
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRu(II)–N-heterocyclic carbene complexes: synthesis, characterization, transfer hydrogenation reactions and biological determination†
Lamia Boubakria,
A. Chakchouk-Mtibaab,
Abdullah S. Al-Ayedc,
L. Mansourd,
Nael Abutahad,
Abdel Halim Harrathd,
L. Melloulib,
I. Özdemir e,
S. Yasare and
Naceur Hamdi*ac
e,
S. Yasare and
Naceur Hamdi*ac
aResearch Laboratory of Environmental Sciences and Technologies (LR16ES09), Higher Institute of Environmental Sciences and Technology, University of Carthage, Hammam-Lif, Tunisia. E-mail: naceur.hamdi@isste.rnu.tn; Tel: +21698503980
bLaboratory of Microorganisms and Biomolecules, Center of Biotechnolgy of Sfax, Road of Sidi Mansour, Km 6 B.P. 1117, 3018, Sfax, Tunisia
cChemistry Department, College of Science and Arts, Qassim University, Al-Rass, Kingdom of Saudi Arabia
dZoology Department, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia
eİnönü University, Faculty of Science and Art, Department of Chemistry, Malatya, Turkey
First published on 25th October 2019
Abstract
A series of ruthenium(II) complexes with N-heterocyclic carbene ligands were successfully synthesized by transmetalation reactions between silver(I) N-heterocyclic carbene complexes and [RuCl2(p-cymene)]2 in dichloromethane under Ar conditions. All new compounds were characterized by spectroscopic and analytical methods. These ruthenium(II)–NHC complexes were found to be efficient precatalysts for the transfer hydrogenation of ketones by using 2-propanol as the hydrogen source in the presence of KOH as a co-catalyst. The antibacterial activity of ruthenium N-heterocyclic carbene complexes 3a–f was measured by disc diffusion method against Gram positive and Gram-negative bacteria. Compounds 3d exhibited potential antibacterial activity against five bacterial species among the six used as indicator cells. The product 3e inhibits the growth of all the six tested microorganisms. Moreover, the antioxidant activity determination of these complexes 3a–f, using 2,2-diphenyl-1-picrylhydrazyl (DPPH) and 2,2′-azinobis-3-ethylbenzothiazoline-6-sulphonic acid (ABTS) as reagent, showed that compounds 3b and 3d possess DPPH and ABTS antiradical activities. From a concentration of 1 mg ml−1, these two complexes presented a similar scavenging activity to that of the two used controls gallic acid (GA) and butylated hydroxytoluene (BHT). From a concentration of 10 mg ml−1, the percentage inhibition of complexes 3b and 3d was respectively 70% and 90%. In addition, these two Ru–NHC complexes exhibited antifungal activity against Candida albicans. Investigation of the anti-acetylcholinesterase activity of the studied complexes showed that compounds 3a, 3b, 3d and 3e exhibited good activity at 100 μg ml−1 and product 3d is the most active. In a cytotoxicity study the complexes 3 were evaluated against two human cancer cell lines MDA-MB-231 and MCF-7. Both 3d and 3e complexes were found to be active against the tested cell lines showing comparable activity with examples in the literature.
1. Introduction
Transfer hydrogenation, a potentially useful protocol for the reduction of ketones and aldehydes to their corresponding alcohols, has been extensively studied.1 A large number of studies have appeared with transition metal-catalyzed enantioselective hydrogenation of ketones using 2-propanol or formic acid under basic conditions,2,3 because the method has advantages, such as low cost, ease of handling, and high solubility of 2-propanol or formic acid as a hydrogen-donor reagent. The most active and versatile homogeneous catalytic sec-alcohol/ketone oxidation/reduction systems are the metal–ligand bifunctional ruthenium complexes developed by Noyori and co-workers.4–6 A variety of related ligands and transition metal complex catalysts have been developed for this purpose.7,8 One of these ruthenium complexes is Ru(NHC) (NHC: N-heterocyclic carbene). N-heterocyclic carbene ligands appear particularly suited to this purpose, in that their ruthenium(II) complexes possess high thermal and hydrolytic stability.9Ligand effects are extremely important in homogeneous catalysis by metal complexes. Carbenes are both reactive intermediates and ligands in catalysis. After the first stable N-heterocyclic carbene ligands were reported by Arduengo et al., N-heterocyclic carbenes (NHCs) have been indispensable ligands for transition metals and homogeneous catalysis.10–12 Due to their topological and electronic versatility, N-heterocyclic carbene-based ligands have widespread applications not only in organometallic chemistry but also in industrial applications of homogeneous catalysis. Strong σ-donation from NHCs increases the stability of the complexes and gives complexes with advantageous properties in catalytic reactions.13–15 The first catalytic applications of NHC complexes were reported by Herrmann in 1995, together with the recognition that NHCs are excellent ligands for many homogeneous catalysts.11 Different NHC complexes and their catalytic applications have been reported by many research groups.16–24 Continued research has focused on synthesis of new functional NHC complexes. Functionality of the complexes depends on the steric and electronic effect of the ligands on the metal center. Ru–NHC complexes can be functionalized by N-substituted NHC ligands. In this context, our research group has focused on synthesis, characterization, and catalytic activity of functional N-heterocyclic carbene ligands and their metal complexes.25–28
2. Results and discussion
2.1. Preparation of Ru–NHC complexes (3a–f)
Silver complexes of N-heterocyclic carbenes were prepared by the addition of Ag2O to 5,6-dimethylbenzimidazolium salts solutions (1a–f) in CH2Cl2 under exclusion of light at room temperature (Scheme 1 and Fig. 1). White solid (in 75–90% yield) were isolated.29 When the white solid were left in air, they began to slowly turn black. Low field signals attributed to acidic NCHN protons were not observed in the 1H NMR spectra of the new Ag–NHC complexes (2a–f). This observation confirmed that the 5,6-dimethylbenzimidazolium salts were deprotonated. However, coordination of N-heterocyclic carbene ligands to the metal center resulted in low field resonances for carbene carbon.29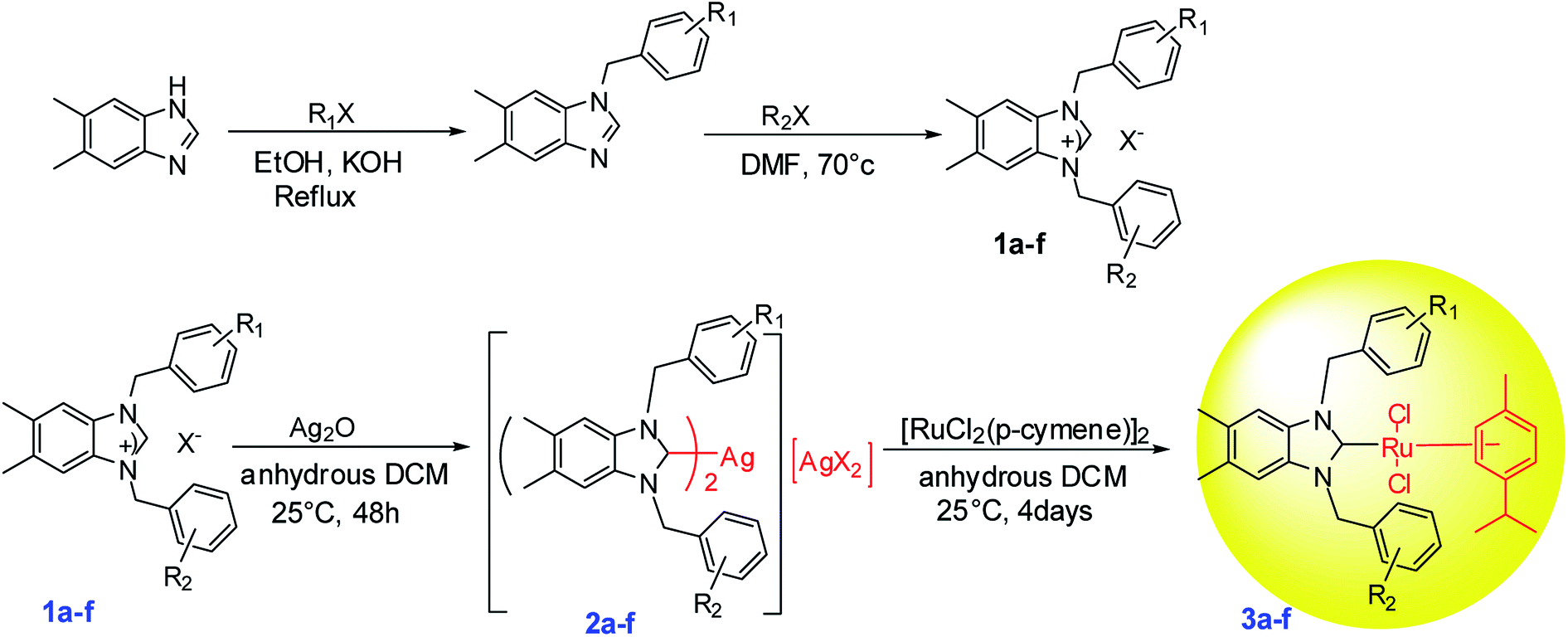 | ||
| Scheme 1 Synthesis of ruthenium–NHC carbene complexes (3a–f). | ||
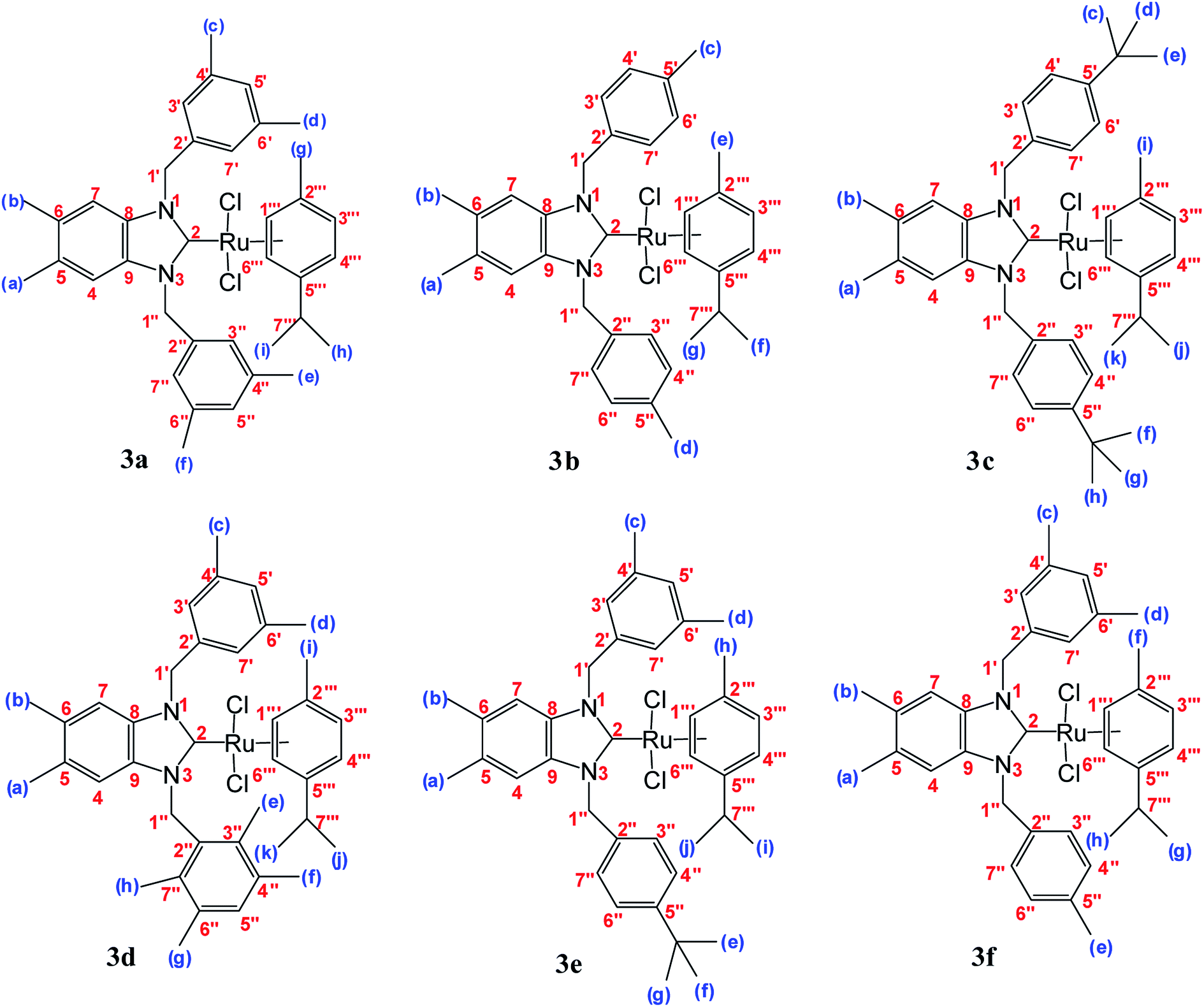 | ||
| Fig. 1 Structure of ruthenium–NHC carbene complexes 3(a–f). | ||
All Ag–NHC complexes are widely used for the next reaction as transfer compounds. To show the feasibility of this approach for 2a–f, a series of Ru(II)–NHC complexes were synthesized in good yield as stable red solids. 3a–f were obtained by reaction of [RuCl2(p-cymene)]2 with a solution of 2a–f in the dark at room temperature (Scheme 1). The air and moisture-stable ruthenium N-heterocyclic carbene complexes (3a–f) isolated in high purity after recrystallization with DCM/ether, were soluble in solvents such as dichloromethane, chloroform, toluene, and tetrahydrofurane and insoluble in nonpolar solvents (Fig. 1).30
The ruthenium–NHC complexes 3a–f were characterized by NMR (1H and 13C), IR, and DART-TOF mass spectrometry. The 1H-NMR spectra of complex 3a was recorded in CDCl3 at room temperature. Here, the proton H7′′′ of the isopropyl group of p-cymene was seen as a multiplet at 2.62 ppm (Fig. 2).
