DOI:
10.1039/C9RA05687D
(Review Article)
RSC Adv., 2019,
9, 30835-30867
Bicyclic 6 + 6 systems: the chemistry of pyrimido[4,5-d]pyrimidines and pyrimido[5,4-d]pyrimidines
Received
23rd July 2019
, Accepted 17th September 2019
First published on 30th September 2019
Abstract
The present study provides an overview of the chemistry and biological significance of pyrimido[4,5-d]pyrimidine and pyrimido[5,4-d]pyrimidine analogs as types of bicyclic [6 + 6] systems. The main sections include: (1) synthesis methods; (2) the reactivities of the substituents linked to the ring carbon and nitrogen atoms; and (3) biological applications. A discussion demonstrating the proposed mechanisms of unexpected synthetic routes is intended. The aim of this study is to discuss the synthetic significance of the titled compounds and to establish the biological characteristics of this class of compounds as studied to date, where the compounds have been applied on a large scale in the medical and pharmaceutical fields. This survey will help researchers in the fields of synthetic organic and medicinal chemistry to undertake and improve new approaches for the construction of new standard biological components.
 M. Monier | Dr Mohammed Monier Mohammed Ibrahim received a BSc in Chemistry (2000), a MSc in Organic Chemistry (2005) and a PhD in Organic and Polymer Chemistry (2011) from Faculty of Science, Mansoura University, Egypt. His main research field is bioengineering and applied chemistry and his main research interests include the uses of natural and synthetic polymers in biomedical and environmental applications. Dr M. Monier is currently an Associate professor at Mansoura University, Faculty of Science, Chemistry Department, Egypt (Tenure-Permanent) and at Taibah University, Chemistry Department, Yanbu, KSA (Contracted). Previously he was a Demonstrator in the Chemistry Department (2000–2005), an Assistant Lecturer (2005–2009), a research assistant in the Chemistry Department at Drexel University, Philadelphia (2009–2011), an assistant professor in the Chemistry Department (2011–2014), and a Fulbright Visiting Scholar in the Chemistry Department at Drexel University (2015). |
 Doaa Abdel-Latif | Dr Doaa Ahmed Abdel-Latif received a BSc in Chemistry (2000), a MSc in Inorganic Chemistry (2005), and a PhD in Inorganic Chemistry (2011) from Faculty of Science, Mansoura University, Egypt. Her main research field is metal complexes and their applications and her main research interests include the synthesis of metal complexes and their uses in biomedical and environmental applications. Dr D. Abdel-Latif is currently an Assistant professor at Mansoura University, Faculty of Science, Chemistry Department, Egypt (Tenure-Permanent) and at Taibah University, Chemistry Department, Yanbu, KSA (Contracted). She was a Demonstrator in the Chemistry Department (2000–2005), an Assistant Lecturer (2005–2009), a research assistant in the Chemistry Department at Drexel University in Philadelphia (2010–2011), and is an assistant professor in the Chemistry Department (2011 to date). |
 Ahmed El-Mekabaty | Dr Ahmed El-Mekabaty Mohamed was born in 1982 in Salamoon El-Komash, Mansoura, Egypt. He received his BSc 2003 from the Faculty of Science, Mansoura University, and his MSc in 2007 (his thesis was on the synthesis of some novel heterocyclic compounds of pharmaceutical interest) from the same University. He obtained a PhD in Organic and Petroleum Chemistry in 2011 from Faculty of Science, Mansoura University, Egypt (PhD thesis advisor: Prof. O. M. O. Habib, Mansoura University). He is an Associate Professor of Organic and Petroleum Chemistry at the Faculty of Science at Mansoura University, Egypt. He is a member of the American Chemical Society and acts as a reviewer for international journals. His research focuses on the synthesis of heterocyclic compounds of pharmaceutical interest and also evaluating their efficiency as oxidation and corrosion inhibitors for lubricating oils. |
 Khaled M. Elattar | Dr Khaled M. Elattar was born in Menyet Sammanoud, Aga, El-dakahlia, Egypt (1979). He received his BSc in 2002 from the Faculty of Science, Mansoura University, Egypt and his MSc in 2006 from the Faculty of Science, Benha University, Egypt. He obtained his PhD in organic chemistry in 2011 from the Faculty of Science at Mansoura University (PhD supervisors: Prof. A. A. Fadda and Prof. A. S. Fouda). He was a Lecturer in Organic Chemistry at the Faculty of Education at Sert University from 2012–2015. He is a member of the Egyptian Chemical Society and is a reviewer for scientific international journals. His main research interests are in the field of organic synthesis, in particular the synthesis of heterocyclic compounds of pharmaceutical and medicinal chemistry interest. He joined the editorial board of the OA Journal - Pharmaceutics in 2018 (http://oa.enpress-publisher.com/index.php/Pha/about/editorialTeam). |
1. Introduction
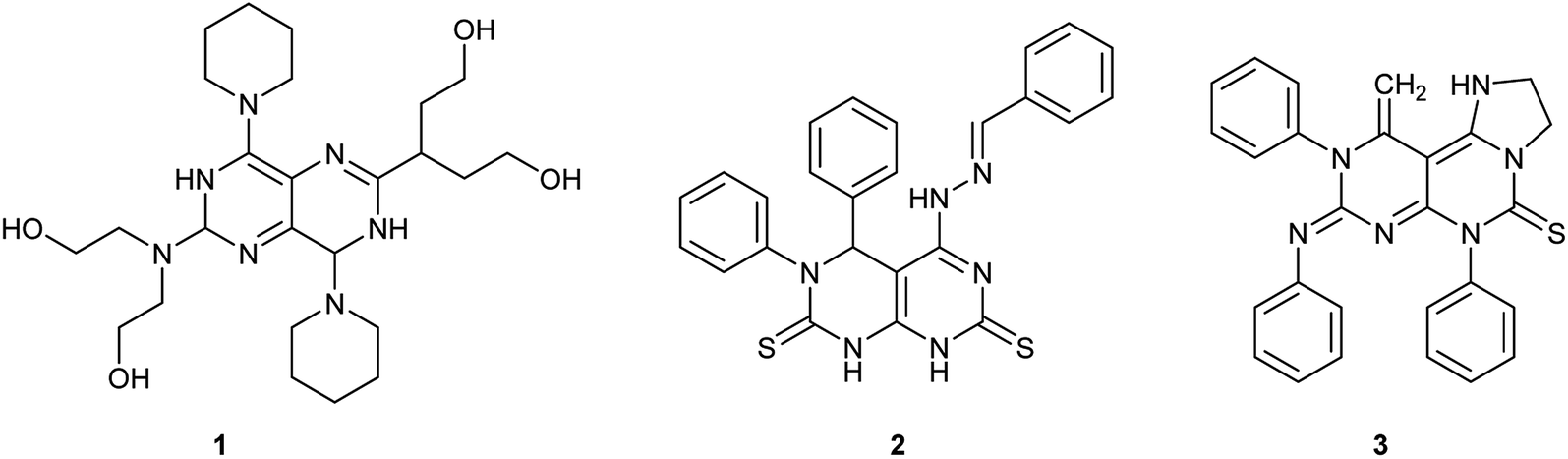
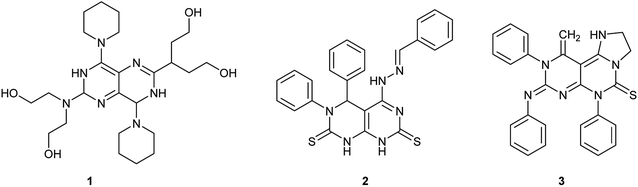
Pyrimidopyrimidines or tetra-azanaphthalenes are two fused pyrimidine rings with four possible structural isomers. Consequently, they are bicyclic compounds with two nitrogen atoms in each ring and a possible single nitrogen atom at the ring junction. Recently, the chemistry of pyrimidopyrimidines has gained great importance as a result of the diverse biological applications and the synthetic importance on the large scale. Accordingly, this class of compounds inhibits the growth of cancer cells,1 the deamination of adenosine caused by blood2 and the formation of thrombi in rabbits.3 Dipyridamole (Fig. 1) is a drug that prevents the formation of blood clots when given chronically but causes vasodilation in high doses over a short period of time. It is one of the most important biochemical agents, and was applied in vivo on a large scale, by synergistically improving synergistically the biochemical paths: (1) it acts as an inhibitor for cAMP-phosphodiesterase platelets; (2) supports adenosine inhibition of thrombocytopenia by preventing the absorption of vascular and blood cells; (3) strengthens PGI2 anti-aggregation activity and enhances the bio-synthesis of PGI2; and (4) decreases pulmonary hypertension.4–7
 |
| | Fig. 1 Structures of the potent bioactive components. | |
Furthermore, pyrimido[4,5-d]pyrimidines (Fig. 1) revealed potent activities as anticancer,8 antioxidant,9 dihydrofolate reductase,10 type 2 diabetes treatment,11 antiangiogenic,12 resistance-modifying,13 anti-myco-bacterium tuberculosis,14 antibacterial,15 antihypertensive,16 anti-inflammatory,17 anti-allergic,18,19 antitumor, antiviral,20–22 and hepatoprotective agents.23 Moreover, pyrimidopyrimidines are conveyed as receptors for tyrosine kinase24 and have been applied in the biosynthesis of 5-phosphoribosyl-1-pyrophosphate.25
Presently, the synthetic procedures of compounds that have a pyrimido[4,5-d]pyrimidine skeleton are variable using different routes, including: (1) the multicomponent condensation reaction of formaline with uracil analogs and amines which gives a product that has antidepressant activity;22 and (2) several procedures including the three-component condensation of aryl aldehydes, amines such as aniline, aminouracil, thiocyanate salts or aminopyrimidine derivatives and active methylenes under catalytic conditions in a basic or acidic medium,26–38 in addition to other reported synthetic approaches.39,40 On the other hand, pyrimidopyrimidines were synthesized using a regiospecific one-pot reaction under solvent-free and microwave irradiation conditions.41 The reported methods have several disadvantages, for example, the starting materials are not readily accessible or complex techniques, harsh conditions or multistep processes are required.42,43
The synthesis of the pyrimido[4,5-d]pyrimidines was first reported by Taylor et al.18 who reported the reaction of the 4-aminopyrimidine-5-carboxamides with 4-amino-5-cyanopyrimidines and the application of the products as diuretic agents. Burch et al.44 synthesized a series of N,N-disubstituted-2-(5-nitrofuran-2-yl)pyrimido[4,5-d]pyrimidin-4-amines by treating N-(5-cyanopyrimidin-4-yl)-5-nitrofuran-2-carboxamide with sulfuric acid followed by heating, intramolecular cyclization and subsequent chlorination with phosphorous pentasulfide/thionyl chloride and nucleophilic substitution with amines. Sharma et al. also reported the synthesis of 4,6-diaryl-3-(aryldiazenyl)-7-thioxo-4,6,7,8-tetra-hydro-pyrimido[4,5-d]pyrimidine-2,5-(1H,3H)-diones as antimicrobial agents37 or from a regioselective route by a Diels–Alder type cycloaddition from the reaction of a tetrahydropyrimidine derivative as a Biginelli type compound with N-arylidine-N′-methylformamidines and N-arylidine guanidine in toluene.45 In addition, Saravanan et al. reported the synthesis of a series of pyrimido[4,5-d]pyrimidines as nucleoside transport inhibitors with improved in vivo pharmacokinetic properties and reduced α1-acid glycoprotein (AGP) binding relative to dipyridamole.46 Recently, the investigated compounds were prepared from the reaction of 2-amino-4,6-dihydroxypyrimidine with 4,6-dihydroxy-pyrimidine using a multi-step synthesis process, in which the compounds have potential for use against in vitro HBV DNA replication inhibition and as nucleoside transport inhibitors.47 Patil et al.48a reported the synthesis of substituted dihydro-2H-dipyrimido[1,2-a,4,5-d]pyrimidine-2,4-(3H)-diones through a convenient route using the multicomponent reaction of barbituric acid with aryl aldehydes and 2-aminopyrimidine in refluxing ethanol in the absence of a catalyst. In addition, Delia48b reported the hydration and ring-opening of a series of 2,4-disubstituted-pyrimido[4,5-d]pyrimidines with the formation of 4-amino-5-formyl-2,6-disubstituted-pyrimidines. Recently, Hao et al.48c reported the synthesis of a series of pyrimido[4,5-d]pyrimidine-2,4(1H,3H)-diones as selective inhibitors for EGFR. Therefore, the multistep-synthesis was initiated by the protection of benzene-1,3-diamine followed by reaction with ethyl 2,4-dichloropyrimidine-5-carboxylate, arylamines, intramolecular cyclization, acid hydrolysis and substitution with acryloyl chloride under catalytic conditions.
The present review is an extension of our studies on the chemistry of heterocyclic compounds49–51 with the aim of establishing the synthetic and biological importance of the pyrimido[4,5-d]pyrimidines and pyrimido[5,4-d]pyrimidines that have been widely applied in the fields of medicinal and pharmaceutical chemistry.
2. Synthetic methods
2.1. Synthesis of substituted pyrimidopyrimidines
2.1.1. Synthesis from alkyl pyrimidine-carboxylates.
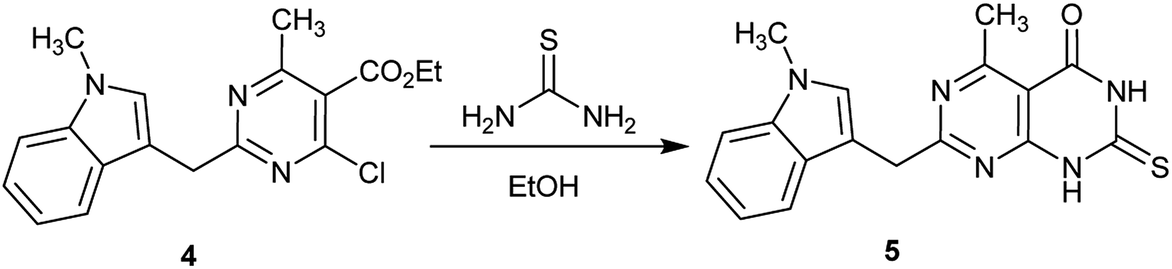
The reaction of ethyl 2-(1-methyl-1H-indol-3-yl)pyrimidine-5-carboxylate 4 with thiourea in refluxing ethanol containing triethylamine yielded 1H-indolyl-dihydropyrimido[4,5-d]pyrimidinone 5 (Scheme 1). The activity of compound 5 was evaluated as an analgesic and antiparkinsonian agent, and the compound had moderate results in both tests.52
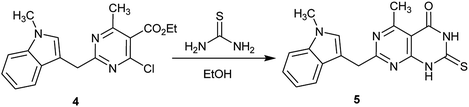 |
| | Scheme 1 Synthesis of 2-thioxo-2,3-dihydropyrimido[4,5-d]pyrimidin-4(1H)-one. | |
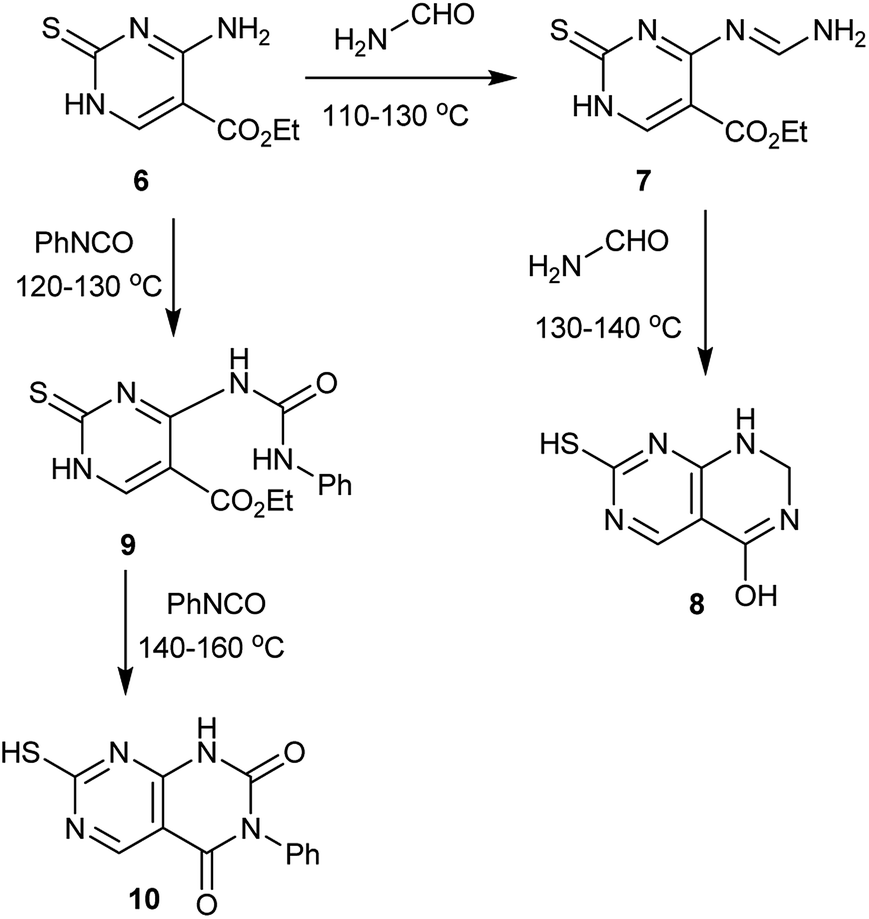
In another route, the temperature was the critical factor, below 110 °C no reaction takes place and above 140 °C, the products decompose with the formation of aminomercaptopyrimidine. Thus, heating ethyl 4-amino-2-thioxo-1,2-dihydropyrimidine-5-carboxylate (6) with formamide at 110–130 °C leads to the formation of the Schiff base intermediate 7, which reacted with another mole of formamide 130–140 °C to give 7-mercapto-1,2-dihydropyrimido[4,5-d]pyrimidin-4-ol (8). On the other hand, heating of 6 with phenyl isocyanate at 120–130 °C generates the amide intermediate 9, which reacted with another mole of phenyl isocyanate at 140–160 °C to give the desired compound 7-mercapto-3-phenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (10) (Scheme 2).53
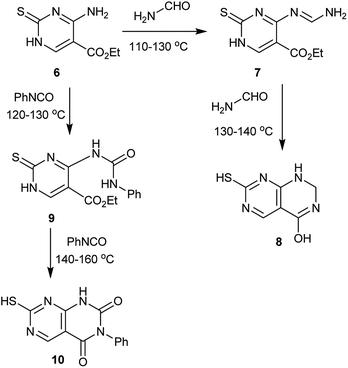 |
| | Scheme 2 Synthesis route from ethyl 4-amino-2-thioxo-1,2-dihydropyrimidine-5-carboxylate. | |
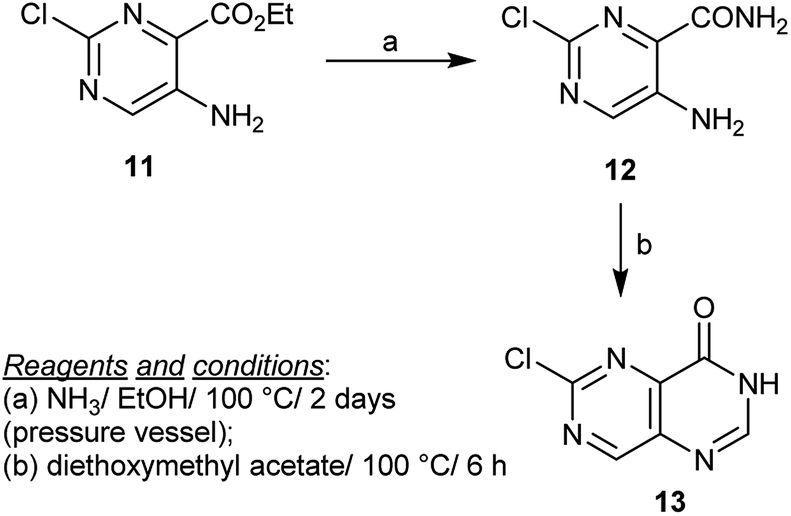
Heating of ethyl 5-amino-2-chloropyrimidine-4-carboxylate (11) with ammonia in ethanol yielded the respective amide 12. The amide 12 reacted with triethyl orthoformate or with diethoxymethyl acetate to give the cyclization product 13 in a much higher yield, in the case of diethoxymethyl acetate two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds and one C–N bond were formed (Scheme 3).24
N bonds and one C–N bond were formed (Scheme 3).24
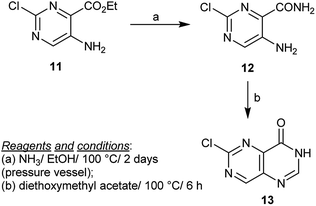 |
| | Scheme 3 Synthesis of 6-chloropyrimido[5,4-d]pyrimidin-4(3H)-one. | |
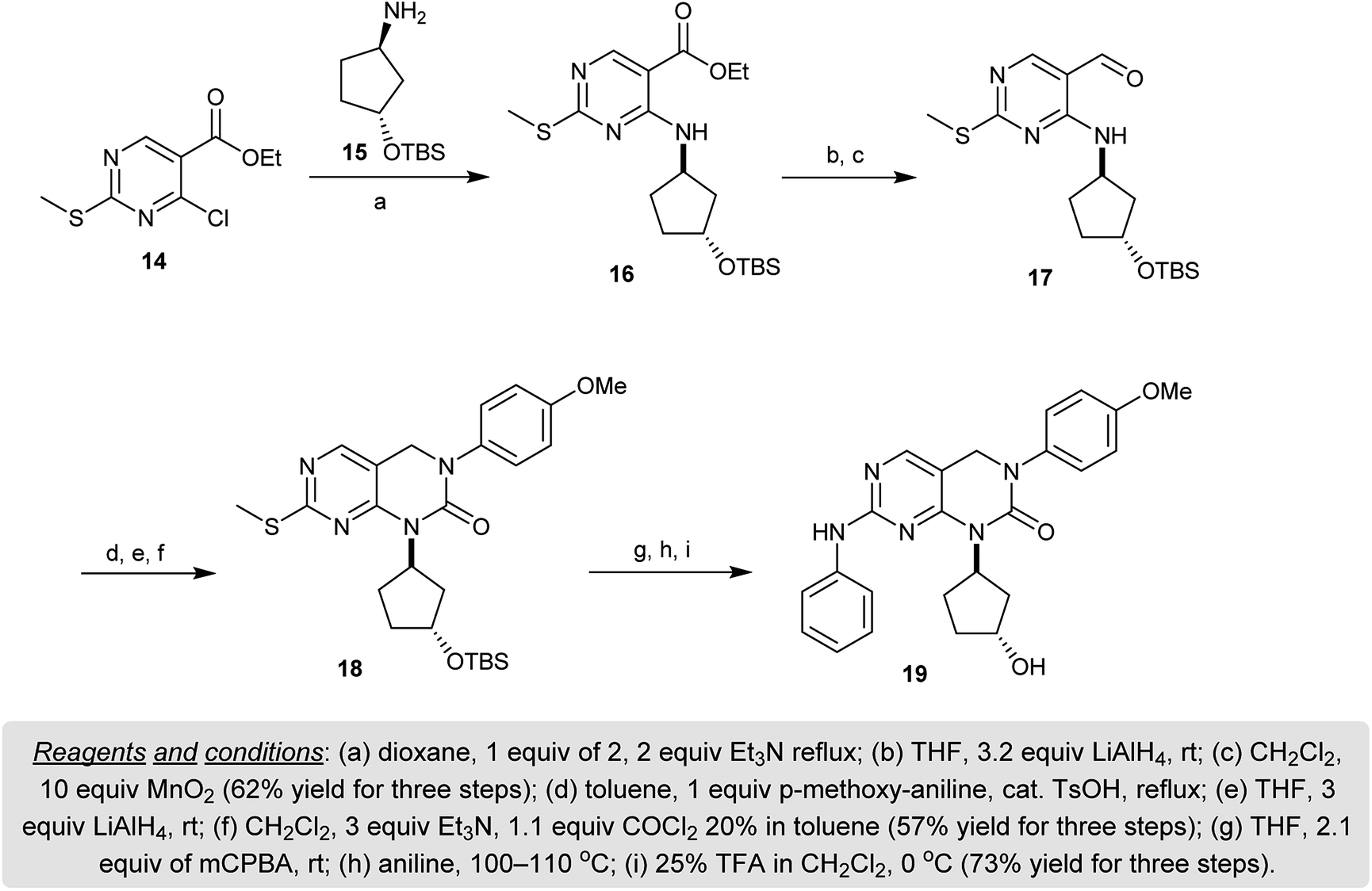
Refluxing of ethyl 4-chloro-2-(methylthio)pyrimidine-5-carboxylate (14) with the racemic protected-(1R,3R)-3-((tert-butyl-dimethyl-silyl)oxy)cyclopentan-1-amine (15) in dioxane containing triethylamine gave the imine 16. Reduction of 16 with lithium aluminum hydride followed by subsequent oxidation with manganese(IV) oxide furnished the respective aldehyde 17. Condensation of the aldehyde 17 with p-anisidine yielded the imine intermediate which was reduced with lithium aluminum hydride and cyclized upon reaction with phosgene to give the respective pyrimidopyrimidinone 18. Oxidation of 18 followed by reaction with aniline and hydrolysis of the protected group with trifluoroacetic acid gave the desired dihydro-pyrimido[4,5-d]pyrimidin-2(1H)-one 19 (Scheme 4). Compound 19 was evaluated as a tyrosine kinase inhibitor against KDR, FGFR, and PDGFR and found to be effective with a high bioavailability and plasma exposure at 25 mg kg−1.54
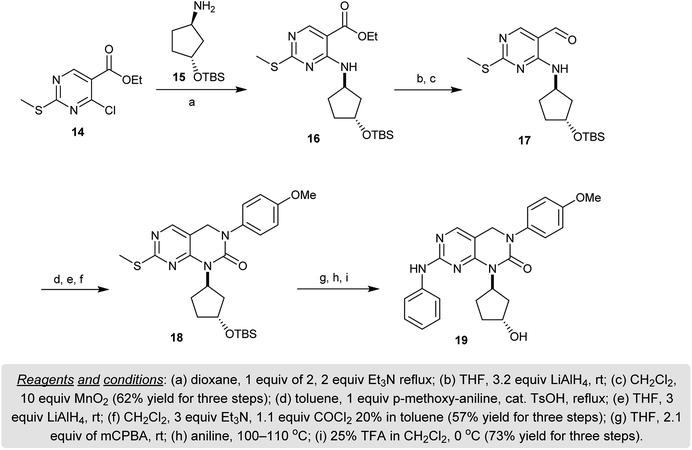 |
| | Scheme 4 Synthesis of 1,3,7-trisubstituted-3,4-dihydro-pyrimido[4,5-d]pyrimidin-2(1H)-one (19). | |
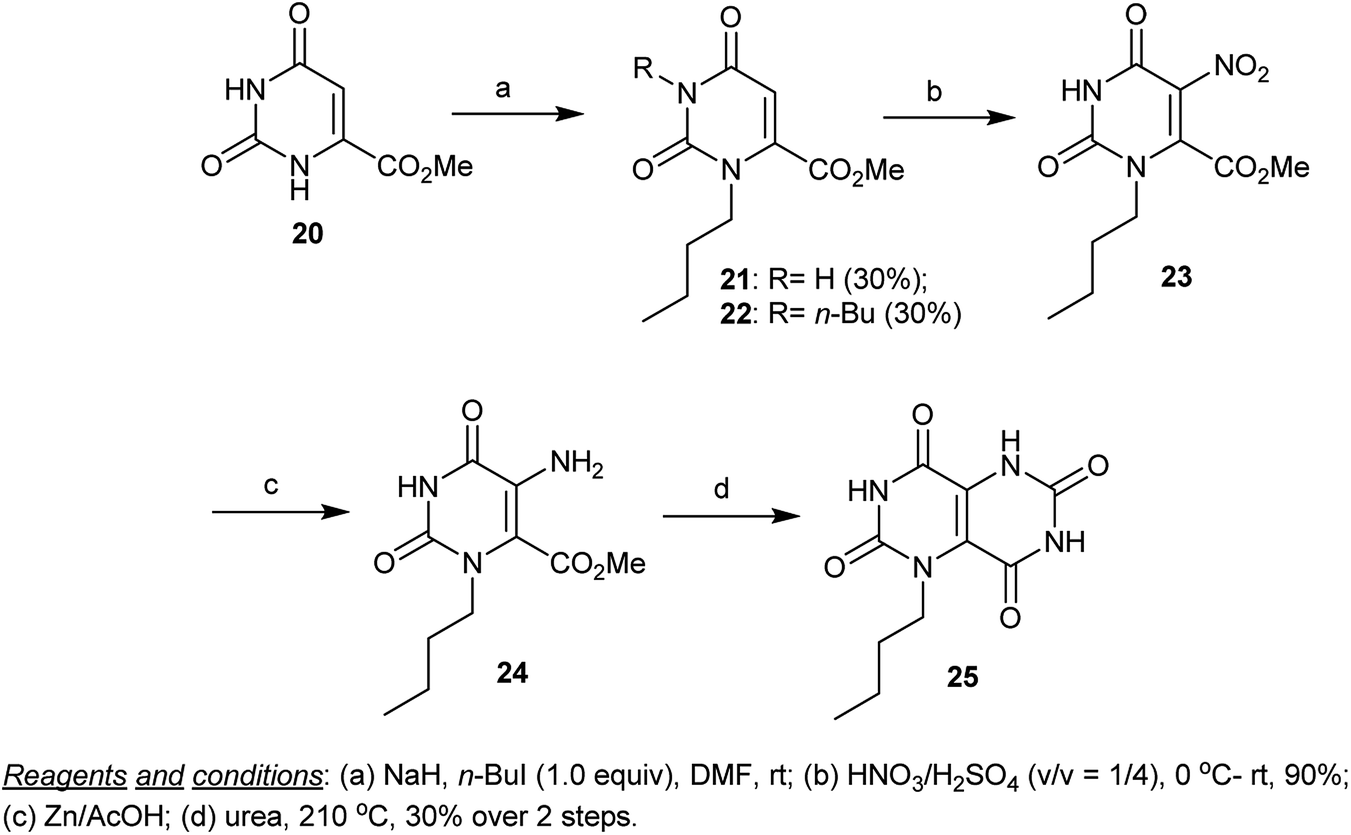
A regioselective synthetic route to prepare pyrimido[5,4-d]pyrimidine-2,4,6,8(3H,7H)-tetraones 9 was reported by Huang et al.55 In this route, methyl 2,6-dioxo-1,2,3,6-tetrahydropyrimidine-4-carboxylate (20) was alkylated with n-butyl iodide in the presence of sodium hydride to give the mono- and dialkylated products 21 and 22. The alkylation with an alkyl halide took place at the N1 and N3 atoms. The nitration of 21 with HNO3/H2SO4 (v/v = 1/4) yielded the nitro derivative 23 in a 90% yield. Reduction of 23 with Zn/AcOH gave the corresponding amine 24, which was cyclized by reacting with urea to give the desired 1-butyl-1,5-dihydropyrimido[5,4-d]pyrimidine-2,4,6,8(3H,7H)-tetraone (25) through the nucleophilic attack of the amino group of urea on the carbonyl ester of 24 and a subsequent cyclocondensation process (Scheme 5).
