
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGuanine deaminase provides evidence of the increased caffeine content during the piling process of pu'erh tea†
Si-an Pan‡
 ,
Ying Sun‡,
Mengmeng Li,
Wei-Wei Deng* and
Zheng-Zhu Zhang*
,
Ying Sun‡,
Mengmeng Li,
Wei-Wei Deng* and
Zheng-Zhu Zhang*
State Key Laboratory of Tea Plant Biology and Utilization, Anhui Agricultural University, 130 Changjiang West Road, Hefei, Anhui 230036, China. E-mail: dengweiwei@ahau.edu.cn; zzz@ahau.edu.cn; Fax: +86 551 65785471; Tel: +86 551 65785471
First published on 7th November 2019
Abstract
Wet piling is a key process for producing pu'erh tea because various components change under the action of microorganisms. Among these components, caffeine content is increased. Evidence has indicated a salvage pathway for caffeine biosynthesis in microbes, in which xanthine is methylated in the order of N-3 → N-1 → N-7. In addition, guanine can be used to synthesize xanthine through guanine deaminase (EC: 3.5.4.3). In this study, we investigated the variation in caffeine content during piling fermentation with supplementary guanine, 15N-labeled guanine and xanthine. We cloned the guanine deaminase gene (GUD1) from Saccharomyces cerevisiae (one dominant strain in piling fermentation). The results revealed that [15N]xanthine could be synthesized from [15N]guanine, and [15N]caffeine was also detected during piling with supplementary [15N]xanthine. Furthermore, ScGUD1 could catalyze the conversion of guanine to xanthine, which is likely to be methylated for caffeine synthesis under microorganism action. The obtained results revealed the mechanism underlying the increased caffeine content during piling of pu'erh tea.
1. Introduction
Currently, pu'erh teas are highly popular in China because of their attractive flavor and health promoting functions. The processing of pu'erh tea differs considerably from that of other teas such as green tea, black tea, white tea, yellow tea, and oolong tea. Pu'erh tea is post-fermented by bacterial and fungal fermentation in a warm and humid environment under controlled conditions; this process is called piling or wet piling. During the piling process, many compounds are varied to ultimately result in the specific smell and taste of pu'erh tea.1 Many microorganisms are propagated in large quantities during piling. Taxonomic analysis of the microbial community in pile fermentation of pu'erh tea revealed three dominant bacterial phyla and one dominant eukaryotic phylum, namely Ascomycota. Further taxonomic analysis of Eukaryota showed that Saccharomycetes is the most abundant class and Yarrowia, Saccharomyces, and Aspergillus are the three most abundant genera.2 A previous study supported the occurrence of fungal diversity during piling for pu'erh tea; Aspergillus niger, Saccharomyces cerevisiae, and Penicillium glabrum were found to be the dominant strains.3Microbes can alter the content of many tea components through their respiratory and metabolic effects.4 One report showed that polyphenol content decreased significantly during the pile up process.5 In addition, amino acid content decreases dramatically during the piling process.6 However, caffeine content increases during pile fermentation because of not only its own stable properties but also microbial metabolism.7,8
Caffeine content in different teas varies because of the different processing methods and because differing fresh leaves used.9 In nonfermented or lightly fermented tea, caffeine content remains fairly stable, whereas that in pu'erh tea increases substantially.10,11 The primary pathway of caffeine synthesis in tea plants involves one nucleoside hydrolysis reaction and three methylation reactions; first, xanthosine is methylated to 7-methylxanthosine, hydrolyzed to 7-methylxanthine, and then further methylated to theobromine and caffeine.12,13 However, some previous studies have posited that a salvage pathway for caffeine biosynthesis is likely present in microbes, with the methylation order of N-3 → N-1 → N-7,5 which is distinctive from caffeine biosynthesis in plants. This pathway was subsequently confirmed in Escherichia coli (E. coli).14 Massive accumulation of xanthine provides sufficient substrate for a salvage caffeine biosynthetic pathway. In addition, guanine—one of the basic nucleotides present in both DNA and RNA—can be used to synthesize xanthine through guanine deaminase.
The mechanism of increased caffeine content during piling in pu'erh tea has not been clearly illustrated until now. In this study, we investigated the variation in caffeine content during piling fermentation with supplementary guanine, 15N-labeled guanine, and 15N-labeled xanthine. Furthermore, we cloned the guanine deaminase gene GUD1 from S. cerevisiae15 and constructed recombinant pMAL-ScGUD1, pRSF-ScGUD1, and pZ8-ScGUD1 for enzyme activity determination. The obtained results may reveal the mechanism underlying the increased caffeine content during the piling process in pu'erh tea.
2. Materials and methods
2.1. Caffeine content variation during piling fermentation with supplementary guanine
Crude green teas were purchased from a tea shop. For imitating pu'erh tea, piling fermentation was conducted according to a previously described method with a slight modification.16 To promote microbial activity at the onset of piling, sterilized distilled water was scattered on the leaves until their water content reached 30–35%. Then, tea leaves were placed in an incubator at constant temperature (60 °C) and constant humidity (80%) to imitate the piling process for pu'erh tea production. Before piling, multiple concentrations of exogenous guanine (0–1 mg g−1) were added to tea samples. The tea leaves were turned over every two days to ensure homogeneity in the piling process. Moreover, none piled green tea were stored at 4 °C and used as control; sterilized tea leaves were undergone same conditions with the piled tea samples and also treated as control. The imitated pu'erh tea samples with 10- and 20 day piling were used for caffeine content determination.Caffeine was extracted from the samples by using the method described by Deng et al.17 with minor modifications. In brief, 1.00 g of the dried tea leaf sample was ground and extracted in 200 mL boiling water at 100 °C for 20 min. After centrifugation, the precipitate was re-extracted with 200 mL boiling water. The supernatants were combined and then finally metered to 500 mL. The samples were then filtered through a 0.22 μm membrane and used for high-performance liquid chromatography (HPLC) analysis.
