem. Soc., 2016, 138, 3294 CrossRef CAS.
- (a) J. Clayden, W. J. Moran, P. J. Edwards and S. R. LaPlante, Angew. Chem., Int. Ed., 2009, 48, 6398 CrossRef CAS; (b) S. R. LaPlante, L. D. Fader, K. R. Fandrick, D. R. Fandrick, O. Hucke, R. Kemper, S. P. F. Miller and P. J. Edwards, J. Med. Chem., 2011, 54, 7005 CrossRef CAS.
- (a) K. Friedrich and K. Wallenfels, in The Chemistry of the Cyano Group, ed. Z. Rappaport, John Wiley & Sons, 1970, ch. 2 Search PubMed; (b) A. J. Fatiadi, in The Chemistry of Triple-Bonded Functional Groups, ed. S. Patai and Z. Rappaport, John Wiley & Sons, 1983, ch. 26, vol. 2 Search PubMed.
- The replacement of the cyano group with an ethynyl group in 1a gave much lower enantioselectivity in the presence of 3c; the replacement of the NH group with an NMe group in 3c gave much lower enantioselectivity and catalytic activity in the reaction of 1a; the replacement of the OH group with an OMe group in 1a resulted in no reaction in the presence of 3c. These results suggest that the cyano and OH groups of 1a and the NH group of 3c are involved in hydrogen bonding between 1a and 3c during catalysis. See Scheme S2 in the ESI† for details..
- Results of further catalyst screening are described in the ESI (Table S1†)..
- Slow addition of 4a over hours resulted in low yields, probably because 4a was decomposed in its CH2Cl2 solution in the syringe while addition after being left for a while..
- It was difficult to prepare substrates bearing substituted 2-naphthonitriles..
- The reaction of 1h using 0.5 equiv. of 4a afforded an ortho-monobrominated product in 43% yield with 81% ee (see Scheme S3 in the ESI† for details), which is not higher than the result (84% ee) obtained in Scheme 2. These results also imply that aliphatic substituents increase the efficiency of dynamic kinetic resolution..
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, analytical and spectroscopic data for synthetic compounds, and copies of NMR. CCDC 1936455. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra05532k |
| ‡ Present address: Graduate School of Pharmaceutical Sciences, Kyoto University, Yoshida-Shimoadachi, Sakyo, Kyoto 606-8501, Japan. |
| This journal is © The Royal Society of Chemistry 2019 |