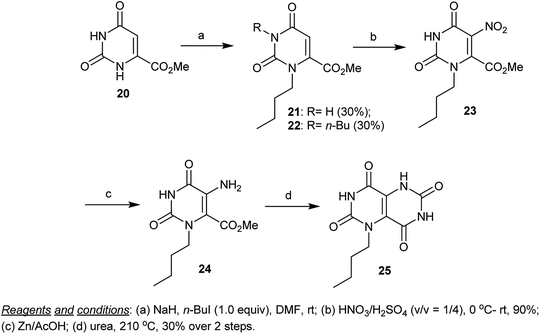 |
| | Scheme 5 Synthesis of 1-butyl-1,5-dihydropyrimido[5,4-d]pyrimidine-2,4,6,8(3H,7H)-tetraone. | |
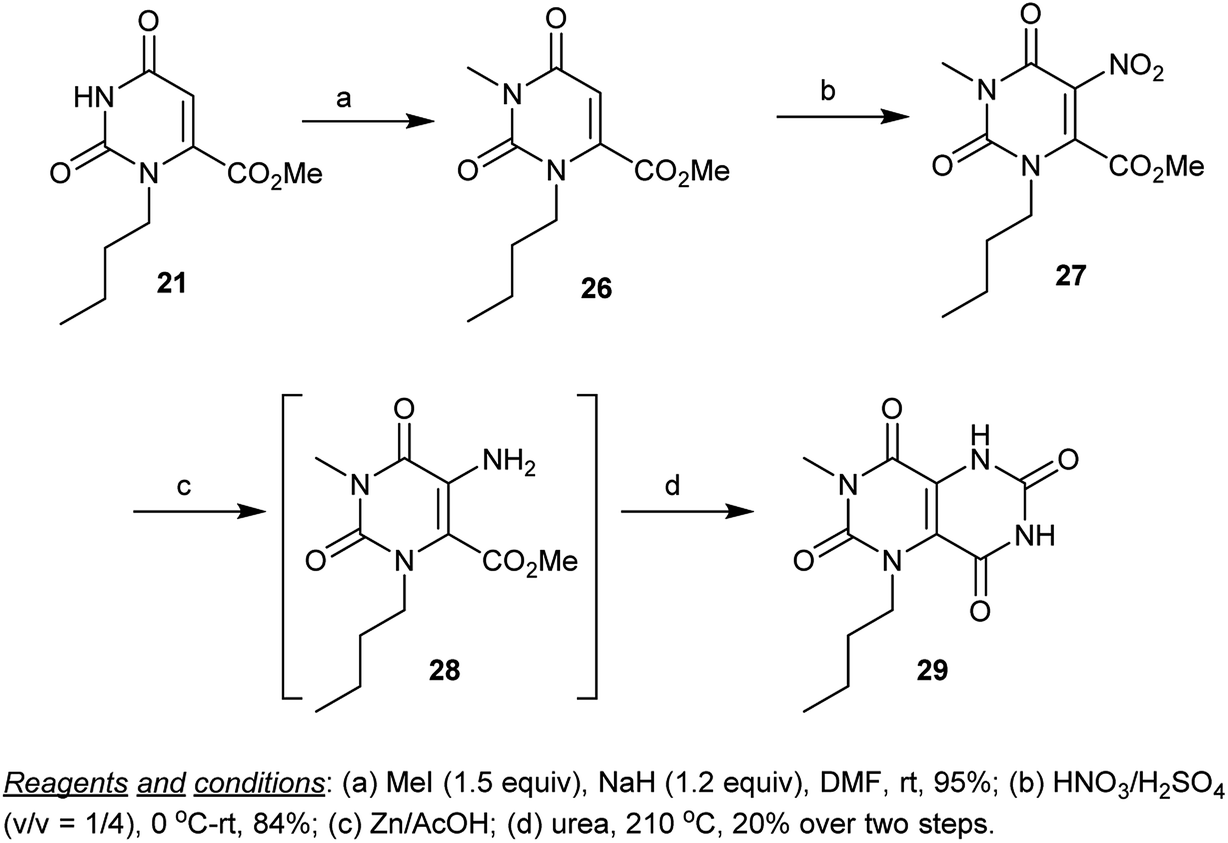
Similarly, methylation of methyl carboxylate 21 with methyl iodide in the presence of sodium hydride yielded the alkylation product 26, which underwent nitration with HNO3/H2SO4 to give the corresponding nitro derivative 27. Reduction of 27 by treatment of zinc in acetic acid, followed by cyclization by heating with urea afforded the respective 1-butyl-3-methyl-1,5-dihydropyrimido[5,4-d]pyrimidine-2,4,6,8(3H,7H)-tetraone (29) through the formation of the intermediate 28 (Scheme 6).55
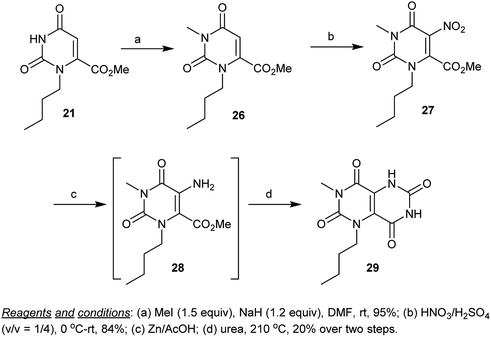 |
| | Scheme 6 Synthesis of 1,3-dialkyl-1,5-dihydropyrimido[5,4-d]pyrimidine-2,4,6,8(3H,7H)-tetraone. | |
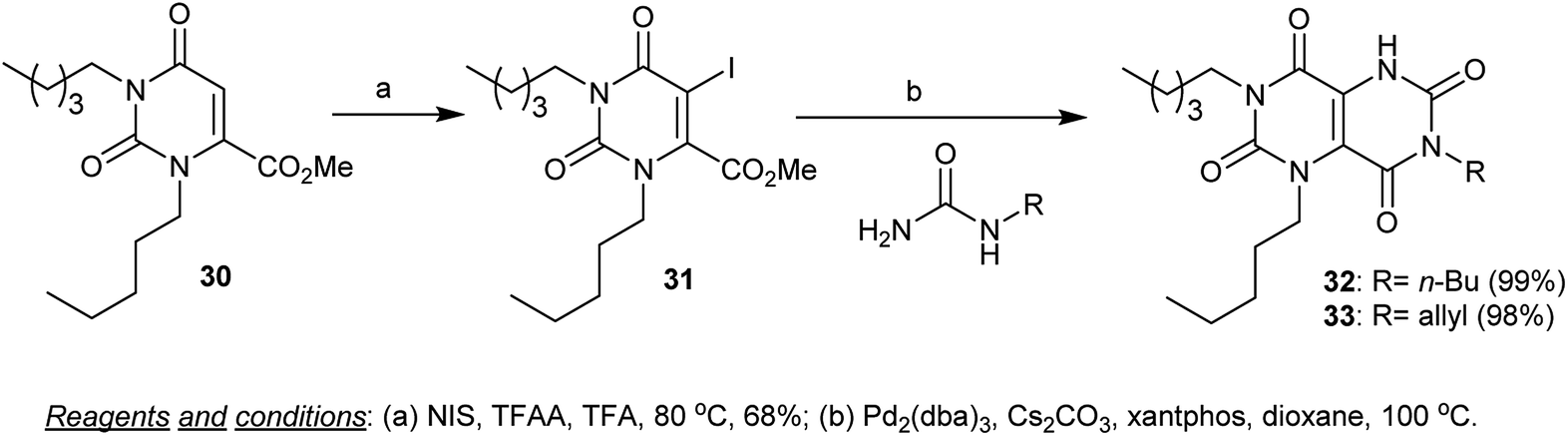
Furthermore, the iodination of 1-alkyl-methyl 2,6-dioxo-3-pentyl-1,2,3,6-tetrahydropyrimidine-4-carboxylate (30) was achieved by heating 30 in trifluoroacetic acid with NIS and TFAA to yield the iodo derivative 31. The halogenation took place at C5. The palladium catalyzed cyclization reaction of 31 with alkyl urea derivatives in dioxane in the presence of cesium carbonate yielded 3,7-dialkyl-1-pentyl-1,5-dihydropyrimido[5,4-d]pyrimidine-2,4,6,8(3H,7H)-tetraones 32 and 33 (Scheme 7).55
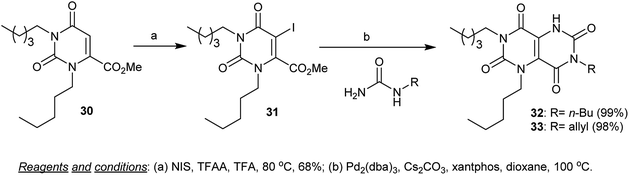 |
| | Scheme 7 Synthesis of 7-alkyl-1,3-dipentyl-1,5-dihydropyrimido[5,4-d]pyrimidine-2,4,6,8(3H,7H)-tetra-ones. | |
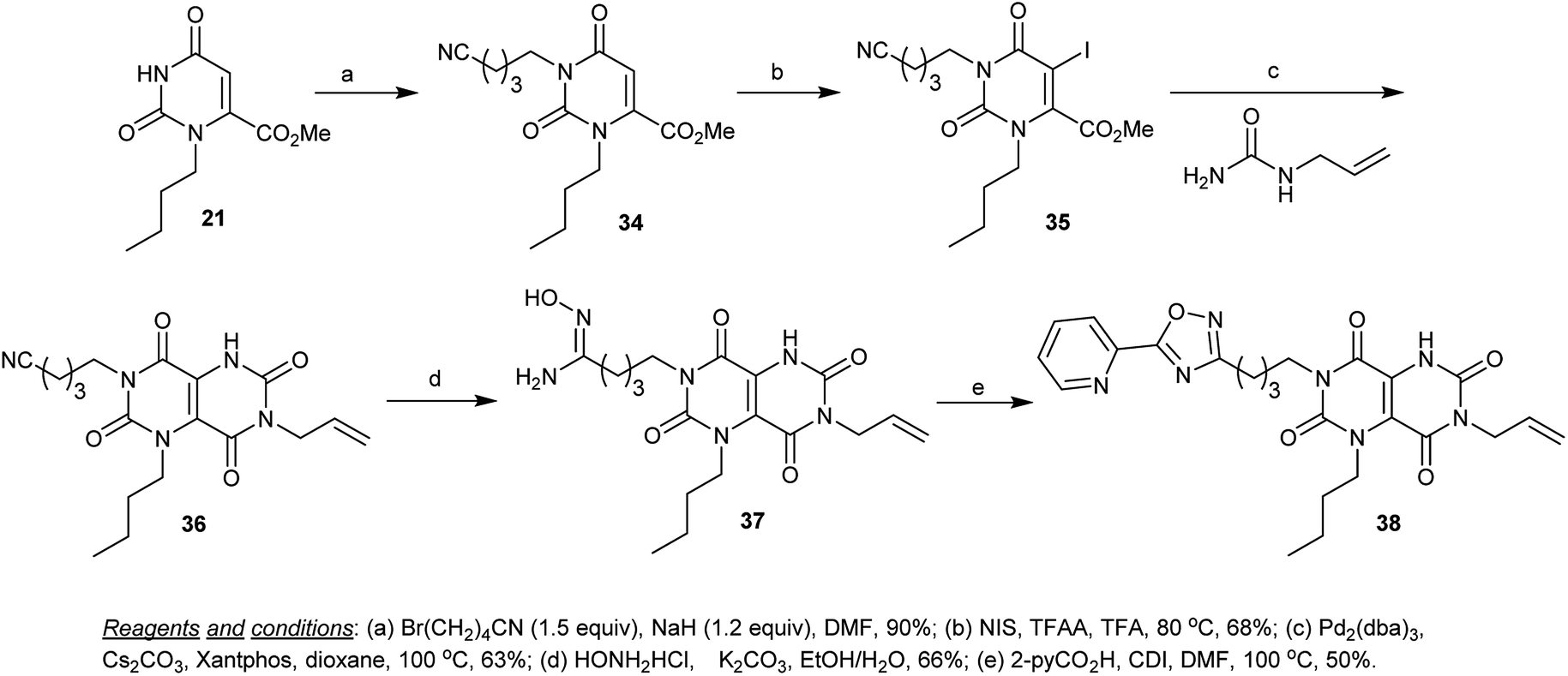
Additionally, alkylation of methyl 3-butyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidine-4-carboxylate (21) with 5-bromopentanenitrile in DMF containing sodium hydride gave the cyanoalkylation product 34. The alkylation process took place at N1. Similar to the iodination of previous products (30 → 31), iodination of 34 with NIS in the presence of TFA and TFAA yielded the iodo derivative 35. The cyclization step was proceeded by the reaction of 35 with allyl urea under palladium catalyzed conditions to give the cyclization product 36. Nucleophilic addition of hydroxyl amine to the cyano function of tetraoxo-1,4,5,6,7,8-hexahydro-pyrimido[5,4-d]pyrimidine 36 in a mixture of ethanol/water containing potassium carbonate yielded the intermediate 37, which was cyclized by the reaction with picolinic acid by heating in DMF containing catalytic CDI to furnish the respective 1,2,4-oxadiazole 38 in 50% yield (Scheme 8).55
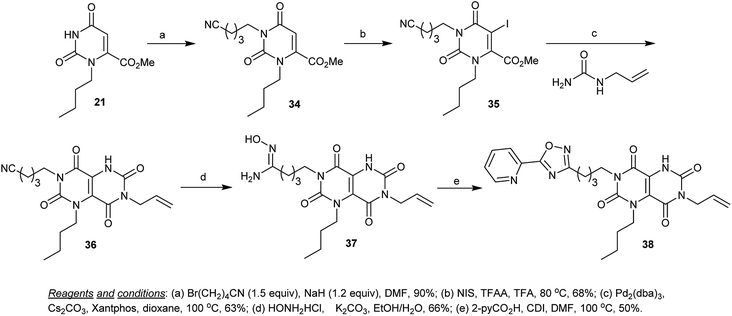 |
| | Scheme 8 Synthesis of N7-alkyl-heteryl-1,5-dihydropyrimido[5,4-d]pyrimidine-2,4,6,8(3H,7H)-tetraone. | |
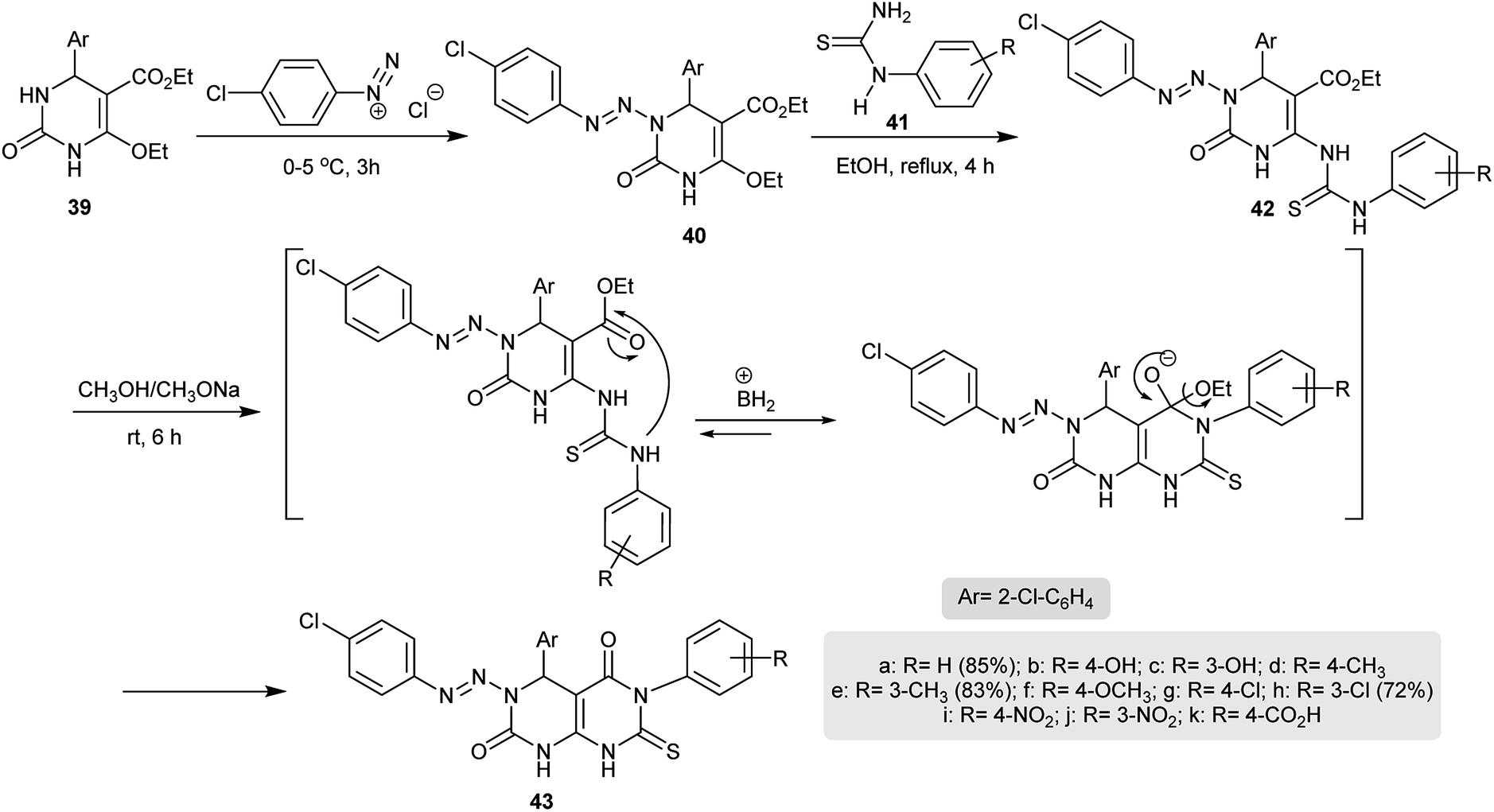
In another route, excellent yields of the desired 4,6-diaryl-3-(aryldiazenyl)-7-thioxo-4,6,7,8-tetrahydro-pyrimido[4,5-d]pyrimidine-2,5(1H,3H)-diones 43 were obtained through a multistep synthesis. Thus, diazocoupling of pyrimidinone 39 with the diazonium salt of p-chloroaniline under diazotization conditions gave the arylazo derivative 40. Refluxing of 40 with N-aryl-thiourea derivatives in ethanol yielded compound 42. Cyclization of 42 took place by treatment in sodium methoxide solution at room temperature by nucleophilic attack of the aryl-NH group on the carbonyl ester to afford the corresponding tetrahydropyrimido[4,5-d]pyrimidine-2,5(1H,3H)-diones 43 (Scheme 9). Compounds 43 were assessed in vitro as antimicrobial agents against E. coli, P. diminuta, S. aureus, B. subtilis, A. niger, and C. albicans species. The best results were noted for the compounds having hydroxy, chloro, nitro or methyl substituents in the meta position.37
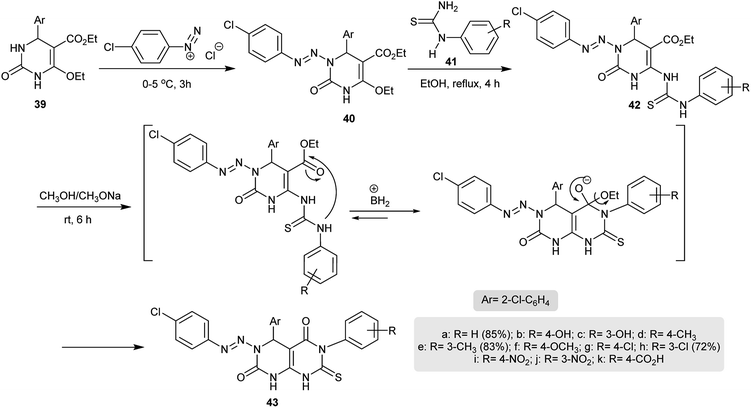 |
| | Scheme 9 Synthesis of 7-thioxo-4,6,7,8-tetrahydropyrimido[4,5-d]-pyrimidine-2,5(1H,3H)-diones. | |
