DOI:
10.1039/C9RA05572J
(Paper)
RSC Adv., 2019,
9, 28775-28782
Retracted Article: Aclarubicin regulates glioma cell growth and DNA damage through the SIRT1/PI3K/AKT signaling pathway
Received
19th July 2019
, Accepted 6th September 2019
First published on 12th September 2019
Abstract
Aclarubicin (ACR), an anthracycline anti-tumor agent, is known to play important roles in cancer. Evidence has suggested that ACR has therapeutic effects on rats intracranially implanted with C6 glioma cells. However, the function and mechanism of ACR in glioma cells remain elusive. In this study, we examined the effects of ACR on glioma cell growth, apoptosis, and DNA damage. Our results showed that treatment with different concentrations of ACR (1, 2, and 5 μM) markedly impeded glioma cell survival, significantly decreased cell proliferation, and increased cell apoptosis and caspase-3 activity. Furthermore, ACR treatment promoted DNA damage through phosphorylation of ATM and CHK1 in U87 and U251 cells. Treatment with ACR also increased sirtuin 1 (SIRT1) expression and inhibited phosphatidylinositol 3′-kinase (PI3K)/AKT pathway activation. Interestingly, we found that AKT overexpression reversed the effects of ACR on glioma cell survival, proliferation, apoptosis, and DNA damage. Thus, our data suggest that ACR induces apoptosis and DNA damage in U87 and U251 cells through the SIRT1/PI3K/AKT signaling pathway.
1. Introduction
Glioblastoma is one of the most prevalent malignant tumors and commonly occurs in any part of the brain and spinal cord, accounting for 80% of all brain tumors.1,2 Currently, surgical techniques, followed by radiotherapy and chemotherapy, have been used to treat patients with glioblastoma. Surgery is the first therapeutic option, which is used for tissue diagnosis and reduction of tumor volume, while radiotherapy and chemotherapy have been shown to prolong patient survival and preserve quality of life.3 However, these treatments are not satisfactory as the median survival for patients with glioblastoma has been reported to be less than 15 months.4 Due to its poor prognosis, glioblastoma remains an unsolved problem in neuro-oncology.5 Thus, the development of effective methods or medicines to treat gliomas is critical.
Anthracyclines, including aclarubicin (ACR; the newer N,N-dimethylated trisaccharide anthracycline) and doxorubicin (DOX; the classical monosaccharide daunomycinone anthracycline) have been shown to exert strong anti-tumor activity in solid tumors and act as effective antineoplastic agents.6,7 ACR, also known as aclacinomycin A, has a trisaccharide chain, and was characterized and discovered in the 1970s by Oki et al.8 It has been reported to play anti-tumor roles through catalytically inhibiting topoisomerase I and interacting with topoisomerase II.9,10 Additionally, ACR functions in regulation of tumor progression and various cellular processes, such as cell growth, differentiation, and apoptosis.11 Recently, Wei Lu et al. suggested that ACR-loaded cationic albumin-conjugated pegylated nanoparticles may serve as potential therapeutic agents for the treatment of glioblastoma.12 Although the anti-cancer role of ACR has been demonstrated in various types of cancers and gliomas, its physiological function and mechanism in glioma pathogenesis and progression remain largely unknown.
Sirtuin 1 (SIRT1), a nicotinamide-adenine dinucleotide (NAD)-dependent protein deacetylase, has been shown to be associated with glioma progression and tumorigenesis.13 Previous evidence also confirms the biological functions of SIRT1 in cell apoptosis, cell viability, cellular stress, and metabolism.14 In addition, SIRT1 plays an important role in cancer cell biology, such as cancer cell apoptosis, differentiation, and proliferation.15 However, the expression and function of SIRT1 in different cancer types remain controversial. SIRT1 may be up- or down-regulated in cancer, and thus may act as an oncogene or exert anti-proliferative and pro-apoptotic effects.16–18 The phosphatidylinositol 3′-kinase (PI3K)/AKT pathway has been shown to be associated with cancer cell survival.19 Furthermore, Wang et al. showed that ACR treatment inhibited PI3K/AKT signaling in acute myeloid leukemia cells.20 Here, we found that ACR is involved in glioma cell growth and apoptosis via the SIRT1/PI3K/AKT signaling pathway.
In conclusion, the findings of our study revealed that ACR exerts anti-tumor activity against gliomas through modulating cancer cell growth and apoptosis. Our results showed that ACR induced ATM and CHK1 kinase phosphorylation in response to DNA damage in glioma cells. Additionally, we reported that ACR plays a role in glioma tumor biology through the SIRT1/PI3K/AKT signaling pathway.
2. Methods
2.1. Cell culture and treatment
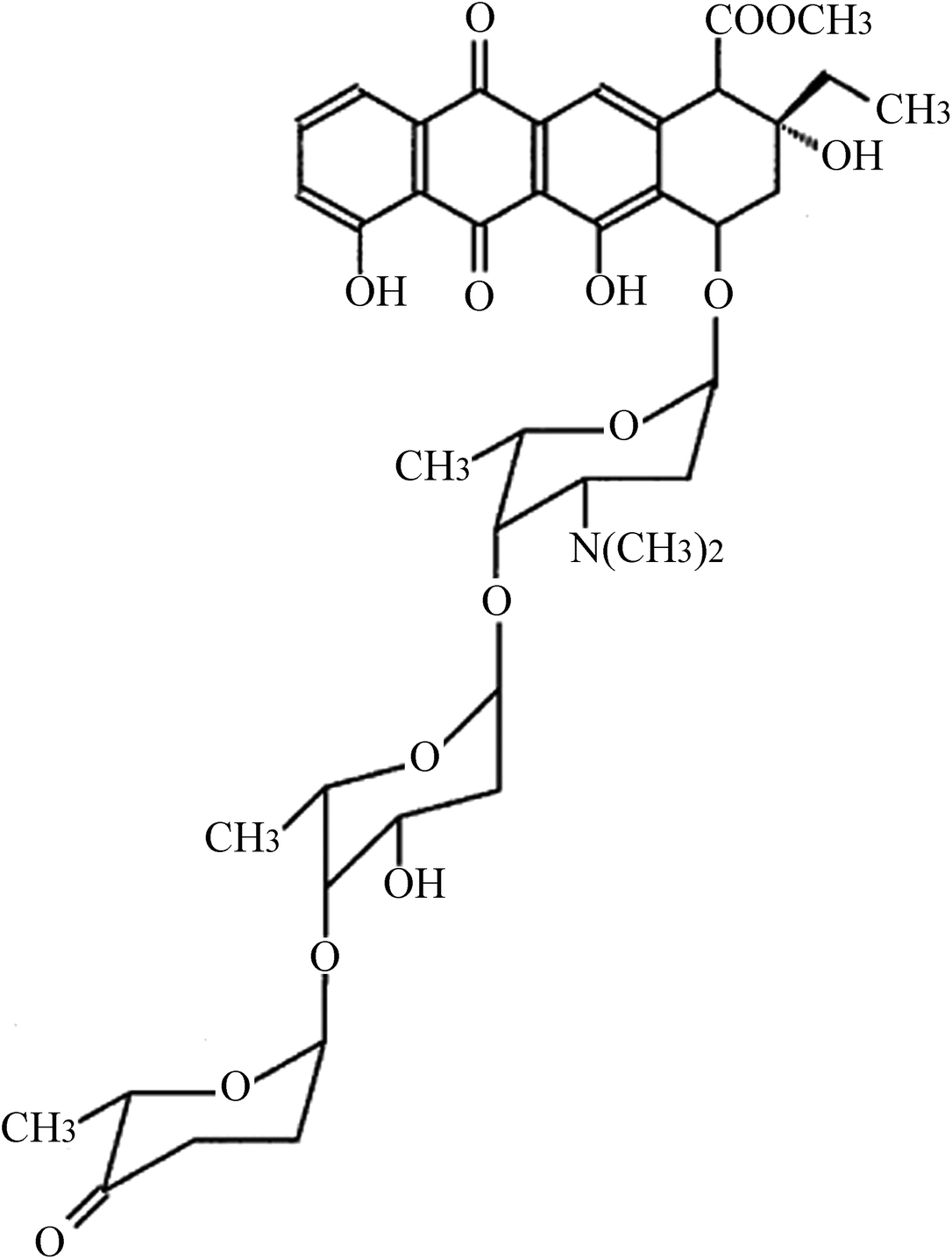
Glioma U87 and U251 cells were obtained from American Type Culture Collection (ATCC, Rockville, USA). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 100 μg mL−1 streptomycin, 100 U mL−1 penicillin, and 10% fetal bovine serum (Gibco, Life Technologies, Carlsbad, CA, USA) at 37 °C with 5% CO2. ACR (structure shown in Fig. 1) was purchased from Sigma-Aldrich (St. Louis, MO, USA; purity ≥ 98.5%) and dissolved in dimethyl sulfoxide. U87 and U251 cells were treated with 0, 1, 2, and 5 μM ACR for 1, 6, 12, and 24 h. Cell survival, proliferation, and apoptosis were then assayed.