The standards and sample extracts were analyzed using a Waters HPLC system (Waters, Milford, MA, USA) through a reversed-phase column (Waters; 100 Å, 5 μm, 4.6 mm × 250 mm), and the detection method was described previously.14
2.2. Tracer experiment of [15N]guanine and [15N]xanthine
[15N]guanine (15N5, 98%, Cambridge Isotope Laboratories, Woburn, MA, USA) and [15N]xanthine (15N2, 98%, Cambridge Isotope Laboratories, Woburn, MA, USA) were added to the tea samples at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000 (g g−1). Pile fermentation and sample preparation were performed as described in Section 2.1. Ultra-HPLC coupled with quadrupole time-of-flight mass spectrometry (MS) was performed to identify [15N]guanine and [15N]xanthine, and [15N]caffeine was analyzed using ultra-HPLC coupled with triple quadrupole MS through a reversed-phase column (Waters; 1.7 μm, 2.1 mm × 100 mm) maintained at 35 °C. The detection wavelength was set at 274 nm. The samples were eluted at a flow rate of 0.3 mL min−1, with an injection volume of 5 μL for the filtrate. The mobile phase consisted of 0.2% (v/v) acetic acid (A) in water and 100% methyl alcohol (B), and the gradient elution was as follows: 0–2 min, A: 94–94%; 2–3.5 min, A: 94–92%; 3.5–4.5 min; A: 92–90%; 4.5–7.5 min, A: 90–82%; 7.5–10.5 min, A: 82–74%; 10.5–13.5 min, A: 74–20%; 13.5–14.5 min, A: 20–20%; 14.5–16 min, A: 20–94%; 16–17 min, A: 94–94%.
:1000 (g g−1). Pile fermentation and sample preparation were performed as described in Section 2.1. Ultra-HPLC coupled with quadrupole time-of-flight mass spectrometry (MS) was performed to identify [15N]guanine and [15N]xanthine, and [15N]caffeine was analyzed using ultra-HPLC coupled with triple quadrupole MS through a reversed-phase column (Waters; 1.7 μm, 2.1 mm × 100 mm) maintained at 35 °C. The detection wavelength was set at 274 nm. The samples were eluted at a flow rate of 0.3 mL min−1, with an injection volume of 5 μL for the filtrate. The mobile phase consisted of 0.2% (v/v) acetic acid (A) in water and 100% methyl alcohol (B), and the gradient elution was as follows: 0–2 min, A: 94–94%; 2–3.5 min, A: 94–92%; 3.5–4.5 min; A: 92–90%; 4.5–7.5 min, A: 90–82%; 7.5–10.5 min, A: 82–74%; 10.5–13.5 min, A: 74–20%; 13.5–14.5 min, A: 20–20%; 14.5–16 min, A: 20–94%; 16–17 min, A: 94–94%.
2.3. Cloning of ScGUD1 and construction of recombinant of pMAL-ScGUD1 in E. coli
The previous report showed guanine deaminase catalyzed the conversion of guanine to xanthine in wild-type Saccharomyces cerevisiae,18 and GUD1 (YDL238c) encoded guanine deaminase.15 The sequence of ScGUD1 (GUD1 from S. cerevisiae) was acquired from GenBank (accession no. NM_001180298). General DNA manipulation in S. cerevisiae was performed using standard methods.19 Polymerase chain reaction (PCR)-specific primers with the BamHI restriction enzyme site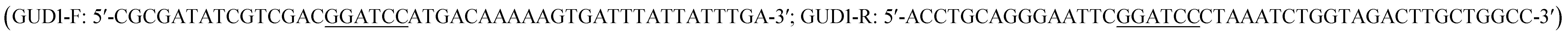 used for cloning were designed by CE Design V1.04 after bioinformatics analysis; the primers were then synthesized by General Biosystems, Inc. (Anhui, China).
used for cloning were designed by CE Design V1.04 after bioinformatics analysis; the primers were then synthesized by General Biosystems, Inc. (Anhui, China).
To obtain the recombinant pMAL-ScGUD1, we purified this amplified open reading frame (ORF) of ScGUD1, using the AxyPrep DNA Gel Extraction Kit (Axygen, USA). Later, ScGUD1 was linked with pMAL-c5X vector. And the recombinant expression plasmid was constructed and transformed into the expression host bacteria E. coli (BL21) (TransGen Biotech) since the ligated cloned fragment had been confirmed.
2.4. In vitro and in vivo enzyme assay of pMAL-ScGUD1 in E. coli
For comparison, the pMAL-c5X vector served as a control. The cells of transformants were cultured overnight at 37 °C in lysogeny broth media which contained ampicillin (100 μg mL−1). The cells were further cultured at 37 °C to an OD600 of 0.6–0.8; subsequently, in order to induce the expression of recombinant protein, 1.0 M isopropyl β-D-1-thiogalactopyranoside (IPTG) was added (final concentration, 1.0 mM). The samples without IPTG induction were treated as controls. The cells were incubated overnight at 16 °C for 20 h. Bacterial cells were harvested through centrifugation at 3381 × g for 10 min at 4 °C and resuspended in 1× phosphate-buffered saline (PBS; pH 7.4–7.6). Subsequently, the strains were disrupted on ice through ultrasonication, and then the supernatant and precipitate were collected and boiled for 8 min for sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) analysis; the cells without induction were used as controls. Moreover, the cells with or without induction were resuspended in PBS (pH 7.0) and boiled for 8 min to obtain total crude proteins.Supernatants from the induced cells were performed to measure the activity of recombinant pMAL-ScGUD1 after purification on a maltose-binding protein (MBP) column. The mixture for the enzyme activity reaction was prepared using the following components: 10 mM MgCL2; 3.3 mM guanine solution; and 200 μL purified protein. The mixture was adjusted to a final volume of 5.0 mL. The reaction was incubated at 37 °C for 0, 5, 10, 30, and 60 min before enzymatic product determination using HPLC.