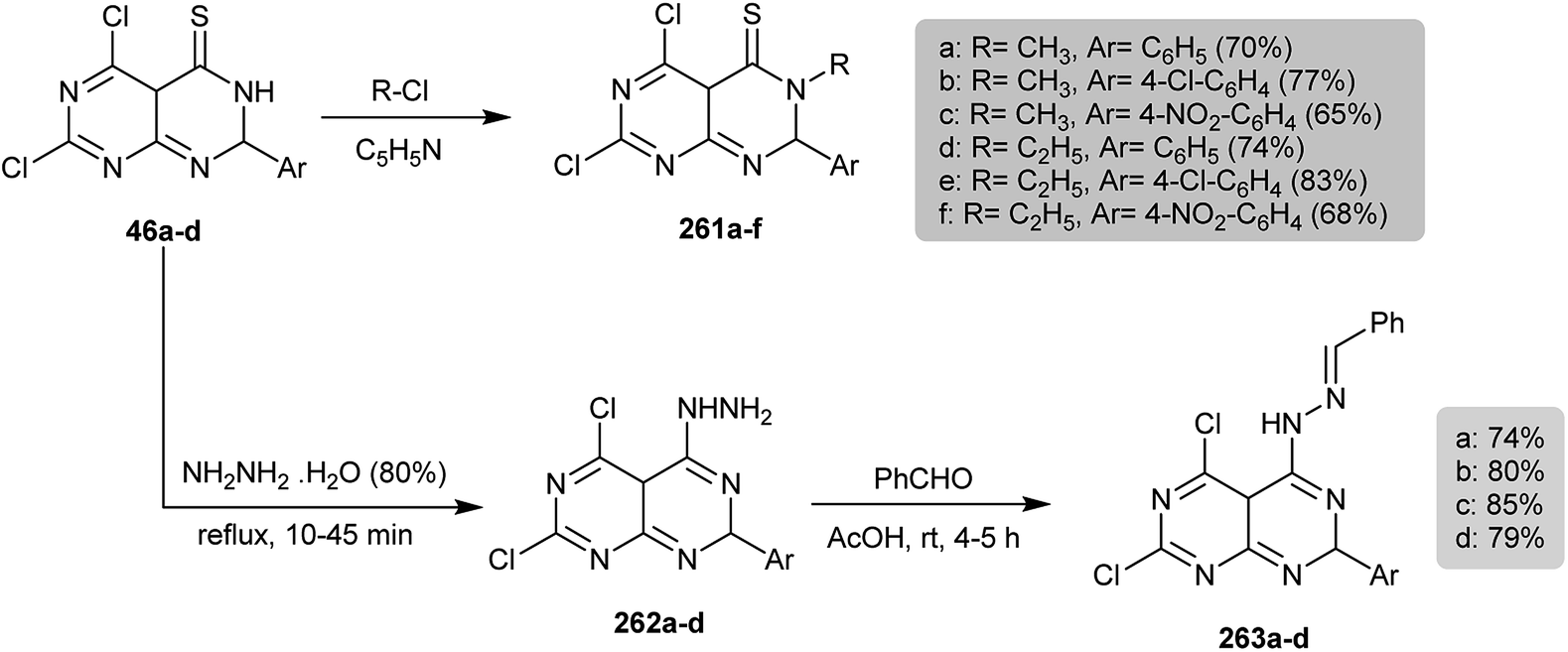
2.1.2. Synthesis from 4-amino-2,6-dichloropyrimidine.
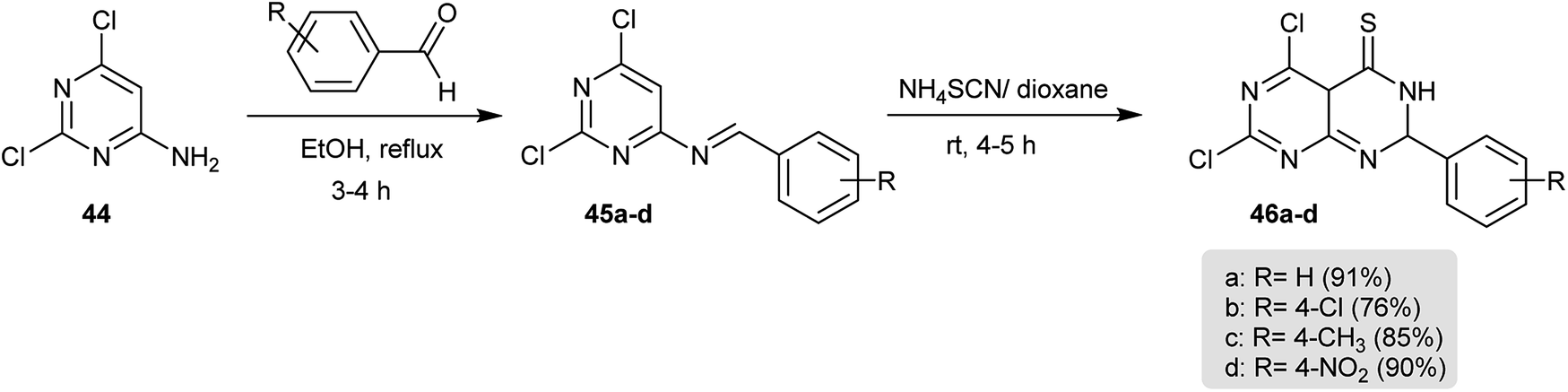
The Schiff bases 45a–d were prepared by condensation of 2,6-dichloropyrimidin-4-amine (44) with aromatic aldehydes in refluxing ethanol. Successive treatment of 45a–d with ammonium thiocyanate in 1,4-dioxane yielded the cyclization products 46a–d, respectively (Scheme 10).56
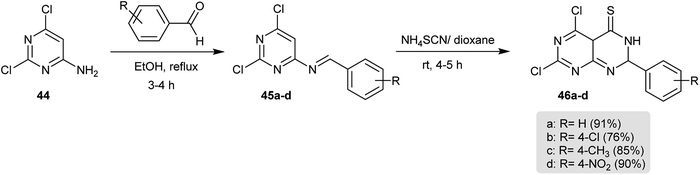 |
| | Scheme 10 Synthesis of 5,7-dichloro-2-aryl-2,4a-dihydropyrimido[4,5-d]pyrimidine-4(3H)-thiones. | |
2.1.3. Synthesis from 2,6-diaminopyrimidin-4(3H)-one.
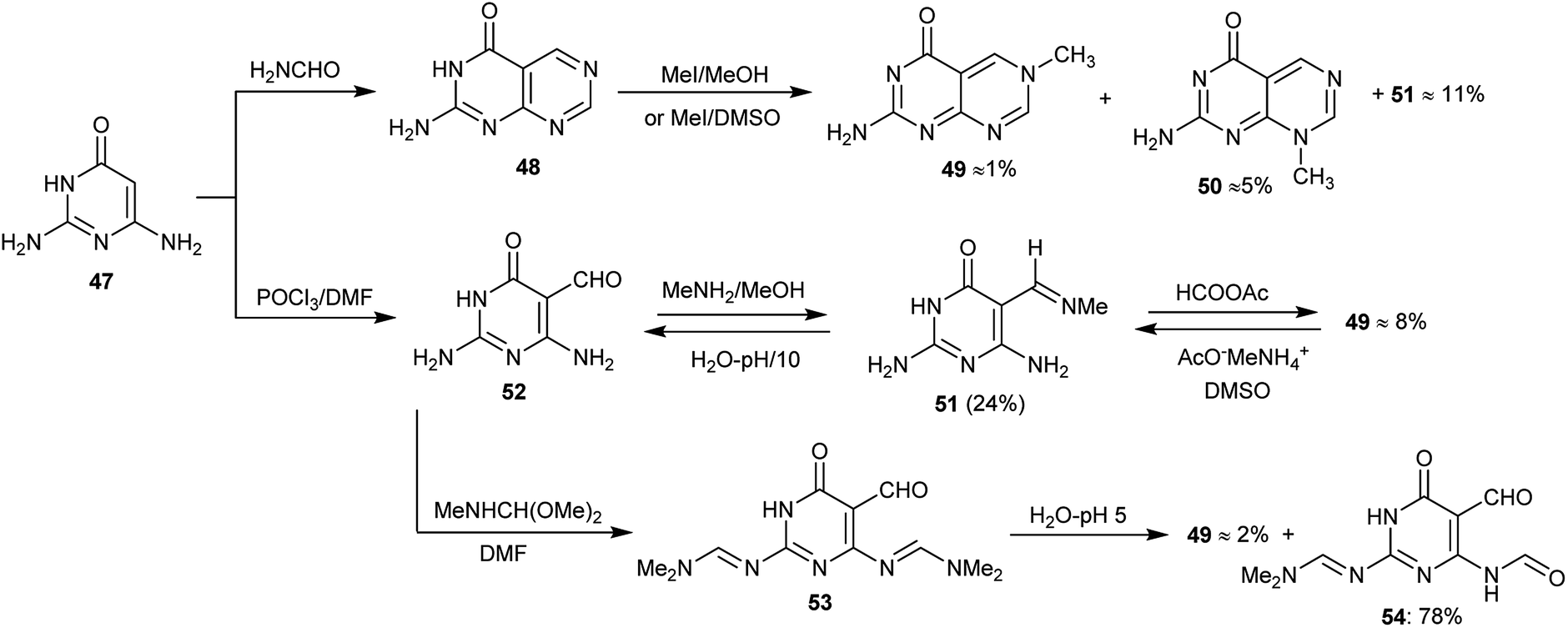
Three methods for the preparation of 2-aminopyrimidopyrimidinones have been reported by Gebauer et al.10 although the products 48, 49 and 50 were obtained with very low yields. The 2-aminopyrimido-pyrimidinones are analogs of 8-alkylpterins (N8-alkyl-2-aminopteridin-4(8H)-ones), which are well-known inhibitors for dihydrofolate reductase. The low yield of the products is due to the deficiency of the π-electrons of the heterocycles 49 and 50 which make the quaternized pyrimidine ring susceptible to the addition of a nucleophile with the promotion of the pyrimidopyrimidine ring-opening (Scheme 11). Compounds 49 and 50 showed remarkable activities as inhibitors of dihydrofolate reductase.
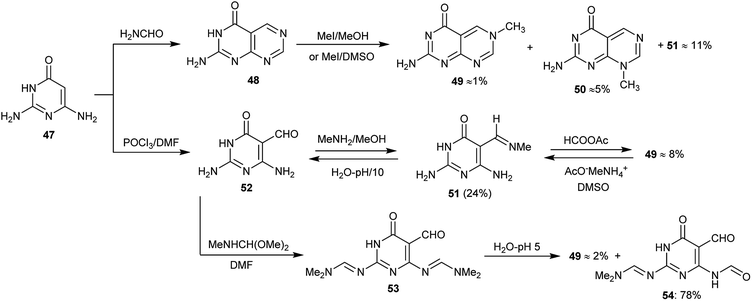 |
| | Scheme 11 Synthesis of 2-aminopyrimidopyrimidinones. | |
2.1.4. Synthesis from barbituric and thiobarbituric acids.
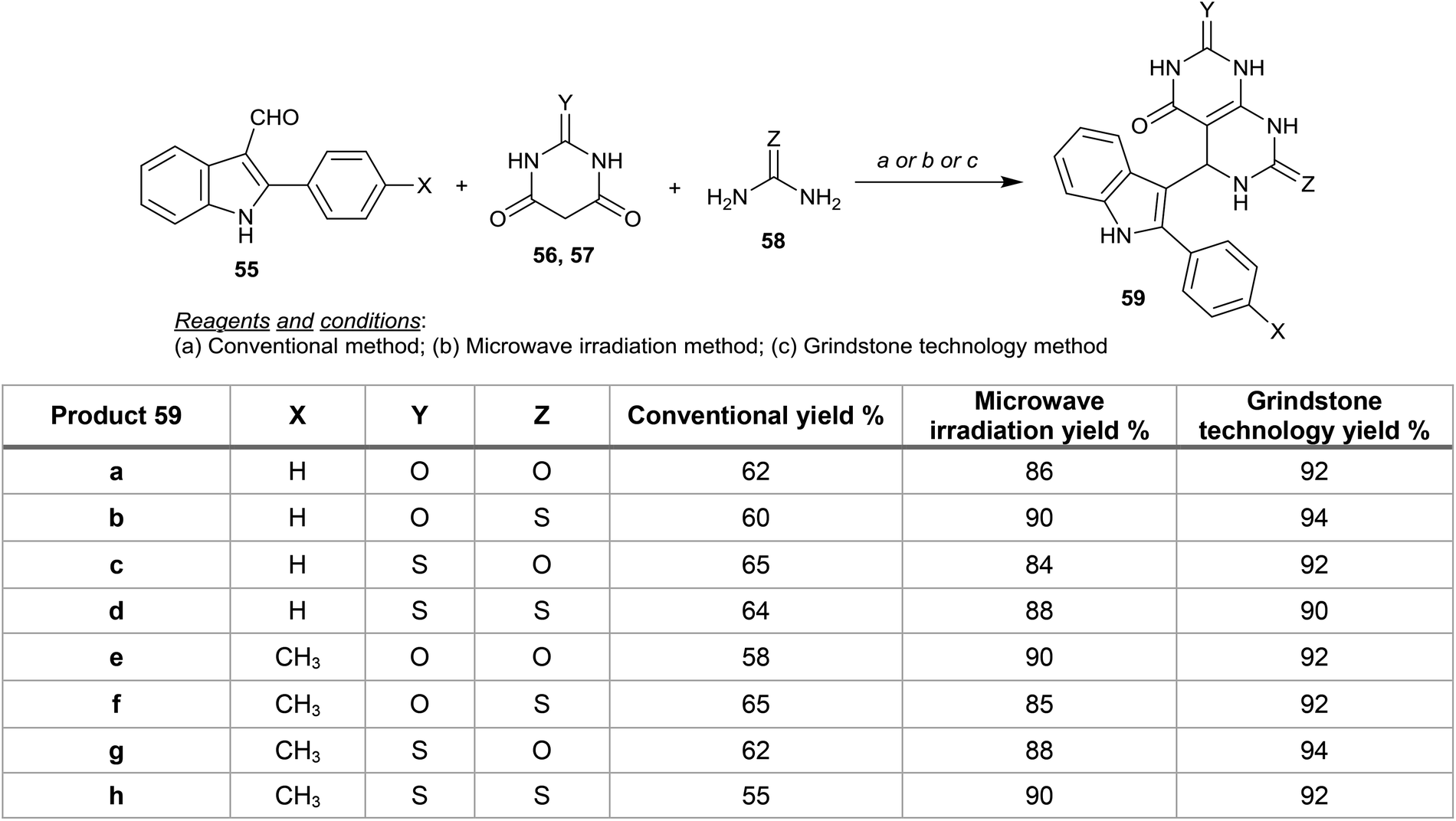
Gupta et al.57 reported proficient procedures for the synthesis of 5-(1H-indol-3-yl)-5,8-dihydropyrimido[4,5-d]pyrimidines 59 in dry media through multicomponent reactions of 2,3-disubstituted indoles 55 with barbituric or thiobarbituric acids 56, 57 and urea or thiourea 58 under microwave irradiation conditions. Compounds 59 were also prepared following conventional and grinding methods in good yields (Scheme 12). In addition, compounds 59a–h were evaluated as antimicrobial agents against well-known microbial species, the results prove that compound 59d has the ability to protect against microbial growth and that compound 59e has a good activity against F. oxysporum at concentrations of 600, 800 and 1000 ppm. On the other hand, compounds 59a and 59g are potent agents against P. aeruginosa at 800 and 1000 ppm.57
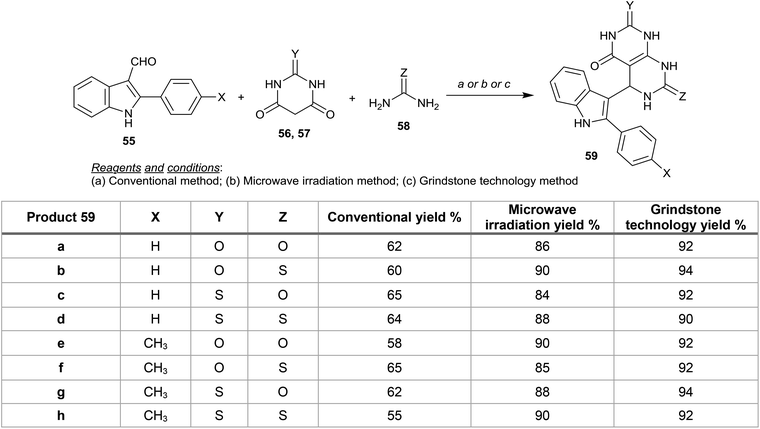 |
| | Scheme 12 Synthesis of 5-(1H-indol-3-yl)-5,8-dihydropyrimido[4,5-d]pyrimidines. | |
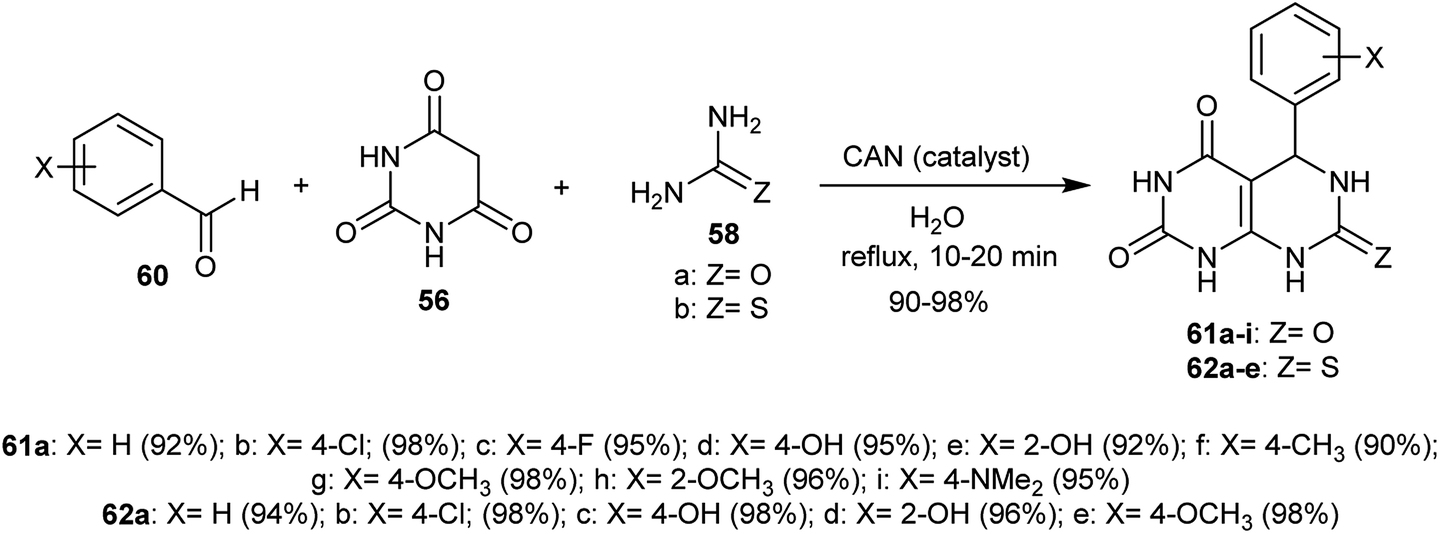
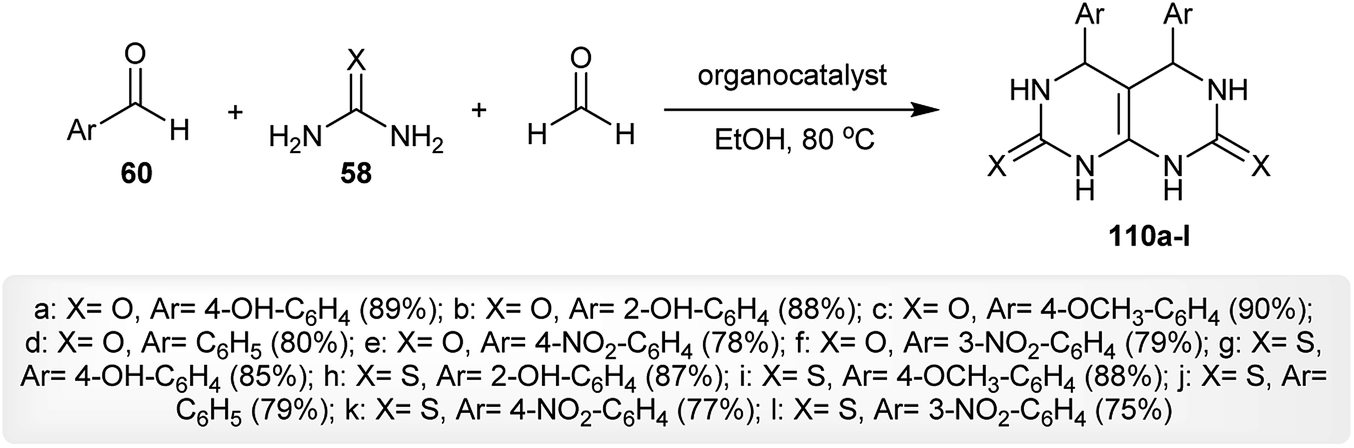
An efficient procedure for the synthesis of two series of tetrahydropyrimido[4,5-d]pyrimidine-diones 61 and 62 has been reported through a Biginelli-type reaction of the aryl aldehydes 60 with barbituric acid 56 and urea or thiourea 58 in the presence of ceric ammonium nitrate (CAN) as a catalyst. The reactions proceeded in a one-pot multicomponent step in refluxing water and the products 61 and 62 were obtained in excellent yields (Scheme 13).58
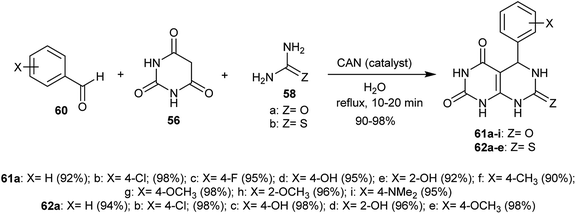 |
| | Scheme 13 Synthesis of 5-aryl-(7-thioxo and 7-oxo)-5,6,7,8-tetrahydropyrimido[4,5-d]pyrimidine-diones. | |
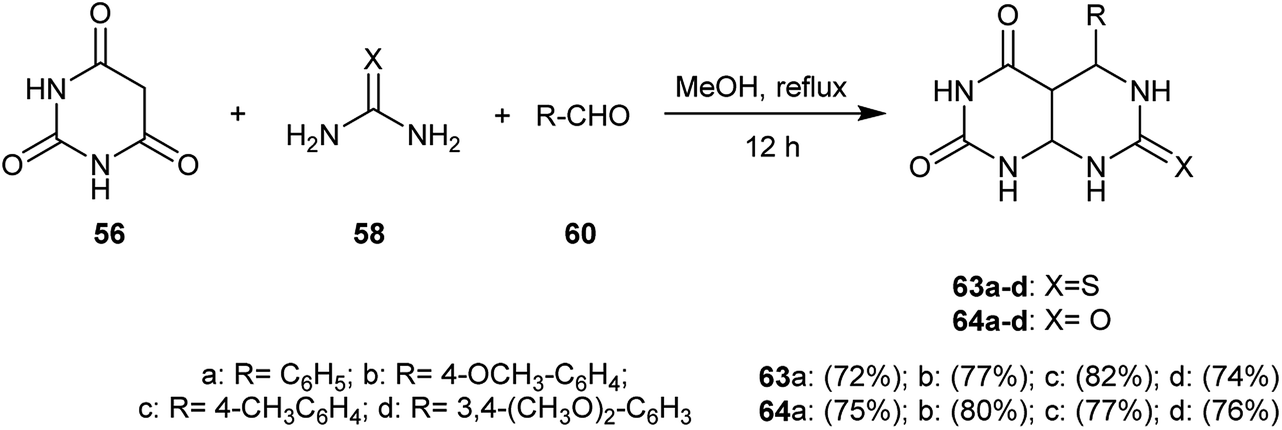
Multicomponent condensation reactions of barbituric acid (56) with urea or thiourea (58) and aromatic aldehydes 60 in equivalent amounts in refluxing methanol yielded a series of 5-aryl-7-(thioxo and oxo-)-hexa-hydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-diones 63a–d and 64a–d (Scheme 14).59
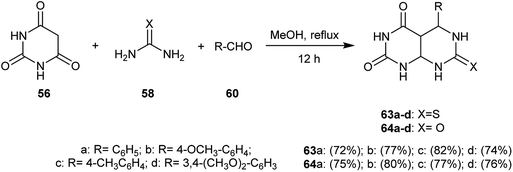 |
| | Scheme 14 Synthesis of 5-aryl-7-(thioxo and oxo)-hexahydropyrimido[4,5-d]pyrimidine-2,4-diones. | |
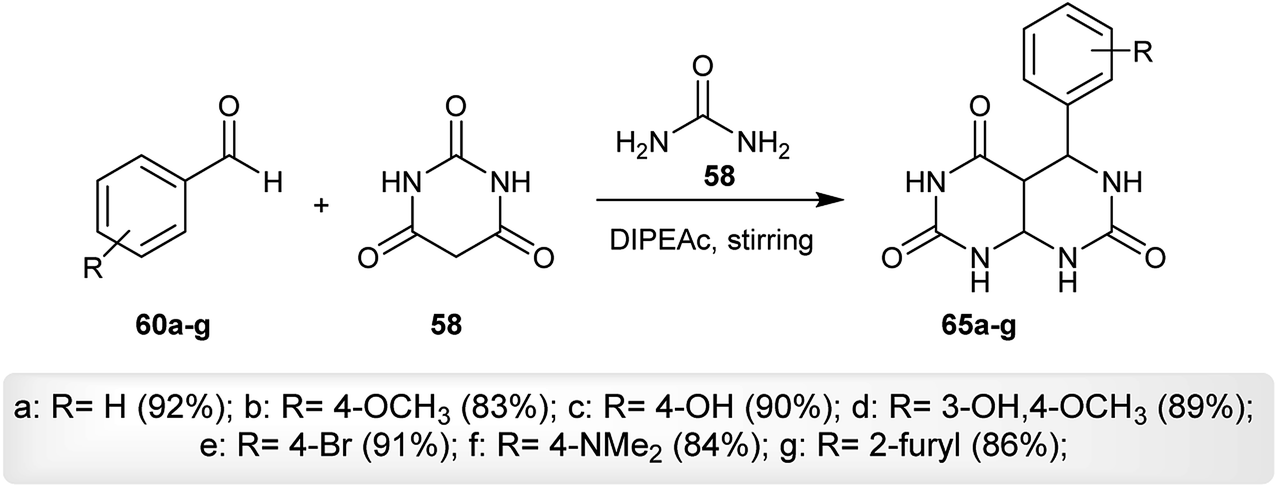
Three component reactions were employed for the preparation of a series of 5-aryl-tetrahydro-pyrimido[4,5-d]pyrimidine-2,4,7(1H,3H,4aH)-triones (65a–g) by reacting aryl aldehydes (60a–g) with barbituric acid (56) and urea (58) at room temperature via a one-pot procedure using diisopropyl-ethyl-ammonium acetate (DIPEAc) as an alternative solvent and a catalyst, which promotes the cyclocondensation step-reaction providing high yields from the reactions and reusability of the catalyst. The procedure provided high yields in a short time (Scheme 15). The desired tetrahydro-pyrimido[4,5-d]pyrimidine-triones (65a–g) were assessed in vitro as anticancer agents against MCF-7, HeLa, A-549, and MCF-10A cancer cell lines relative to the standard Adriamycin and tyrosinase inhibitors. The compounds showed moderate anticancer activities against all tested cell lines and compound 65f was found to be the most potent agent relative to the results of the other compounds against MCF-7, HeLa, and A-549 cell lines. The compounds are inactive against the MCF-10A cancer cell line. The presence of nitrogen substitution at position 4 of the aryl ring is essential to achieving potency. On the other hand, compounds 65c, 65d and 65f are tyrosinase inhibitors, while the other compounds are inactive agents. The presence of phenyl-substituted groups with stable radicals enhances the biological results.60
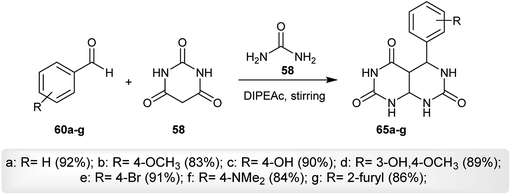 |
| | Scheme 15 Synthesis of 5-aryl-tetrahydro-pyrimido[4,5-d]pyrimidine-2,4,7(1H,3H,4aH)-triones. | |