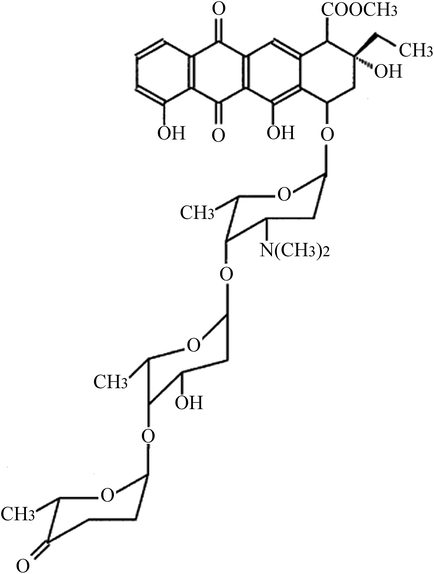 |
| | Fig. 1 Structure of ACR. | |
2.2. Cell transfection
U87 and U251 cells were transfected with control small interfering RNA (siRNA) vector, SIRT1 siRNA, control pcDNA3.1 vector or pcDNA3.1-AKT for 48 h using Lipofectamine 2000, according to the manufacturer's instructions. The transfection efficiencies of SIRT1 siRNA and AKT plasmid were about 83 and 80%, respectively. The transfected cells were then treated with 5 μM ACR for 12 h.
2.3. Cell survival
U87 and U251 cells were subjected to transfection and ACR treatment as described above. Cells were then cultured in DMEM in 96-well plates in triplicates. Cell survival was measured with the CellTiter-Blue kit (G8080; Promega, Madison, WI, USA) according to the manufacturer's instructions. Fluorescence was detected at 560 nm using a microplate reader.
2.4. Cell proliferation
Cell Counting Kit-8 (CCK-8) assay was used to assess U87 and U251 cell proliferation. Cells were plated in 96-well plates at a density of 1 × 104 cells per well. Cells were transfected and treated as described earlier. Next, CCK-8 solution (10 μL) was added to each well and incubated for 2 h. Absorbance was recorded at 450 nm using a microplate reader (Bio-Rad, Hercules, CA, USA).
2.5. Cell apoptosis assay
U87 and U251 cell apoptosis was measured using the cell death detection ELISA kit (Roche, Indianapolis, IN, USA). U87 and U251 cells were seeded in 96-well plates, and transfected and treated as described above. Cells were then treated with 100 μL of lysis buffer for 30 min, and the cell lysate was transferred to a streptavidin-coated plate. Absorbance was recorded at 405 nm to measure the amount of histone-coupled DNA fragments, which is an indicator of cell apoptosis.
2.6. Caspase-3 activity
Caspase-3/CPP32 colorimetric assay kit was used to determine caspase-3 activity in U87 and U251 cells according to the manufacturer's protocol. Briefly, U87 and U251 cells were transfected and treated as described above. Cells were then treated with reaction buffer containing 10 mM DTT, followed by treatment with 4 mM DEVD-pNA substrate solution (5 μL). Absorbance was measured at 405 nm.
2.7. Western blot
Cultured U87 and U251 cells were lysed in RIPA buffer. The extracted proteins were separated on 10% sodium dodecyl sulfate-polyacrylamide gels and then transferred onto PVDF membranes. Membranes were then incubated with the following primary antibodies: mouse monoclonal SIRT1 antibody (0.5 μg mL−1), rabbit monoclonal ATM (phospho S1981) antibody (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50000), rabbit polyclonal ATM antibody (1:10000), rabbit polyclonal CHK1 (phospho S345) antibody (1 μg mL−1), rabbit polyclonal CHK1 antibody (1:1000), rabbit polyclonal pAKT antibody (1:1000), rabbit polyclonal AKT antibody (1:500), and mouse monoclonal β-actin antibody (1:5000). Next, membranes were treated with the following secondary antibodies: horseradish peroxidase (HRP)-conjugated rabbit anti-mouse IgG (1:5000) and HRP-conjugated goat anti-rabbit IgG (1:10000). All antibodies were obtained from Abcam (Cambridge, MA, USA). The bands were detected using enhanced chemiluminescence reagent. Results were normalized to the loading control β-actin.
:50000), rabbit polyclonal ATM antibody (1:10000), rabbit polyclonal CHK1 (phospho S345) antibody (1 μg mL−1), rabbit polyclonal CHK1 antibody (1:1000), rabbit polyclonal pAKT antibody (1:1000), rabbit polyclonal AKT antibody (1:500), and mouse monoclonal β-actin antibody (1:5000). Next, membranes were treated with the following secondary antibodies: horseradish peroxidase (HRP)-conjugated rabbit anti-mouse IgG (1:5000) and HRP-conjugated goat anti-rabbit IgG (1:10000). All antibodies were obtained from Abcam (Cambridge, MA, USA). The bands were detected using enhanced chemiluminescence reagent. Results were normalized to the loading control β-actin.
2.8. Statistical analysis
Data are presented as the mean ± standard deviation (S.D). Three biological replicates were analyzed and three technical repeats were performed per sample. Statistical analyses were performed using GraphPad Prism 5.0 and SPSS 13.0 softwares. Significant differences between groups were evaluated by analysis of variance (ANOVA) followed by Bonferroni post-hoc test. A p-value of <0.05 was considered to be statistically significant.