For in vivo enzymatic activity determination, strains were cultured at 37 °C with shaking at 200 rpm for 120 h to determine the in vivo activities of the ScGUD1 with supplementary guanine (10 μM). The samples of the cultures were centrifuged at 13680 × g for 10 min, and culture supernatants were analyzed through HPLC, as described previously.14
2.5. Recombinant pRSF-ScGUD1 in E. coli and its enzyme assay
PCR-specific primers with BamHI and PstI restriction enzyme sitesemployed for cloning were designed by CE Design V1.04. The purified PCR product was ligated into the pRSF-Duet-1 vector (BioVector NTCC Inc., Beijing, China). The plasmid containing recombinant pRSF-ScGUD1 was introduced into E. coli (BL21) for enzyme activity determination. The enzyme activity reaction was conducted as described above, and the HPLC determination was described previously.14
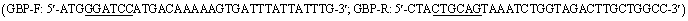
2.6. Recombinant pZ8-ScGUD1 in E. coli and its enzyme assay
Specific primers with PstI and SalI restriction enzyme sites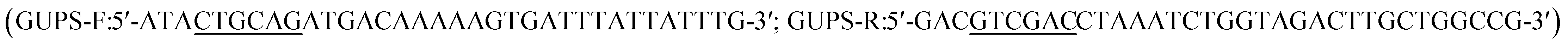 employed for cloning were designed by CE Design V1.04. The purified PCR product was ligated into the pZ8-1 vector (provided by Dr Oliver Yu at Wuxi NewWay Biotechnology, China). The plasmid containing recombinant pZ8-ScGUD1 was transformed into E. coli trans-T1 and then to Corynebacterium glutamicum ATCC 13032 through electroporation. The enzyme activity reaction was conducted as described above, and the HPLC determination was described previously.14
employed for cloning were designed by CE Design V1.04. The purified PCR product was ligated into the pZ8-1 vector (provided by Dr Oliver Yu at Wuxi NewWay Biotechnology, China). The plasmid containing recombinant pZ8-ScGUD1 was transformed into E. coli trans-T1 and then to Corynebacterium glutamicum ATCC 13032 through electroporation. The enzyme activity reaction was conducted as described above, and the HPLC determination was described previously.14
2.7. Statistical analysis
Data represent the mean ± standard deviations of three independent experiments. The significance differences between different groups were measured by two-way ANOVA post hoc Bonferroni test and one-way ANOVA post hoc Bonferroni test, as appropriate. All statistical analyses were considered statistically significant, which p values of <0.05.3. Results and discussion
3.1. Caffeine content variation during piling fermentation with exogenous guanine
Green teas were used to pile pu'erh tea under specified conditions in an incubator. During piling, caffeine content increased in the imitated pu'erh tea samples (Fig. 1). In the sterilized tea leaves undergone same conditions with the piled tea samples, caffeine content was almost consistent after 20 days of piling. A higher concentration of guanine yielded higher caffeine content in the piled tea samples. In the control sample without supplementary guanine, caffeine content slightly increased after 10 days of piling (from 2.55% ± 0.03% to 2.57% ± 0.04%) but did not continue to increase until 20 days of piling (2.57% ± 0.08%). After piling for 20 days, caffeine content increased in the guanine-supplied samples (0.01–1 mg g−1) higher than without guanine supplement; the highest caffeine content (2.65% ± 0.03%) was observed in the 1 mg g−1 guanine-supplied sample; 2.63% ± 0.03% and 2.58% ± 0.02% caffeine were found in the 0.1 mg g−1 and 0.01 mg g−1 guanine-supplied samples, respectively. The caffeine content showed significant correlation to the fermentation time (p = 0.007); caffeine content did not differ significantly at different guanine concentrations (p = 0.428) (P < 0.05).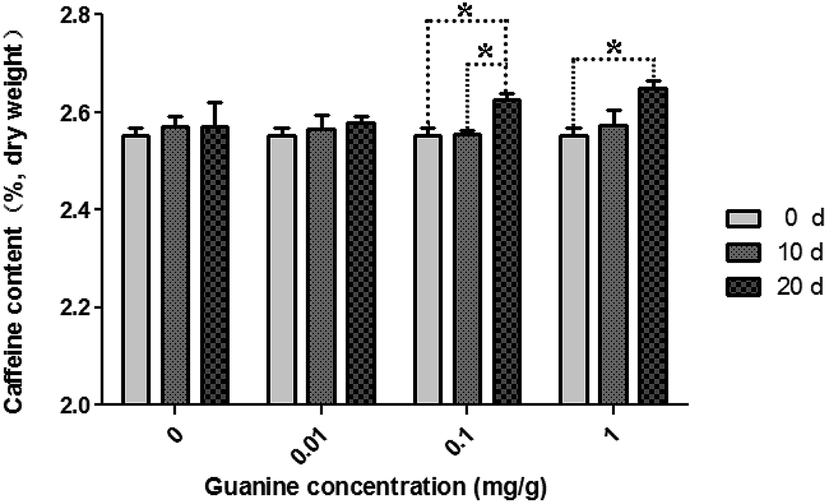 | ||
| Fig. 1 Variation in caffeine content during piling fermentation with multiple concentrations of exogenous guanine. The significances of differences were compared between the groups of fermentation time on different guanine concentrations (*p < 0.05). Data represent the mean ± standard deviation of three independent experiments. | ||
In a previous study, after fermentation by microorganisms (piling), the level of caffeine in ripened pu'erh tea was elevated compared with that in aged pu'erh tea.8 Ripened pu'erh tea is artificially fermented for 6 months to 1 year by microorganisms from raw pu'erh, whereas aged pu'erh tea is pressed into cake from raw pu'erh and stored in natural conditions. Our investigation revealed that caffeine content increased during the piling process for ripened pu'erh tea, especially in the samples with supplementary guanine. This finding indicated that caffeine may be synthesized from guanine under microorganism action during the process of pile fermentation for ripened pu'erh tea because the endogenous enzymes in tea leaves had already been deactivated by the traditional process referred to as “kill green” in raw pu'erh tea processing (or green tea processing).
3.2. [15N]xanthine and [15N]caffeine were detected in tea samples supplied with [15N]guanine and [15N]xanthine during piling
Each of [15N]guanine and [15N]xanthine was separately added to the tea samples (1:1000, g g−1) for piling fermentation (20 days). After sample preparation, [15N]xanthine (m/z, 153 + 4) was detected through LC-MS-MS (Fig. 2), as was [15N]caffeine (m/z, 195 + 2) (Fig. 3). This finding showed that the possible pathway of guanine → xanthine →→→ caffeine occurred during pile fermentation by microorganisms for ripened pu'erh tea. We have reported “guanine-to-caffeine” conversion in E. coli.14 Moreover, some previous studies have revealed that a salvage pathway for caffeine biosynthesis is likely present in microbes, with the methylation order of N-3 → N-1 → N-7.7 The obtained results suggested that guanine—a basic nucleotide present in both DNA and RNA—can be used to synthesize xanthine, and xanthine can be used for further caffeine biosynthesis during the piling process for pu'erh tea (Fig. 4).