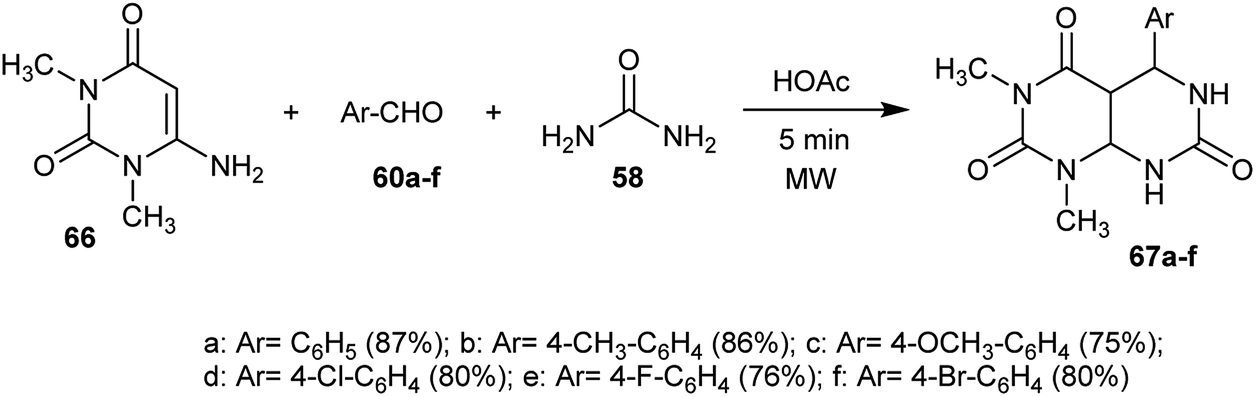
2.1.5. Synthesis from 6-amino-1,3-dialkyl-pyrimidine-2,4(1H,3H)-dione.
The reaction of 6-amino-1,3-dimethylpyrimidine-2,4(1H,3H)-dione (66) with aryl aldehydes 60 and urea (58) in the presence of a catalytic amount of acetic acid under microwave irradiation conditions yielded the respective tetrahydropyrimido[4,5-d]pyrimidine-triones 67. The best yield was obtained using acetic acid as a catalyst and the use of the electron-withdrawing substituents of the aldehydes. The reaction mechanism was proceeded by the initial condensation of one mole of the aryl aldehydes with two moles of pyrimidindione 66 followed by nucleophilic attack of the amino group of urea on the formed intermediate and subsequent intramolecular cyclization after the evolution of ammonia to give the tetrahydropyrimido[4,5-d]pyrimidine-2,4,7(1H,3H,4aH)-triones 67 (Scheme 16).26
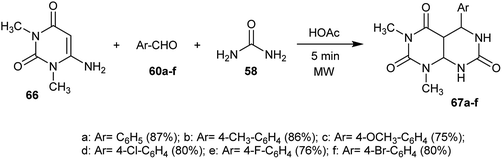 |
| | Scheme 16 Synthesis of 1,3-dimethyl-5-aryl-tetrahydropyrimido[4,5-d]pyrimidine-2,4,7(1H,3H,4aH)-triones. | |
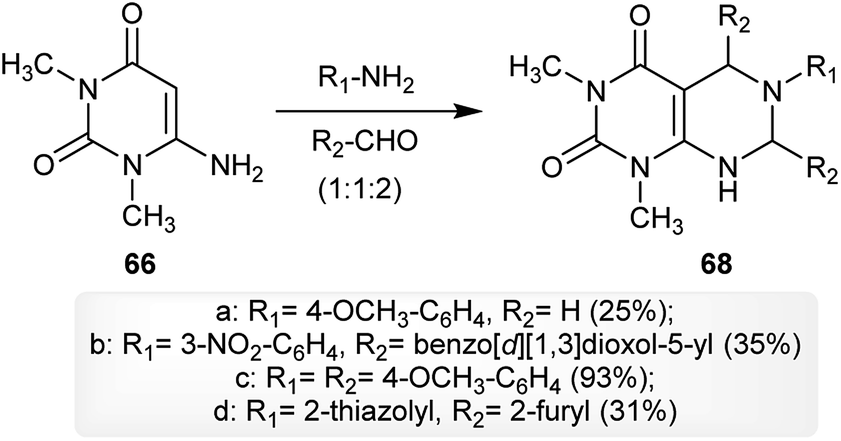
The multicomponent reaction of 6-amino-1,3-dimethylpyrimidine-2,4(1H,3H)-dione (66) with arylamines and aldehydes in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 yielded the respective 5,6,7-trisubstituted-1,3-dimethyl-5,6,7,8-tetrahydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-diones 68 in good to moderate yields. The best yield was noticed for the reaction of one equivalent of 66 with p-anisidine and 4-methoxybenzaldehyde (2 equivalents) for the preparation of compound 68c (93%) and the other reactions gave the products with moderate to low yields. The use of this technique provided the best product yields in each case depending on the substituents attached to the amines and aldehydes (Scheme 17).61
:1:2 yielded the respective 5,6,7-trisubstituted-1,3-dimethyl-5,6,7,8-tetrahydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-diones 68 in good to moderate yields. The best yield was noticed for the reaction of one equivalent of 66 with p-anisidine and 4-methoxybenzaldehyde (2 equivalents) for the preparation of compound 68c (93%) and the other reactions gave the products with moderate to low yields. The use of this technique provided the best product yields in each case depending on the substituents attached to the amines and aldehydes (Scheme 17).61
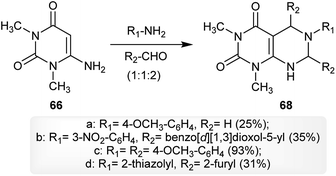 |
| | Scheme 17 Multicomponent synthesis of 1,3-dimethyl-5,6,7-triaryl-5,6,7,8-tetrahydropyrimido[4,5-d]-pyrimidine-2,4(1H,3H)-diones. | |
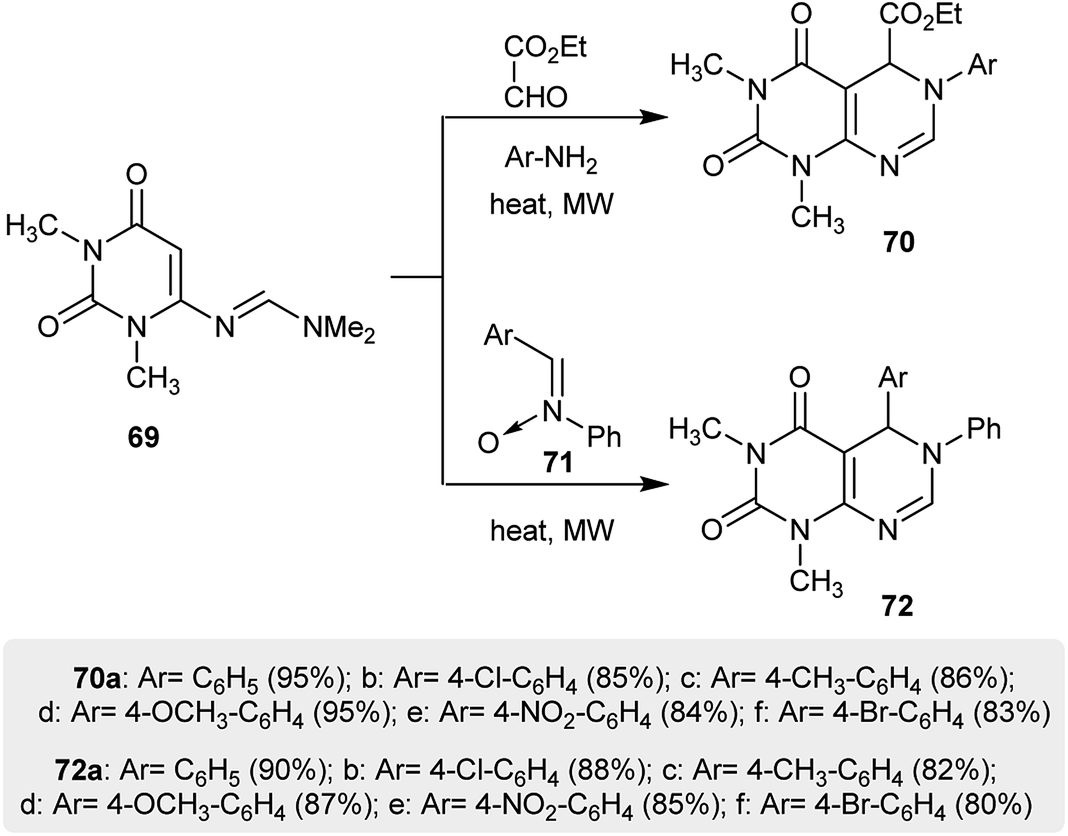
A procedure reported by Prajapati et al.41 provided a proficient synthetic route for the synthesis of pyrimidopyrimidines 70 in a one-pot step with exceptional yields. Thus, [4 + 2] cycloaddition reactions of uracil 69 with various glyoxylate imines (which were in situ generated by the reaction of ethyl 2-oxoacetate with aryl amines) and imine oxides 71 were used to afford ethyl hexahydro-pyrimido-[4,5-d]pyrimidine-carboxylates 70 and 5,6-dihydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-diones 72 under microwave irradiation conditions (Scheme 18).
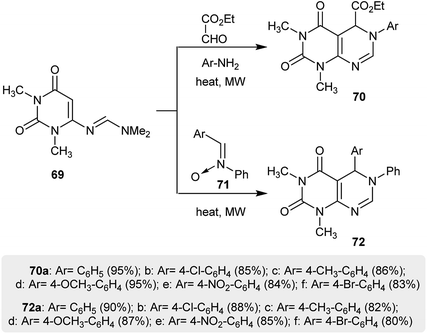 |
| | Scheme 18 Synthesis of hexahydro- and 5,6-dihydro-pyrimido[4,5-d]pyrimidine-2,4(1H,3H)-diones. | |
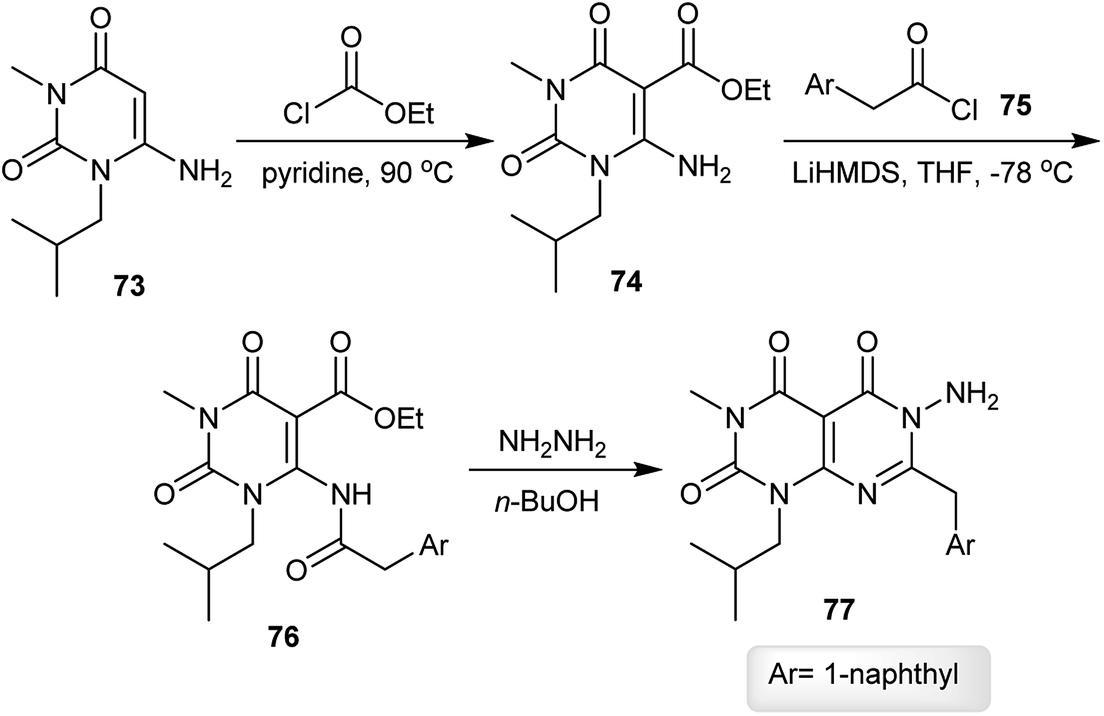
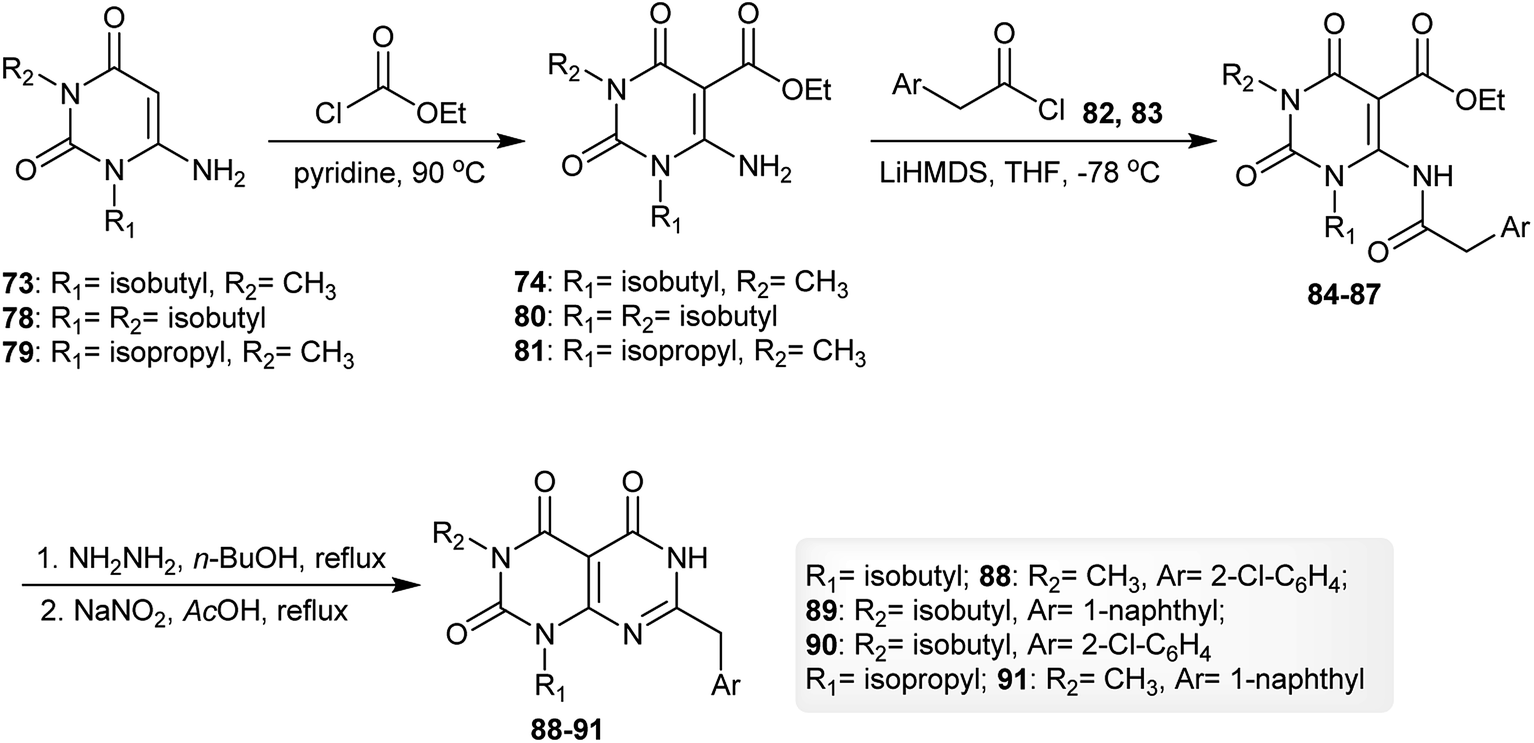
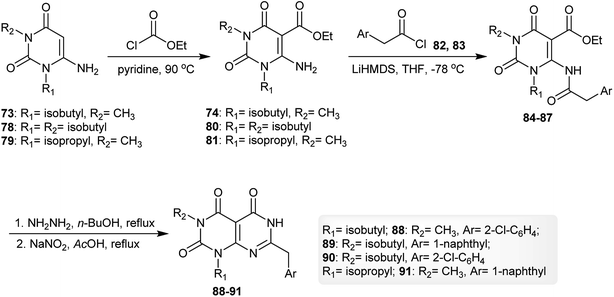
The reaction of 6-amino-1-isobutyl-3-methylpyrimidine-2,4(1H,3H)-dione (73) with ethyl carbono-chloridate by heating in pyridine gave the respective ester 74, which reacted with 2-(naphthalen-1-yl)acetyl chloride (75) in THF in the presence of LiHMDS to give the desired amide 76. Cyclization of the amide 76 was achieved by reaction with hydrazine hydrate in n-butanol to afford pyrimido[4,5-d]pyrimidine-2,4,5-(1H,3H,6H)-trione 77 (Scheme 19).62
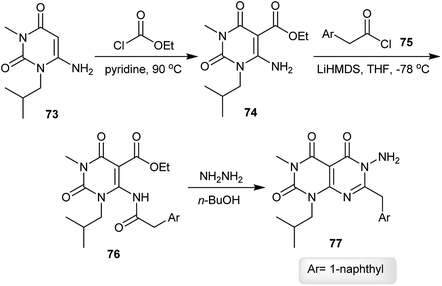 |
| | Scheme 19 Synthesis of 1,3-dialkyl-6-amino-(naphthalen-1-ylmethyl)pyrimido[4,5-d]pyrimidine-2,4,5-(1H,3H,6H)-trione. | |
Similarly, using three aminouracils, pyrimido[4,5-d]pyrimidine-2,4,5(1H,3H,6H)-triones 88–91 were prepared in four step-reactions under the same conditions described for the synthesis of pyrimido[4,5-d]pyrimidine-2,4,5-(1H,3H,6H)-trione 77. Compounds 74, 80 and 81 were obtained with yields of 60–70%, compounds 84–87 were obtained with yields of 47–52% and compounds 88–91 were obtained with yields of 31–54% (Scheme 20).62
 |
| | Scheme 20 Synthesis of 7-alkylaryl-1,3-dialkyl-pyrimido[4,5-d]pyrimidine-2,4,5(1H,3H,6H)-triones. | |
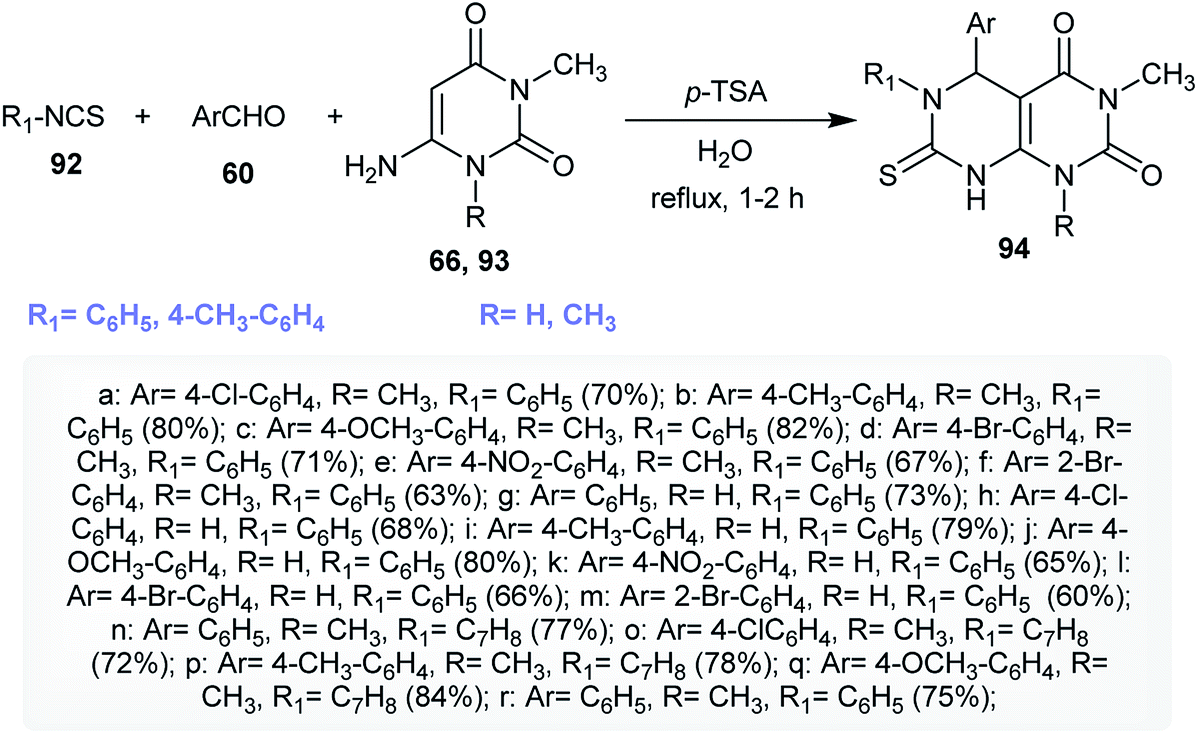
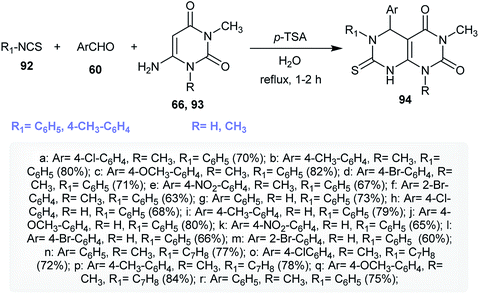
An efficient multicomponent synthetic route for the synthesis of 5,6-diaryl-1-alkyl-3-methyl-7-thioxo-5,6,7,8-tetrahydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-diones 94 was reported by Majumder and coworkers.36 A regioselective one-pot multicomponent reaction of isothiocyanates 92 with aryl aldehydes 60 and 1-alkyl-6-amino-3-methylpyrimidine-2,4(1H,3H)-diones 66 and 93 yielded the desired products 94 in good yields. The reactions proceeded in water under catalytic conditions using p-TSA as a Lewis acid catalyst (Scheme 21). The use of 20 mol% of p-TSA was found to give the best yield of 93 (75%). The mechanism of the reactions progressed through the initial nucleophilic attack of the amino group of 93 into the CN of the isothiocyanates with the formation of the NH–C bond. Next, the CC bond of the formed intermediate attack the carbonyl group of the aldehyde, in which the presence of the catalyst supports this step, and consecutive intramolecular cyclization and condensation furnished the anticipated products 94.36
 |
| | Scheme 21 Synthesis of 5,6-diaryl-1-alkyl-3-methyl-7-thioxo-5,6,7,8-tetrahydropyrimido[4,5-d]-pyrimidine-2,4(1H,3H)-diones. | |
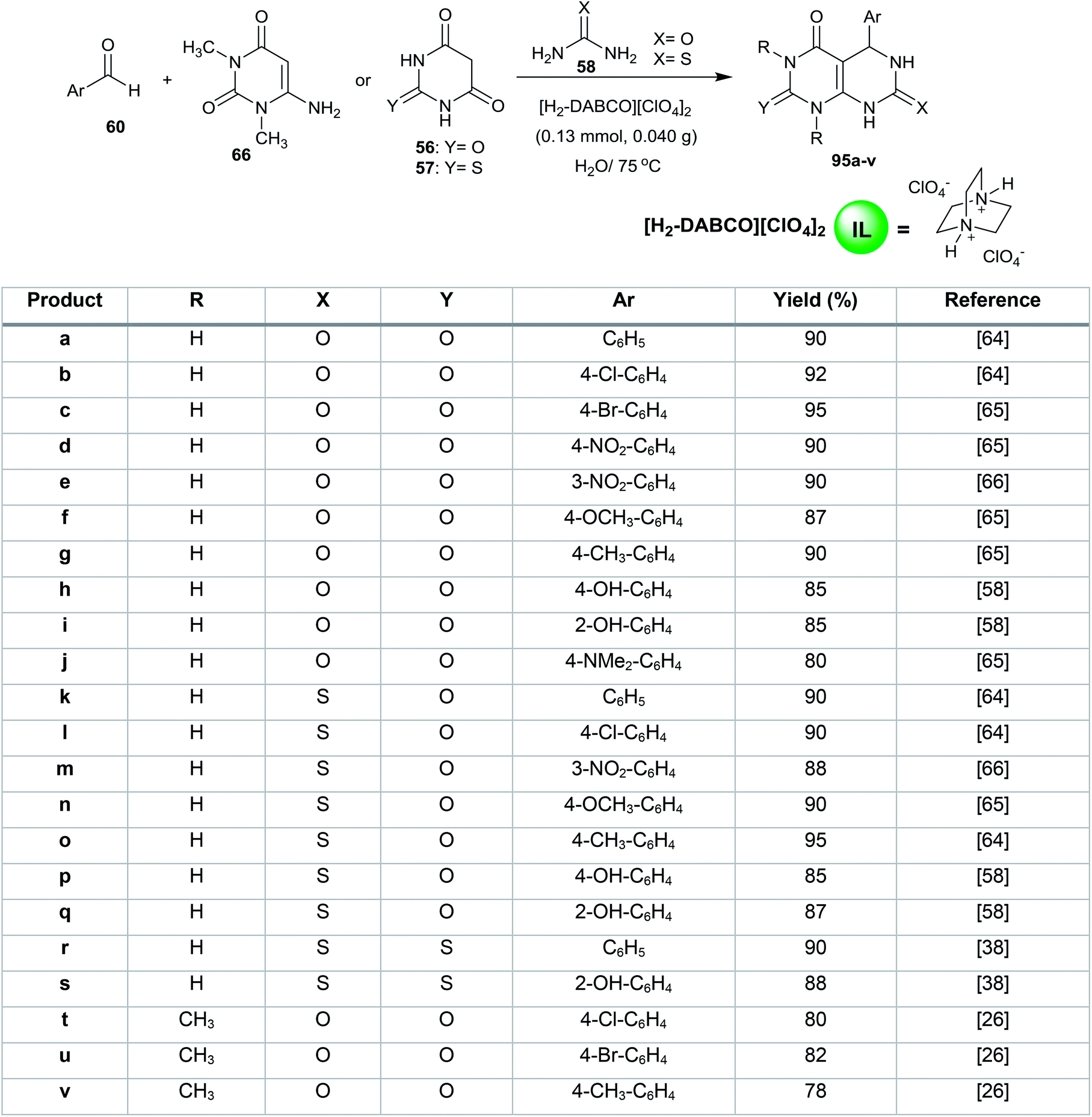
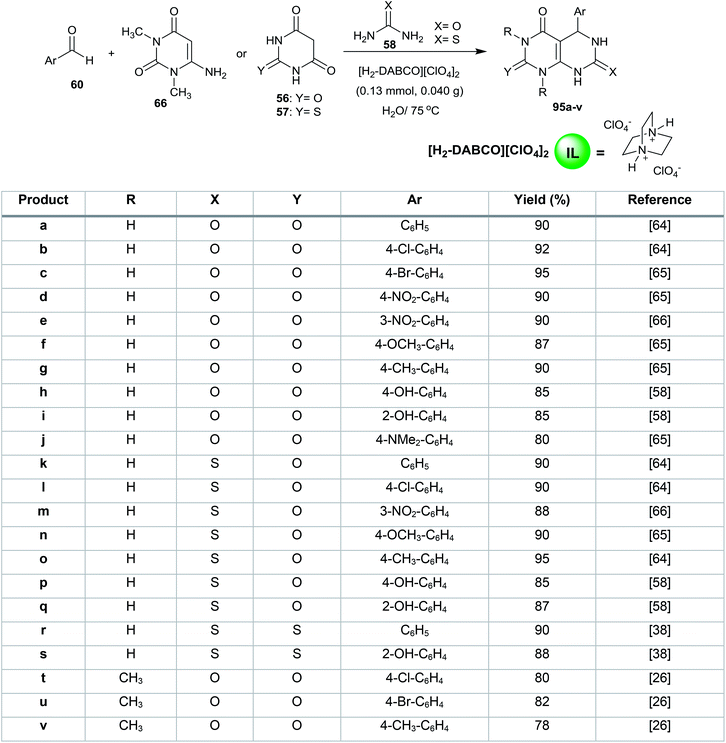
Shirini et al.63 reported the synthesis of 1,4-diazabicyclo[2.2.2]octane-1,4-diium perchlorate ([H2-DABCO][ClO4]2) as an efficient base ionic liquid in 95% yield by stirring a mixture of 1,4-diazabicyclo[2.2.2]octane (DABCO) in dry dichloromethane in an ice-bath followed by the addition of perchloric acid (70%, 2 equiv.). The catalyst ([H2-DABCO][ClO4]2) was used in the multicomponent synthesis of 1,3-disubstituted-5-aryl-2,7-dithioxo/dioxo-2,3,5,6,7,8-hexahydropyrimido[4,5-d]pyrimidin-4(1H)-ones 95a–v for the reaction of aryl aldehydes with 6-amino-1,3-dimethyluracil 66, barbituric acid 56 or thiobarbituric acid 57 with urea or thiourea 58 (Scheme 22).26,38,58,64–66 The procedure provides short reactions times, saves costs, allows catalyst reusability, is moisture resistant and gives high yields.