3. Results
3.1. ACR reduced the survival and proliferation of U87 and U251 cells
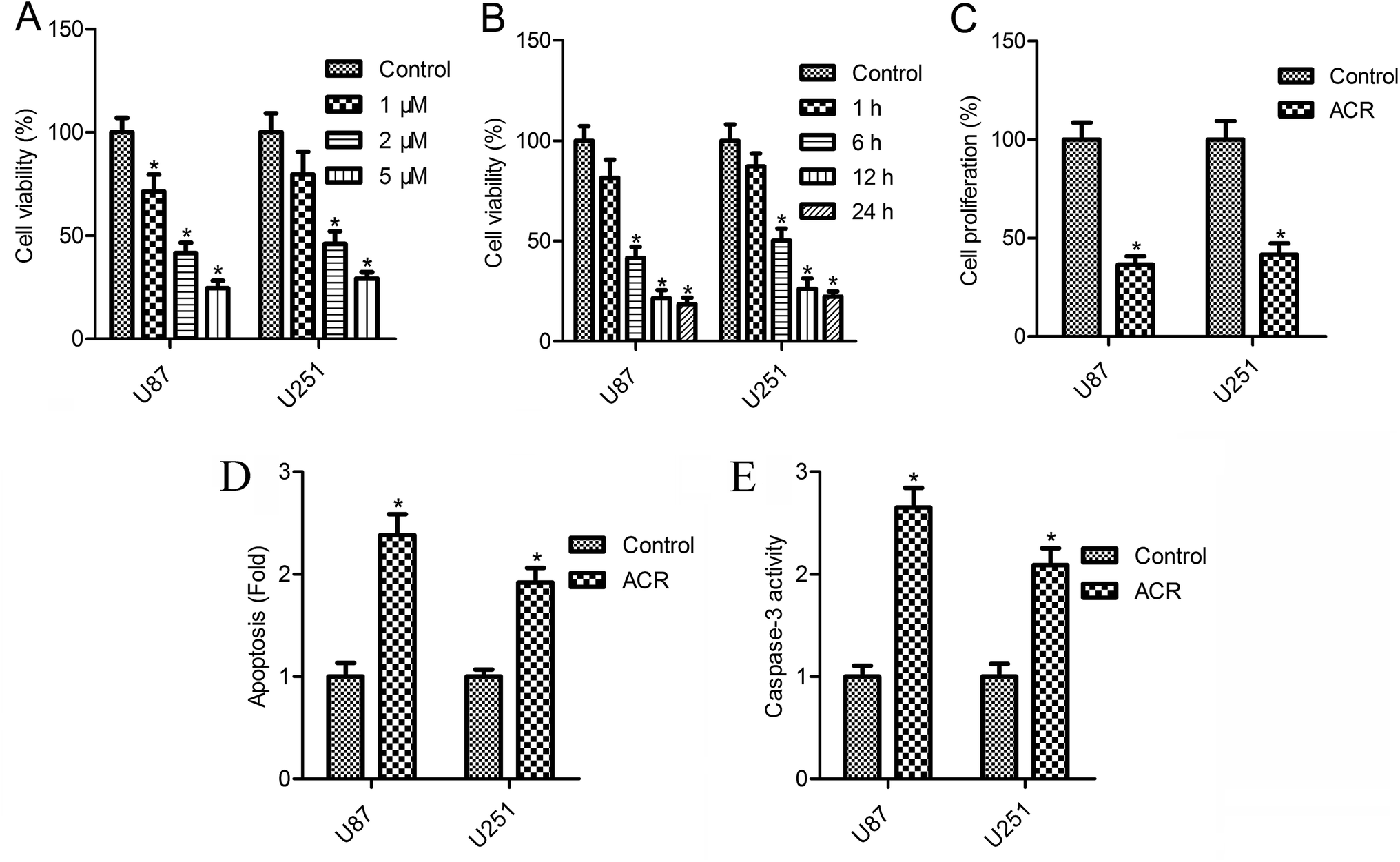
The CellTiter-Blue assay results showed that cell survival decreased gradually with the increasing concentrations of ACR (0–5 μM) (Fig. 2A and B). U87 cell survival was reduced to 71.3, 41.5, and 24.6%, while U251 cell survival was reduced to 79.5, 46.1, and 29.3% after 12 h of treatment with 1, 2, and 5 μM ACR, respectively (Fig. 2A). Treatment with 5 μM ACR for 1, 6, 12, and 24 h significantly decreased U87 and U251 cell survival (Fig. 2B). Furthermore, treatment with 5 μM ACR for 12 h markedly altered the proliferation of U87 and U251 cells (Fig. 2C). These results suggested that ACR suppressed glioma cell survival in a time- and dose-dependent manner.
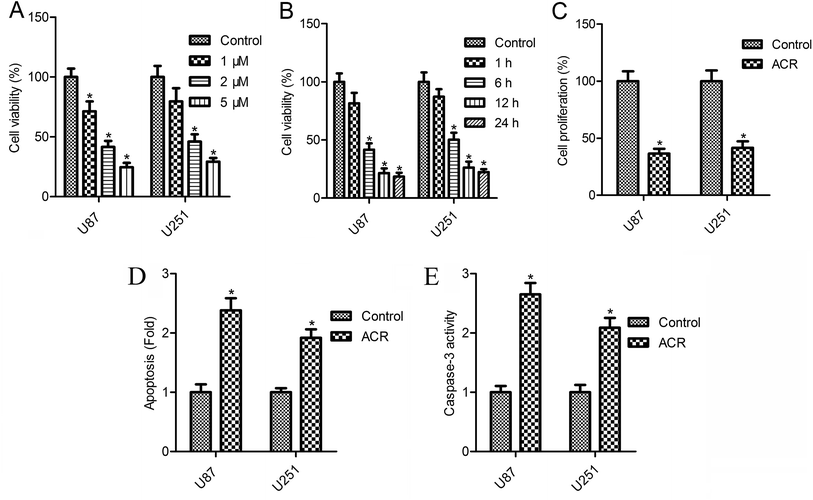 |
| | Fig. 2 ACR suppressed cell growth and promoted cell apoptosis in U87 and U251 cells. (A and B) The CellTiter-Blue kit was used to detect cell survival. (C) CCK-8 assay was used to measure cell proliferation. (D) Cell death detection ELISA assay was used to detect cell apoptosis. (E) Caspase-3/CPP32 colorimetric assay kit was used to measure caspase-3 activity. For cell survival analysis, U87 and U251 cells were treated with 1, 2, and 5 μM ACR for 1, 6, 12, and 24 h. For evaluation of cell proliferation, apoptosis, and caspase-3 activity, U87 and U251 cells were treated with 5 μM ACR for 12 h. Control cells were left untreated. *p < 0.05 compared with the control group. | |
3.2. ACR enhanced cell apoptosis and caspase-3 activity in U87 and U251 cells
Changes in cell apoptosis after ACR treatment are shown in Fig. 2D. Compared with control treatment, a significant increase in cell apoptosis was observed after ACR treatment in both the cell lines (Fig. 2D, p < 0.05). To further confirm the effect of ACR on glioma cell apoptosis, caspase-3 activity was measured. The results of caspase-3/CPP32 colorimetric assay showed that after 12 h of ACR treatment, the caspase-3 activity of U87 and U251 cells was dramatically increased (Fig. 2E, p < 0.05).