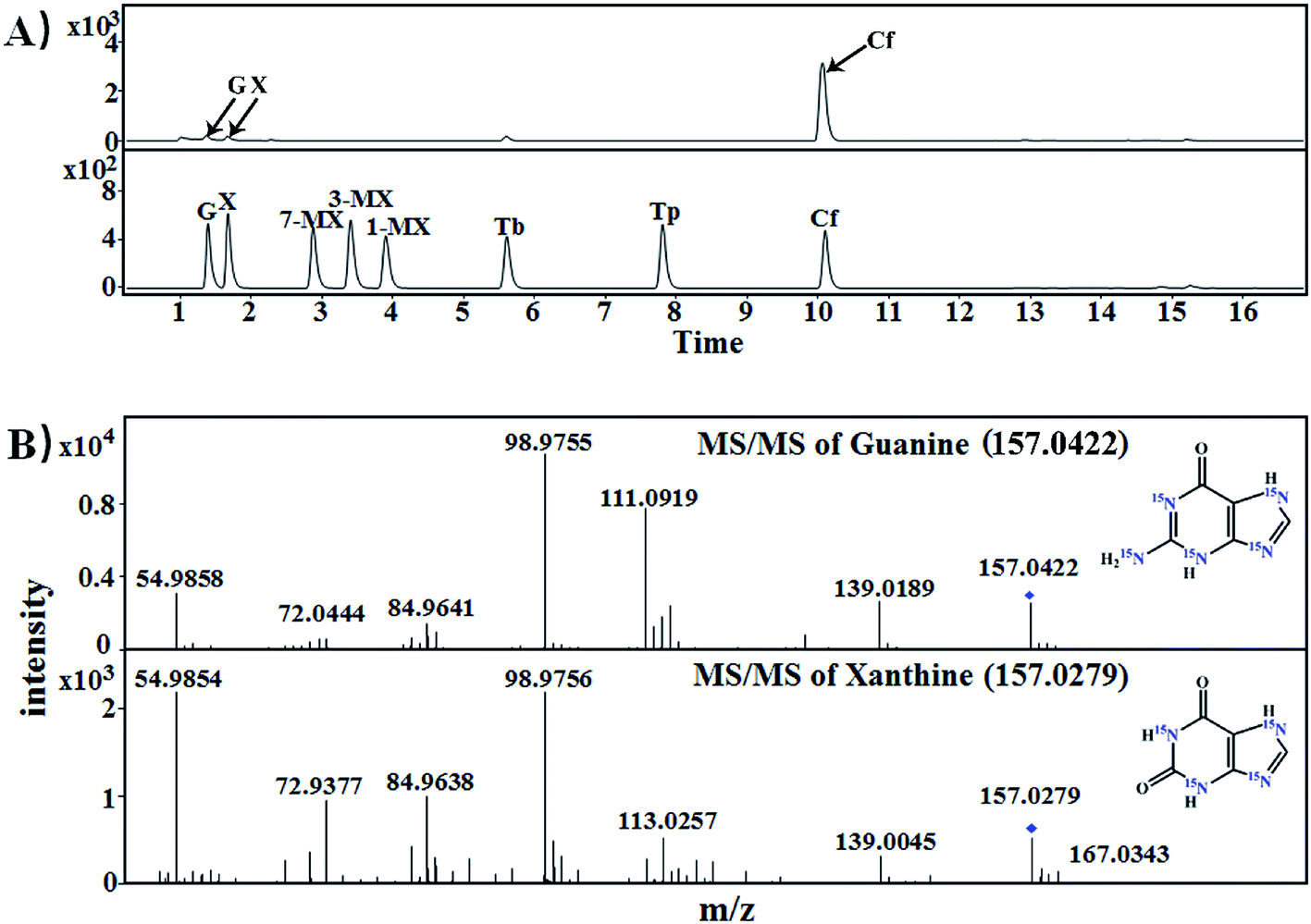 | ||
| Fig. 2 Mass spectra of [15N]xanthine in samples of imitated pu'erh tea for piling with exogenous [15N]guanine. (A) LC-MS chromatograms of purine standards and piling fermented tea samples; (B) identification of [15N]guanine (m/z: 157.0422) and [15N]xanthine (m/z: 157.0279) in imitated pu'erh tea samples. G, guanine; X, xanthine; 1-MX, 1-methylxanthine; 3-MX, 3-methylxanthine; 7-MX, 7-methylxanthine; Tb, theobromine; Tp, theophylline; Cf, caffeine. | ||
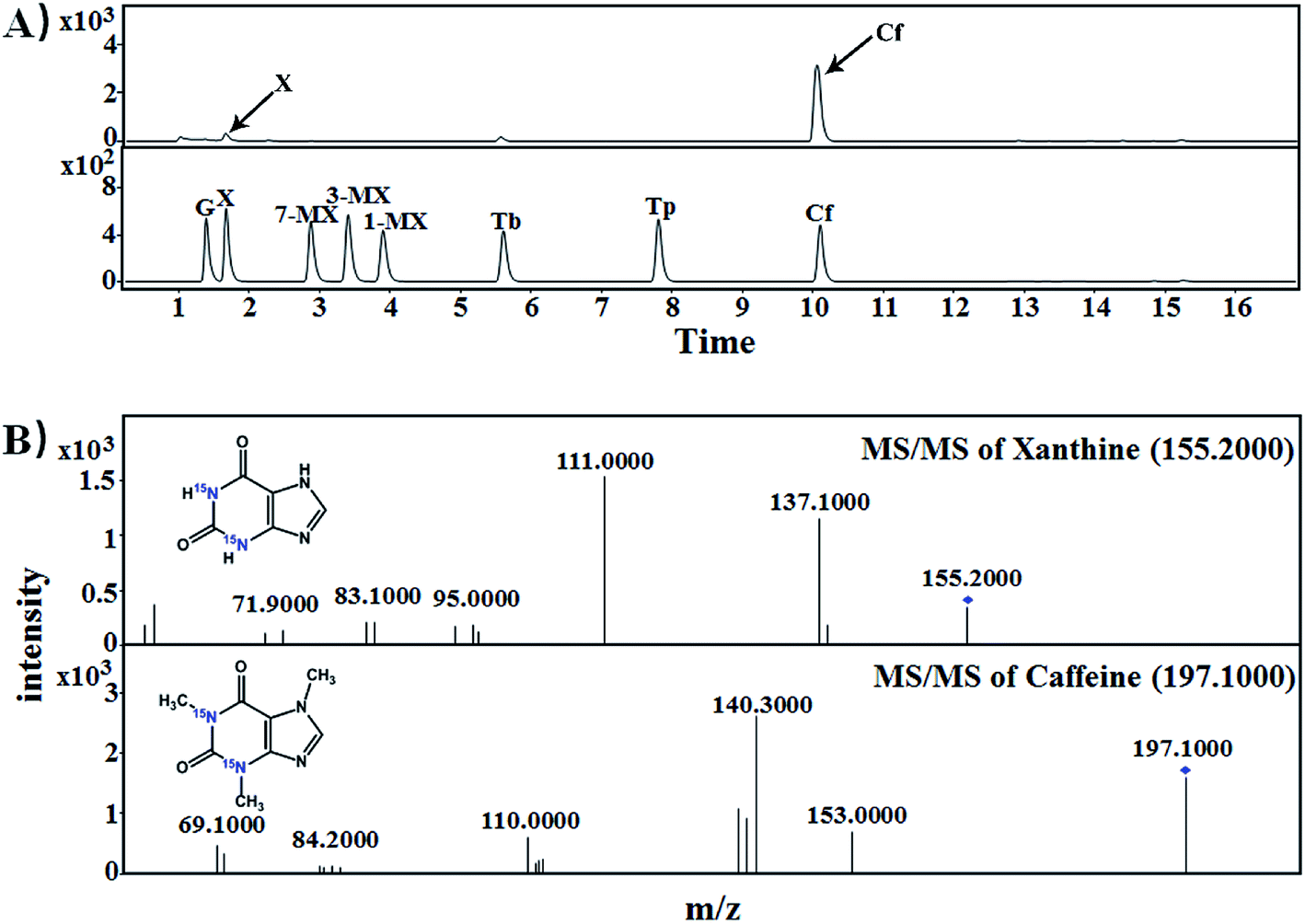 | ||
| Fig. 3 Mass spectra of [15N]caffeine in samples of imitated pu'erh tea samples for piling with exogenous [15N]xanthine. (A) LC-MS chromatograms of purine standards and piling fermented tea samples; (B) MS-MS spectra identification of [15N]xanthine (m/z: 155.2000) and [15N]caffeine (m/z: 197.1000). G, guanine; X, xanthine; 1-MX, 1-methylxanthine; 3-MX, 3-methylxanthine; 7-MX, 7-methylxanthine; Tb, theobromine; Tp, theophylline; Cf, caffeine. | ||
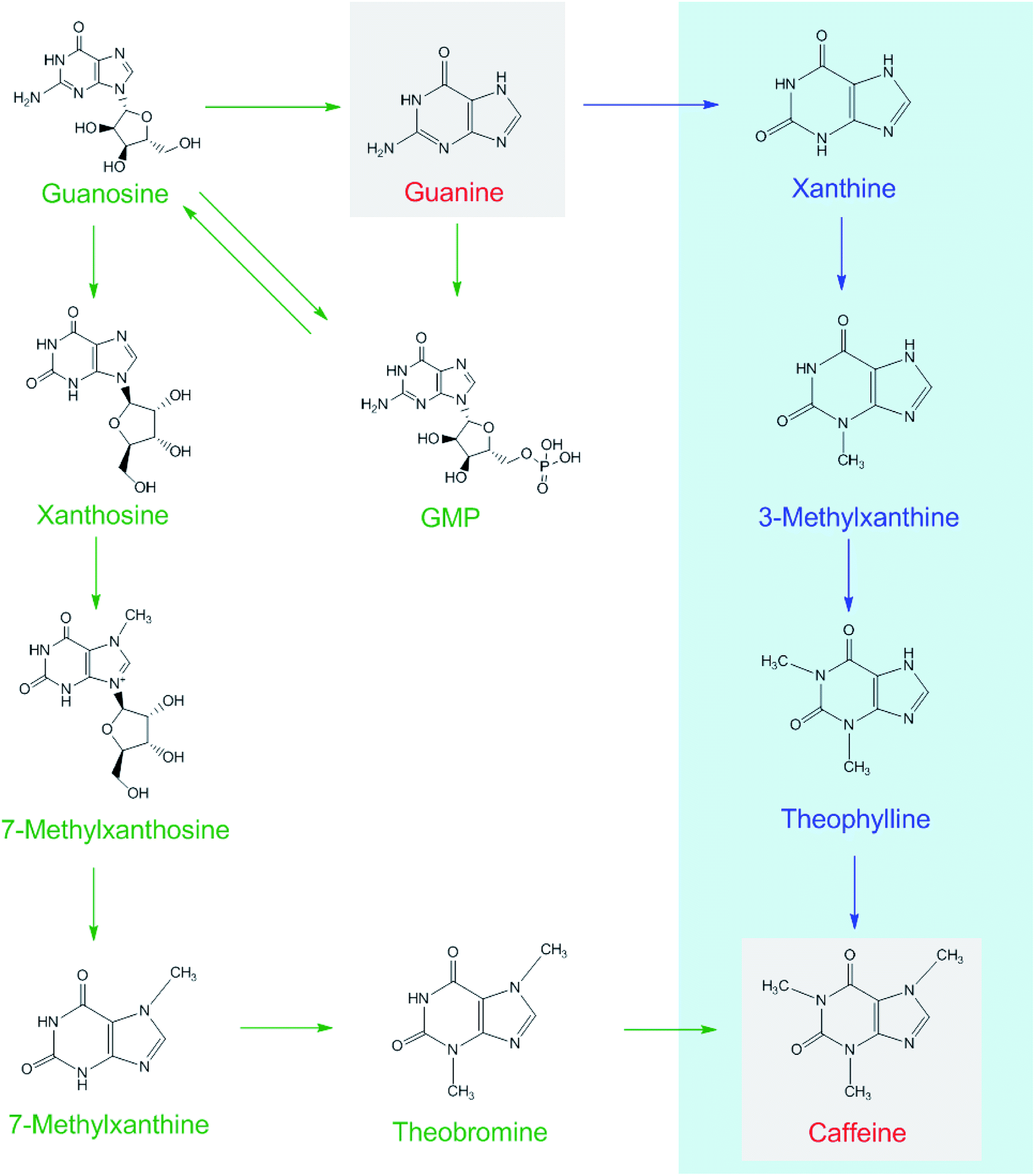 | ||
| Fig. 4 Possible caffeine biosynthetic pathways in tea plants (shown as green arrows) and in the piling process of pu'erh tea under microorganism action (blue arrows). | ||
3.3. Enzyme activity of recombinant BL21/pMAL-ScGUD1