 |
| | Scheme 22 Catalytic multicomponent synthesis of 2,3,5,6,7,8-hexahydropyrimido[4,5-d]pyrimidinones. | |
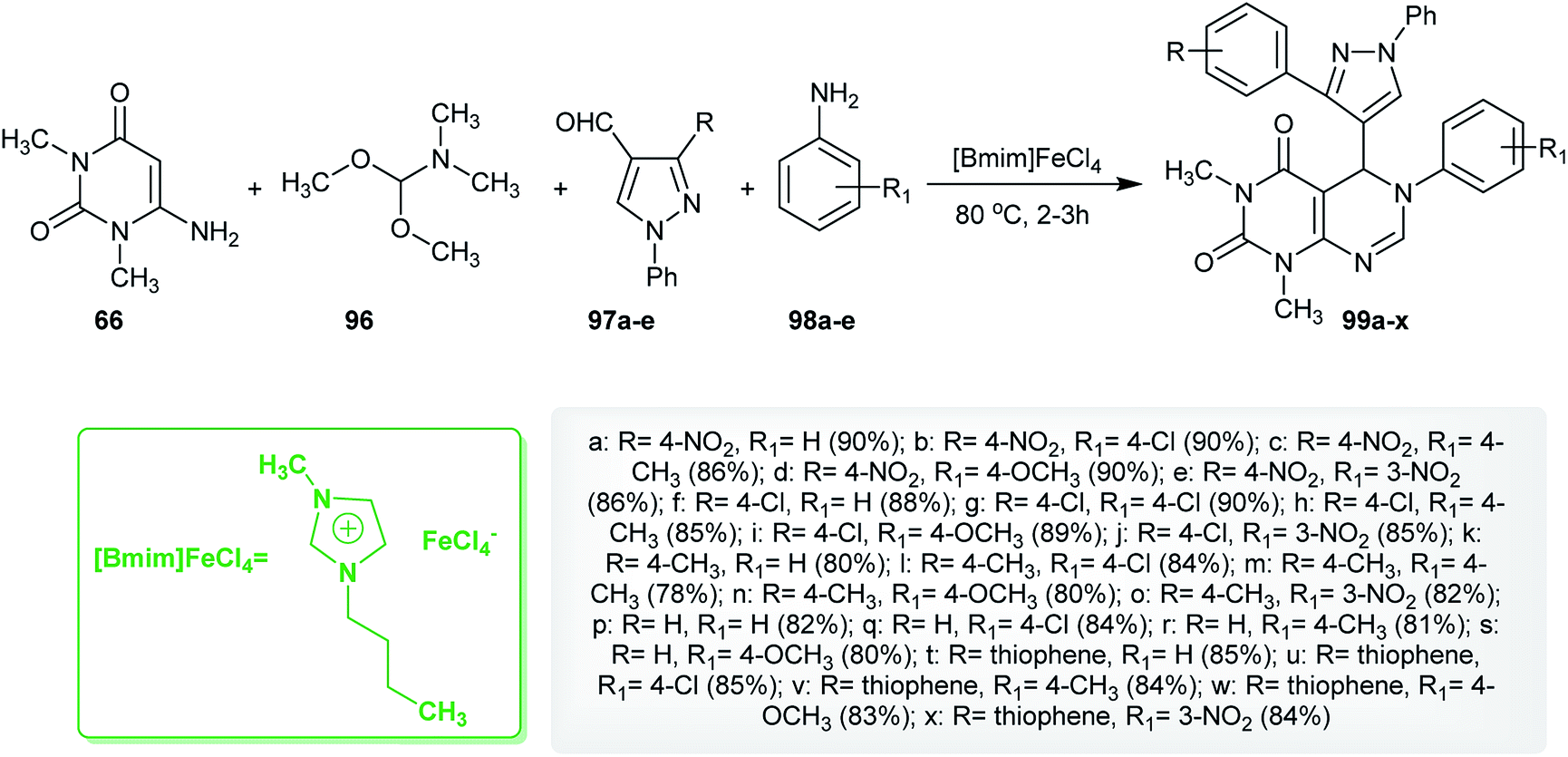
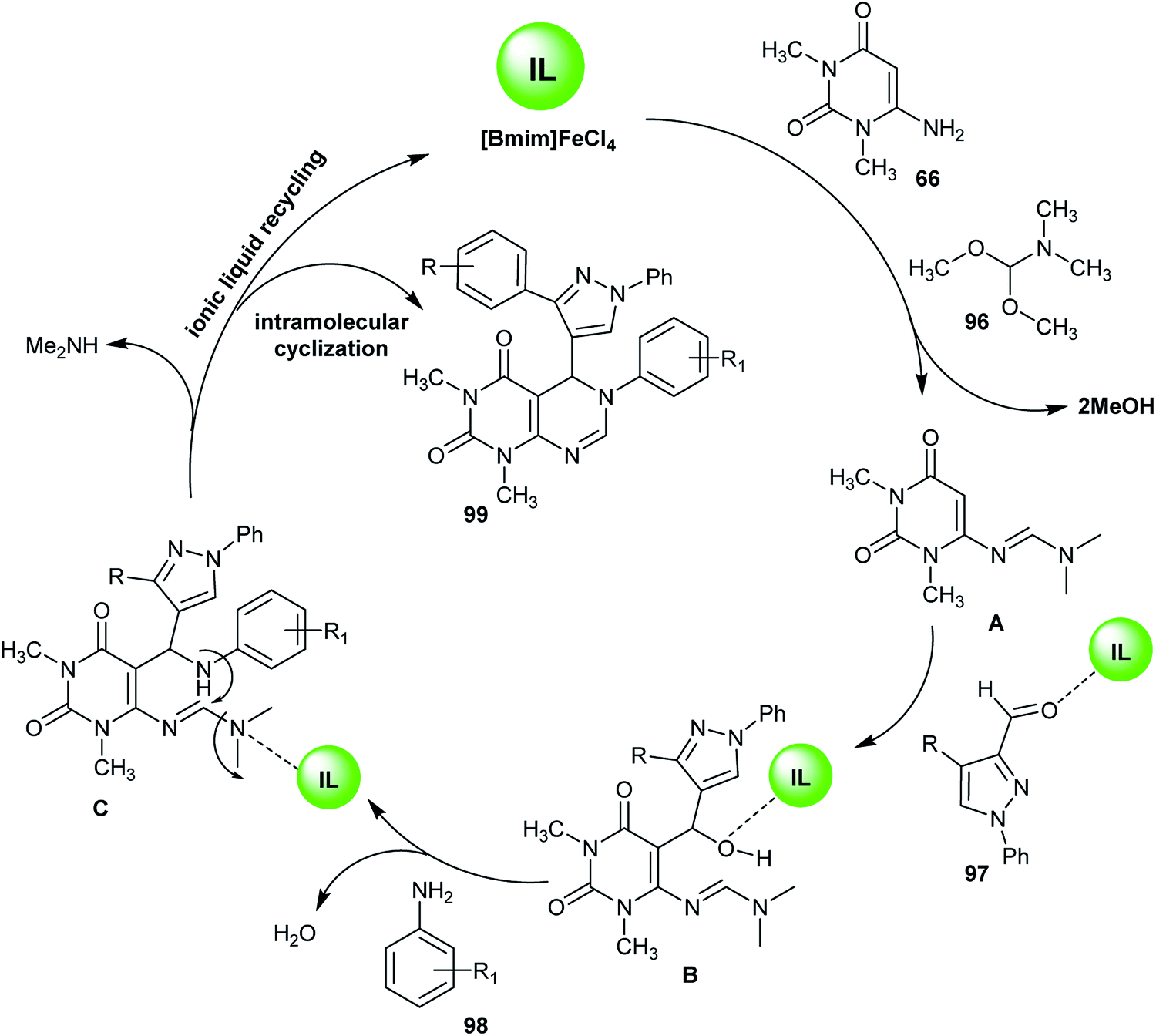
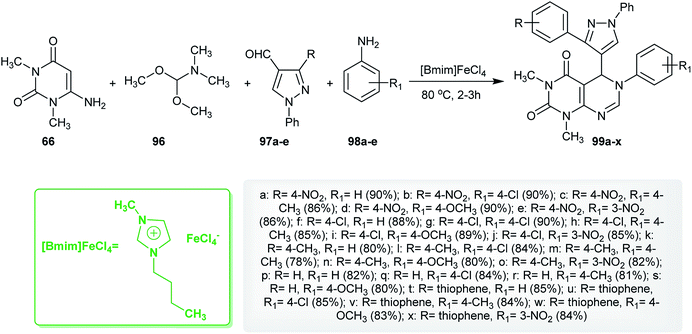
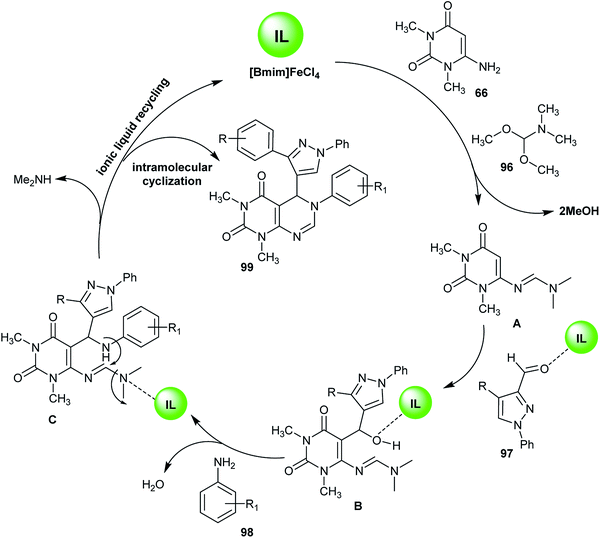
Similarly, an efficient method for the synthesis of 1,3-dimethyl-6-aryl-5-heteroaryl-5,6-dihydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-diones 99 using 1-butyl-3-methyl-1H-imidazol-3-ium iron(III) chloride ([Bmim]FeCl4) as an ionic liquid was reported by Suresh et al.67 by applying four-component reactions of 6-amino-1,3-dimethyluracil (66), DMF-DMA (96), 3-substituted-1-phenyl-1H-pyrazole-4-carbaldehydes 97 and aryl-amines under reflux conditions. The synthesis of compound 99f was established using X-ray crystallography (Scheme 23). The investigated analogs were estimated as antibacterial agents, in which compounds 99c, 99i, 99l, and 99m have potent results with minimum inhibitory concentration (MIC) values ranging between 3.9–15.6 μg mL−1 and the minimum bactericidal concentration (MBC) was found to be 2-fold greater than the antibacterial results. In addition, the compounds of the series 99 were assessed as biofilm inhibitors. Compounds 99l and 99m revealed potent results with half maximal inhibitory concentration (IC50) values ranging between 1.8–8.2 μg mL−1. Compound 99l increases the level of intracellular reactive oxygen species (ROS) when treating biofilms of Micrococcus luteus (MTCC 2470) cultured at a concentration of 0.5 μg mL−1 and increases the membrane permeability allowing protein leakage and the successive death of the bacterial cell.67
 |
| | Scheme 23 Four-component synthesis of 5,6-dihydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-diones. | |
The ionic liquid catalyst ([Bmim]FeCl4) was recycled from the previous reaction using a method similar to that reported by Shirini et al.63 using 1,4-diazabicyclo[2.2.2]octane-1,4-diium perchlorate ([H2-DABCO][ClO4]2) as an ionic liquid catalyst. The reaction mechanism proceeded through the initial formation of the amidine intermediate A by reaction of aminopyrimidindione 66 with DMF-DMA (96). The addition of formyl pyrazole 97 led to reaction with intermediate A and the formation of intermediate B. Dehydration of intermediate B with arylamines 98 resulted in the formation of intermediate C. Subsequent intramolecular cyclization on the intermediate C yielded the respective 1,3-dimethyl-6-aryl-5-heteroaryl-5,6-dihydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-diones 99 with recycling of the ionic liquid catalyst and elimination of the dimethylamine molecule (Scheme 24).67
 |
| | Scheme 24 The proposed mechanistic route for the synthesis of 5,6-dihydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-diones. | |
2.2. Ring synthesis from acyclic compounds
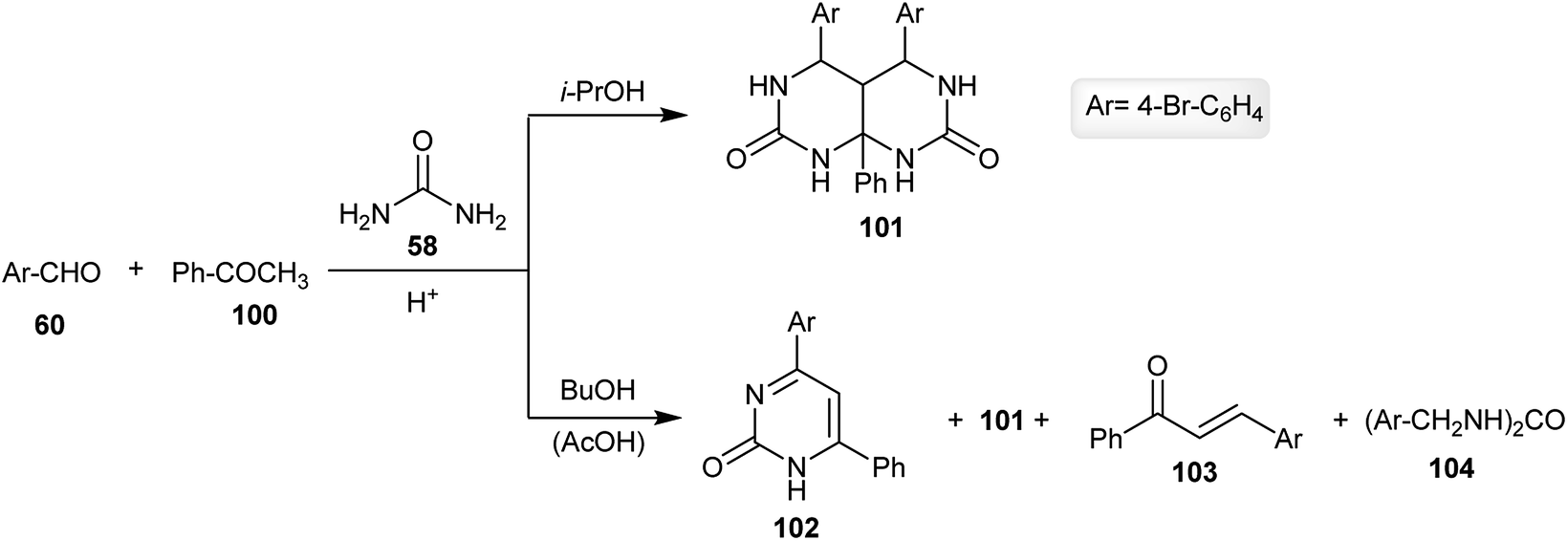
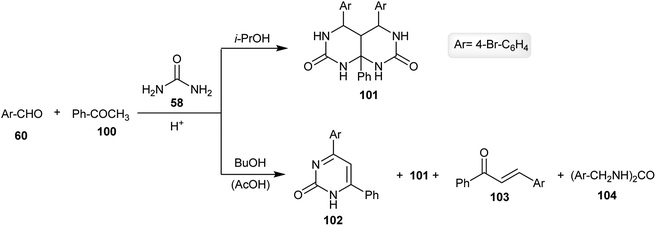
The multicomponent one-pot reaction of the aromatic aldehyde 60 with acetophenone (100) and urea (58) in an excess amount (1:1:4) in isopropanol yielded the respective hexahydropyrimido[4,5-d]pyrimidine-2,7(1H,3H)-dione 101 (63% yield) instead of the expected products 102 or its tautomeric form. The product 101 was obtained in a good yield through a cyclocondensation step reaction. The ratio of the reactants (1.5:1:3) allowed the formation of compound 102 with the formation of hexahydro-pyrimidopyrimidine-dione 101. On the other hand, changing the solvent in the previous reaction to butanol gave rise to the possibility of the formation of by-products 102, 103 and 104 in addition to the formation of the main product 101 (Scheme 25).68
 |
| | Scheme 25 Synthesis of hexahydropyrimido[4,5-d]pyrimidine-2,7(1H,3H)-dione. | |
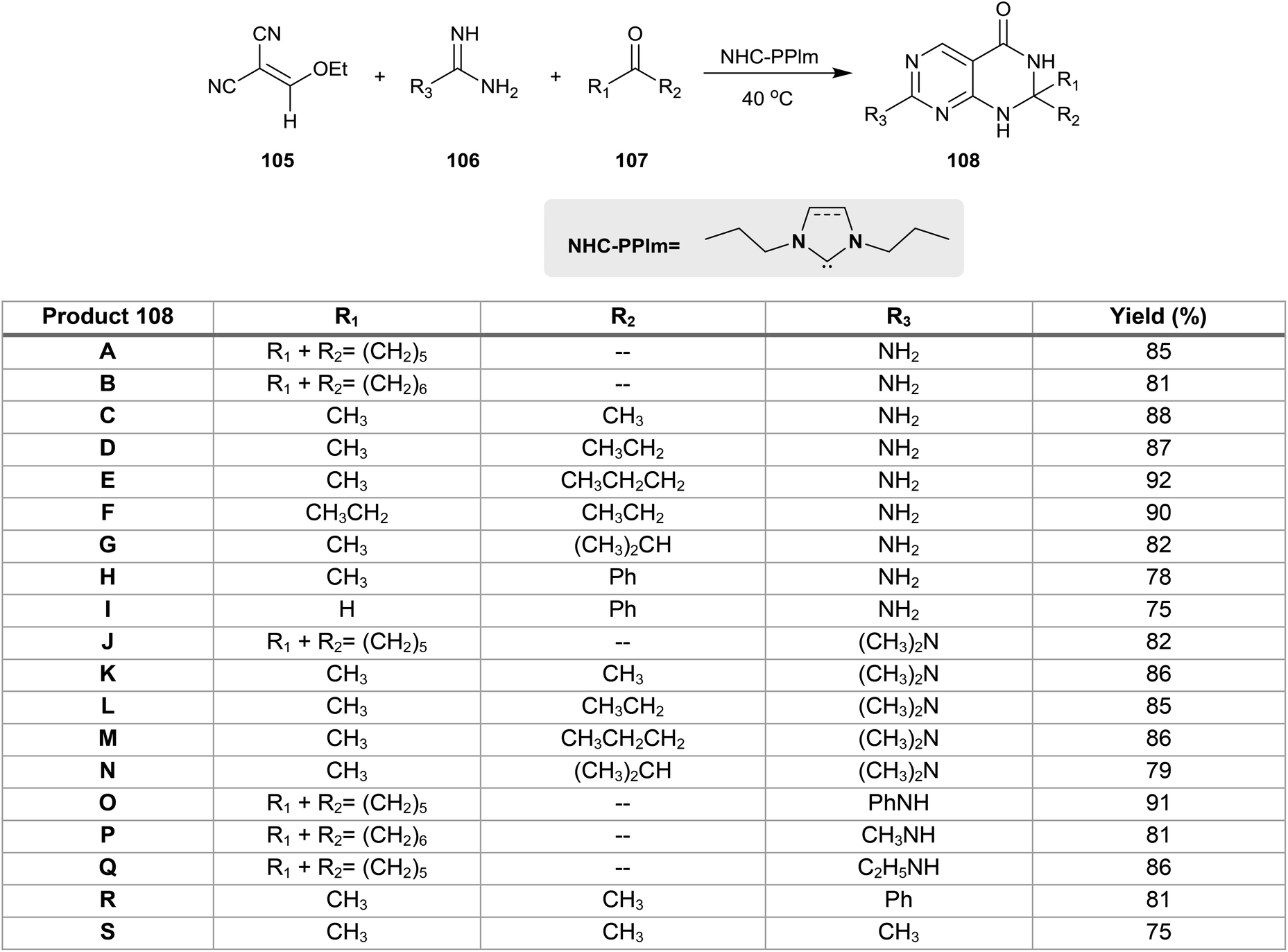
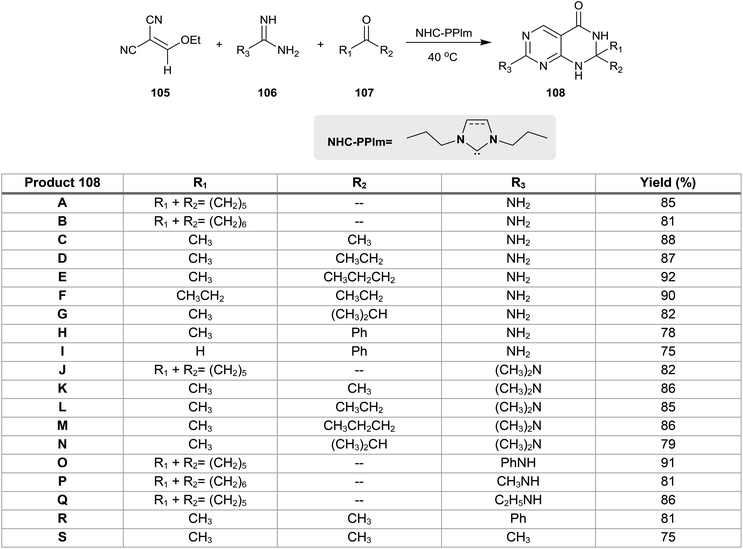
The multicomponent one-pot reactions of 2-(ethoxymethylene)malononitrile (105) with guanidine or amidines derivatives 106 and ketones or aldehydes 107 in the presence of catalytic N-heterocyclic carbenes gave the respective 2,2,7-trisubstituted-2,3-dihydropyrimido[4,5-d]pyrimidin-4(1H)-ones 108. The reaction mechanism involved the Michael addition of compound 105 with guanidine followed by subsequent cyclization, isomerization and aromatization to form the intermediate 4-amino-2-substituted-pyrimidine-5-carbonitrile. Next, nucleophilic attack of different nucleophiles (formed from the attack of the catalytic NHC-PPIm on the ketones or aldehydes) on the nitrile function of the recently formed intermediates took place, followed by release of the catalyst and then the Dimroth rearrangement to afford 108. The best yield of 108a were obtained by heating the reactants in ethanol containing N-heterocyclic carbenes (NHCs)-assisted “NHC-PPlm” (0.4 equiv.) at 40 °C (Scheme 26).27
 |
| | Scheme 26 Synthesis of 2,2,7-trisubstituted-2,3-dihydropyrimido[4,5-d]pyrimidin-4(1H)-ones. | |
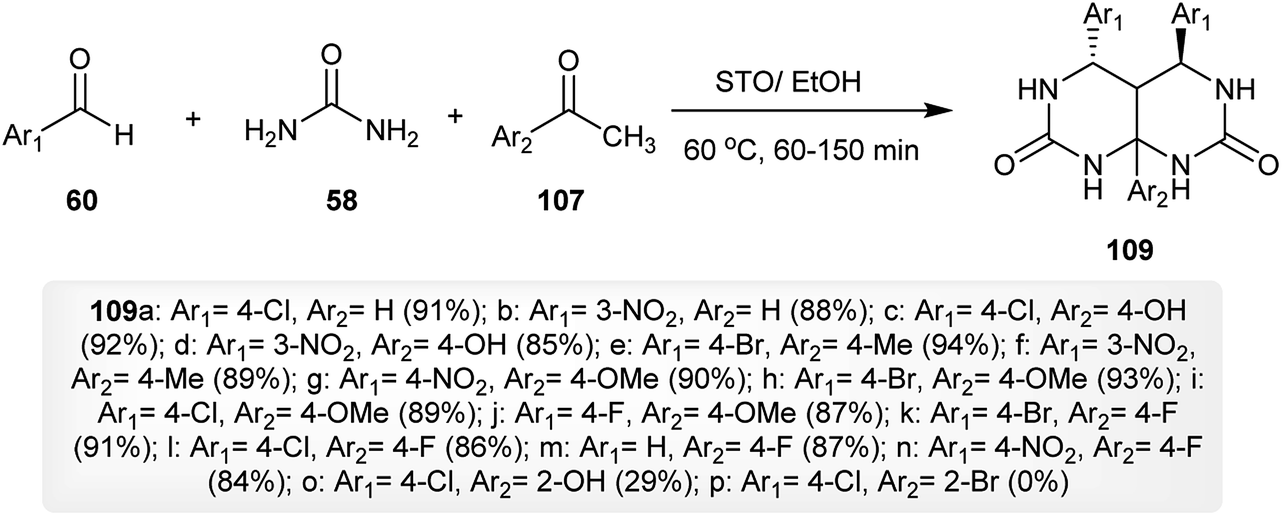
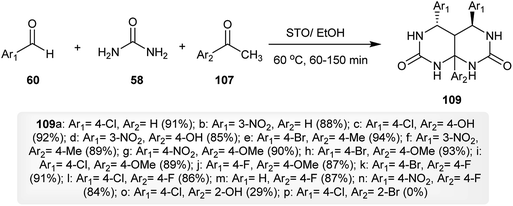
The procedure involved the synthesis of the Biginelli-type, 4,5,8a-trisubstituted-hexahydro-pyrimido-[4,5-d]pyrimidine-diones 109 was described by Magar and coworkers.40 The process specified a multicomponent one-pot synthesis from the reaction of aromatic aldehydes 60 with urea (58) and acetophenones 107 under optimum conditions in the presence of a recyclable catalyst, sulfated tin oxide (STO), to give the hexahydro-pyrimido-pyrimidine-diones 109. The catalyst was prepared by heating a solution of stannous chloride with a few drops of nitric acid and the subsequent addition of ammonia solution at pH = 8. The procedure allows the preparation of pyrimidopyrimidines with high yields after a short reaction time (Scheme 27).
 |
| | Scheme 27 Synthesis of 4,5,8a-trisubstituted-hexahydropyrimido[4,5-d]pyrimidine-2,7(1H,3H)-diones. | |
Multicomponent one-pot reactions of aromatic aldehydes 60 with urea or thiourea 58 and formaldehyde in boiling ethanol containing the organocatalyst gave the respective tetrahydropyrimido[4,5-d]pyrimidine-diones and thiones 110a–l. The best yield of compound 110a was achieved by treating the reactants with ethanol as a solvent and (S)-N-(4-fluorophenyl)-1-tosylpyrrolidine-2-carboxamide as a catalyst after 15 h (Scheme 28).69
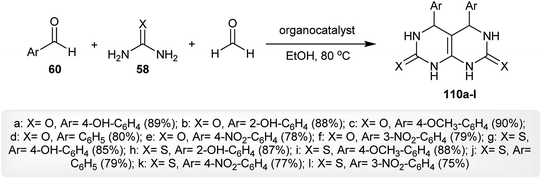 |
| | Scheme 28 Multicomponent synthesis of 4,5-diaryl-4,5,6,8-tetrahydropyrimido[4,5-d]pyrimidine-2,7(1H,3H)-diones and thiones. | |
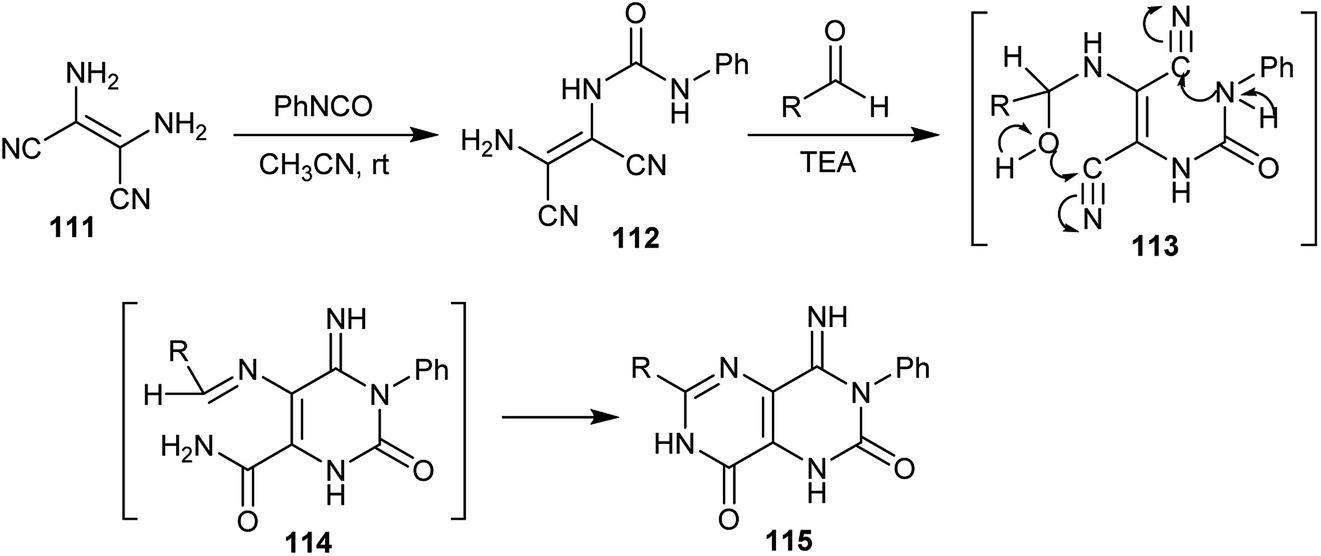
A series of 4-imino-6-substituted-3-phenyl-1,3,4,7-tetrahydropyrimido[5,4-d]pyrimidine-2,8-diones (115) was first synthesized by Ohtsuka in 1978,70 through the reaction of phenyl isocyanate with 2,3-diaminomaleonitrile (111) via a condensation process to give the respective 1-(2-amino-1,2-dicyanovinyl)-3-phenylurea (112) followed by reaction with diverse aldehydes in the presence of triethylamine as a basic medium. The reaction involved the formation of two intermediates, 113 and 114 (Scheme 29).