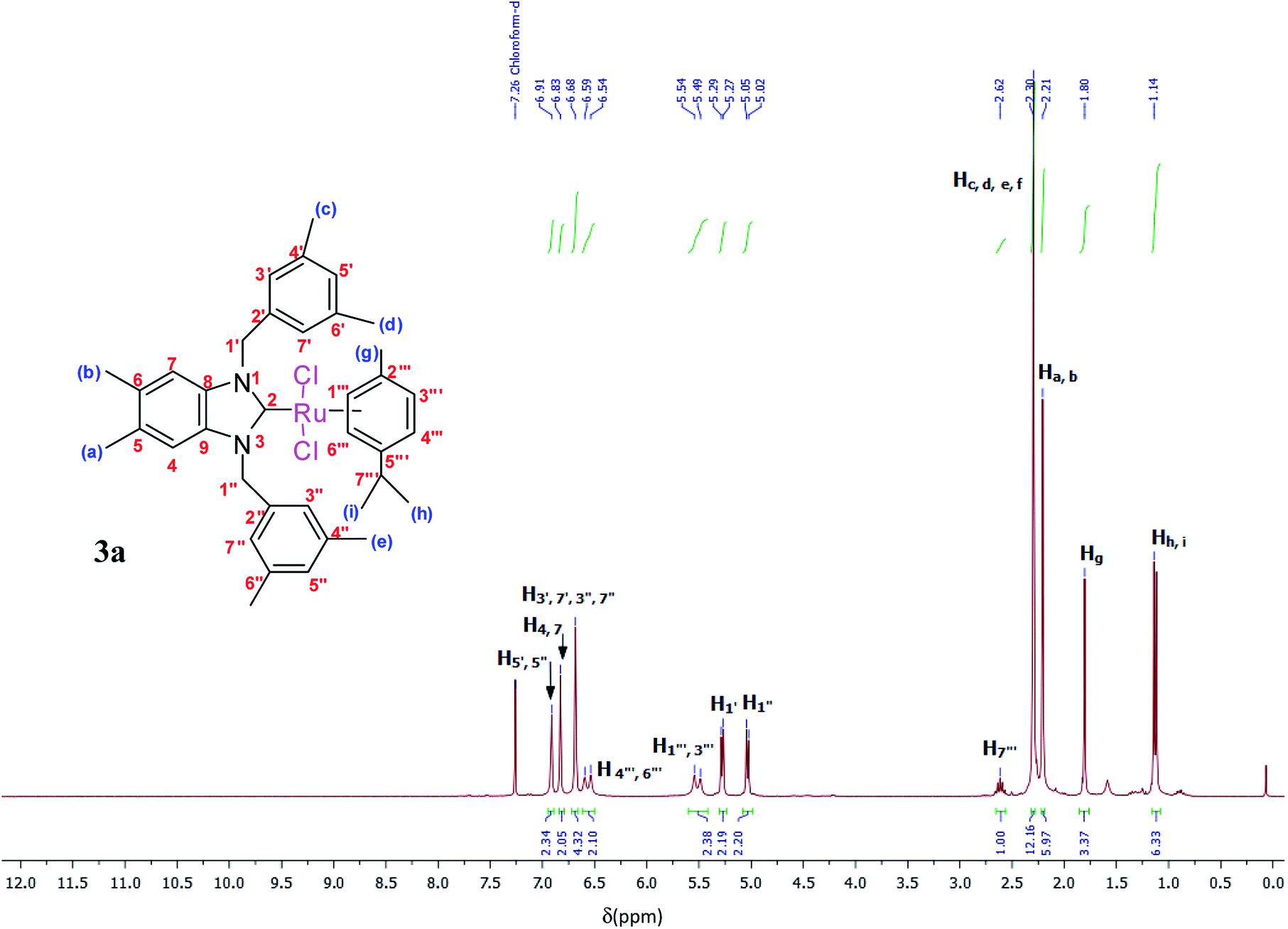 | ||
| Fig. 2 1H NMR spectra of Ru(II)–N-heterocyclic carbene complexes 3a (CDCl3, 300 MHz). | ||
The aromatic protons appeared between 5.54 and 6.91 ppm as a multiplet while methylic protons appeared between 1.14 and 2.30 ppm as singlets. In the 1H-NMR spectra of 3a, (NCH2) was resonated at 5.29 and 5.05 ppm as a doublet.
The spectra of these products in CDCl3 supported the successful formation of the ruthenium complexes because of the absence of NCHN signal proton in a downfield for 2–3.
The 1H and 13C NMR spectra of these new complexes (3a–f) are consistent with the proposed formula. 13C{1H} NMR spectra prove an increasing downfield shift of the characteristic peak of the carbenic carbon resonance from 2a–f to 3a–f: for example, the 13C{1H}N–C–N shifts of 3a displays as a singlet at 188.8 ppm which confirmed the transmetallation between silver and ruthenium (Fig. 3).31
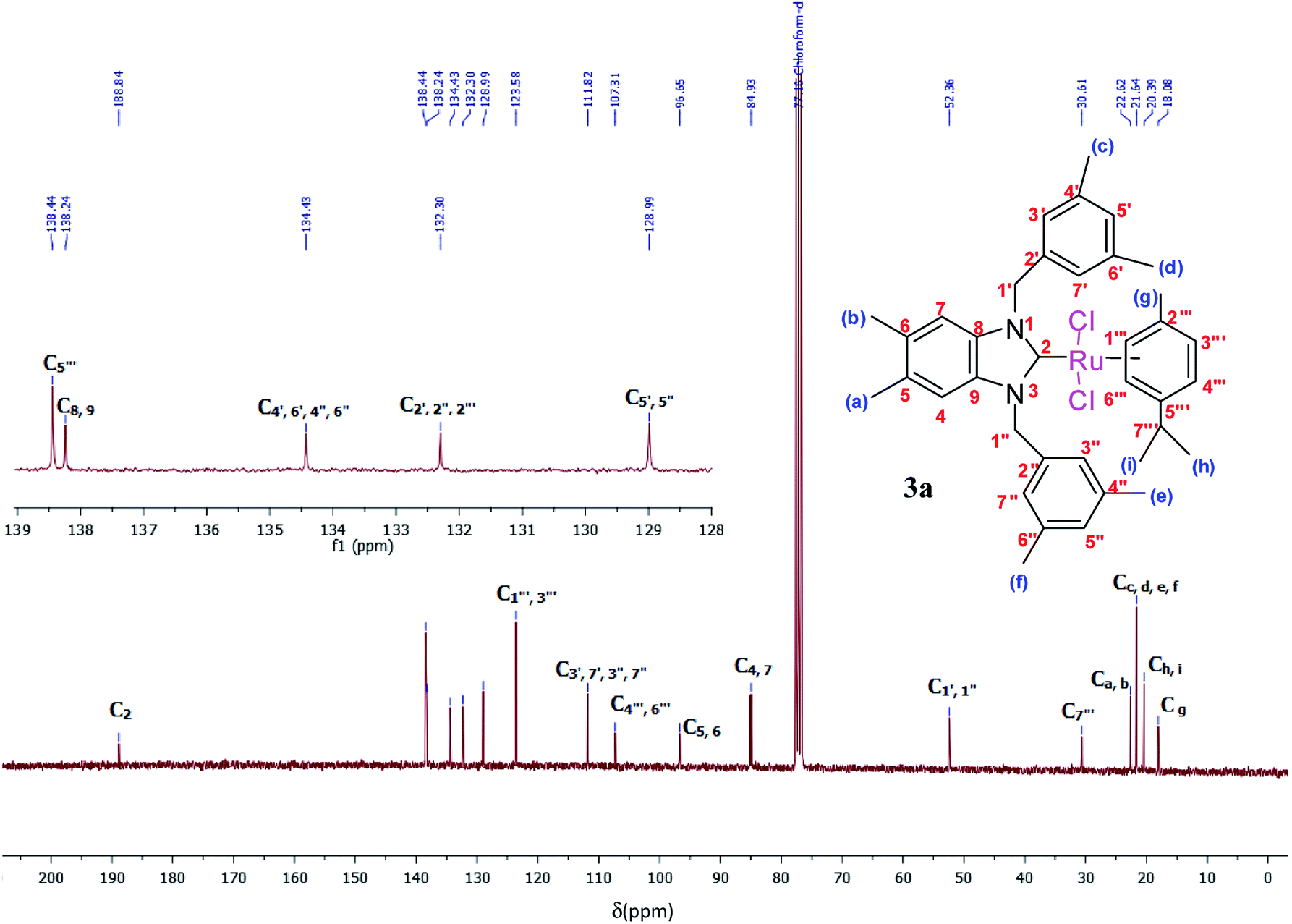 | ||
| Fig. 3 13C NMR spectra of Ru(II)–N-heterocyclic carbene complexes 3a (CDCl3, 75 MHz). | ||
The results of the mass spectrometry analyses are proof verification of the synthesized compounds (Fig. 4).