3.3. ACR promoted DNA damage in U87 and U251 cells
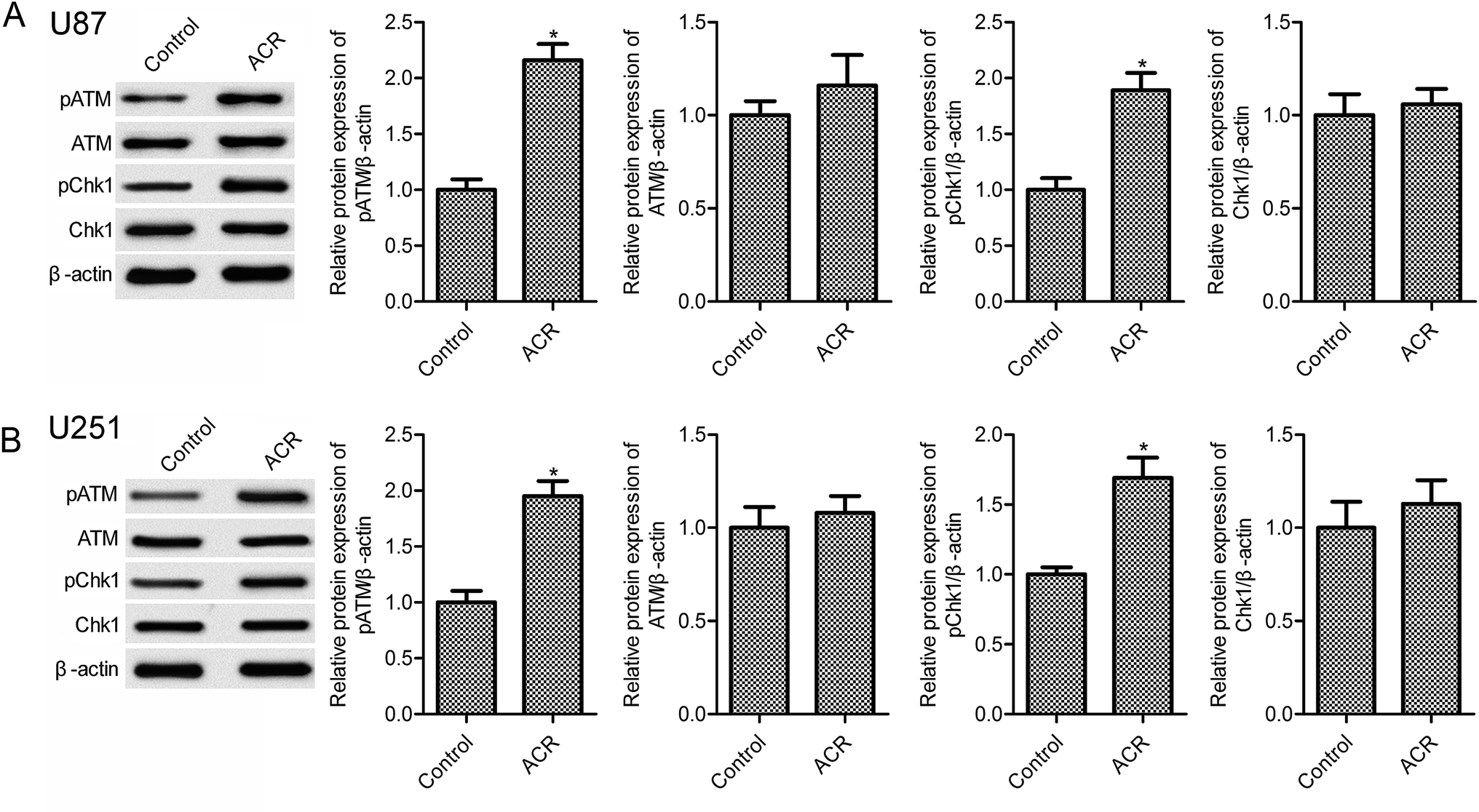
DNA damage has been associated with cell apoptosis. Previous evidence suggests that the DNA damage response pathway is involved in human tumors and may serve as a novel therapeutic target for cancer treatment.21 The sensor kinases ATM and CHK1 are markers of DNA damage response.22 In the present study, ACR treatment significantly increased pATM and pCHK1 protein expression in U87 and U251 cells compared with control treatment (Fig. 3, p < 0.05); however, no obvious changes in ATM and CHK1 expression were observed (Fig. 3, p > 0.05). These results suggested that ACR promoted DNA damage in glioma cells.
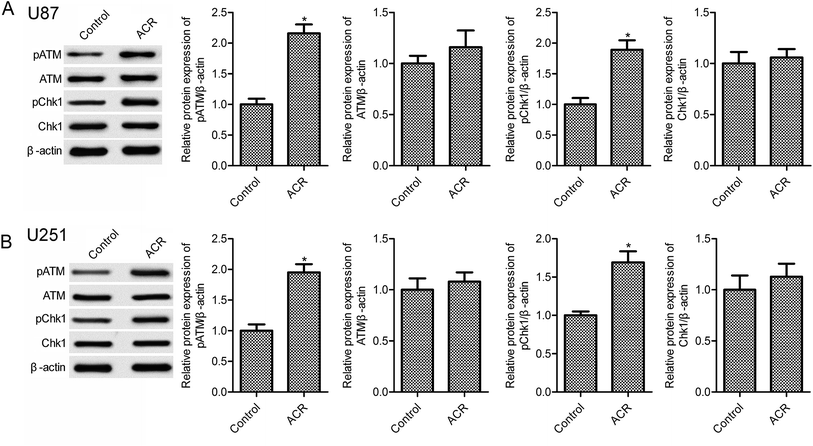 |
| | Fig. 3 ACR increased U87 and U251 cell DNA damage. Western blotting was used to measure the protein expression of pATM, pCHK1, ATM, and CHK1 in U87 (A) and U251 (B) cells. U87 and U251 cells were treated with 5 μM ACR for 12 h. Control cells were left untreated. *p < 0.05 compared with the control group. | |
3.4. ACR induced the activation of SIRT1/PI3K/AKT signaling
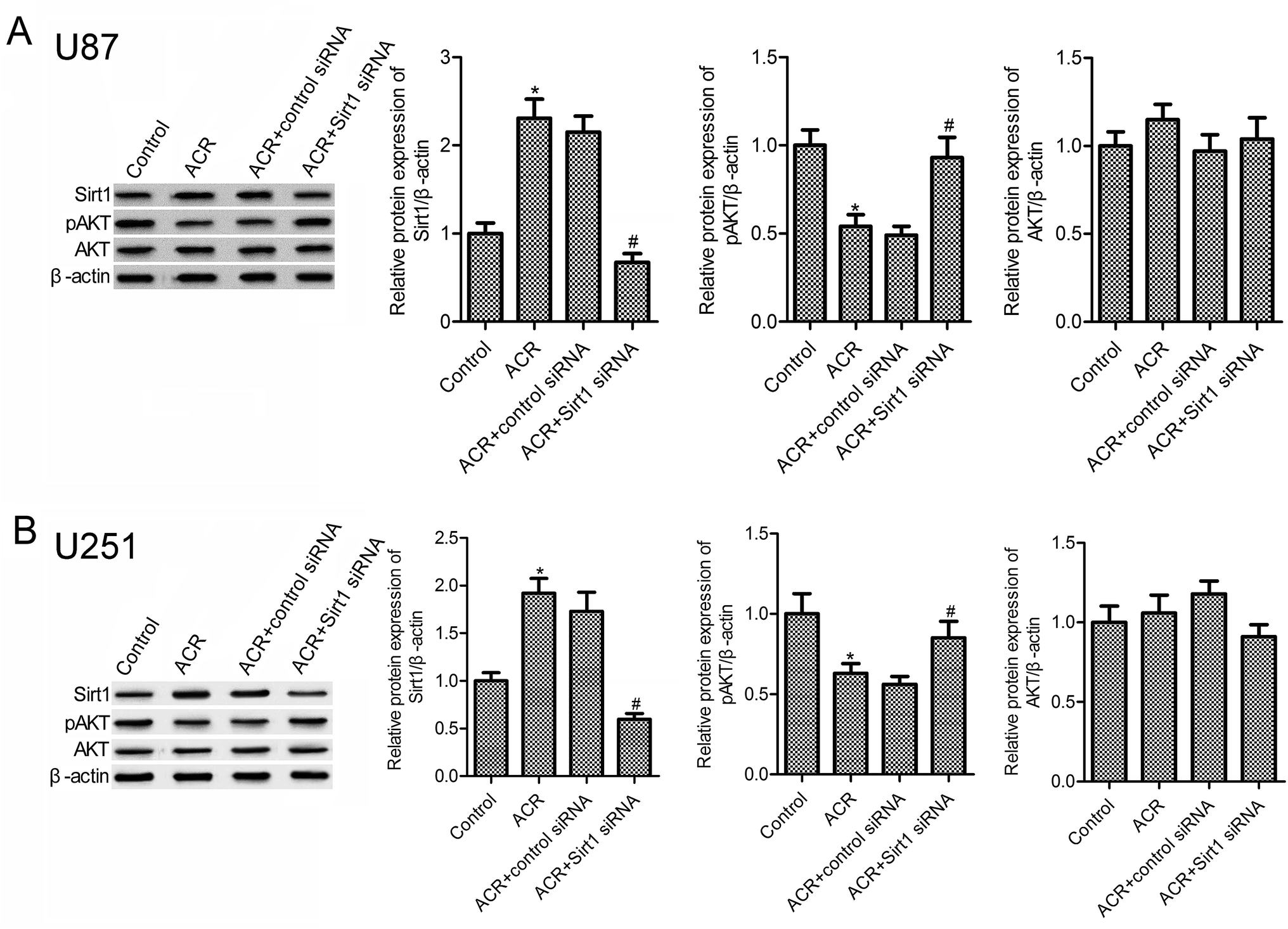
Previous evidences suggest that SIRT1 and PI3K/AKT pathways are important regulators of DNA damage response and cell apoptosis.23–25 Hence, we next examined the molecular mechanisms underlying the role of ACR in glioma cell growth, apoptosis, and DNA damage. Results showed that, compared to control treatment, ACR treatment markedly increased the protein expression of SIRT1 in U87 and U251 cells (Fig. 