Guanine can be used to synthesize xanthine through guanine deaminase. The crystal structure of guanine deaminase in Bacillus subtilis was reported previously.20 A 50.2 kDa protein encoded guanine deaminase in E. coli; it had ability to catalyze guanine to synthesize xanthine and had a Km (15 μM) with the substrate of guanine.21We cloned the guanine deaminase gene GUD1 from S. cerevisiae (Fig. 5A) and constructed recombinant pMAL-ScGUD1 for enzyme activity determination. Expression of the recombinant pMAL-ScGUD1 in E. coli was induced through addition of IPTG (1.0 mM) and incubation at two temperatures (16 °C and 30 °C). The recombinant protein was detected in the supernatant with a molecular weight of 90 kDa, which was evaluated through SDS-PAGE analysis of bacterial lysates. The analysis revealed that inducing the expression of recombinant pMAL-ScGUD1 resulted in production of a protein with a molecular weight of approximately 50 kDa. The molecular weight of protein was consistent with that predicted by DNAStar software (Fig. 5B). Moreover, compared with induction for 8 h at 30 °C, more soluble protein expression was observed after induction for 20 h at 16 °C (Fig. 5B). The supernatants from the cells were used to examine the activity of recombinant pMAL-ScGUD1 after purification on an MBP column (Fig. 5C). The purified protein was used for further in vitro enzymatic assays. The reactions were terminated and measured using HPLC. As shown in Fig. 5D, xanthine was detected in a reaction catalyzed by recombinant pMAL-ScGUD1 with guanine as the substrate but was not detected in the control strain with pMAL only.