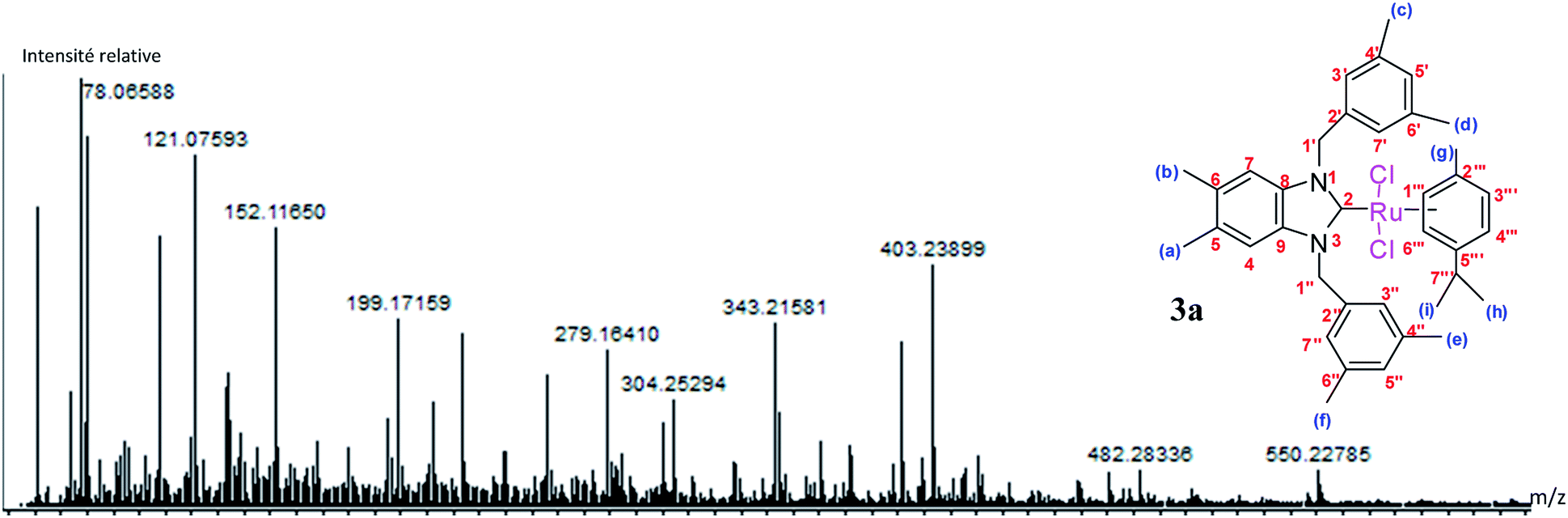 | ||
| Fig. 4 DART-TOF-MS of Ru(II)–N-heterocyclic carbene complexes 3a. | ||
The fragmentation leading to the m/z = 279 and m/z = 343 ions can occur by the mechanisms shown in Scheme 2.
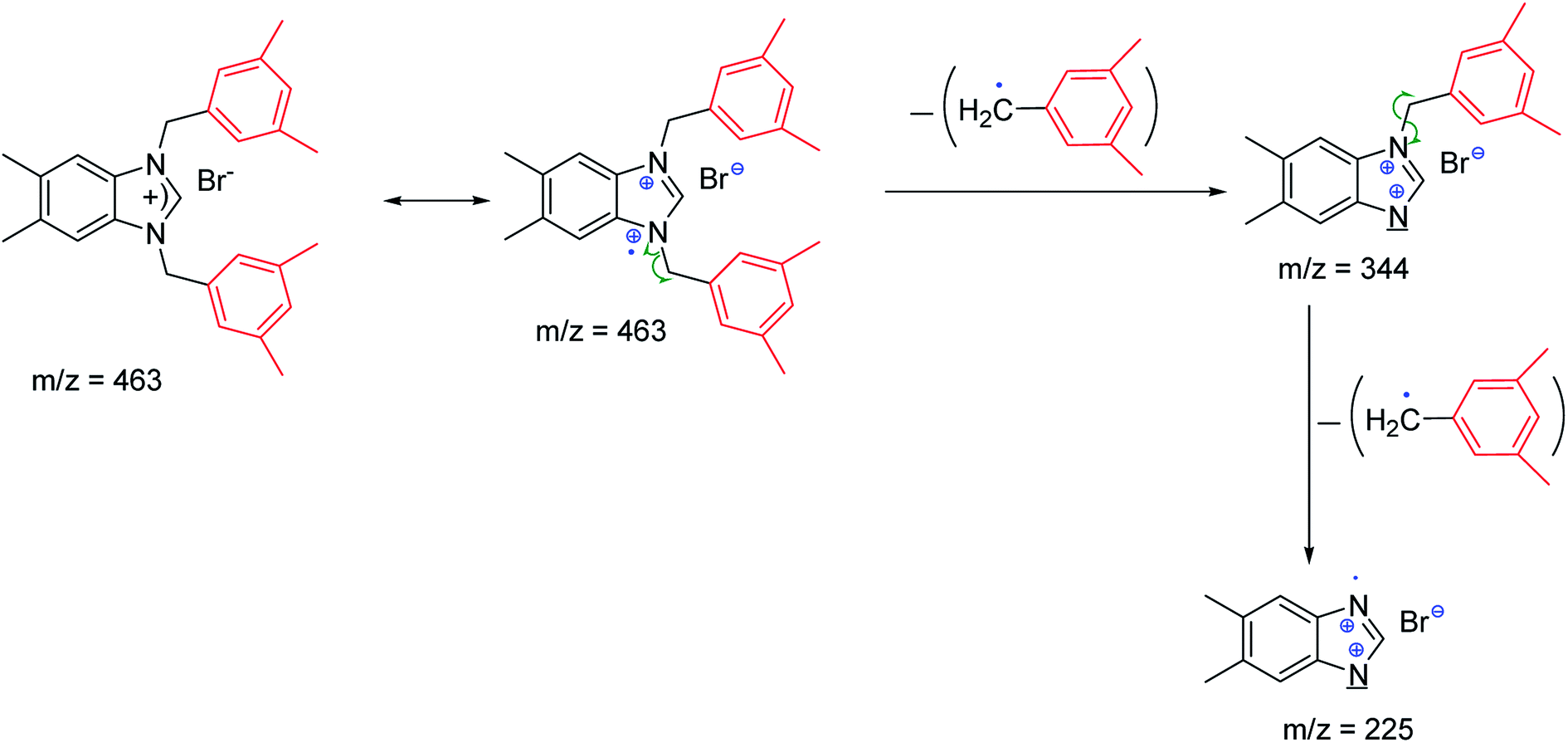 | ||
| Scheme 2 Mechanism of fragmentation leading to peaks m/z = 225. | ||
2.2. Transfer hydrogenation
The transfer hydrogenation process, which involves hydrogen transfer from a donor to an acceptor, has become one of the most important synthetic routes for producing alcohols from ketones.32 Non-toxic reagents, green solvents and mild reaction conditions are remarkable features of transfer hydrogenation reactions.33 Transition-metal catalyzed transfer hydrogenation using 2-propanol and KOH as a hydrogen source and promoter respectively, has become an efficient method in organic synthesis as illustrated by several useful applications reported in recent years.34 The reaction conditions for this important process are easy to handle (moderate boiling point 82 °C), inexpensive, relatively mild and environmentally benign.30
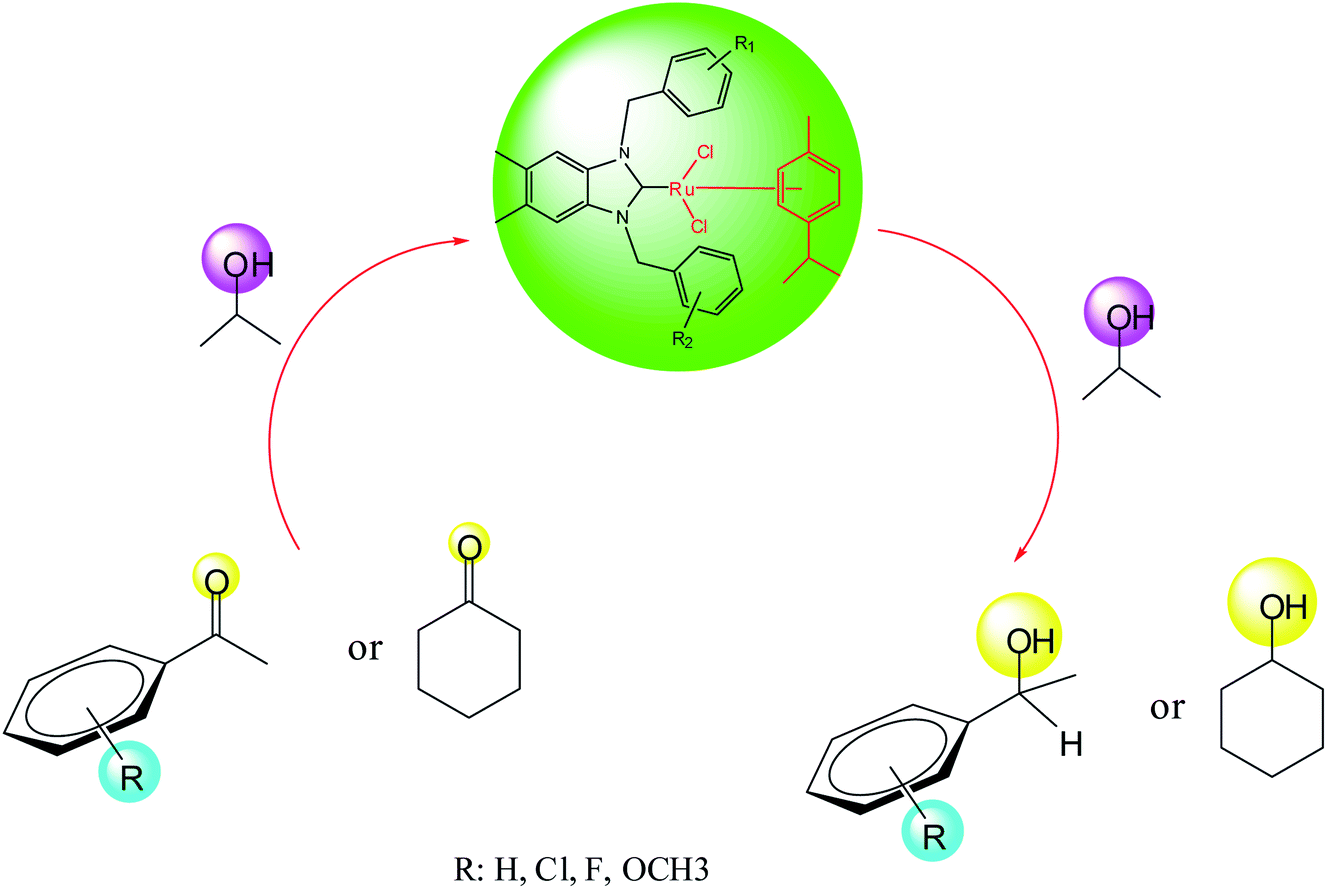
As part of our ongoing investigation into novel functionalized NHC ligands as the supporting environment for metal complexes and their application in catalysis. We herein report a series of catalytic transfer hydrogenation reactions employing the ruthenium–NHC complexes to discover their benefits. The transfer hydrogenation reactions are very susceptible to the type of the base and its concentration.31 As the starting point, the performance of the catalysts in the transfer hydrogenation was screened by using acetophenone as a model substrate.30,31,33 Previously, our laboratory reported the application of ruthenium–NHC complexes in the transfer hydrogenation of ketones.30 According to the literature, in a typical experiment, we dissolved the preformed, isolated crystalline catalyst (0.005 mmol) in 2-propanol. After the catalysts had completely dissolved, ketone (0.5 mmol) and a KOH (2 mmol) were added and the reaction was performed at 80 °C in 1 h.