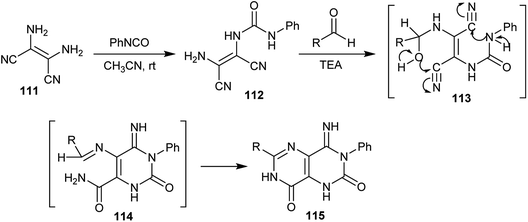 |
| | Scheme 29 Synthesis of 1,3,4,7-tetrahydropyrimido[5,4-d]pyrimidine-2,8-diones. | |
Under mild conditions, a proficient method for the synthesis of a series of 4a,8a-disubstituted-1,3,6,8-tetramethylhexahydropyrimido[4,5-d]pyrimidine-2,7(1H,3H)-diones 118 from the reactions of ketones 116 with para-formaldehydes 117 following the use of l-(+)-tartaric acid-dimethylurea as a catalyst, reaction medium, and solvent at the same time. The catalyst l-(+)-tartaric acid-DMU (70:30) was used to achieve the best yields from the reactions by reacting 1 mole of the ketone with 4 moles of paraformaldehyde 117. The method is an eco-friendly technique for the preparation of the products 118 in excellent yields (Scheme 30).71
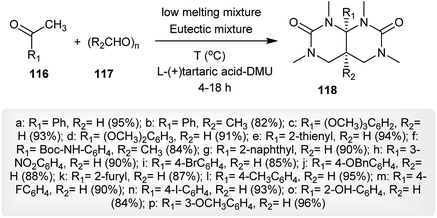 |
| | Scheme 30 Synthesis of hexahydropyrimido[4,5-d]pyrimidine-2,7(1H,3H)-diones. | |
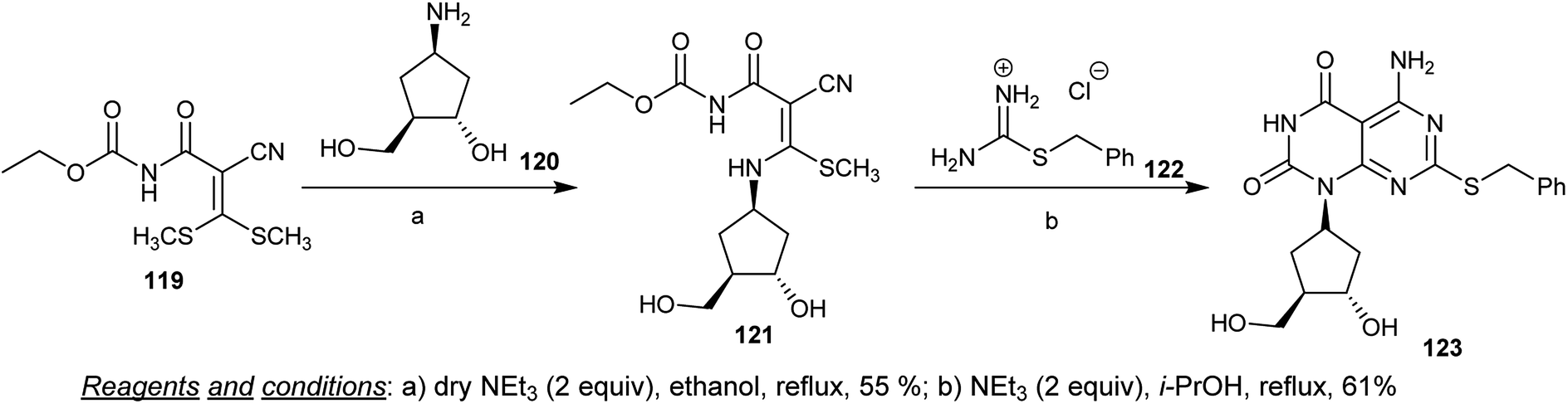
Condensation of ethyl (2-cyano-3,3-bis(methylthio)acryloyl)carbamate (119) with racemic 4-amino-2-(hydroxymethyl)cyclopentan-1-ol (120) in refluxing ethanol containing triethylamine yielded the condensation intermediate 121. The cyclization of the respective ethyl carbamate intermediate 121 did not occur using different bases and catalysts. The cyclization of 121 through reaction with 2-benzyl-isothiouronium chloride (122) was achieved by refluxing isopropanol in the presence of triethylamine to give the desired cyclopentyl-pyrimido[4,5-d]pyrimidine-dione 123 (Scheme 31).72
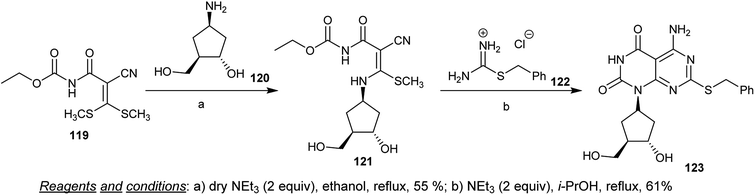 |
| | Scheme 31 Synthesis of cyclopentyl-pyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione. | |
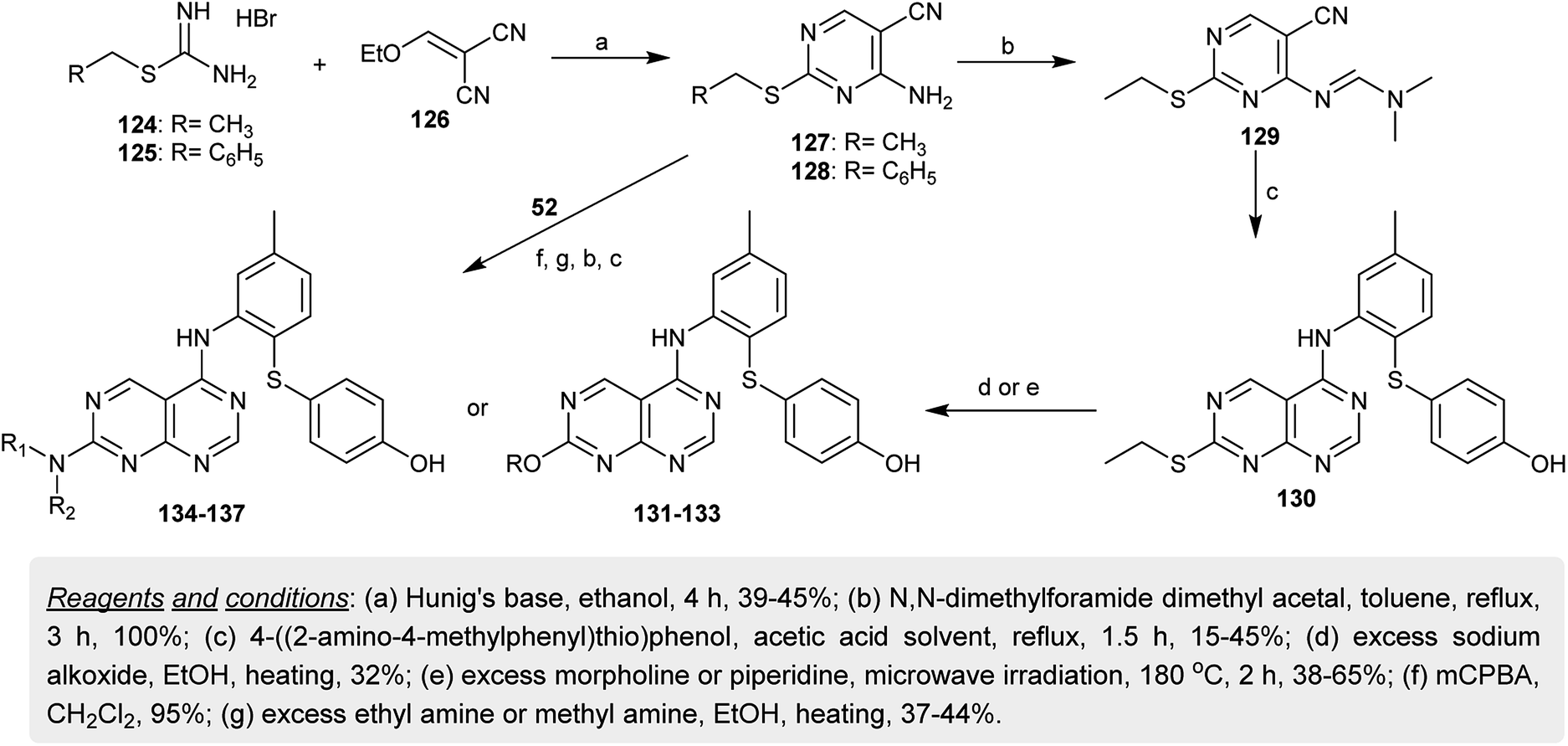
In another synthetic route, substituted-methyl carbamimidothioate hydrobromides 124 and 125 both reacted with 2-(ethoxymethylene)malononitrile (126) in ethanol containing Hunig's base to give the analogous aminopyrimidines 127 and 128. In refluxing toluene, the amine 127 was transformed into the respective amidine 129 by reaction with DMF-DMA. Heating of 129 with 4-((2-amino-4-methylphenyl)-thio)phenol in acetic acid gave the cyclization product 130. In a strong basic medium, the ethylsulfanyl moiety of 130 was displaced by nucleophiles such as morpholine or piperidine under microwave irradiation conditions to give the respective ether 131–133 and amine 134, 135 derivatives. Consecutively, the oxidation of the mercapto moiety of 128 into the respective sulfone followed by nucleophilic displacement with amines yielded the amines 136 and 137 (Scheme 32). The compounds were evaluated as inhibitors for hepatitis C virus replication using two assays (genotype 1a and 1b cell culture HCV replicon), in which compound 130 demonstrated the highest potency against 1b replication (half maximal effective concentration (EC50) = 10 μM) and the other compounds exhibited activities with an EC50 greater than 2 μM.73
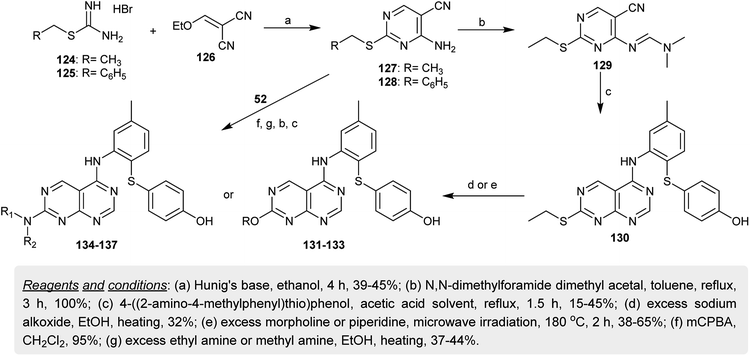 |
| | Scheme 32 Synthesis of 2,5-disubstituted-pyrimido[4,5-d]pyrimidines. | |
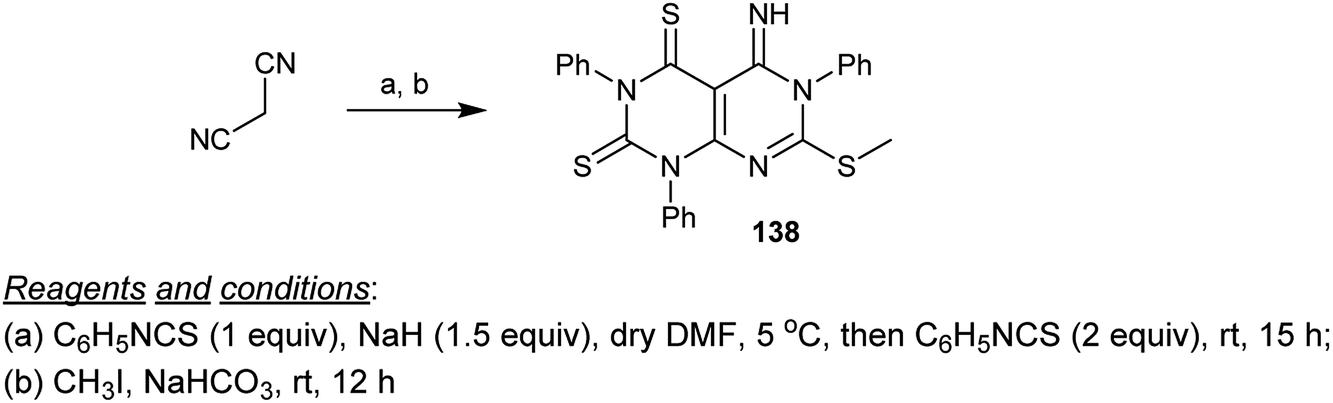
Condensation of malononitrile with three equivalents of phenyl isothiocyanate in DMF/sodium hydride and subsequent methylation of the SH group of the formed intermediate at C7 gave the respective products incorporating a dihydropyrimido[4,5-d]pyrimidine core 138 in good yield (Scheme 33).74
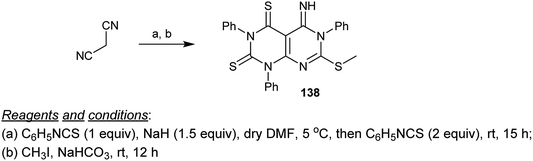 |
| | Scheme 33 Synthesis of 5-imino-7-(methylthio)-1,3,6-triphenyl-5,6-dihydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dithione. | |
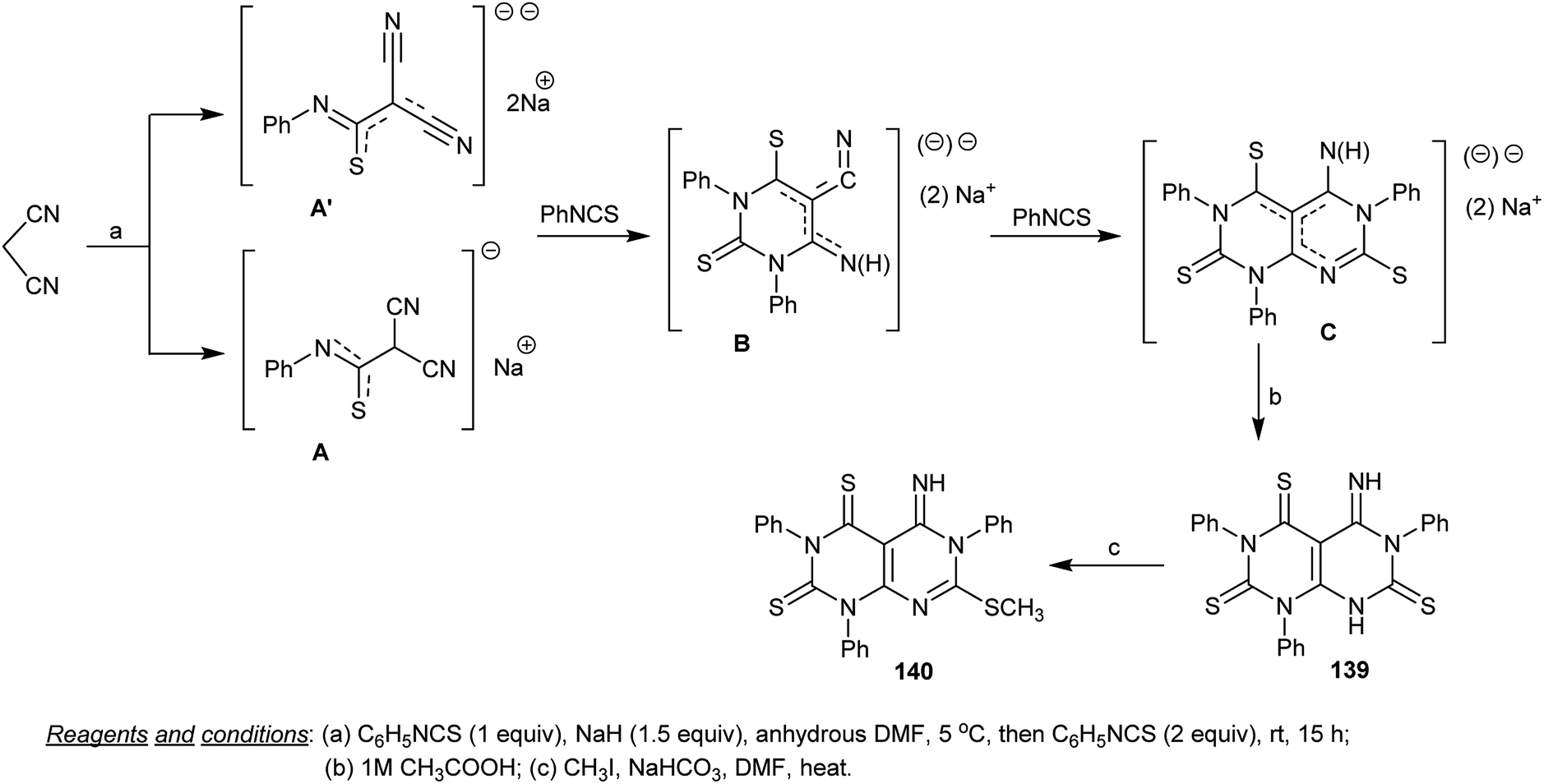
The intermediate, 5-imino-1,3,6-triphenyl-5,8-dihydropyrimido[4,5-d]pyrimidine-2,4,7-(1H,3H,6H)-trithione (139) was synthesized through the reaction of malononitrile with one equivalent of phenyl isothiocyanate in DMF containing sodium hydride followed by the addition of two equivalents of phenyl isothiocyanate to the generated mono and dianion intermediates A and A′ and subsequent treatment of the intermediate C with acetic acid. The presence of sodium hydride in the first step facilitates the acidic proton abstraction from malononitrile to form the anion which attacks the CS of phenyl isothiocyanate. Methylation of 139 was accessed by reaction with methyl iodide in DMF containing sodium bicarbonate to give the methylation product 140 (Scheme 34).75
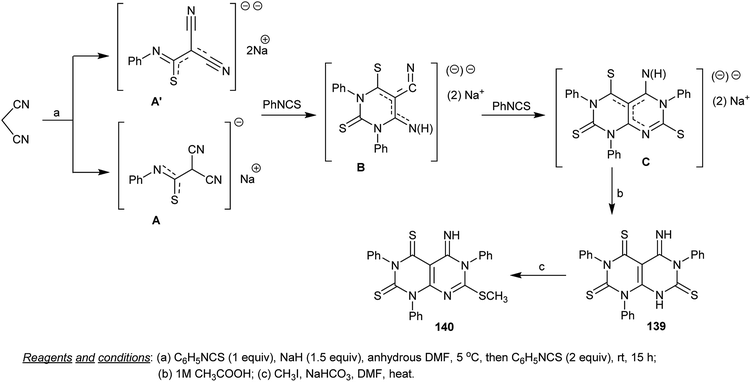 |
| | Scheme 34 Synthesis of 5-imino-7-(methylthio)-1,3,6-triphenyl-5,6-dihydropyrimido[4,5-d]pyrimidine-2,4-(1H,3H)-dithione. | |
2.3. Synthesis of tricyclic systems
2.3.1. Multicomponent synthesis.
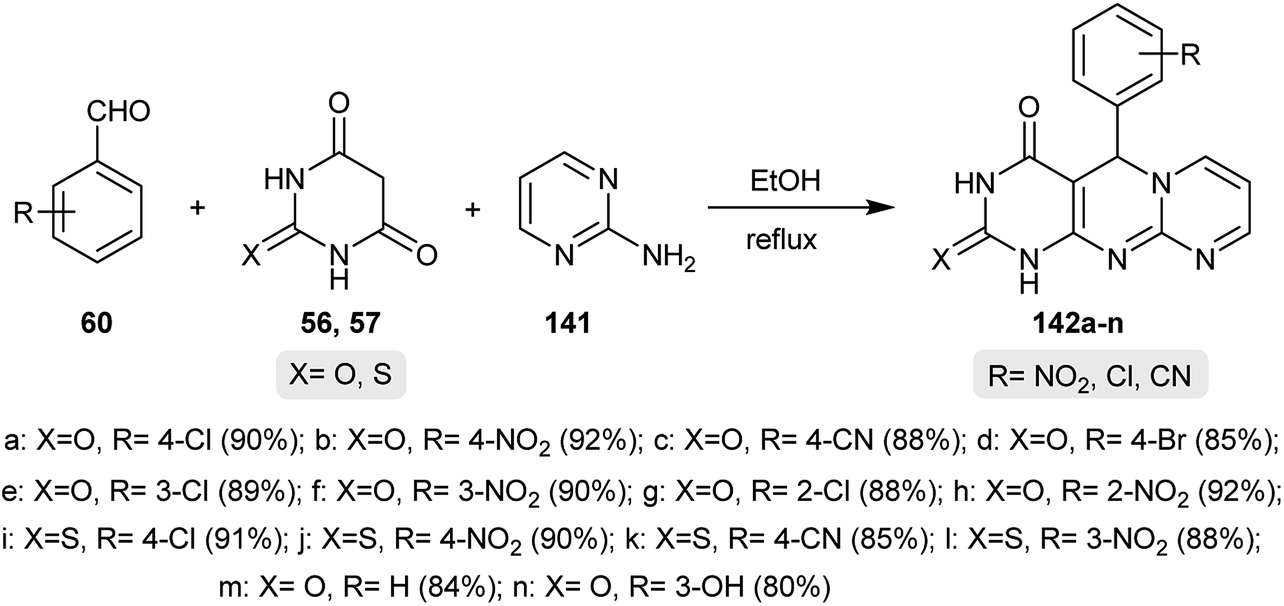
Patil and coworkers48 developed a new method for the synthesis of tetrahydro-4H-dipyrimido[1,2-a:4′,5′-d]pyrimidin-4-ones. The reactions involved one-pot multicomponent reactions of aryl aldehydes 60, barbituric or thiobarbituric acids 56 and 57 and 2-aminopyrimidine (141). The respective tetrahydro-4H-dipyrimido[1,2-a:4′,5′-d]pyrimidin-4-ones 142a–n were obtained by refluxing the reactants in ethanol under non-supported catalytic conditions (Scheme 35). The mechanism for the formation of products 142a–n was proposed to be an initial Knoevenagel condensation of the aryl aldehydes 60 with barbituric or thiobarbituric acids 56 and 57 to form a conjugate enone intermediate. 2-Aminopyrimidine (141) followed an amine-imine rearrangement resulting in the nucleophilic attack of the pyrimidine nitrogen at the enone intermediate double bond and the subsequent Michael addition led to the formation of the pyrimidinyl-iminopyrimidine intermediate. Cyclization and oxidation of the recently formed intermediate gave the anticipated products 142a–n.