4A and B), but decreased that of pAKT (Fig. 4B). Further experiments showed that silencing of SIRT1 attenuated ACR-induced inhibition of PI3K/AKT signaling activation. These results suggested that ACR action is mediated via the SIRT1/PI3K/AKT signaling pathway, and SIRT1 silencing restrained the effects of ACR on PI3K/AKT signaling.
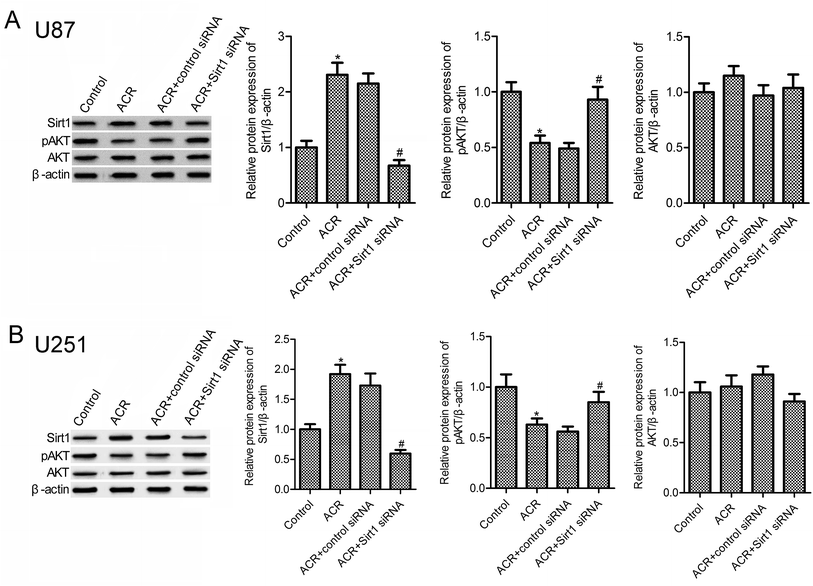 |
| | Fig. 4 ACR induced the activation of SIRT1/PI3K/AKT signaling pathway in U87 and U251 cells. Western blotting was used to measure the protein expression of SIRT1, pAKT, and AKT in U87 (A) and U251 (B) cells. U87 and U251 cells were transfected with control or SIRT1 siRNA for 48 h and then treated with 5 μM ACR for 12 h. Control cells were left untreated. *p < 0.05 compared with the control group, #p < 0.05 compared with the ACR group. | |
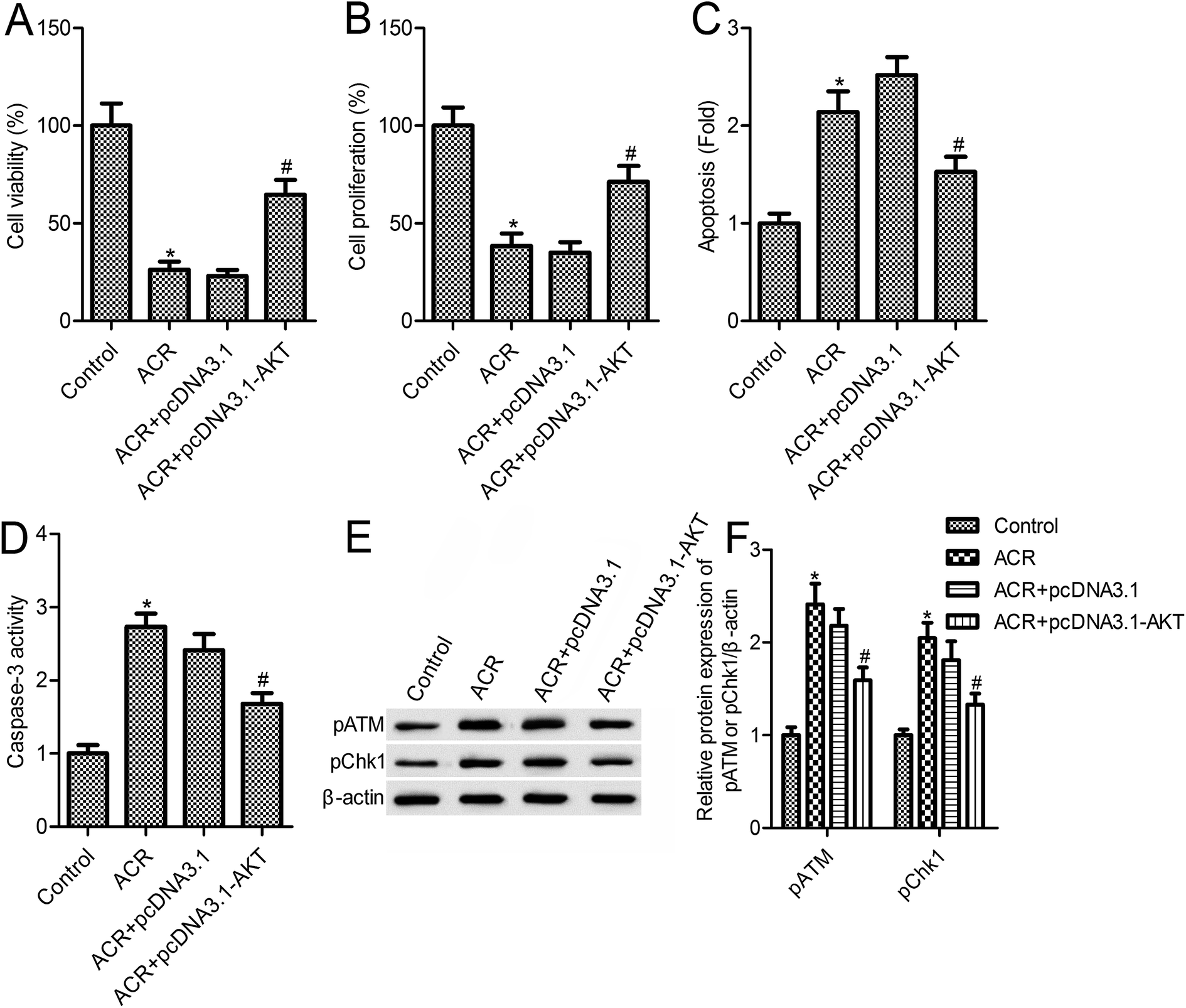
3.5. ACR regulated U87 cell growth and DNA damage via PI3K/AKT signaling
The above results suggested that ACR treatment induced the PI3K/AKT signaling pathway in U87 cells. To further confirm the role of PI3K/AKT signaling in mediating the action of ACR, U87 cells were transfected with pcDNA3.1-AKT vector for overexpression of PI3K/AKT (Fig. 5A). Results showed that, compared with the ACR treatment group, AKT overexpression increased cell survival and proliferation (Fig. 5B and C), and reduced apoptosis, caspase-3 activity, and DNA damage in U87 cells (Fig. 5D–F). These results suggested that AKT overexpression inhibited the ACR-induced increase in pATM and pCHK1 expression (Fig. 5F).