In this context, we investigated the nature of the catalysts and the substrate that could impact on yield in the transfer hydrogenation reaction. Catalyst performance depends on the nature of substrates, the behavior of the ligands in the complexes, and concentration of the catalyst. Electrophilic effect of the substrate, nature of the catalysts, donor function, and flexibility of the backbone in the NHC ligands can be responsible for activity of the complexes. Steric and electronic properties of the substrates also affect conversions. Acetophenone and p-chloroacetophenone reacted very cleanly and turned into corresponding alcohol in good yields (Table 1, entries 1–6). However, 3b complex exhibited the lowest catalytic activity with all ketones. This low catalytic activity was attributed to structure of complex 3b, presumably due to decreased steric bulk around the metal or lower electron donation provided by the carbene ligands (Table 1, entries 2, 5, 8, 11 and 14). When p-methoxyacetophenone was used as substrate, low yield was observed on the transfer hydrogenation reaction catalysed by 3a–c (Table 1, entries 10–12). We are of the opinion that the nature of substrate can also give rise to low yield. Substrates bearing electron-withdrawing groups (Table 1, entries 4–9) were reduced to secondary alcohols in better yield than substrates bearing electron-donating groups (Table 1, entries 10–12). Similarly, with only 0.5 mol% of the same catalysts, it was possible to hydrogenate cyclohexanone and obtain the corresponding alcohols with the highest yields (Table 1, entries 13–15).
|
||||
|---|---|---|---|---|
| Entry | Substrate | Catalyst | Product | Yieldb (%) |
| a Reaction conditions: 0.005 mmol Ru–NHC, i-PrOH (5 ml), KOH (2 mmol), substrate (0.5 mmol), 80 °C, 1 h.b Conversions are based on corresponding ketones. | ||||
| 1 |  |
3a | 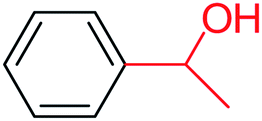 |
56 |
| 2 | 3b | 45 | ||
| 3 | 3c | 60 | ||
| 4 | 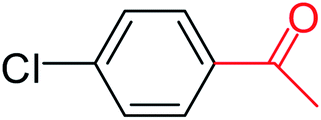 |
3a | 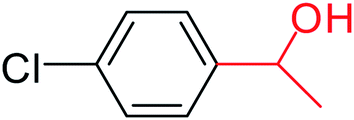 |
64 |
| 5 | 3b | 48 | ||
| 6 | 3c | 57 | ||
| 7 | 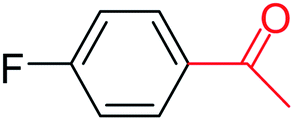 |
3a | 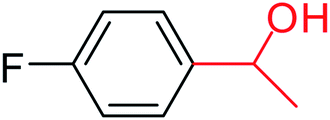 |
35 |
| 8 | 3b | 25 | ||
| 9 | 3c | 40 | ||
| 10 | 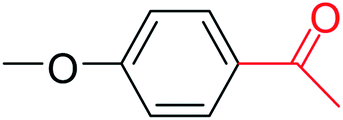 |
3a | 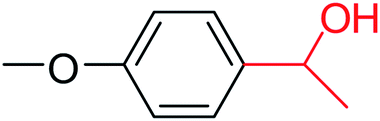 |
32 |
| 11 | 3b | 15 | ||
| 12 | 3c | 28 | ||
| 13 | 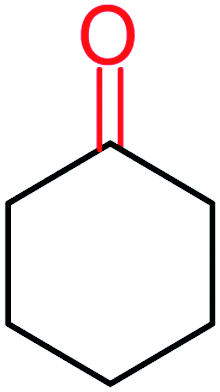 |
3a | 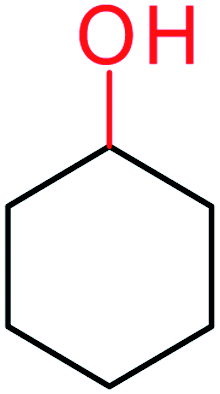 |
70 |
| 14 | 3b | 64 | ||
| 15 | 3c | 66 | ||
3. Biological activities
3.1. Antibacterial activity
The synthesized complexes were examined in vitro for their antibacterial activity using the well diffusion method (Fig. 5). Ampicillin (AMC) was used as standard control drugs for antibacterial activity. The ruthenium–NHC complexes (3a–f) were evaluated against three Gram-positive bacteria (Micrococcus luteus LB14110, Listeria monocytogenes ATCC19117, Staphylococcus aureus ATCC6538) and three Gram-negative bacteria (Salmonella typhimurium ATCC14028, Pseudomonas aeruginosa ATCC 9189, Escherichia coli). The obtained antibacterial activity is presented in Fig. 6 and Table 2.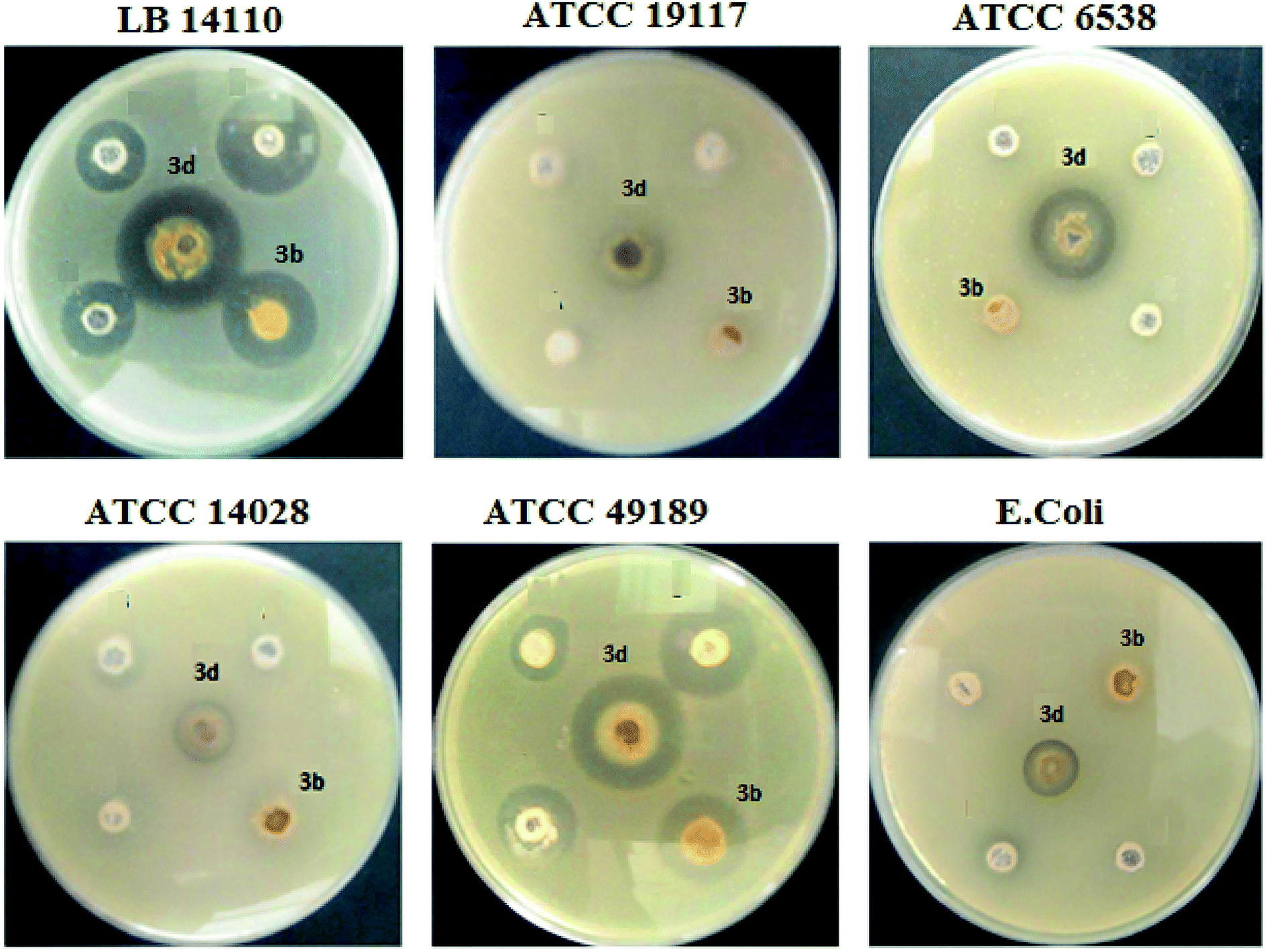 | ||
| Fig. 5 Zone of inhibition of Ru–NHC complexes (3a–f). | ||
 | ||
| Fig. 6 The method of determining the MIC. | ||
| Compounds | Microorganism indicator | |||||
|---|---|---|---|---|---|---|
| Micrococcus luteus | Listeria monocytogenes | Staphylococcus aureus | Salmonella typhimurium | Pseudomonas aeruginosa | E. coli | |
| LB 14110 | ATCC 19117 | ATCC 6538 | ATCC 14028 | ATCC 49189 | ||
| 3a | 30 ± 0.5 | 16 ± 0.2 | — | 16 ± 0.2 | — | — |
| 3b | 20 ± 0.4 | — | — | — | 20 ± 0.2 | — |
| 3c | 18 ± 0.2 | 20 ± 0.4 | — | 14 ± 0.7 | — | — |
| 3d | 30 ± 0.32 | 18 ± 0.1 | 22 ± 0.4 | 18 ± 0.1 | 26 ± 0.11 | — |
| 3e | 23 ± 0.3 | 16 ± 0.5 | 17 ± 0.3 | 16 ± 0.6 | 16 ± 0.22 | 15 ± 0.2 |
| 3f | — | — | — | — | — | — |
Globally, all complexes tested showed an important antibacterial activity against the Micrococcus luteus LB14110. Although complex 3d presents the highest inhibitory effect. In addition, 3e inhibit the growth of Gram-positive and Gram-negative bacteria in high and low concentrations. In contrast, the other complexes show no activity against E. coli.