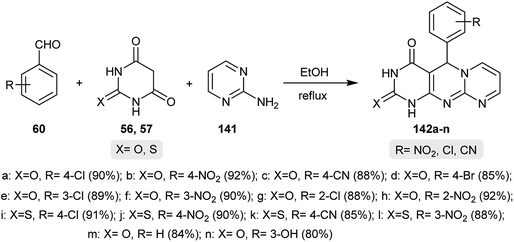 |
| | Scheme 35 Synthesis of tetrahydro-4H-dipyrimido[1,2-a:4′,5′-d]pyrimidin-4-ones. | |
2.3.2. Synthesis of pyrimidoquinazoline-diones.
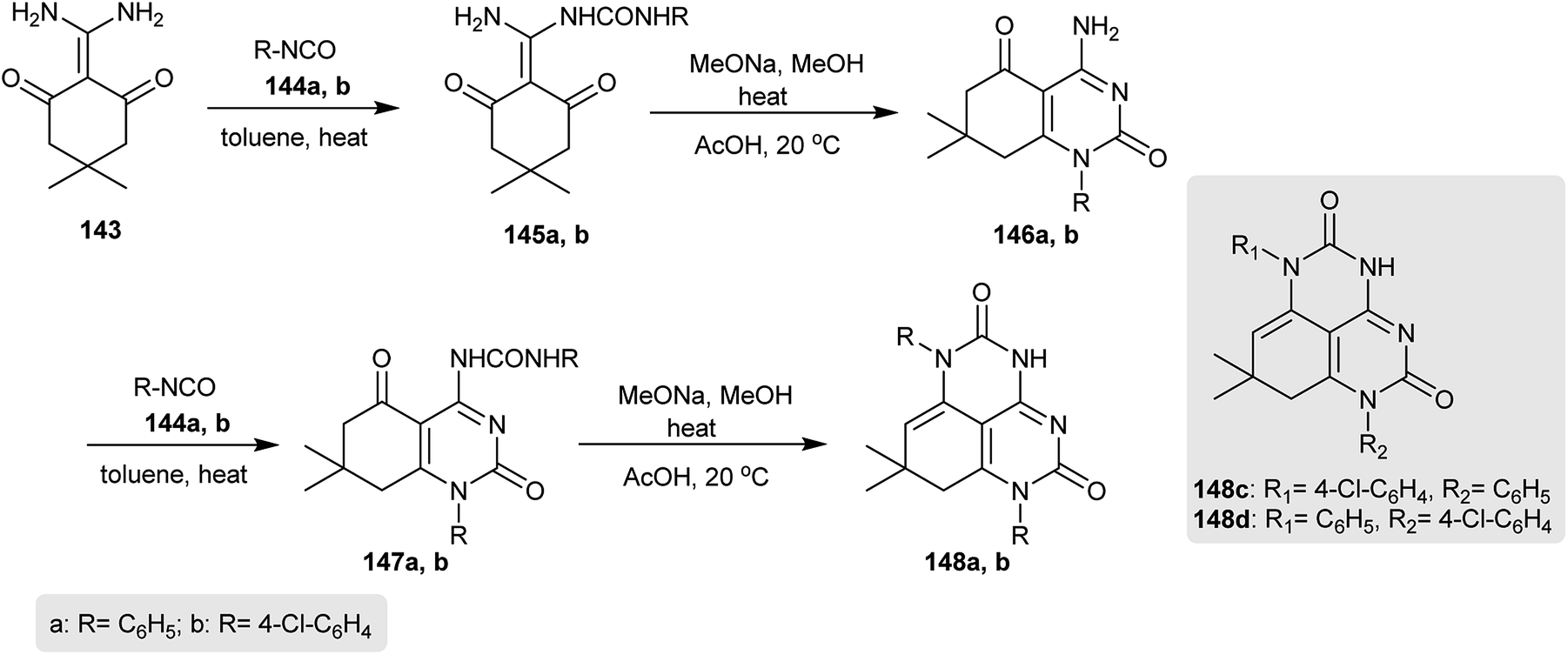
The reaction of 2-(diaminomethylene)-5,5-dimethylcyclohexane-1,3-dione (143) with excess aryl isocyanates 144 in refluxing toluene gave the urea derivatives 145. Cyclization of 134 by heating in sodium methoxide solution yielded the respective aminopyrimidinones 146, in which the enamine moiety was involved in the heterocyclization. In addition, heating the aryl isocyanates 144 with aminopyrimidinones 146 gave urea derivatives 147a and 147b in 87–89% yields. Heterocyclization of 147 by heating in a sodium methoxide solution afforded the desired 7,8-dihydro-1H-pyrimidoquinazoline-2,5(3H,6H)-diones 148a and 148b with yields of 80 and 65%, respectively. Changing the aryl isocyanates allowed the preparation of 148c and 148d with yields of 67 and 80% following similar preceding synthetic approaches (Scheme 36).76
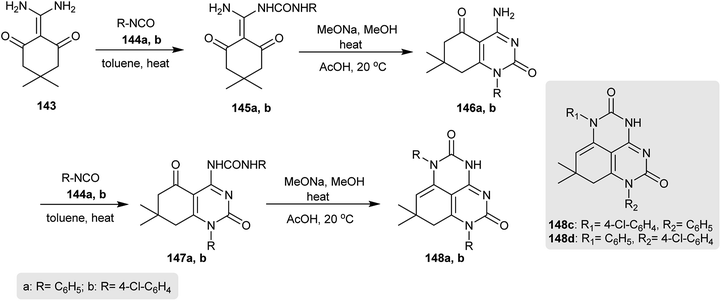 |
| | Scheme 36 Synthesis of 7,8-dihydro-1H-pyrimidoquinazoline-2,5(3H,6H)-diones. | |
2.3.3. Synthesis of pyridopyrimidopyrimidinones.
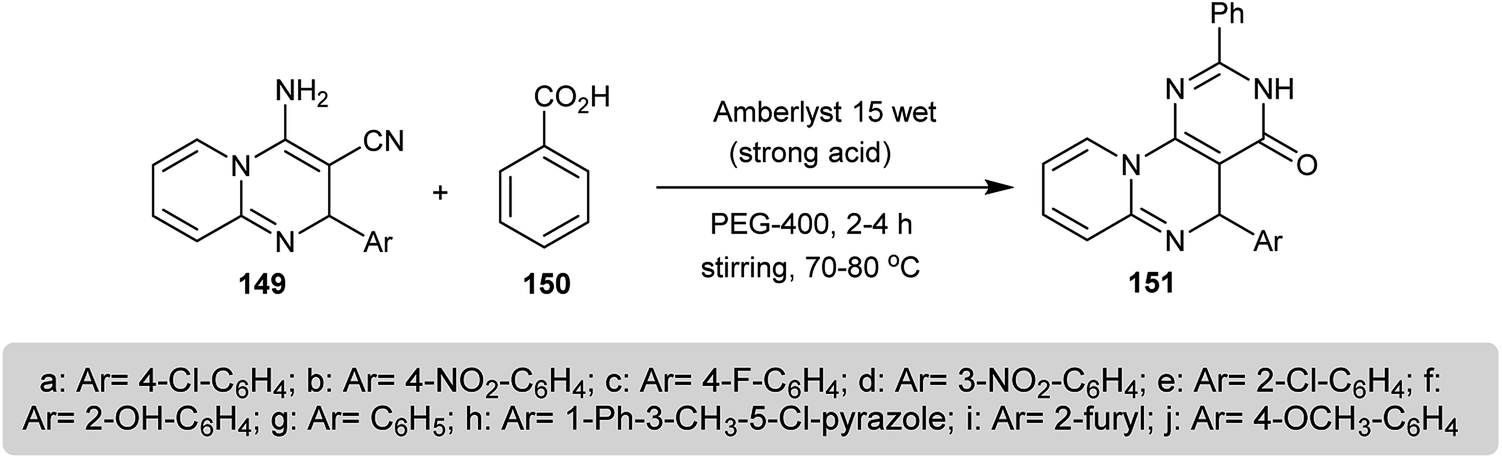
A series of 5-aryl-2-phenyl-3,5-dihydro-4H-pyrido[1,2-a]pyrimido[5,4-e]pyrimidin-4-ones 151 were prepared by the reaction of benzoic acid (150) with 2-aryl-3-cyano-4-amino-2H-pyrido[1,2-a]pyrimidines 149 through a green technique in PEG-400 (polyethylene glycol) in the presence of a catalytic amount of Amberlyst 15-wet. The use of PEG-400 enabled the availability and low toxicity and the catalyst Amberlyst 15-wet provided a strong acid medium (Scheme 37). The mechanism of the reaction proceeded through condensation between the carboxylic group of the benzoic acid and the amino group of 149 and the subsequent intramolecular cyclization. The compounds 151 were assessed as good antifungal agents against A. niger, C. albicans, and A. flavus species and the results were compared to the standard antibiotic Nystatin at 50 μg mL−1.77
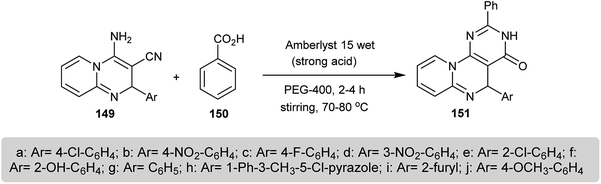 |
| | Scheme 37 Synthesis of 5-aryl-2-phenyl-3,5-dihydro-4H-pyrido[1,2-a]pyrimido[5,4-e]pyrimidin-4-ones. | |
2.4. Synthesis of tetracyclic systems
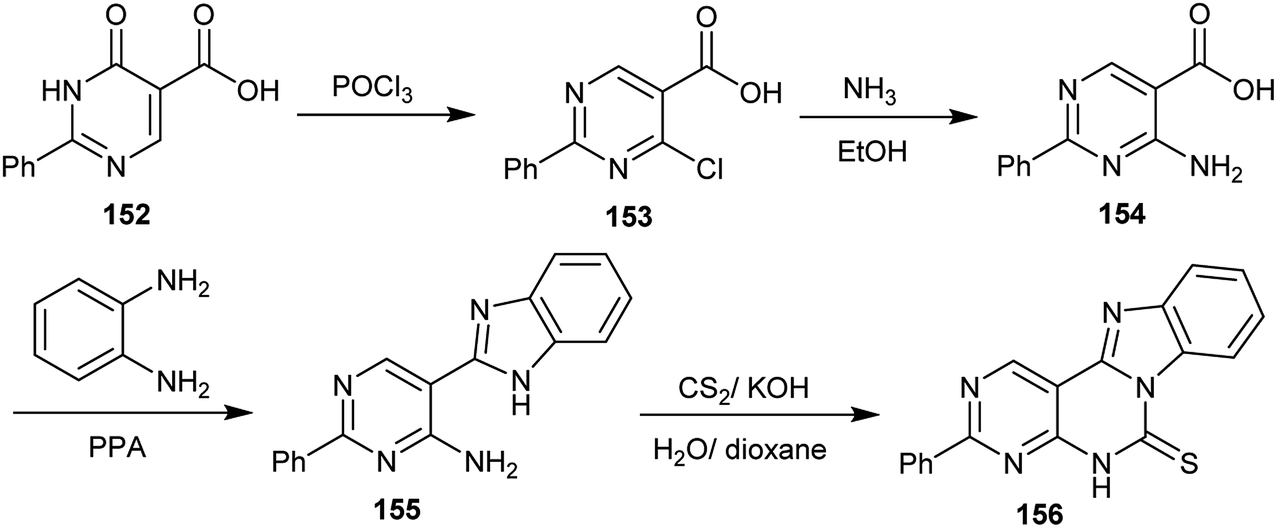
Chlorination of 6-oxo-2-phenyl-1,6-dihydropyrimidine-5-carboxylic acid (152) with phosphorus oxychloride yielded the respective 4-chloro-2-phenylpyrimidine-5-carboxylic acid (153). Treatment of 153 with ammonia in ethanol gave the amine 154, which was cyclized to the desired 5-(1H-benzo[d]imidazol-2-yl)-2-phenylpyrimidin-4-amine (155) upon heating with o-phenylenediamine in polyphosphoric acid. Compound 155 reacted with carbon disulfide in a mixture of water and dioxane containing potassium hydroxide to afford benzimidazo-pyrimido-pyrimidine-thione 156 in a 34% yield (Scheme 38).78
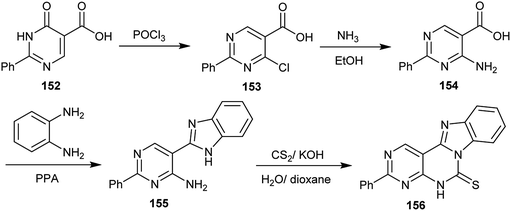 |
| | Scheme 38 Synthesis of 3-phenylbenzo[4,5]imidazo[1,2-c]pyrimido[5,4-e]pyrimidine-6(5H)-thione. | |
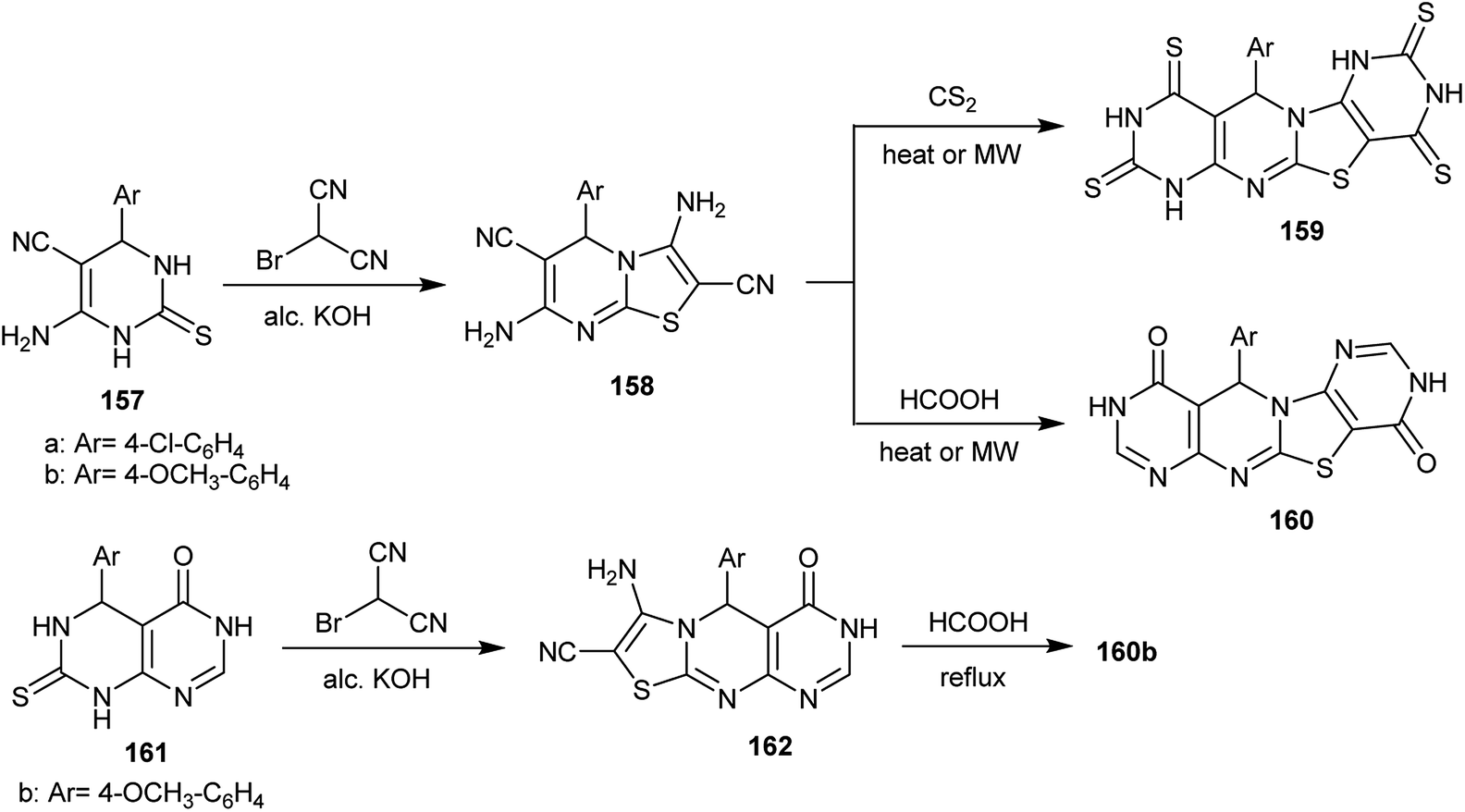
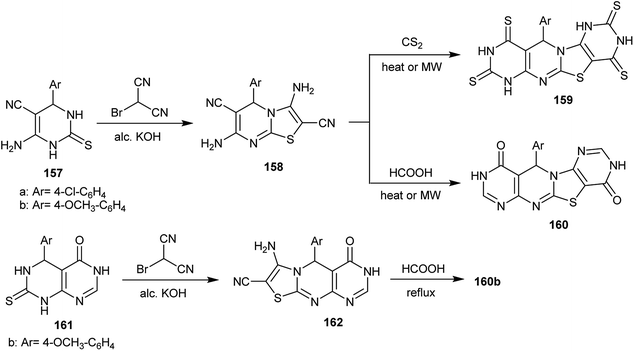
The reaction of 6-amino-4-aryl-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carbonitriles 157 with 2-bromo-malononitrile in an alcoholic potassium hydroxide solution yielded the respective 3,7-diamino-5-aryl-5H-thiazolo[3,2-a]pyrimidine-2,6-dicarbonitriles 158. The thiazolopyrimidines 158 reacted with both carbon disulfide and formic acid under microwave-assisted conditions to afford the corresponding polycyclic pyrimido[4,5-d]pyrimidines 159 and 160, respectively. The mechanism of this reaction involved the initial condensation of the two amino groups of 158 with two moles of formic acid, the partial hydrolysis and the cyclocondensation step. Compound 160b could be obtained by another route, the reaction of 5-aryl-7-thioxo-5,6,7,8-tetrahydropyrimido[4,5-d]pyrimidin-4(3H)-one 161 with 2-bromo-malononitrile in alcoholic potassium hydroxide solution and subsequent cyclization by reaction of the formed dihydro-4H-pyrimidothiazolo[3,2-a]pyrimidine 162 with formic acid (Scheme 39).79
 |
| | Scheme 39 Synthesis of polycyclic pyrimido[4,5-d]pyrimidines. | |
2.5. Ring synthesis by transformation of another ring
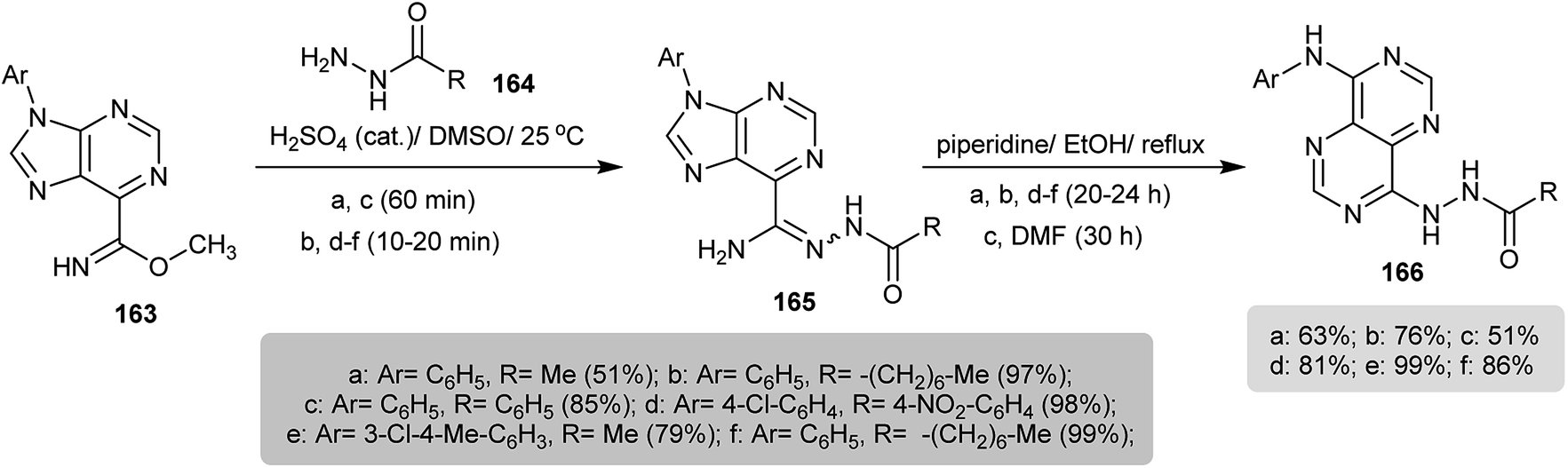
The reaction of methyl 9-aryl-9H-purine-6-carbimidates 163 with hydrazides 164 in DMSO containing a catalytic amount of sulfuric acid yielded N′-acetyl-9-aryl-9H-purine-6-carbohydrazonamides 165 in good to excellent yields. Subsequent refluxing of 165 in ethanol containing catalytic drops of piperidine resulted in imidazole ring cleavage followed by intramolecular cyclization of the formed intermediate to afford the respective N′-(8-(aryl-amino)pyrimido[5,4-d]pyrimidin-4-yl)substituted-hydrazides 166 (Scheme 40).80
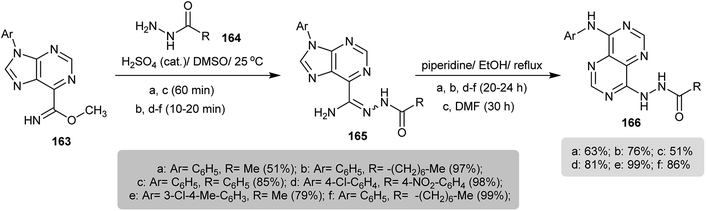 |
| | Scheme 40 Synthesis of N′-(8-(aryl-amino)pyrimido[5,4-d]pyrimidin-4-yl)substituted-hydrazides. | |
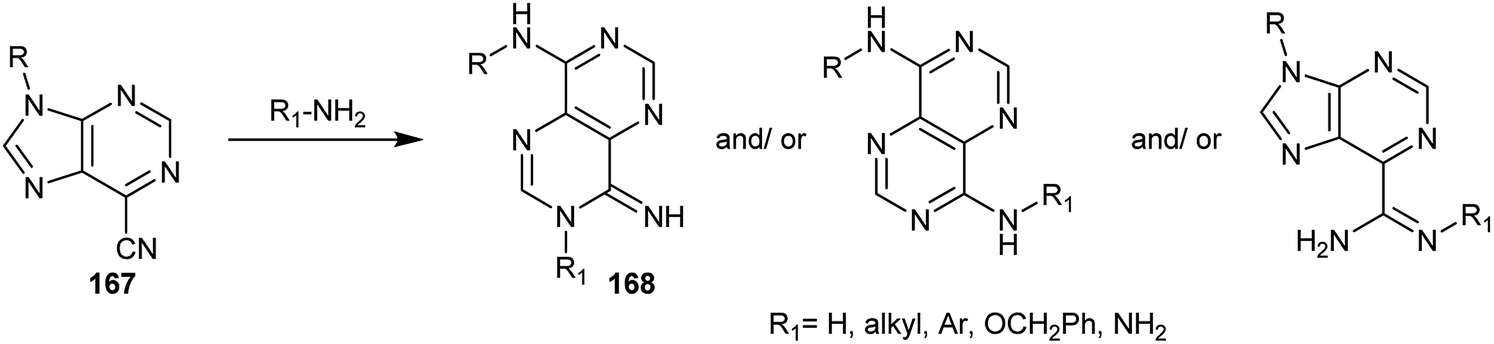
Furthermore, 9-substituted-9H-purine-6-carbonitriles 167 were used as synthetic intermediates for the synthesis of pyrimido[5,4-d]pyrimidine analogs 168 by reaction with various amines through ring transformations (Scheme 41).14,24,81–89
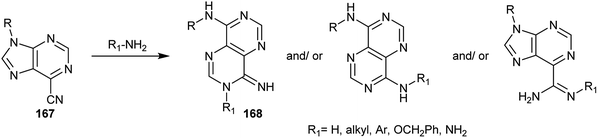 |
| | Scheme 41 Reactions of 9-substituted-9H-purine-6-carbonitriles with different amines. | |
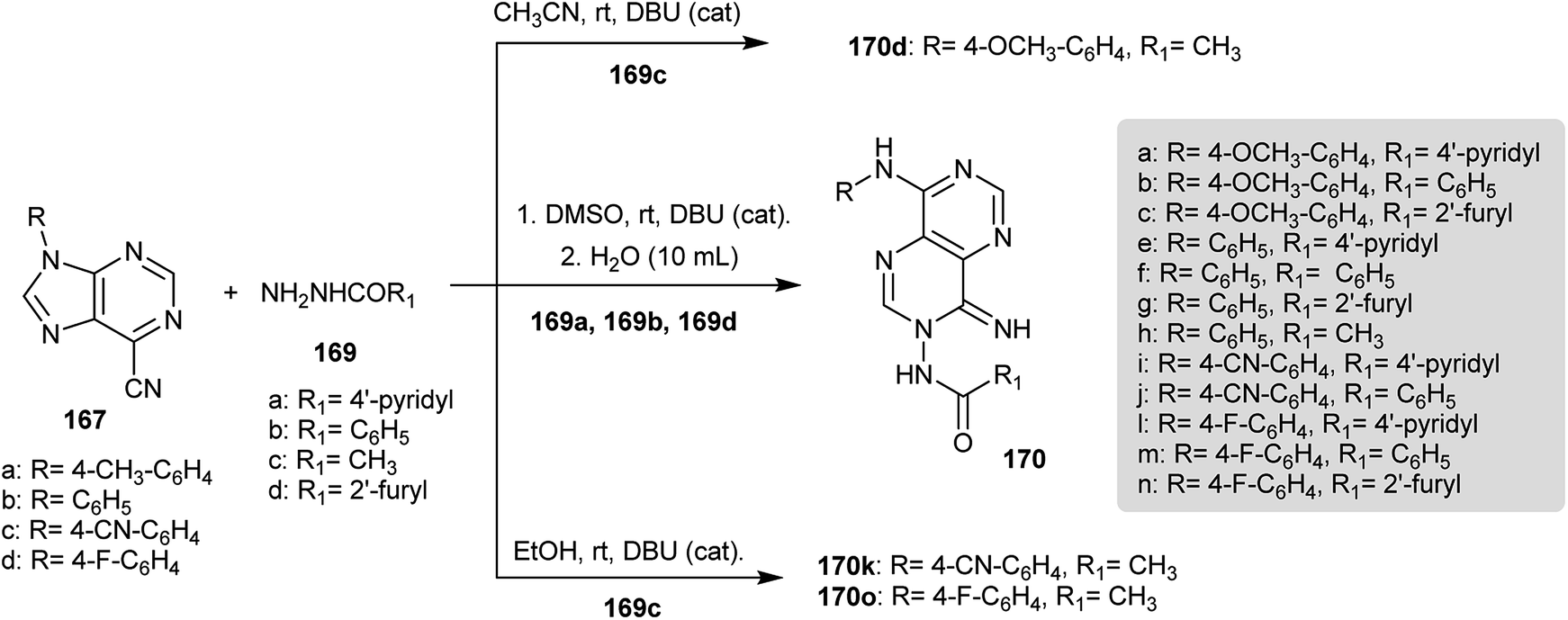
A series of pyrimido[5,4-d]pyrimidines 170 were prepared by reaction of 9-substituted-9H-purine-6-carbonitriles (167) with various hydrazides 169 by treatment in DMSO and water using catalytic DBU. Compound 170d was obtained by the reaction of 9-aryl-9H-purine-6-carbonitriles 167 with acetohydrazide (169c) in acetonitrile instead of DMSO and compounds 170k and 170o were prepared in ethanol (Scheme 42). The compounds were tested as antibacterial agents against Mycobacterium tuberculosis strains in which the biological impact is based on the substituents at the C8 and N3 atoms of the pyrimidopyrimidine skeleton. The incorporation of 4-F-aryl and 4-OCH3-aryl substituents into C8 and the heteroaryl substituents into N3 enhance the activity. Compound 170l has a high antitubercular activity at IC90 = 3.58 μg mL−1.14
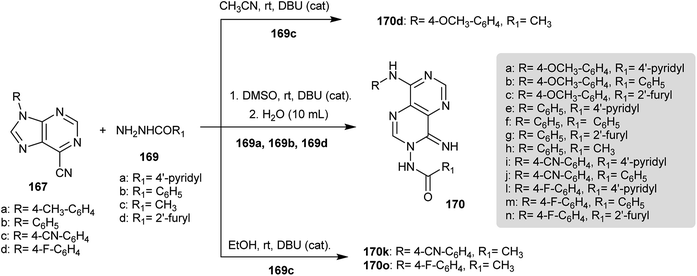 |
| | Scheme 42 Synthesis of 3,8-disubstituted-amino-4-iminopyrimido[5,4-d]pyrimidines. | |
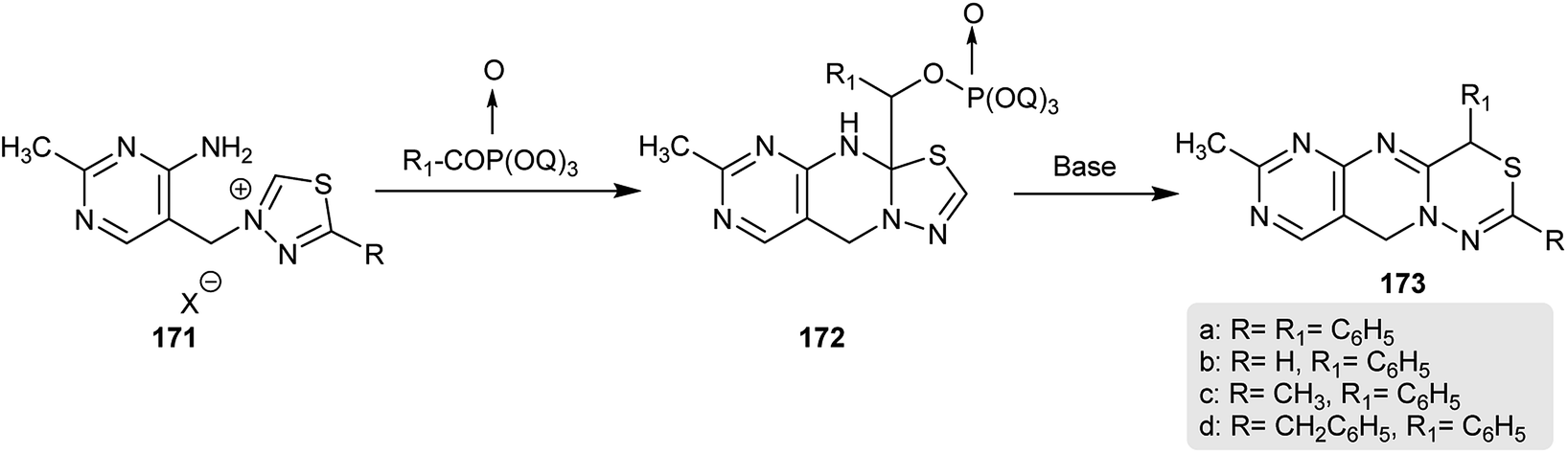
In 1974, Takamizawa and Sato90 patented a different technique for the production of dihydro-pyrimido-pyrimido-thiadiazines 173. Therefore, treatment of 3-((4-amino-2-methylpyrimidin-5-yl)-methyl)-5-substituted-1,3,4-thiadiazol-3-ium salts 171 with diethyl benzoyl phosphonate followed by treating the formed intermediates 172 with a base afforded the respective dihydro-pyrimido-[4′,5′:4,5]-pyrimido[1,2-d]1,3,4-thiadiazines 173 (Scheme 43).