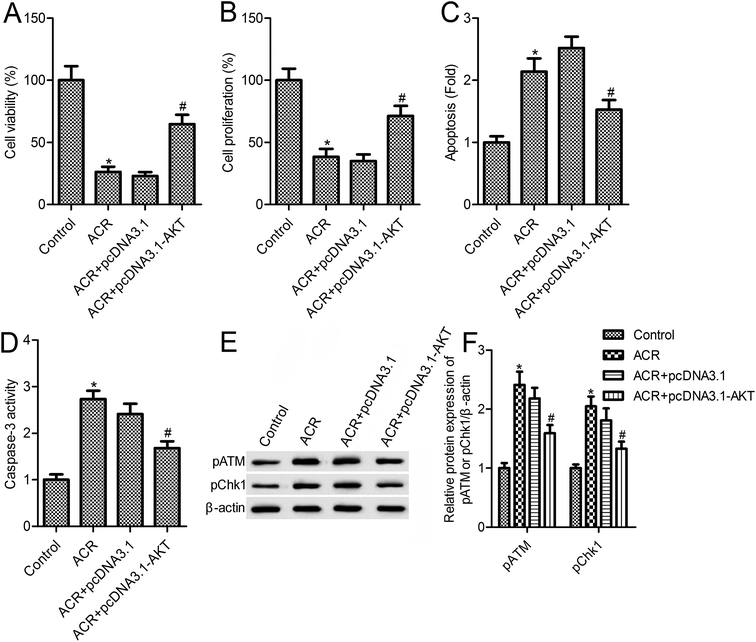 |
| | Fig. 5 Overexpression of PI3K/AKT reversed the effects of ACR in U87 cells. (A) Western blotting was used to measure the protein expression of pAKT and AKT. (B) CellTiter-Blue kit was used to detect cell survival. (C) CCK-8 assay was used to measure cell proliferation. (D) Cell death detection ELISA assay was used to detect cell apoptosis. (E) Caspase-3/CPP32 colorimetric assay kit was used to measure caspase-3 activity. (F) Western blotting was used to measure the protein expression of pATM and pCHK1. U87 cells were transfected with pcDNA3.1 or pcDNA3.1-AKT vector for 48 h and then treated with 5 μM ACR for 12 h. Control cells were left untreated. *p < 0.05 compared with the control group, #p < 0.05 compared with the ACR group. | |
4. Discussion
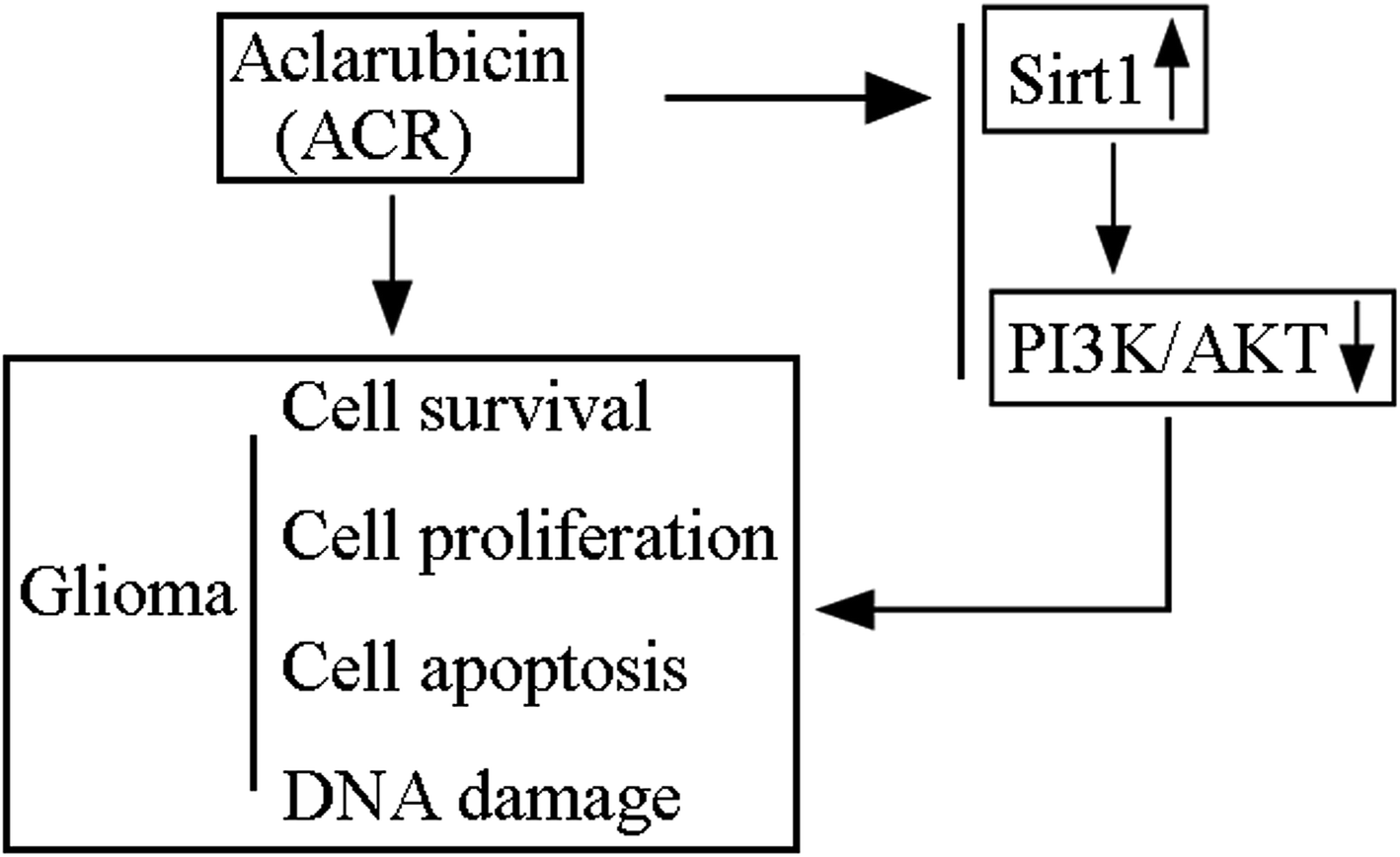
ACR has been shown to act as an effective anthracycline chemotherapeutic agent against solid tumors and hematologic cancers. It has lesser cytotoxic effects and can perturb chromatin function through DNA topoisomerase inhibition and histone eviction in the nucleus.26 ACR and DOX, both of which belong to the family of anthracyclines, have anti-cancer effects; however, ACR has been reported to be less cardiotoxic than DOX.27,28 A previous study showed that ACR exerted anti-tumor activity in rats intracranially implanted with glioma cells via significantly increasing rat survival time and inducing tumor cell apoptosis.12 Additionally, it showed that ACR-loaded cationic albumin-conjugated pegylated nanoparticles could penetrate the blood–brain barrier and target glioma cells, thus having a therapeutic effect.12 However, the function and significance of ACR in glioma cell growth and apoptosis remain unclear. In this regard, the present study is the first to provide evidence and basic insights into the function and mechanism of ACR in glioma cells. Our results showed that ACR administration significantly reduced glioma cell survival and proliferation, and increased glioma cell apoptosis. Moreover, our findings confirmed that ACR treatment enhances DNA damage in glioma cells (Fig. 6).