The antibacterial activity of ruthenium–N-heterocyclic carbene complexes (3a–f) was reported in terms of the MIC values, defined as the lowest concentration of an antibacterial that visibly inhibits the growth of the bacteria after an overnight incubation. The MICs values complexes 3 were determined against the two Gram positive bacteria Micrococcus luteus LB 14110 and Listeria monocytogenes ATCC 19117 and the Gram-negative bacterium Salmonella typhimurium ATCC 14028. As shown in Table 3, the MICs values range from 0.0195–0.625 mg ml−1 for Micrococcus luteus LB 14110; 0.0781–5 mg ml−1 for Listeria monocytogenes ATCC 19117 and Salmonella typhimurium ATCC 14028. The most active compound was 3d which presents against Salmonella typhimurium ATCC 14028 the lowest MIC value (0.0781 mg ml−1) than the used standard (ampicillin). Thus, the best activity was recorded for this complex with MIC values of 0.0195 mg ml−1 against Micrococcus luteus LB14110. Complex 3e also has MIC values of 0.0781 mg ml−1 against Listeria monocytogenes ATCC 19117.
| Compounds | Minimum inhibitory concentrations (mg ml−1) | ||
|---|---|---|---|
| Micrococcus luteus | Listeria monocytogenes | Salmonella typhimurium | |
| LB 14110 | ATCC 19117 | ATCC 14028 | |
| 3a | 0.039 | 0.3125 | 2.5 |
| 3b | 0.625 | 5 | 5 |
| 3c | 0.039 | 1.25 | 2.5 |
| 3d | 0.0195 | 0.1562 | 0.0781 |
| 3e | 0.0195 | 0.0781 | 1.25 |
| 3f | — | — | — |
| Ampicillin | 0.004 | 0.002 | 0.625 |
3.2. Antioxidant activity
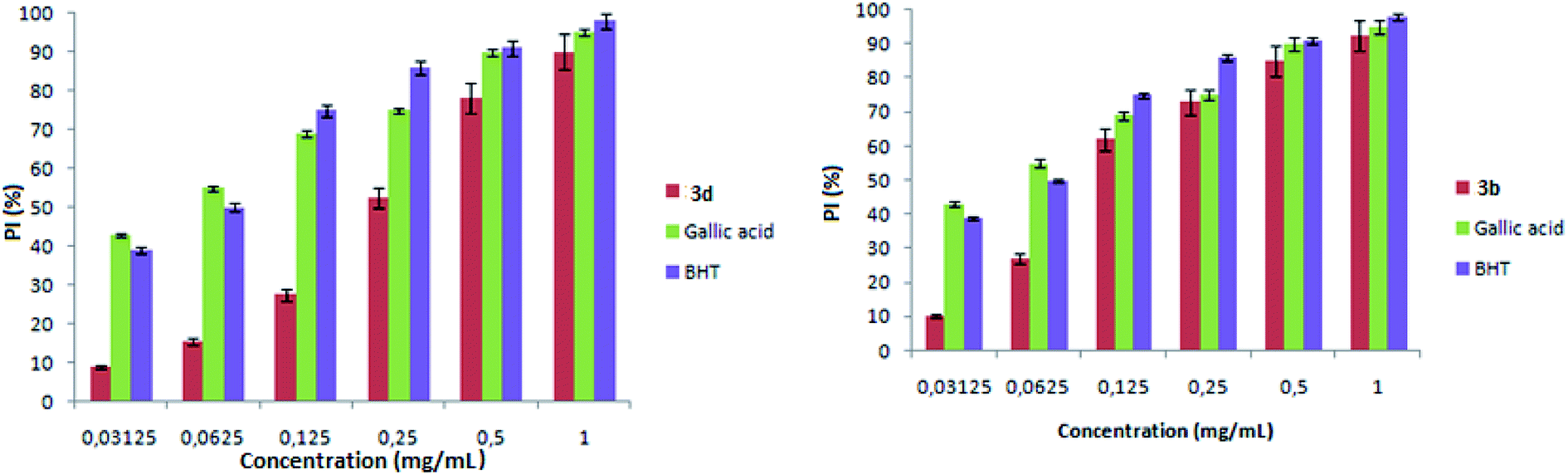 | ||
| Fig. 7 Scavenging activity of compounds 3b and 3d on DPPH radicals. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) v/v. Tubes containing volumes of equivalent amounts of the different complexes are incubated under incubation and controlled for 6 min in the dark. After a sweep, the wavelength of the work chosen for the evaluation of the antiradical activity was set at 734 nm. In order to explore the experimental results obtained, it is necessary to calculate the percentages of inhibition of the free radical ABTS˙+ according to the following formula:
:5) v/v. Tubes containing volumes of equivalent amounts of the different complexes are incubated under incubation and controlled for 6 min in the dark. After a sweep, the wavelength of the work chosen for the evaluation of the antiradical activity was set at 734 nm. In order to explore the experimental results obtained, it is necessary to calculate the percentages of inhibition of the free radical ABTS˙+ according to the following formula:| AAOx% = [(Ac − Ae)/Ac] × 100 |
As shown in Fig. 8, complexes 3b and 3d present a high antioxidant activity. Concerning the compound 3b, from a concentration of 10 mg ml−1 the percentage inhibition was in order to 70%. For compound 3d, the percentage inhibition was in order to 90%.
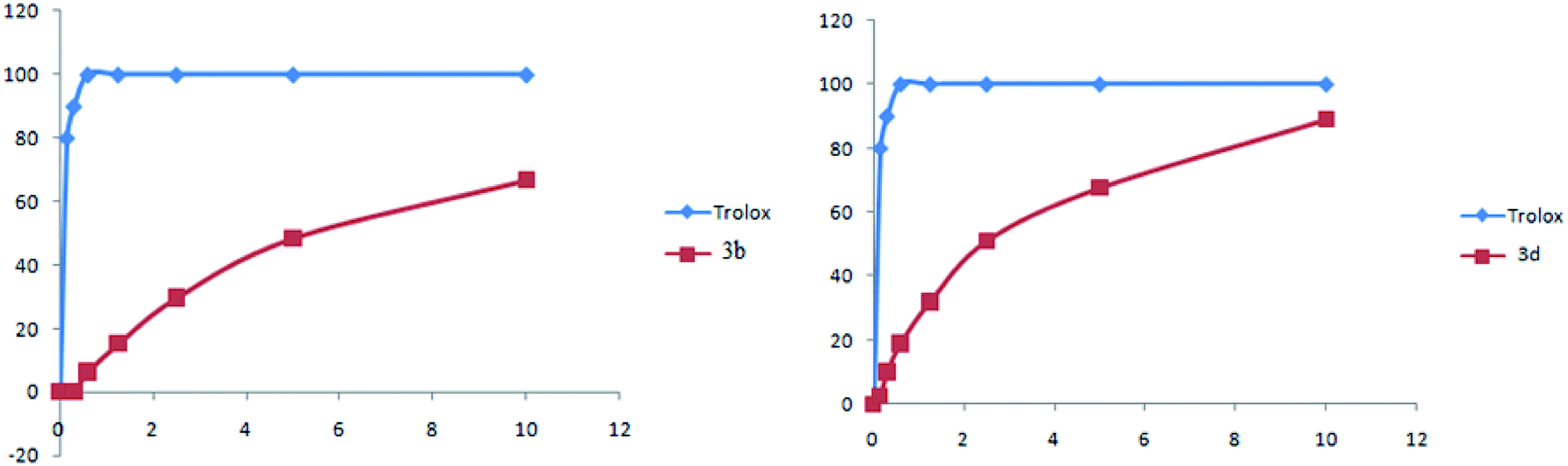 | ||
| Fig. 8 Percent inhibition of complexes 3b and 3d as a function of concentration. | ||
3.3. Antifungal activity
Fungal pathology has increased in recent years due to the increase in deep, visceral and septicemic mycotic sites. The former are caused by fungi that are widespread in nature, usually saprophytic, becoming pathogenic only under certain conditions: increasing use of broad-spectrum antibiotics, patients with AIDS; we talk about iatrogenic mycoses. They are caused by so-called opportunistic fungi that are either levuriform (Candida, Cryptococcus) or filamentous (Aspergillus, Cephalosporium).37 The development of antifungals comes up against obstacles such as the weak fungicidal activity in vivo compared to the results in vitro and the poor diffusion through the very thick wall of the mushrooms because of its composition (chitin, phospholipids, and sterols); these constituents are absent in bacteria, which explains why most antibacterial antibiotics are not antifungals.To evaluate the antifungal activity of the Ru–NHC complexes (3a–f) with respect to the Candida albicans strain, the solid medium diffusion method was carried out (Fig. 9). As shown in Table 4, the complexes 3a, 3c, 3e and 3f are completely inactive against Candida albicans. The compounds 3b and 3d present a high antifungal activity. The most active compound was 3d which presents against Candida albicans the highest diameter of growth inhibition zone with a value of the order of 34 ± 0.18 mm.