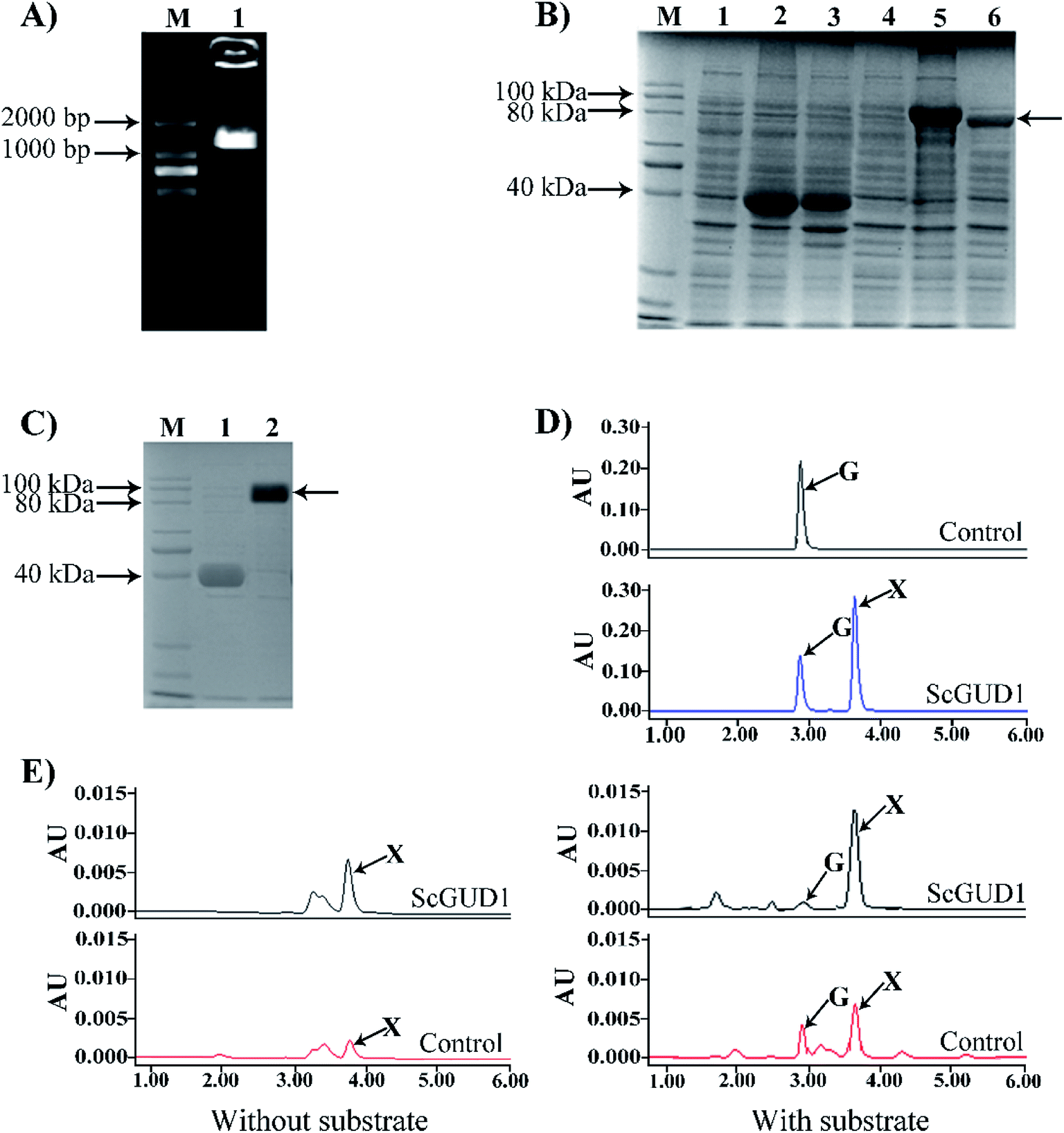 | ||
| Fig. 5 Cloning of ScGUD1, construction of recombinant of pMAL-ScGUD1, and enzyme assay in E. coli. (A) Agarose gel electrophoresis of PCR-amplified ScGUD1. M, DNA marker; (1) PCR product of ScGUD1. (B) SDS-PAGE electrophoresis of recombinant pMAL-ScGUD1. M, Protein marker; (1) total crude protein from the induced cells was transformed using the pMAL-c5X vector alone; (2) suspension of total crude protein from the induced cells using the pMAL-c5X vector by 16 °C; (3) suspension of total crude protein from the induced cells using the pMAL-c5X vector by 30 °C; (4) total crude protein from the induced cells was transformed using the pMAL-ScGUD1; (5) suspension of total crude protein from the induced cells using the pMAL-ScGUD1 by 16 °C; (6) suspension of total crude protein from the induced cells using the pMAL-ScGUD1 by 30 °C. (C) Purification of recombinant pMAL-ScGUD1. M, Protein marker; (1) purified MBP from suspension of total crude protein; (2) purified pMAL-ScGUD1 from suspension of total crude protein. (D) Enzyme assay of recombinant of pMAL-ScGUD1 in vitro. The reactions of recombinant pMAL-ScGUD1 and the control strain were measured through HPLC. (E) In vivo enzyme activity determination of the recombinant pMAL-ScGUD1. The reactions of recombinant pMAL-ScGUD1 and the control strain were measured with and without exogenous guanine through HPLC. | ||
For in vivo enzyme activity determination, strains were cultured at 37 °C and 200 rpm for 120 h to determine the in vivo activities of ScGUD1 with or without guanine supplementation. Xanthine was detected in the strains with pMAL only and pMAL-ScGUD1 (Fig. 5E), but the recombinant strain with pMAL-ScGUD1 consumed guanine far more quickly and synthesized xanthine to a far greater extent than did the control strains. When exogenous guanine was added, 312 μg mL−1 of xanthine was observed in the recombinant strain with pMAL-ScGUD1, and 180 μg mL−1 xanthine was observed in the control strain. However, when no guanine was added, 225 μg mL−1 of xanthine was observed in the recombinant strain with pMAL-ScGUD1, and 110 μg mL−1 of xanthine was observed in the control strain. Both the in vitro and in vivo enzyme assays of pMAL-ScGUD1 in E. coli demonstrated that ScGUD1 catalyzed the conversion of guanine to xanthine.