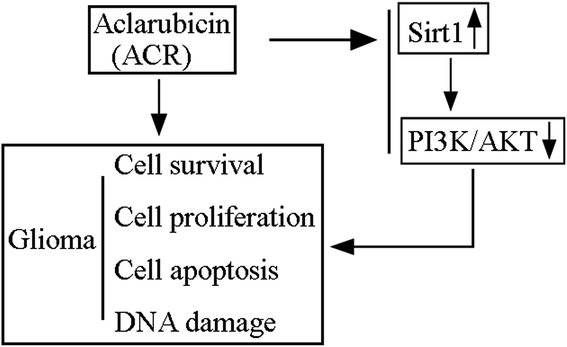 |
| | Fig. 6 Schematic model of ACR-mediated regulation of glioma cell growth and DNA damage via the SIRT1/PI3K/AKT signaling pathway. | |
DNA is susceptible to sustained endogenous and exogenous (genotoxic cancer therapeutics, anti-inflammatory drugs, and dietary and environmental carcinogens) damage, and maintaining the genome integrity of cancer cells requires the coordinated activity of DNA repair signals under normal cellular conditions.29,30 A constitutive level of endogenous DNA damage, in part, contributes to genomic instability, and is thus involved in tumorigenesis.31 Multiple evidences have suggested that cellular DNA damage is associated with cell apoptosis.32,33 A previous study reported that anthracyclines exert anti-cancer activity via inducing DNA damage.34 Furthermore, due to its characteristic DNA-binding property, ACR was shown to induce DNA damage through inhibiting topoisomerase I and II.35,36
The DNA damage response pathway consists of effectors, sensors, and transducers.37,38 Several small molecule kinases, including ATR, WEE1, DNA-PKcs, CHEK2, CHEK1, and ATM are involved in DNA damage response. These molecules have been detected in various DNA repair-proficient and-deficient tumor cell lines.39,40 A recent study showed that ACR induced DNA damage and apoptosis in leukemia cells in the presence of H2O2 and Cu(I).41 In the current study, we found that ACR treatment induced the expression of the DNA damage response biomarkers, pATM and pCHK1, in both U87 and U251 cells. Hence, we believe that ACR is involved in DNA damage in glioma cells. SIRT1 has been shown to play a role in a variety of cancers, including prostate cancer, hepatocellular carcinoma,15 lung cancer,23 and glioma.13 However, it plays different roles in different cancers, and may act as a tumor promoter or tumor suppressor.42 Previous evidence suggests that SIRT1 silencing contributes to tumor suppression in various cancers, such as breast cancer43 and colon cancer.44 Qu et al. showed that SIRT1 promoted proliferation and inhibited apoptosis in glioma cells, thereby promoting glioma tumorigenesis.13 Moreover, SIRT1, which catalyzes the deacetylation of non-histone proteins, has been shown to regulate mammalian cell survival under oxidative stress and DNA damage conditions.45 Our study demonstrated that SIRT1 was involved in ACR-induced DNA damage in U87 and U251 cells. The tumor growth-associated signaling pathways, like Wnt and PI3K/AKT pathways have been previously implicated in regulating the action of SIRT1 in tumor progression.19,20 In this present study, we found that ACR treatment suppressed the activation of the PI3K/AKT signaling pathway and increased SIRT1 expression.
5. Conclusion
In summary, our findings demonstrated that ACR treatment inhibited glioma cell survival and proliferation, and increased glioma cell apoptosis, caspase-3 activity, and DNA damage. Hence, we propose that ACR exerts anti-survival, anti-proliferation, pro-apoptosis, and pro-DNA damage effects in glioma cells. Mechanistically, our data revealed that ACR suppressed glioma cell growth via the SIRT1/PI3K/AKT signaling pathway. However, further in vivo animal studies are required to elucidate the specific role and mechanism of ACR in glioma.
Conflicts of interest
There is no conflict of interest to be declared by the authors.
Abbreviations
| ACR | Aclarubicin |
| NAD | Nicotinamide-adenine dinucleotide |
| SIRT1 | Sirtuin 1 |
| PI3K | Phosphatidylinositol 3′-kinase |
| CCK-8 | Cell counting kit-8 |
References
- R. Chen, M. Smith-Cohn, A. L. Cohen and H. Colman, Neurotherapeutics, 2017, 14, 284–297 CrossRef PubMed.
- N. Sharma, S. Saxena, I. Agrawal, S. Singh, V. Srinivasan, S. Arvind, S. Epari, S. Paul and S. Jha, Sci. Rep., 2019, 9, 8480 CrossRef PubMed.
- M. Weller, E. Le Rhun, M. Preusser, J. C. Tonn and P. Roth, ESMO Open, 2019, 4, e000520, DOI:10.1136/esmoopen-2019-000520.
- R. Stupp, M. E. Hegi, W. P. Mason, M. J. van den Bent, M. J. Taphoorn, R. C. Janzer, S. K. Ludwin, A. Allgeier, B. Fisher, K. Belanger, P. Hau, A. A. Brandes, J. Gijtenbeek, C. Marosi, C. J. Vecht, K. Mokhtari, P. Wesseling, S. Villa, E. Eisenhauer, T. Gorlia, M. Weller, D. Lacombe, J. G. Cairncross and R. O. Mirimanoff, Lancet Oncol., 2009, 10, 459–466 CrossRef CAS PubMed.
- L. M. DeAngelis, N. Engl. J. Med., 2005, 352, 1036–1038 CrossRef CAS PubMed.
- G. Minotti, P. Menna, E. Salvatorelli, G. Cairo and L. Gianni, Pharmacol. Rev., 2004, 56, 185–229 CrossRef CAS PubMed.
- A. Rogalska, A. Koceva-Chyla and Z. Jozwiak, Chem.-Biol. Interact., 2008, 176, 58–70 CrossRef CAS PubMed.
- T. Oki, Y. Matsuzawa, A. Yoshimoto, K. Numata and I. Kitamura, J. Antibiot., 1975, 28, 830–834 CrossRef CAS PubMed.
- N. Hajji, S. Mateos, N. Pastor, I. Dominguez and F. Cortes, Mutat. Res., 2005, 583, 26–35 CAS.