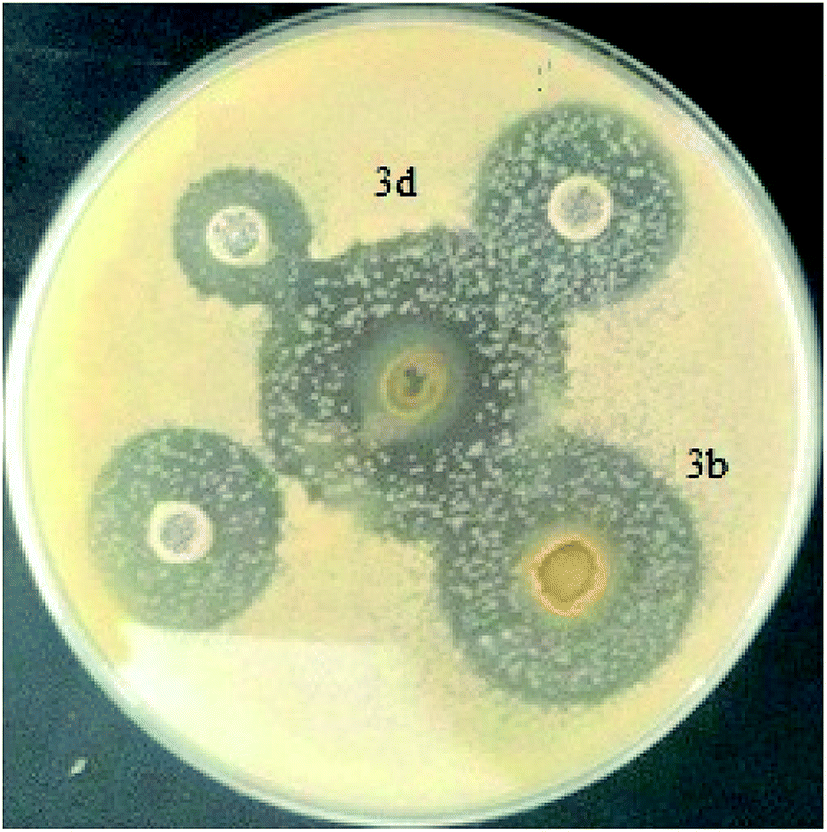 | ||
| Fig. 9 Antifungal activity against Candida albicans. | ||
| Strain | Compounds | |||||
|---|---|---|---|---|---|---|
| 3a | 3b | 3c | 3d | 3e | 3f | |
| Candida albicans | — | 22 ± 0.5 | — | 34 ± 0.18 | — | — |
3.4. Acetylcholinesterase inhibition
The acetylcholinesterase enzyme (AChE) is an attractive target for the rational drug design and for the discovery of mechanism-based inhibitors because of its role in the hydrolysis of the neurotransmitter acetylcholine (ACh). The pathogenesis of Alzheimer's disease (AD) has been linked to a deficiency in the brain neurotransmitter acetylcholine. Subsequently, acetylcholinesterase inhibitors (AchEIs) were introduced for the symptomatic treatment of AD,38,39 and other possible therapeutic applications in the treatment of Parkinson's disease, senile dementia, and ataxia, among others.40 The prevailing view has been that the efficacy of AchEIs is attained through their augmentation of acetylcholine-medicated neuron to neuron transmission. However, AchEIs also protect cells from free radical toxicity and β-amyloid induced injury and increased production of antioxidants.The results presented in Table 5 show that the 3d complex is the most potent inhibitor against AchE. This good activity is confirmed by a value of the percentage inhibition of the order of 47.1%. However, the 3c and 3f complexes are completely inactive and the other Ru–NHC complexes exhibit anti-AchEI activity.
| Compounds | 3a | 3b | 3c | 3d | 3e | 3f |
|---|---|---|---|---|---|---|
| (AChEI) (%) | 32.8 | 45.3 | — | 47.1 | 38 | — |
3.5. Anticancer activity
| Relative cell viability (%) = OD treated C/OD control × 100% |
The compounds were screened calorimetrically using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) cell assay in two human cancer cell lines namely MCF7 and MDA-MB-231 to evaluate their cytotoxicity. As shown in Table 6, many of the compounds affected cell viability of cancer cell lines and showed promising level of cytotoxicity however, the cytotoxicity of 3e and 3d was much stronger in MCF7 with IC50 values 0.68 and 0.6 μg ml−1, respectively as compared to their activity on MDA-MB-231 cells. The cytotoxicity of 3e and 3d on MDA-MB-231 cells was 1.93 and 3.1 μg ml−1. The cytotoxicity of compounds 3c in MCF7 and MDA-MB-231 cells was 1.7 and 16 μg ml−1, while the IC50 values of compound on 3f were 1.3 and 3.3 μg ml−1 against MCF7 and MDA-MB-231, respectively.
| Compounds | Anticancer activity IC50 in μg ml−1 | Antimicrobial activity (50 μg per disc) | |||
|---|---|---|---|---|---|
| MCF7 | MDA-MB-231 | E. coli | MRSA | C. albicans | |
| 3a | 4.2 ± 3.6 | 2.5 ± 4.3 | 21.0 ± 1 | 22.5 ± 2.5 | 25.0 ± 00 |
| 3b | 3.1 ± 3.1 | 2.6 ± 5.9 | 23.3 ± 1.5 | 25.5 ± 1.5 | 28.5 ± 1.5 |
| 3c | 1.7 ± 3.1 | 16 ± 2.8 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| 3d | 0.6 ± 1.8 | 3.1 ± 00 | 25.0 ± 00 | 25.5 ± 0.5 | 30.5 ± 0.5 |
| 3e | 0.68 ± 3.2 | 1.93 ± 2.6 | 100.0 ± 2.0 | 11.0 ± 1.0 | 10.0 ± 0 |
| 3f | 1.3 ± 4.1 | 3.3 ± 2.9 | 12.0 ± 1.0 | 13.0 ± 00 | 13.0 ± 00 |
| Tetracycline | NT | NT | 22 ± 0.3 | 25.5 | 0.0 ± 00 |
The antimicrobial activities of the synthesized compounds against the human pathogenic microorganisms are presented in Table 6. The results revealed that the highest antimicrobial activity was reported in 3e (against E. coli) followed 3d (against E. coli) and 3b (against MRSA) however the highest anti Candida albicans activity was found in 3d (30.5 mm) followed by 3b (28.0 mm) and 3a (25.0 mm). Compound 3c was completely inactive against the three strains (Table 6).
4. Conclusion
In summary, a new series of ruthenium–NHC complexes (3a–f) have been synthesized from silver–NHC complexes with [RuCl2(p-cymene)]2. All these compounds were characterized by spectroscopic and analytic techniques. To demonstrate the efficiency of 3a–c as catalyst precursors, transfer hydrogenations of ketones were performed. Excellent activity and good catalyst stability were observed under mild reaction conditions. Structural differences in 3a–c did not affect yields significantly. The Ru–NHC catalysts described here gave one of the highest conversion rates to date for transfer hydrogenation reaction catalyzed by Ru–NHC complexes with low base loading.The complexes 3a–f were tested for their antibacterial activity against Micrococcus luteus LB 14110, Staphylococcus aureus ATCC 6538, Listeria monocytogenes ATCC 19117, Salmonella typhimurium ATCC 14028, Pseudomonas aeruginosa ATCC 49189 and E. coli. Obtained results show that the complexes 3d and 3e have an effective antibacterial activity against the used indicator bacteria. With regard to the scavenging activity of the synthesized complexes on the DPPH radical, we noted that compounds 3b and 3d, from a concentration of 1 mg ml−1, present a very similar scavenging activity to that of the two used controls butylated hydroxytoluene (BHT) and gallic acid (GA). Interestingly, from a concentration of 10 mg ml−1, 3b and 3d complexes exhibited the percentage inhibition in order to 70% and 90% respectively. Concerning acetylcholinesterase inhibition activity (AChEI), according to our findings, compound 3d presents an interesting AChEI activity with 47.1% of inhibition. In addition, this complex presents against Candida albicans the highest diameter of growth inhibition zone with a value of the order of 34 ± 0.18 mm. The cytotoxic activity of the complexes (3a–f) was also determined against a panel of cell lines such us MDA-MB-231 and T47D. The most actives compounds are 3d and 3e.
5. Experimental section
5.1. Materials and methods
All procedures were carried out under an inert atmosphere using standard Schlenk line techniques. Chemicals and solvents were purchased from Sigma Aldrich. The solvents used were purified and dried by MBraun SPS 800 solvent purification system. Column chromatography was performed by using silica gel 60 (70–230 mesh).1H NMR and 13C NMR spectra were recorded at 300 MHz (1H), 75 MHz (13C) in CDCl3 with tetramethylsilane as an internal reference. The NMR studies were carried out in high-quality 5 mm NMR tubes. Signals are quoted in parts per million as δ downfield from tetramethylsilane (δ = 0.00) as an internal standard. NMR multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, m = multiplet. GC analyses of reactions mixtures have been done using a Shimadzu GC 2010-Plus GCFID system. Column: Teknokroma TRB-5 capillary column, 30 m × 0.32 mm × 0.25 μm. Initial temperature at 50 °C, held for 1 min, ramp 2 °C min−1 next 90 °C, held for 3 min, ramp 40 °C min−1 next 240 °C held for 10 min. The temperature of the injector and detector were held at 240 °C. Melting points were determined with Kofler bench at Isste of Borj Cedria (Hammam Lif, University of Carthage, Borj Cedria, Tunisia).5.2. General procedure for preparation of the ruthenium–NHC complexes (3a–f)
The ruthenium–NHC complexes were synthesized through transmetalation via silver–NHC complexes (2a–f). We reacted one equivalent of silver–NHC complexes and 0.5 equivalent of [RuCl2(p-cymene)]2 in anhydrous dichloromethane (30 ml). The reaction mixture was stirred for 4 days at room temperature in the dark. Then the solution was filtered through Celite and the filtrate was concentrated to dryness in vacuum. The red solid obtained recrystallized from DCM-diethyl ether or DCM-pentane. The structure of Ru–NHC complexes were determined by IR and NMR analysis.5.3. Dichloro-[1,3-bis(3,5-dimethylbenzyl)-benzimidazole-2-ylidene](p-cymene)ruthenium(II), 3a
Yield: (85%); mp 236 °C. C37H45Cl2N2Ru, M = 689.75 g mol−1. IR: ν = 1406 (νC–N) cm−1. 1H NMR (300 MHz, CDCl3, δ (ppm)): 6.91 (s, 2H, CH2C6H3(CH3)2-3,5); 6.83 (s, 2H, C6H2(CH3)2-5,6); 6.68 (s, 4H, CH2C6H3(CH3)2-3,5); 6.59 (d, 2H, p-CH3C6H4CH(CH3)2); 5.54 (d, 2H, p-CH3C6H4CH(CH3)2); 5.29 (d, 2H, H1′, CH2); 5.05 (d, 2H, H1′′, CH2); 2.62 (m, 1H, H7′′′, p-CH3C6H4CH(CH3)2); 2.30 (s, 12H, Hc,d,e,f, 4 × CH3); 2.21 (s, 6H, Ha,b, 2 × CH3); 1.80 (s, 3H, Hg, CH3); 1.14 (s, 6H, Hh,i, 2 × CH3) (p-CH3C6H4CH(CH3)2). 13C NMR (CDCl3, 75 MHz) (δ (ppm)):188.8 (C2, NCN); 138.4; 138.2; 134.4; 132.3; 128.9; 123.5; 111.8; 107.3; 96.6; 84.9; 52.3 (C1′,1′′, 2 × CH2); 30.6 (C7′′′, p-CH3C6H4CH(CH3)2); 22.6 (Ca,b, 2 × CH3, C6H2(CH3)2-5,6); 21.6 (Cc,d,e,f, 4 × CH3); 20.3 (Ch,i, 2 × CH3); 18.0 (Cg, CH3).DART-TOF-MS: m/z = 279, 343.