3.4. Enzyme activity of BL21/pRSF-ScGUD1 recombinant strain
The plasmid containing recombinant pRSF-ScGUD1 was transformed into E. coli (BL21) for further enzyme activity determination, and enzyme activity was measured in the control of pRSF-Duet1. For enzyme activity determination, strains were cultured at 37 °C and 200 rpm for 120 h to determine the in vivo activities of the recombinant pRSF-ScGUD1 with or without guanine (Fig. 6A). The concentrations of xanthine were 117 and 216 μg mL−1 in the BL21/pRSF-Duet1 control strain without or with exogenous guanine, respectively. However, 233 and 429 μg mL−1 of xanthine were found in the BL21/pRSF-ScGUD1 recombinant strain without or with exogenous guanine, respectively. Moreover, the recombinant strain with pRSF-ScGUD1 consumed guanine far more quickly than did the control strain.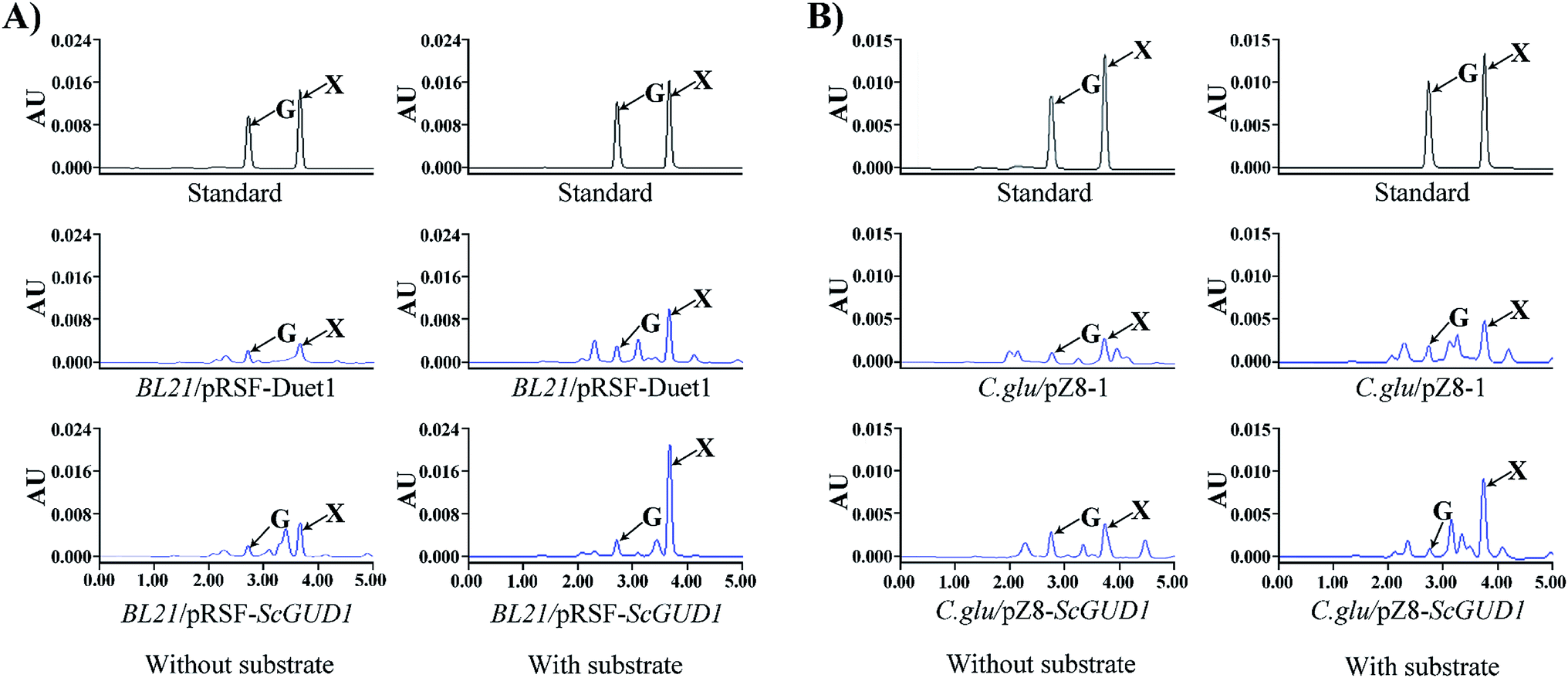 | ||
| Fig. 6 In vivo activity determination of recombinant pRSF-ScGUD1 (A) and pZ8-ScGUD1 (B) with or without exogenous guanine through HPLC. | ||
A comparison of BL21/pRSF-ScGUD1 and BL21/pMAL-ScGUD1 revealed that xanthine formation had increased by 37.5% after 120 h incubation with the same amount of additional substrate and by 23.9% without supplementary guanine. We knew that the pMAL vector contains the lac promoter, and that the pRSF vector contains the T7 promoter. The increased enzyme activities of the recombinant proteins were likely related to differences in promoter efficiency between T7 and Lac. The T7 promoter is a strong promoter that cannot be recognized by E. coli RNA polymerase but can only be specifically recognized by T7 RNA polymerase.22 The starting strain E. coli BL21 (DE3) contains the specifically added T7 polymerase gene; thus, some exogenous genes are more suitable for controlling the T7 expression system. In addition, the PRSF-Duet1 vector used in this study contains two independent T7 promoters and thus is more conducive for studying multiple gene tandem expressions at a later stage.