- J. L. Nitiss, P. Pourquier and Y. Pommier, Cancer Res., 1997, 57, 4564–4569 CAS.
- P. Mayer, M. C. Gorisse, Y. Carpentier and B. Desoize, Bull. Cancer, 1994, 81, 670–676 CAS.
- W. Lu, J. Wan, Q. Zhang, Z. She and X. Jiang, Int. J. Cancer, 2007, 120, 420–431 CrossRef CAS PubMed.
- Y. Qu, J. Zhang, S. Wu, B. Li, S. Liu and J. Cheng, Neurosci. Lett., 2012, 525, 168–172 CrossRef CAS PubMed.
- J. Luo, A. Y. Nikolaev, S. Imai, D. Chen, F. Su, A. Shiloh, L. Guarente and W. Gu, Cell, 2001, 107, 137–148 CrossRef CAS PubMed.
- H. N. Choi, J. S. Bae, U. Jamiyandorj, S. J. Noh, H. S. Park, K. Y. Jang, M. J. Chung, M. J. Kang, D. G. Lee and W. S. Moon, Oncol. Rep., 2011, 26, 503–510 CAS.
- K. Pruitt, R. L. Zinn, J. E. Ohm, K. M. McGarvey, S. H. Kang, D. N. Watkins, J. G. Herman and S. B. Baylin, PLoS Genet., 2006, 2, e40, DOI:10.1371/journal.pgen.0020040.
- F. Yeung, J. E. Hoberg, C. S. Ramsey, M. D. Keller, D. R. Jones, R. A. Frye and M. W. Mayo, EMBO J., 2004, 23, 2369–2380 CrossRef CAS PubMed.
- K. F. Chua, R. Mostoslavsky, D. B. Lombard, W. W. Pang, S. Saito, S. Franco, D. Kaushal, H. L. Cheng, M. R. Fischer, N. Stokes, M. M. Murphy, E. Appella and F. W. Alt, Cell Metab., 2005, 2, 67–76 CrossRef CAS PubMed.
- G. Song, G. Ouyang and S. Bao, J. Cell. Mol. Med., 2005, 9, 59–71 CrossRef CAS PubMed.
- L. Wang, L. S. You, W. M. Ni, Q. L. Ma, Y. Tong, L. P. Mao, J. J. Qian and J. Jin, Leuk. Res., 2013, 37, 1329–1340 CrossRef CAS PubMed.
- L. Carrassa and G. Damia, Cancer Treat. Rev., 2017, 60, 139–151 CrossRef CAS PubMed.
- I. G. Cannell, Y. W. Kong, S. J. Johnston, M. L. Chen, H. M. Collins, H. C. Dobbyn, A. Elia, T. R. Kress, M. Dickens, M. J. Clemens, D. M. Heery, M. Gaestel, M. Eilers, A. E. Willis and M. Bushell, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 5375–5380 CrossRef CAS PubMed.
- G. Chen, B. Zhang, H. Xu, Y. Sun, Y. Shi, Y. Luo, H. Jia and F. Wang, Oncogene, 2017, 36, 6863–6872 CrossRef CAS PubMed.
- A. Juvekar, H. Hu, S. Yadegarynia, C. A. Lyssiotis, S. Ullas, E. C. Lien, G. Bellinger, J. Son, R. C. Hok, P. Seth, M. B. Daly, B. Kim, R. Scully, J. M. Asara, L. C. Cantley and G. M. Wulf, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E4338–E4347 CrossRef CAS PubMed.
- B. Zhang, Y. Liu, Y. Li, X. Zhe, S. Zhang and L. Zhang, Mol. Med. Rep., 2018, 17, 2757–2763 CAS.
- H. Iihoshi, T. Ishihara, S. Kuroda, N. Ishihara and H. Saitoh, Toxicol. Lett., 2017, 277, 109–114 CrossRef CAS PubMed.
- S. A. Mortensen, Eur. J. Haematol., Suppl., 1987, 47, 21–31 CAS.
- R. P. Warrell Jr, Drugs Exp. Clin. Res., 1986, 12, 275–282 CAS.
- J. H. Hoeijmakers, N. Engl. J. Med., 2009, 361, 1475–1485 CrossRef CAS PubMed.
- W. P. Roos, A. D. Thomas and B. Kaina, Nat. Rev. Cancer, 2016, 16, 20–33 CrossRef CAS PubMed.
- L. Fu and C. Xie, Helicobacter, 2019, e12631, DOI:10.1111/hel.12631.
- J. Y. Seo, D. Y. Kim, S. H. Kim, H. J. Kim, H. G. Ryu, J. Lee, K. H. Lee and K. T. Kim, Oncotarget, 2017, 8, 51108–51122 Search PubMed.
- A. R. Delbridge, S. Grabow and A. Strasser, Cell Death Differ., 2017, 24, 1987–1988 CrossRef PubMed.
- H. Mizutani, A. Nishimoto, S. Hotta, K. Ikemura, M. Imai, D. Miyazawa, K. Ohta, Y. Ikeda, T. Maeda, M. Yoshikawa, Y. Hiraku and S. Kawanishi, Anticancer Res., 2018, 38, 2643–2648 CAS.
- A. K. Larsen, A. E. Escargueil and A. Skladanowski, Pharmacol. Ther., 2003, 99, 167–181 CrossRef CAS PubMed.
- S. Ando, K. Kamiya, T. Yoshimura, H. Tsutani, T. Ueda, M. Uchida, T. Nakamura and H. Uchino, Anticancer Res., 1988, 8, 409–415 CAS.
- B. B. Zhou and S. J. Elledge, Nature, 2000, 408, 433–439 CrossRef CAS PubMed.
- A. Sancar, L. A. Lindsey-Boltz, K. Unsal-Kacmaz and S. Linn, Annu. Rev. Biochem., 2004, 73, 39–85 CrossRef CAS PubMed.
- E. H. Stover, P. A. Konstantinopoulos, U. A. Matulonis and E. M. Swisher, Clin. Cancer Res., 2016, 22, 5651–5660 CrossRef CAS PubMed.
- J. S. Brown, B. O'Carrigan, S. P. Jackson and T. A. Yap, Cancer Discovery, 2017, 7, 20–37 CrossRef CAS PubMed.
- H. Mizutani, Y. Hayashi, M. Hashimoto, M. Imai, Y. Ichimaru, Y. Kitamura, K. Ikemura, D. Miyazawa, K. Ohta, Y. Ikeda, T. Maeda, M. Yoshikawa, Y. Hiraku and S. Kawanishi, Anticancer Res., 2019, 39, 3443–3451 CrossRef PubMed.
- Z. Lin and D. Fang, Genes Cancer, 2013, 4, 97–104 CrossRef CAS PubMed.
- X. Jin, Y. Wei, F. Xu, M. Zhao, K. Dai, R. Shen, S. Yang and N. Zhang, J. Cancer, 2018, 9, 2012 CrossRef PubMed.
- A. Ghosh, A. Sengupta, G. P. K. Seerapu, A. Nakhi, E. V. V. Shivaji Ramarao, N. Bung, G. Bulusu, M. Pal and D. Haldar, Biochem. Biophys. Res. Commun., 2017, 488, 562–569 CrossRef CAS PubMed.
- W. Y. Chen, D. H. Wang, R. C. Yen, J. Luo, W. Gu and S. B. Baylin, Cell, 2005, 123, 437–448 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Xiao-bing Chen†
* and
Xiao-bing Chen†