5.4. Dichloro-[1,3-bis(4-methylbenzyl)benzimidazol-2-ylidene](p-cymene)-ruthenium(II), 3b
Yield: (90%); mp 210 °C. C35H41Cl2N2Ru, M = 733.85 g mol−1. IR: ν = 1409 (νC–N) cm−1. 1H NMR (300 MHz, CDCl3, δ (ppm)): 0.14 (d, 4H, CH2C6H4(CH3)-4); 7.00 (d, 4H, CH2C6H4(CH3)-4); 6.79 (s, 2H, C6H2(CH3)2-5,6); 6.46 (d, 2H, p-CH3C6H4CH(CH3)2); 5.68 (d, 2H, p-CH3C6H4CH(CH3)2); 5.29 (s, 2H, H1′, CH2); 5.03 (s, 2H, H1′′, CH2); 2.68 (p, 1H, H7′′′, p-CH3C6H4CH(CH3)2); 2.34 (s, 6H, Hc,d, 2 × CH3); 2.18 (s, 6H, Ha,b, 2 × CH3); 1.84 (s, 3H, He, CH3); 1.16 (d, 6H, Hf,g, 2 × CH3) (p-CH3C6H4CH(CH3)2). 13C NMR (CDCl3, 75 MHz) (δ (ppm)): 189.0 (C2, NCN); 137.0; 134.9; 134.4; 132.3; 129.6; 125.9; 111.9; 107.6; 97.0; 85.30; 52.5 (C1′,1′′, 2 × CH2); 30.6 (C7′′′, p-CH3C6H4CH(CH3)2); 22.6 (Cf,g, 2 × CH3); 21.2 (Cc,d, 2 × CH3); 20.3 (Ce, CH3); 18.3 (Ca,b, 2 × CH3); 14.2 (Ch, CH3).5.5. Dichloro-[1,3-bis(4-tertbutylbenzyl)benzimidazol-2-ylidene](p-cymene)-ruthenium(II), 3c
Yield: (87%); mp 224 °C. C40H53Cl2N2Ru, M = 733.85 g mol−1. IR: ν = 1609 (νC–N) cm−1. 1H NMR (300 MHz, CDCl3, δ (ppm)): 7.36 (d, 4H, CH2C6H4C(CH3)3-4); 7.03 (d, 4H, CH2C6H4C(CH3)3-4); 6.82 (s, 2H, C6H2(CH3)2-5,6); 6.55 (d, 2H, p CH3C6H4CH(CH3)2); 5.62 (d, 2H, p-CH3C6H4CH(CH3)2); 5.31 (s, 2H, H1′, CH2); 5.02 (s, 2H, H1′′, CH2); 2.61 (s, 6H, Hd,g, 2 × CH3); 2.19 (s, 6H, Ha,b, 2 × CH3); 1.81 (s, 3H, Hi, CH3); 1.66 (s, 1H, H7′′′, p-CH3C6H4CH(CH3)2); 1.31 (s, 15H, Hc,e,f,h,l, 5 × CH3); 1.12 (d, 6H, Hj,k, 2 × CH3) (p-CH3C6H4CH(CH3)2). 13C NMR (CDCl3, 75 MHz) (δ (ppm)): 188.9 (C2, NCN); 150.4; 134.9; 134.4; 132.3; 125.8; 125.5; 111.9; 106.8; 97.3; 85.5; 52.3 (C1′,1′′, 2 × CH2); 41.1 (C7′′′, p-CH3C6H4CH(CH3)2); 34.6 (Cd,g, 2 × CH3, CH2C6H4C(CH3)3-4); 31.4 (Cc,e,f,h, 4 × CH3); 30.5 (Ci, CH3); 22.6 (Ca,b, 2 × CH3); 20.3 (Cj,k, 2 × CH3). DART-TOF-MS: m/z = 453, 426, 291.5.6. Dichloro-[1-(3,5-dimethylbenzyl)-3-(2,3,5,6-tetramethylbenzyl)benzimidazol-2-ylidene](p-cymene)ruthenium(II), 3d
Yield: (88%); mp 184 °C. C39H49Cl2N2Ru, M = 0.717.80 g mol−1. IR: ν = 1418 (νC–N) cm−1. 1H NMR (300 MHz, CDCl3, δ (ppm)): 7.69 (d, 2H, p-CH3C6H4CH(CH3)2); 7.54 (d, 2H, p-CH3C6H4CH(CH3)2); 7.26 (s, 1H, CH2C6H3(CH3)2-3,5); 6.89 (s, 2H, CH2C6H3(CH3)2-3,5); 6.63 (s, 2H, C6H2(CH3)2-5,6); 5.88 (s, 1H, CH2C6H(CH3)4-2,3,5,6); 4.21 (s, 4H, H1′,1′′, 2 × CH2); 2.86 (p, 1H, H7′′′, p-CH3C6H4CH(CH3)2); 2.27 (s, 18H, Ha,b,c,d,f,g, 6 × CH3); 2.20 (s, 3H, Hi, CH3); 1.95 (s, 3H, Hl, CH3); 1.32 (s, 6H, He,h, 2 × CH3); 0.92 (s, 6H, Hj,k, 2 × CH3) (p-CH3C6H4CH(CH3)2). 13C NMR (CDCl3, 75 MHz) (δ (ppm)): 187.8 (C2, NCN); 138.4; 135.4; 135.2; 134.5; 133.5; 133.2; 132.3; 131.6; 131.3; 130.0; 128.9; 124.6; 123.4; 107.1; 96.2; 54.0 (C1′, CH2); 51.8 (C1′′, CH2); 31.0 (C7′′′, p-CH3C6H4CH(CH3)2); 21.5 (Cj,k, 2 × CH3, p CH3C6H4CH(CH3)2); 20.8 (Cc,d, 2 × CH3); 20.2 (Ci, CH3); 18.4 (Cf,g, 2 × CH3); 16.3 (Ca,b, 2 × CH3); 15.4 (Ce,h,l, 3 × CH3).5.7. Dichloro-[1-(3,5-dimethylbenzyl)-3-(4-tertbutylbenzyl)benzimidazol-2-ylidene](p-cymene) ruthenium(II), 3e
Yield: (86%); mp 222 °C. C39H49Cl2N2Ru, M = 717.80 g mol−1. IR: ν = 1404 (νC–N) cm−1. 1H NMR (300 MHz, CDCl3, δ (ppm)): 7.34 (d, 2H, p-CH3C6H4CH(CH3)2); 7.03 (d, 2H, p-CH3C6H4CH(CH3)2); 6.92 (s, 1H, CH2C6H3(CH3)2-3,5); 6.84 (s, 4H, CH2C6H4C(CH3)3-4); 6.68 (s, 2H, CH2C6H4C(CH3)3-4); 6.54 (s, 2H, CH2C6H3(CH3)2-3,5); 5.58 (s, 2H, C6H2(CH3)2-5,6); 5.30 (s, 2H, H1′, CH2); 5.01 (s, 2H, H1′′, CH2); 2.58 (p, 1H, H7′′′, p-CH3C6H4CH(CH3)2); 2.30 (s, 6H, Hc,d, 2 × CH3); 2.20 (s, 6H, Ha,b, 2 × CH3); 1.81 (s, 3H, Hf, CH3); 1.57 (s, 3H, Hh, CH3); 1.31 (s, 6H, Hi,j,, 2 × CH3) (p-CH3C6H4CH(CH3)2); 1.12 (s, 6H, He,g, 2 × CH3). 13C NMR (CDCl3, 75 MHz) (δ (ppm)): 189.1 (C2, NCN); 150.4; 138.4; 138.2; 135.0; 134.4; 132.3; 129.0; 127.7; 125.7; 125.6; 123.6; 111.9; 107.1; 97.0; 85.2; 52.3 (C1′, CH2); 52.2 (C1′′, CH2); 34.6 (C7′′′, p-CH3C6H4CH(CH3)2); 31.4 (Cc,d, 2 × CH3, p-CH3C6H4CH(CH3)2); 30.5 (Ca,b, 2 × CH3); 22.7 (Cf, CH3); 21.6 (Ci,j, 2 × CH3); 20.3 (Ch, CH3); 18.1 (Ce,g, 2 × CH3).5.8. Dichloro-[1-(3,5-dimethylbenzyl)-3-(4-methylbenzyl)benzimidazol-2-ylidene](p-cymene) ruthenium(II), 3f
Yield: (80%); mp 204 °C. C36H43Cl2N2Ru, M = 675.73 g mol−1. IR: ν = 1404 (νC–N) cm−1. 1H NMR (300 MHz, CDCl3, δ (ppm)): 7.13 (d, 2H, p-CH3C6H4CH(CH3)2); 7.00 (d, 2H, p-CH3C6H4CH(CH3)2); 6.92 (s, 1H, CH2C6H3(CH3)2-3,5); 6.84 (d, 4H, CH2C6H4(CH3)-4); 6.67 (s, 2H, CH2C6H3(CH3)2-3,5); 6.51 (m, 2H, CH2C6H4(CH3)-4); 5.57 (m, 2H, C6H2(CH3)2-5,6); 5.30 (s, 2H, H1′, CH2); 5.02 (s, 2H, H1′′, CH2); 2.64 (p, 1H, H7′′′p-CH3C6H4CH(CH3)2); 2.34 (s, 3H, He, CH3); 2.30 (s, 6H, Hc,d, 2 × CH3); 2.19 (s, 6H, Ha,b, 2 × CH3); 1.82 (s, 3H, Hf, CH3); 1.58 (s, 3H, Hi, CH3); 1.15 (s, 6H, Hg,h, 2 × CH3) (p-CH3C6H4CH(CH3)2). 13C NMR (CDCl3, 75 MHz) (δ (ppm)): 189.2 (C2, NCN); 138.4; 138.2; 137.0; 135.0; 134.5; 134.3; 132.3; 129.6; 129.0; 125.8; 123.6; 111.9; 107.4; 96.6; 85.2; 52.4 (C1′, CH2); 52.2 (C1′′, CH2); 30.6 (C7′′′, p-CH3C6H4CH(CH3)2); 21.6 (Ch,g, 2 × CH3, p-CH3C6H4CH(CH3)2); 21.2 (Cc,d, 2 × CH3); 22.3 (Ce,f, 2 × CH3); 18.1 (Ca,b,i, 3 × CH3).5.9. Antibacterial activity
These bacterial strains were grown overnight in Luria–Bertani (LB) agar medium (g l−1): peptone 10; yeast extract 5 and NaCl 5 at pH 7.2 under aerobic conditions and constant agitation (200 rpm) at 30 °C for M. luteus LB14110 and L. monocytogenes ATCC 19117 and at 37 °C for S. aureus ATCC 6538, S. typhimurium ATCC 14028 and P. aeruginosa ATCC49189, and then diluted 1:100 in LB media and incubated for 5 h under constant agitation (200 rpm) at the appropriate temperature.
| % ArA = [(absorbance of control − absorbance of test sample)/absorbance of control] × 100. |
All tests are assayed in triplicate and expressed as the average ± standard deviation of the measurements to produce the concentration range of 0.0048–20 mg ml−1. To each test well 10 μl of cell suspension were added to final inoculum concentrations of 106 CFU ml−1 for each microorganisms. Positive growth control wells consisted of microorganisms only in their adequate medium. Cells suspension at the same concentration supplemented with ampicillin was used as control. The plates were then covered with the sterile plate covers and incubated at the appropriate temperature of each microorganism. The MIC was defined as the lowest concentration of the synthesized compound at which the microorganism does not demonstrate.