3.5. Enzyme activity of C. glu/pZ8-ScGUD1 recombinant strain
Corynebacterium glutamicum is a rod-shaped Gram-positive nonpathogenic L-glutamic acid-producing strain that was originally found in soil by Japanese scientists.23 In this study, the plasmid with recombinant pZ8-ScGUD1 was transformed into C. glutamicum for further enzyme activity determination, and enzyme activity was also examined in the control pZ8-1 strain. Strains were cultured at 30 °C and 200 rpm for 120 h to determine the in vivo activities of the recombinant pZ8-ScGUD1 with or without guanine (Fig. 6B). For the C. glu/pZ8-1 control strain, 135 and 237 μg mL−1 of xanthine were detected without and with exogenous guanine supplementation, respectively. However, the concentrations of xanthine were 168 and 291 μg mL−1 in the C. glu/pZ8-ScGUD1 recombinant strain without or with exogenous guanine, respectively.Based on the results, the highest efficiency of xanthine synthesis was observed in the BL21/pRSF-ScGUD1 recombinant strain compared with the BL21/pMAL-ScGUD1 and C. glu/pZ8-ScGUD1 strains. The rate of xanthine accumulation in the C. glu/pZ8-ScGUD1 recombinant strain was lower than that in the BL21/pMAL-ScGUD1 and BL21/pRSF-ScGUD1 strains, likely because the efficiency of the expression system in C. glutamicum was lower than that of the E. coli expression system.
4. Conclusion
In this study, we investigated the variation in caffeine content during piling fermentation with exogenous guanine. We confirmed that caffeine content increased during piling fermentation. The stable isotope of [15N]guanine was added to crude green teas before piling, and [15N]xanthine was detected. In addition, [15N]caffeine was observed in the imitated pu'erh tea samples during piling fermentation with supplementary [15N]xanthine under microorganism action. Guanine can be used to synthesize xanthine through guanine deaminase. Subsequently, we cloned the guanine deaminase gene GUD1 from S. cerevisiae and constructed the recombinants pMAL-ScGUD1, pRSF-ScGUD1, and pZ8-ScGUD1 to investigate ScGUD1 activity. The highest efficiency of xanthine synthesis was observed in the recombinant strain with BL21/pRSF-ScGUD1 compared with the BL21/pMAL-ScGUD1 and C. glu/pZ8-ScGUD1 strains. The obtained results revealed the mechanism underlying the increased caffeine content during the piling process in pu'erh tea.Conflicts of interest
The authors declare that they have no competing interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) project 31570692, and the Changjiang Scholars and Innovative Research Team in University (IRT_15R01).References
- C. S. Chen, H. C. Chan, Y. N. Chang, B. L. Liu and Y. S. Chen, J. Sens. Stud., 2009, 24(4), 534–553 CrossRef.
- C. Lyu, C. Chen, F. Ge, D. Liu, S. Zhao and D. Chen, J. Sci. Food Agric., 2013, 93(13), 3165–3174 CrossRef CAS.
- Y. Zhang, S. Zhao, H. Liang, W. Li, T. Zhao and C. Li, China Brew., 2012, 31(1), 122–125 CrossRef CAS.
- Y. S. Chen, B. L. Liu and Y. N. Chang, J. Biosci. Bioeng., 2010, 109(6), 557–563 CrossRef CAS.
- J. H. Qin, N. Li, P. F. Tu, Z. Z. Ma and L. Zhang, J. Agric. Food Chem., 2012, 60(5), 1213–1217 CrossRef CAS.
- Y. C. Zhu, Y. H. Luo, P. P. Wang, M. Y. Zhao, L. Li, X. S. Hu and F. Chen, Food Chem., 2016, 194, 643–649 CrossRef CAS.
- X. G. Wang, X. C. Wan, S. X. Hu and C. Y. Pan, Food Chem., 2008, 107(3), 1086–1091 CrossRef CAS.
- L. Zhang, N. Li, Z. Z. Ma and P. F. Tu, J. Agric. Food Chem., 2011, 59(16), 8754–8760 CrossRef CAS.
- J. M. Ning, D. X. Li, X. J. Luo, D. Ding, Y. S. Song, Z. Z. Zhang and X. C. Wan, Food Anal. Methods, 2016, 9(11), 3242–3250 CrossRef.
- M. Z. Liang, L. F. Xia, L. B. Chen, C. G. Fang, Z. F. Duan, J. W. Chen, S. M. Yang and R. Q. Sun, Chinese Agricultural Science Bulletin, 2006, 22(10), 321–325 Search PubMed.
- Y. Tian, R. Xiao, K. l. Xu, B. Jiang and C. Y. Shi, Food Sci., 2010, 31(11), 20–24 CAS.
- H. Ashihara, H. Sano and A. Crozier, Phytochemistry, 2008, 69(4), 841–856 CrossRef CAS.
- H. Ashihara and T. Suzuki, Front. Biosci., 2004, 9, 1864–1876 CrossRef CAS.
- M. M. Li, Y. Sun, S. A. Pan, W. W. Deng, O. Yu and Z. Z. Zhang, RSC Adv., 2017, 7(89), 56382–56389 RSC.
- C. Saint-Marc and B. Daignan-Fornier, Yeast, 2004, 21(16), 1359–1363 CrossRef CAS.
- M. Abe, N. Takaoka, Y. Idemoto, C. Takagi, T. Imai and K. Nakasaki, Int. J. Food Microbiol., 2008, 124(2), 199–203 CrossRef CAS.
- W. W. Deng, M. Li, C. C. Gu, D. X. Li, L. L. Ma, Y. Jin and X. C. Wan, Nat. Prod. Commun., 2015, 10(5), 789–792 Search PubMed.
- P. W. Burridge, R. A. Woods and J. F. Henderson, Can. J. Biochem., 1977, 55(9), 935–941 CrossRef CAS.
- L. Chong, Science, 2001, 292(5516), 446 CrossRef CAS.
- S. H. Liaw, Y. J. Chang, C. T. Lai, H. C. Chang and G. G. Chang, J. Biol. Chem., 2004, 279(34), 35479–35485 CrossRef CAS.
- J. T. Maynes, R. G. Yuan and F. F. Snyder, J. Bacteriol., 2000, 182(16), 4658–4660 CrossRef CAS.
- J. G. G. Vethanayagam and A. M. Flower, Microb. Cell Factories, 2005, 4(1), 1–7 CrossRef.
- S. Abe, K. I. Takayama and S. Kinoshita, J. Gen. Appl. Microbiol., 1967, 13(3), 279–301 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05655f |
| ‡ These authors contributed equally in this work. |
| This journal is © The Royal Society of Chemistry 2019 |