:5) v/v to which the previous solution was added. White is ethanol and ultrapure water (5:5) v/v. Samples and control were incubated for 6 min in the dark, then the OD was measured at 734 nm. Gallic acid, ascorbic acid and BHT were used as standards the calculation is done according to the following formula:| AAOx% = [(Ac − Ae)/Ac] × 100 |
5.10. Antifungal activity
Antifungal activity was evaluated against Candida albicans. This strain is a species of yeast. The evaluation of the activity of the compounds synthesized with respect to the strain of C. albicans was carried out using the diffusion method on a solid medium.
C. albicans is cultivated in a YPD medium whose composition is as follows: 10 g of yeast extract; 20 g peptone; 20 g glucose (or dextrose); H2O qsp 1 L; pH = 5.6.
In solid medium: C. albicans is cultivated in Sabouraud medium and incubated for 48 h at 30 °C.
In liquid medium: C. albicans is cultivated in YPD medium in Erlenmeyer flasks. The cultures are incubated at 30 °C for 48 h with constant stirring at 250 rpm. For preservation, 200 μl of each culture is mixed with 800 μl of glycerol in cryotubes and stored at −80 °C.
| % AChEI = 1 − (δAsample/δAcontrol) × 100 |
All synthesised compounds have been tested at 100 μg ml−1 of concentration. This determination was done in triplicate and obtained results were very similar. The reported value is the average of the three tests.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by the Research Supporting Project (RSP-2019/75), King Saud University, Riyadh Saudi Arabia.References
- T. Ikayira and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300 CrossRef.
- G. Zassinovich, G. Mestroni and S. Gladiali, Chem. Rev., 1992, 92, 1051 CrossRef CAS.
- C. F. de Graauw, J. A. Peters, H. van Bekkum and J. Husken, Synthesis, 1994, 10, 1007 CrossRef.
- R. Noyori and S. Hashigushi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS.
- M. Yamakawa, H. Ito and R. Noyori, J. Am. Chem. Soc., 2000, 122, 1466 CrossRef CAS.
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008 CrossRef CAS.
- M. Bierenstiel, M. Dymarska, E. de Jong and M. Schlaf, J. Mol. Catal. A: Chem., 2008, 290, 1 CrossRef CAS.
- M. Zhao, Z. Yu, S. Yan and Y. Li, Tetrahedron Lett., 2009, 50, 4624 CrossRef CAS.
- M. Yigit, B. Yigit, I. Özdemir, E. Çetinkaya and B. Çetinkaya, Appl. Organomet. Chem., 2006, 20, 322 CrossRef CAS.
- (a) N. Marion, S. Diez-Gonzalez and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988 CrossRef CAS; (b) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS; (c) V. Nair, S. Bindu and V. Sreekumar, Angew. Chem., Int. Ed., 2004, 43, 5130 CrossRef CAS.
- (a) S. P. Nolan, N-Heterocyclic Carbenes in Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2006 CrossRef; (b) F. Glorius, N-Heterocyclic Carbenes in Transition Metal Catalysis Topics in Organometallic Chemistry, Springer-Verlag, Berlin, 2007 CrossRef; (c) W. A. Herrmann and C. Kocher, Angew. Chem., Int. Ed., 2007, 36, 2162 CrossRef; (d) W. A. Herrmann, M. Elison, J. Fischer, C. Kocher and G. R. J. Artus, Angew. Chem., Int. Ed. Engl., 1995, 34, 2371 CrossRef CAS.
- A. J. Arduengo, H. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef CAS.
- (a) E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239 CrossRef CAS; (b) M. C. Perry and K. Burgess, Tetrahedron: Asymmetry, 2003, 14, 951 CrossRef CAS.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef CAS.
- C. W. K. Gstottmayr, V. P. W. Bohm, E. Herdtweck, M. Grosche and W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1363 CrossRef CAS.
- V. Lavallo, Y. Canac, A. De Hope, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 7236 CrossRef CAS.
- K. Vehlow, S. Maechling and S. Blechert, Organometallics, 2006, 25, 25 CrossRef CAS.
- (a) J. W. Sprengers, J. Wassenaar, N. D. Clement and K. J. Cavell, Angew. Chem., Int. Ed., 2005, 44, 2026 CrossRef CAS; (b) N. D. Clement and K. J. Cavell, Angew. Chem., Int. Ed., 2004, 43, 3845 CrossRef CAS.
- (a) A. Furstner, O. R. Thiel and C. W. Lehmann, Organometallics, 2002, 21, 331 CrossRef; (b) A. Furstner, L. Ackermann, A. Beck, H. Hori, D. Koch, K. Langemann, M. Liebl, C. Six and W. Leitner, J. Am. Chem. Soc., 2001, 123, 9000 CrossRef CAS.
- G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, J. Am. Chem. Soc., 2001, 126, 15195 CrossRef PubMed.
- (a) T. W. Funk, J. M. Berlin and R. H. Grubbs, J. Am. Chem. Soc., 2006, 128, 1840 CrossRef CAS PubMed; (b) J. P. Morgan and R. H. Grubbs, Org. Lett., 2000, 2, 3153 CrossRef CAS.
- N. Marion, O. Navarro, J. Mei, E. D. Stevens, N. M. Scott and S. P. Nolan, J. Am. Chem. Soc., 2006, 128, 4101 CrossRef CAS.
- (a) M. J. Schultz, S. S. Hamilton, D. R. Jensen and M. S. Sigman, J. Org. Chem., 2005, 70, 3343 CrossRef CAS; (b) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247 CrossRef CAS.
- (a) R. H. Grubbs, Handbook of Olefin Metathesis, VCH-Wiley, Weinheim, 2003 CrossRef; (b) S. Beligny and S. Blechert, in N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2004 Search PubMed.
- (a) I. Ozdemir, B. Yigit, B. Cetinkaya, D. Ulku, M. N. Tahir and C. Arici, J. Organomet. Chem., 2001, 633, 27 CrossRef CAS; (b) S. Yasar, I. Ozdemir, B. Cetinkaya, J. L. Renaud and C. Bruneau, Eur. J. Org. Chem., 2008, 2142 CrossRef CAS.
- (a) I. Ozdemir, S. Yasar and B. Cetinkaya, Transition Met. Chem., 2005, 30, 831 CrossRef; (b) M. Yigit, B. Yigit, I. Ozdemir, E. Cetinkaya and B. Cetinkaya, Appl. Organomet. Chem., 2006, 20, 322 CrossRef CAS.
- (a) S. Yaşar, K. J. Cavell, B. D. Ward and B. Kariuki, Appl. Organomet. Chem., 2011, 25, 374 CrossRef; (b) N. Gurbuz, I. Ozdemir, S. Demir and B. Cetinkaya, J. Mol. Catal. A: Chem., 2004, 209, 23 CrossRef CAS.
- I. Ozdemir, E. O. Ozcan, S. Gunal and N. Gurbuz, Molecules, 2010, 15, 2499 CrossRef CAS.
- L. Boubakri, K. Dridi, A. S. Al-Ayed, I. Ozdemir, S. Yasar and N. Hamdi, J. Coord. Chem., 2019, 72, 2080–2090 CrossRef CAS.
- N. Gürbüz, E. Ö. Özcan, İ. Özdemir, B. Çetinkaya, O. Şahin and O. Büyükgüngör, Dalton Trans., 2012, 41, 2330 RSC.
- S. Yaşar, S. Çekirdek and İ. Özdemir, J. Coord. Chem., 2014, 67(7), 1236 CrossRef.
- E. A. Baude and R. P. Linstead, J. Chem. Soc., 1954, 3544 Search PubMed.
- S. Yaşar, E. O. Karaca, Ç. Şahin, İ. Özdemir, O. Şahin and O. Büyükgüng, J. Organomet. Chem., 2015, 789–790, 1 CrossRef.
- (a) M. Poyatos, J. A. Mata, E. Falomir, R. H. Crabtree and E. Peris, Organometallics, 2003, 22, 1110 CrossRef CAS; (b) R. Corbean, M. Sanau and E. Peris, Organometallics, 2007, 26, 3492 CrossRef; (c) D. Gnanamgari, A. Moores, E. Rajaseelan and R. H. Crabtree, Organometallics, 2007, 26, 1226 CrossRef CAS; (d) R. Castarlenas, M. A. Esteruelas and E. Onate, Organometallics, 2008, 27, 3240 CrossRef CAS; (e) D. Gnanamgari, E. L. O. Sauer, N. D. Schley, C. Butler, C. D. Incarvito and R. H. Crabtree, Organometallics, 2009, 28, 321 CrossRef CAS; (f) S. Kuhl, R. Schneider and Y. Fort, Organometallics, 2003, 22, 4184 CrossRef CAS.
- N. Tabet, Acetylcholinesterase inhibitors for Alzheimer's disease: anti-inflammatories in acetylcholine clothing!, Age Ageing, 2006, 35(4), 336 CrossRef CAS.
- O. Ismail, G. Yetkin, Ö. Özlem, K. Murat, D. Henri and B. Christian, Eur. J. Inorg. Chem., 2010, 1798 Search PubMed.
- (a) M. K. Kathiravan, A. B. Salake, A. S. Chothe, P. B. Dudhe, R. P. Watode, M. S. Mukta and S. Gadhwe, Bioorg. Med. Chem., 2012, 20, 5678 CrossRef CAS; (b) B. Zhai and X. Lin, Curr. Pharm. Biotechnol., 2011, 12, 1255 CrossRef CAS.
- S. K. Kalauni, M. I. Choudhary, A. Khalid, M. D. Manandhar, F. Shaheen and G. M. B. Atta-ur-Rahman, New cholinesterase inhibiting steroidal alkaloids from the leaves of Sarcococca coriacea of Nepalese origin, Chem. Pharm. Bull., 2002, 50, 1423 CrossRef CAS.
- W. A. T. Atta-ur-Rahman, S. A. Nawas and M. I. Choudhary, New cholinesterase inhibiting bisbenzylisoquinoline alkaloids from Cocculus pendulus, Chem. Pharm. Bull., 2004, 52, 802 CrossRef.
- (a) W. Ahmad, B. Ahmad, M. Ahmad, Z. Iqbal, M. Nisar and M. Ahmad, In vitro inhibition of acetylcholinesterase, butyrylcholinesterase and lipoxygenase by crude extract of Myricaria elegans Royle, J. Biol. Sci., 2003, 11, 1046 Search PubMed; (b) R. Re, N. Pellegrini, A. Proteggente, A. Pannala, M. Yang and C. Rice-evans, Free Radical Biol. Med., 1999, 26(10), 1231 CrossRef CAS.
- X. Song, et al. Development of PLGA Nanoparticles Simultaneously Loaded with Vincristine and Verapamil for Treatment of Hepatocellular Carcinoma, J. Pharm. Sci., 2010, 99, 4874 CrossRef CAS.
- E. J. De Beer and M. B. Sherwood, The paper-disc agar plate method for the assay of antibiotic substances, J. Bacteriol., 1945, 50, 459 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05605j |
| This journal is © The Royal Society of Chemistry 2019 |