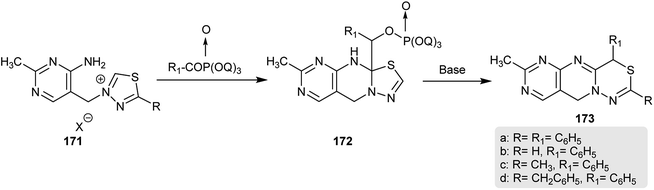 |
| | Scheme 43 Synthesis of 7-methyl-2,4-disubstituted-4,10-dihydropyrimido[4′,5′:4,5]pyrimido[1,2-d]-[1,3,4]-thiadiazines. | |
2.6. Synthesis of bis-pyrimidopyrimidines
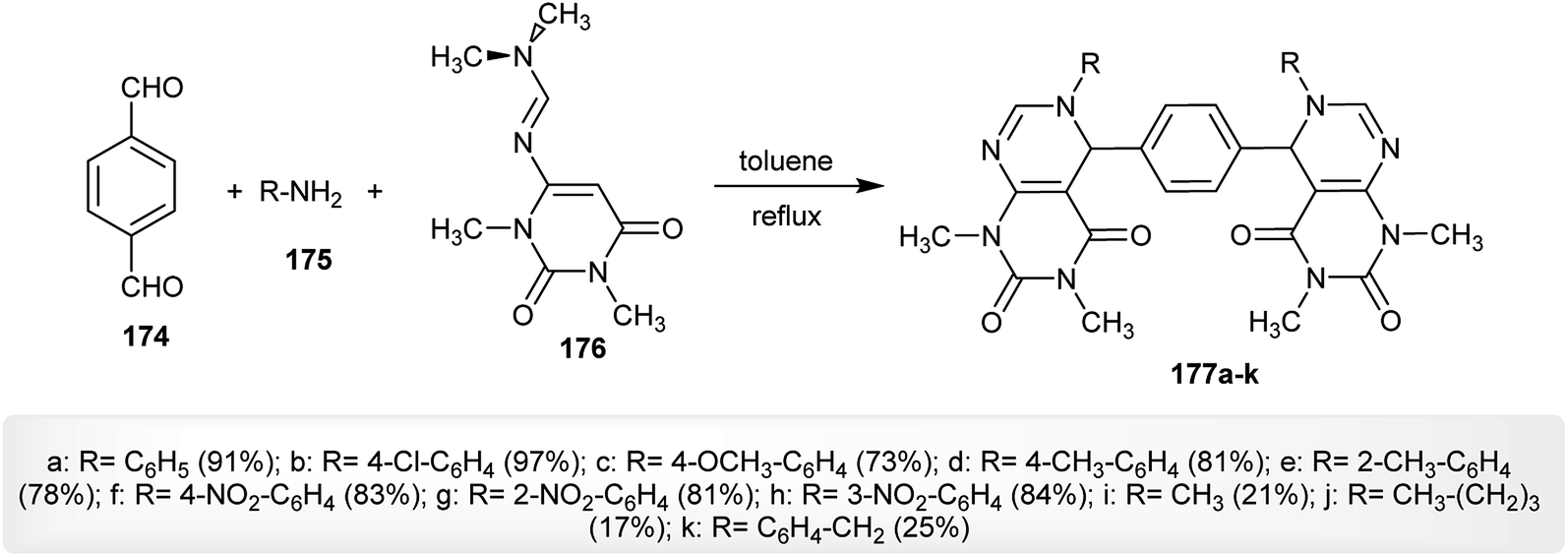
In refluxing toluene and the absence of a catalyst, multicomponent one-pot aza-Diels–Alder reactions of terephthalaldehyde (174) with amines 175 and N′-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)-N,N-dimethylformimidamide (176) gave the anticipated bis-pyrimidopyrimidines 177a–k in a procedure developed by Das et al.30 in another route, a catalytic amount of indium(III)trifluoro-methane-sulfonate [In(OTf)3] was used in chloroform for the successive multicomponent one-pot synthesis of bis-pyrimido-pyrimidines 177a–k through three reaction steps (Scheme 44).
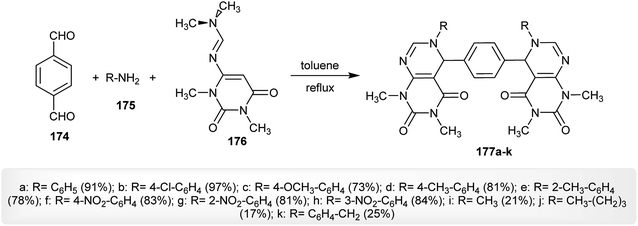 |
| | Scheme 44 Synthesis of bis-pyrimidopyrimidines. | |
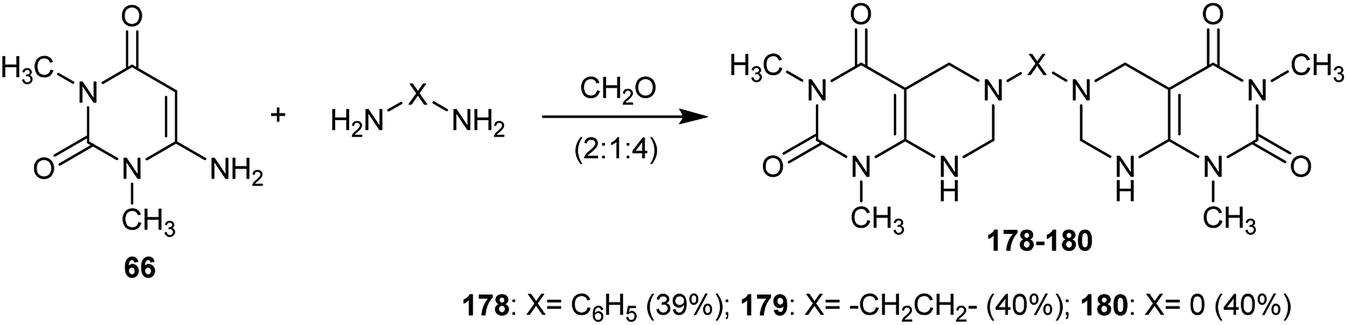
In addition, the respective bis(1,3-dimethyl-5,6,7,8-tetrahydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-diones) 178–180 were synthesized in moderate yields by the reaction of 6-amino-1,3-dimethylpyrimidine-2,4(1H,3H)-dione (66) with diamines and formaldehyde in a ratio of (2:1:4) through multicomponent reactions (Scheme 45).61
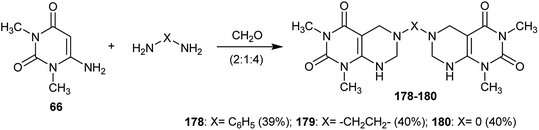 |
| | Scheme 45 Synthesis of bis(1,3-dimethyl-5,6,7,8-tetrahydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-diones). | |
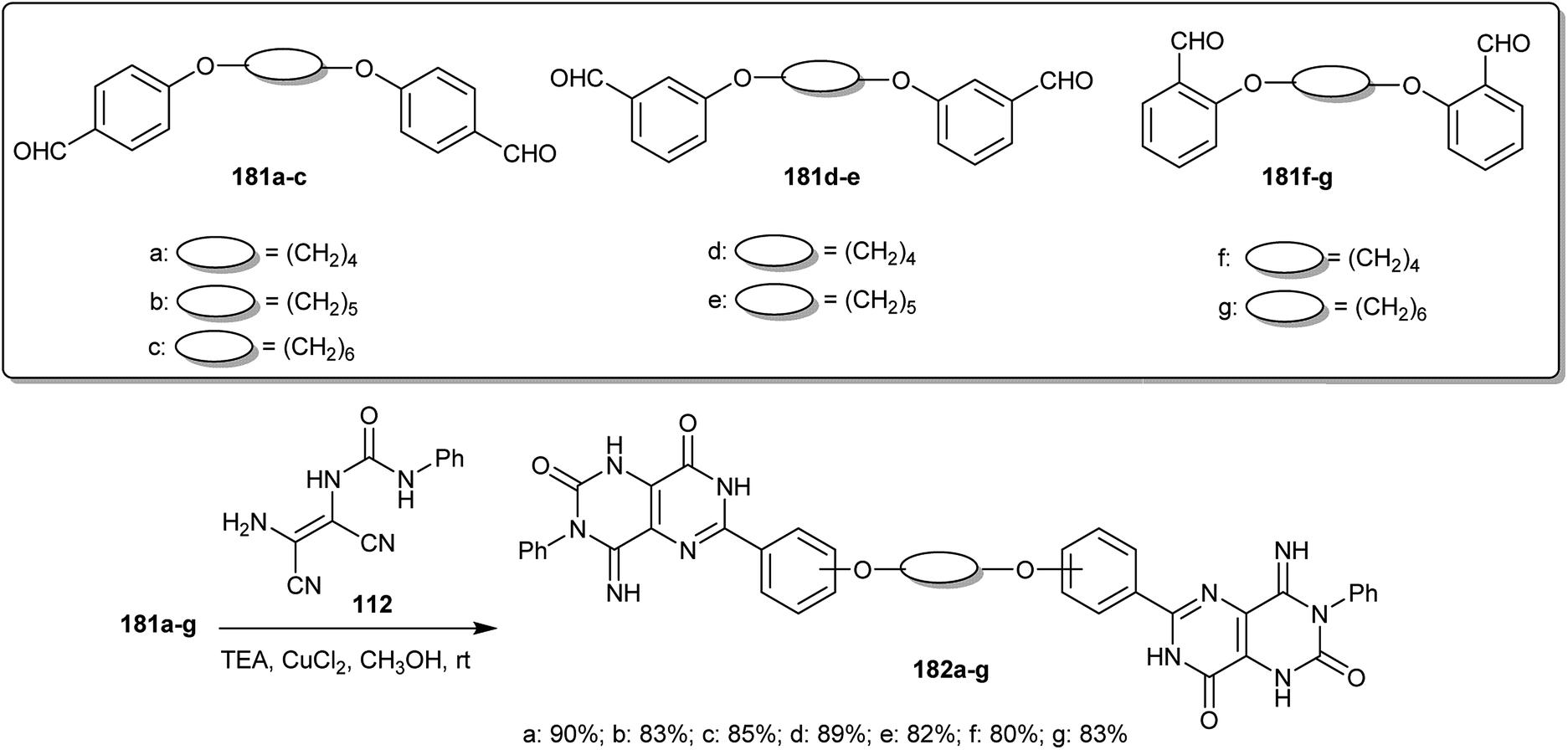
The copper(II) catalyzed reactions of bis-aldehydes (181a–g) “prepared from SN2 reactions of the alkyl dibromides with the respective aryl aldehydes in DMF containing potassium carbonate” with 1-(2-amino-1,2-dicyanovinyl)-3-phenylurea (112) in methanol containing triethylamine at room temperature yielded the desired 4-imino-6-(phenoxy)-3-phenyl-1,3,4,7-tetrahydropyrimido[5,4-d]pyrimidine-2,8-diones 182a–g, respectively, in excellent yields (Scheme 46). The formation of the bis-1,3,4,7-tetrahydropyrimido[5,4-d]pyrimidine-2,8-diones 182 was illustrated in a reported mechanism by the nucleophilic addition of the copper complex formed with phenyl urea 2 to the bis-aldehydes (181) (in a molar ratio of 2:1) and subsequent cyclocondensation of the formed intermediates, a H-shift and an oxidative cyclization step.91
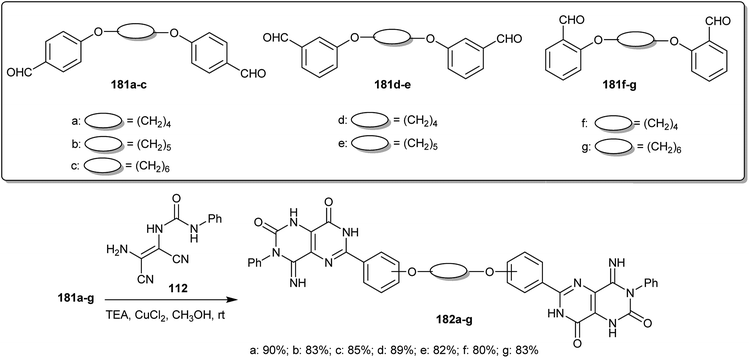 |
| | Scheme 46 Synthesis of bis-1,3,4,7-tetrahydropyrimido[5,4-d]pyrimidine-2,8-diones. | |
3. Reactivity
3.1. Ring cleavage
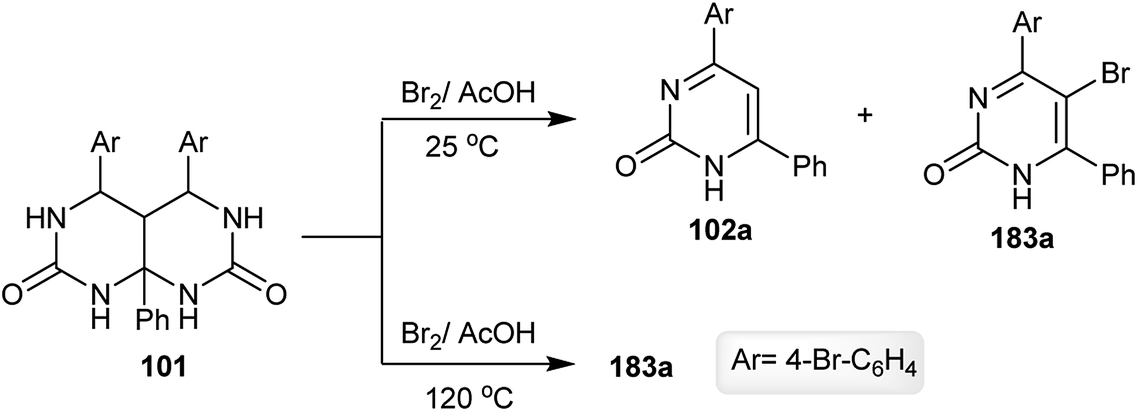
Hexahydropyrimido[4,5-d]pyrimidine-2,7(1H,3H)-dione 101 is unstable against bromination in acetic acid at room temperature, and gave two products of pyrimidinones 102a and 183a. The formation of the product 183a was established by the bromination of 102a. The bromination in acetic acid at 120 °C yielded the desired bromopyrimidinone 183a (24% yield) (Scheme 47).68
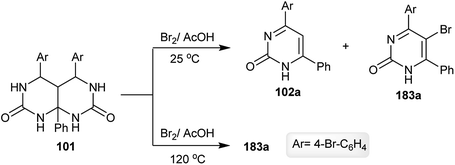 |
| | Scheme 47 Bromination of hexahydropyrimido[4,5-d]pyrimidine-2,7(1H,3H)-dione. | |
3.2. Reactivity of substituents attached to ring carbon and heteroatoms
3.2.1. Condensation reactions.
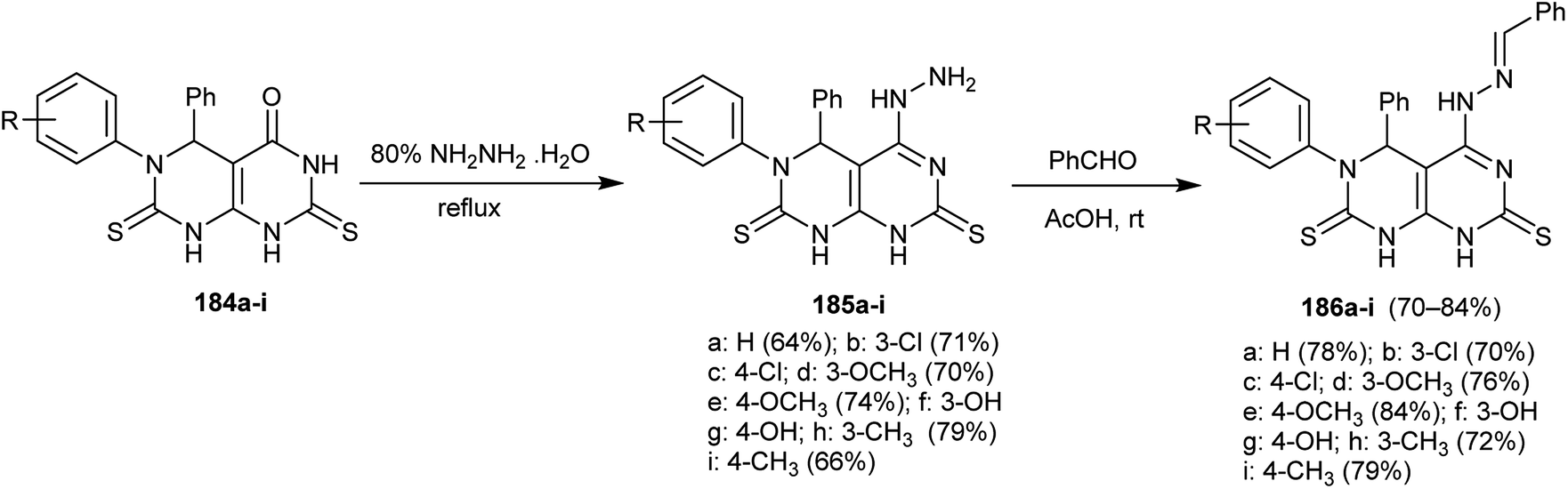
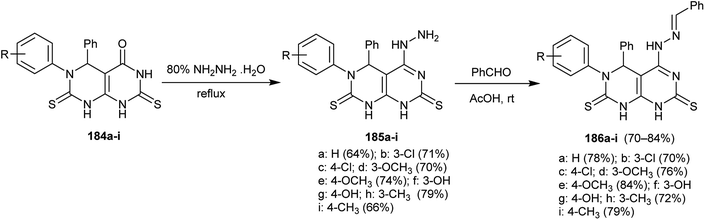
Condensation of 5,6-diaryl-2,7-dithioxo-2,3,5,6,7,8-hexahydropyrimido[4,5-d]pyrimidin-4(1H)-ones 184a–i37 with hydrazine hydrate yielded the corresponding hydrazine analogs 185a–i. In addition, the Schiff bases 186a–i were obtained by condensation of 185a–i with benzaldehyde in acetic acid at room temperature (Scheme 48). The compounds 186a–i were evaluated as antibacterial agents against different types of bacterial strains with potent results against Gram-positive bacterial species mainly, the substituted N-benzylidinehydrazine compounds conveyed an intense effect relative to the results of Ampicillin.92
 |
| | Scheme 48 Synthesis of 4,8-dihydropyrimido[4,5-d]pyrimidine-2,7(1H,3H)-dithiones. | |
3.2.2. Nucleophilic substitution reactions.
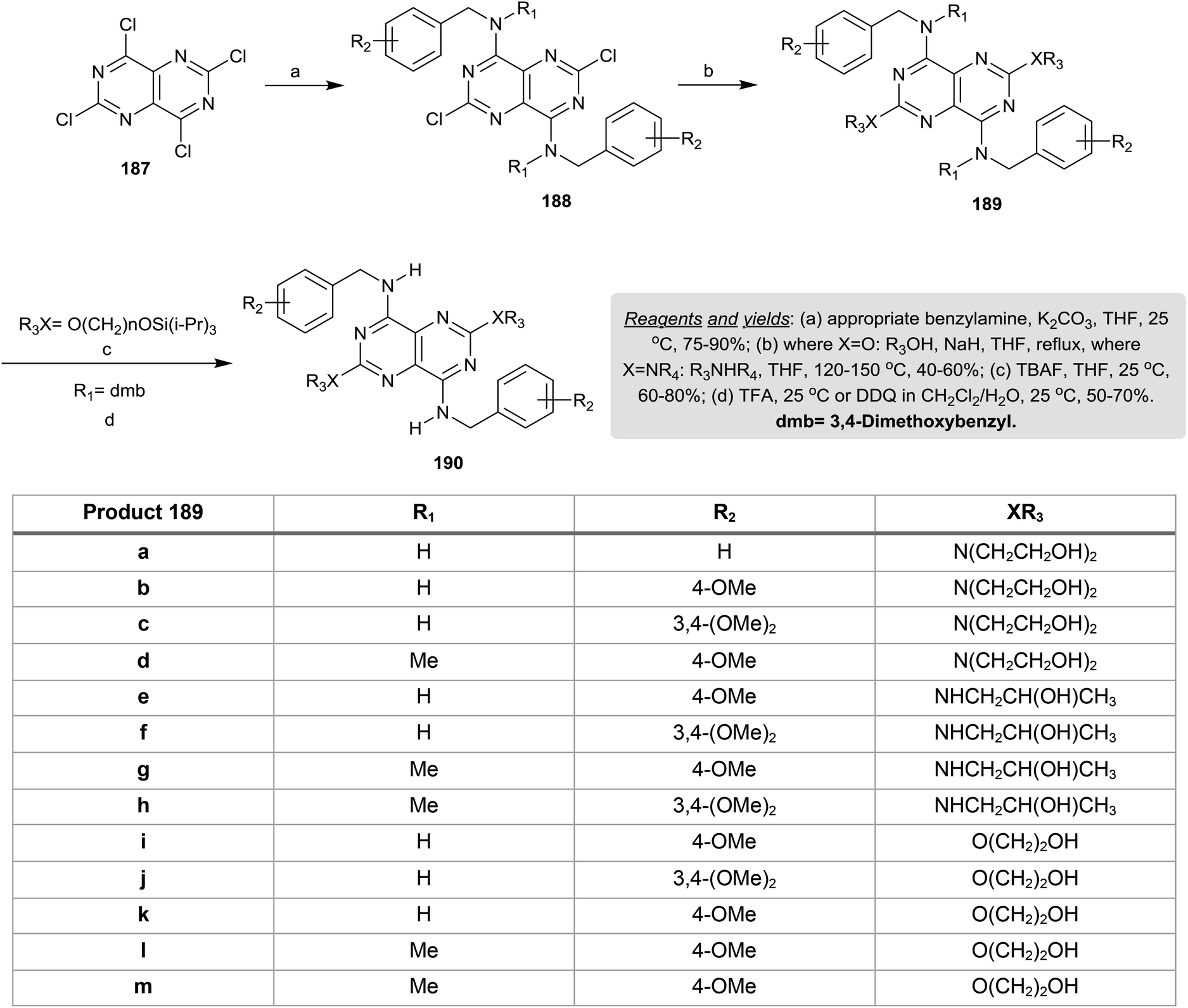
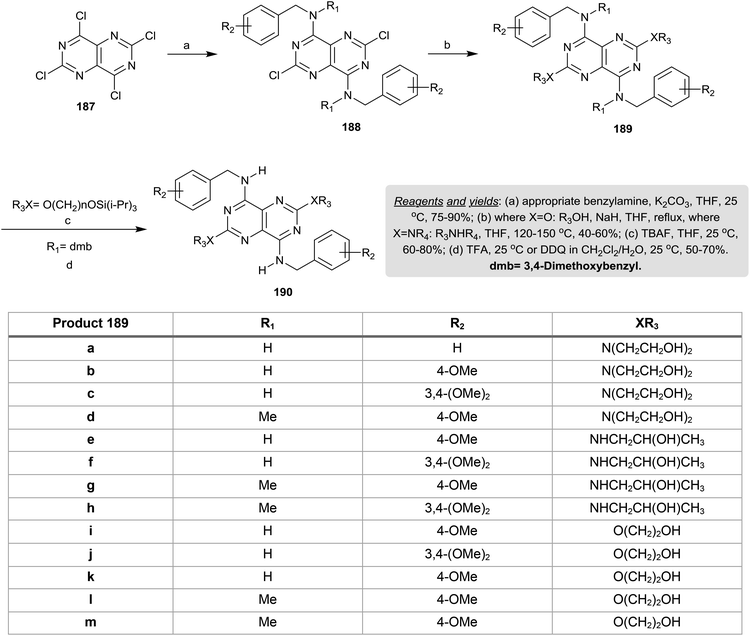
Treatment of perchloropyrimido[5,4-d]pyrimidine (187) with various benzyl amines in THF containing potassium carbonate at room temperature yielded a series of diamines 188 in 75–90% yields through nucleophilic displacement at C4 and C8. Refluxing of 188 with alcohols in THF/sodium hydride or with amines in THF gave the pyrimidopyrimidines 189, in which the substitution took place at C2 and C6. The removal of the triisopropylsilyl groups was achieved by treatment with TBAF and the protecting dmb groups were cleaved by treatment with trifluoroacetic acid or 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) at room temperature to give the anticipated pyrimidopyrimidines 190 in a good yield (Scheme 49). The tetra-substituted-pyrimidopyrimidines 190 were assessed as resistance-modifying agents by inhibiting the transportation of the nucleoside in the presence of the α1-acid glycoprotein.13
 |
| | Scheme 49 Nucleophilic substitution reactions. | |
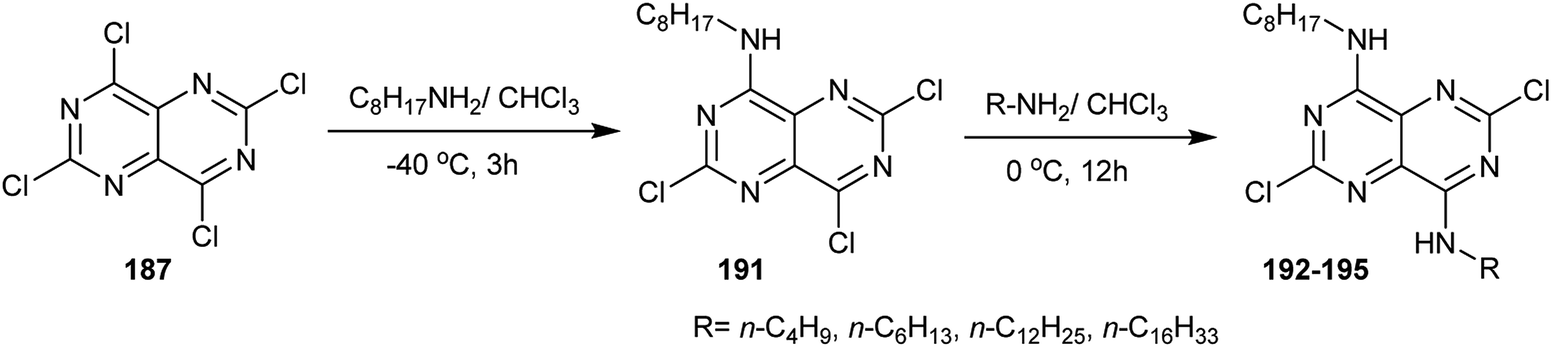
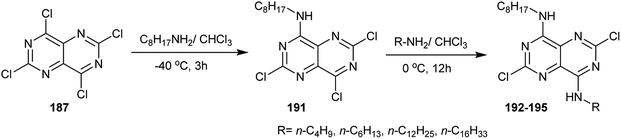
Treatment of perchloropyrimido[5,4-d]pyrimidine (187) with long chain alkyl amines in chloroform afforded the monoalkylamines 191 through nucleophilic displacement at C4. Subsequent treatment of 191 with alkyl amines, such as n-butylamine, n-hexylamine, n-dodecylamine and n-hexadecylamine in chloroform, afforded the diamines 192–195, in which the substitution took place at C8 (Scheme 50).93
 |
| | Scheme 50 Synthesis of diamines. | |
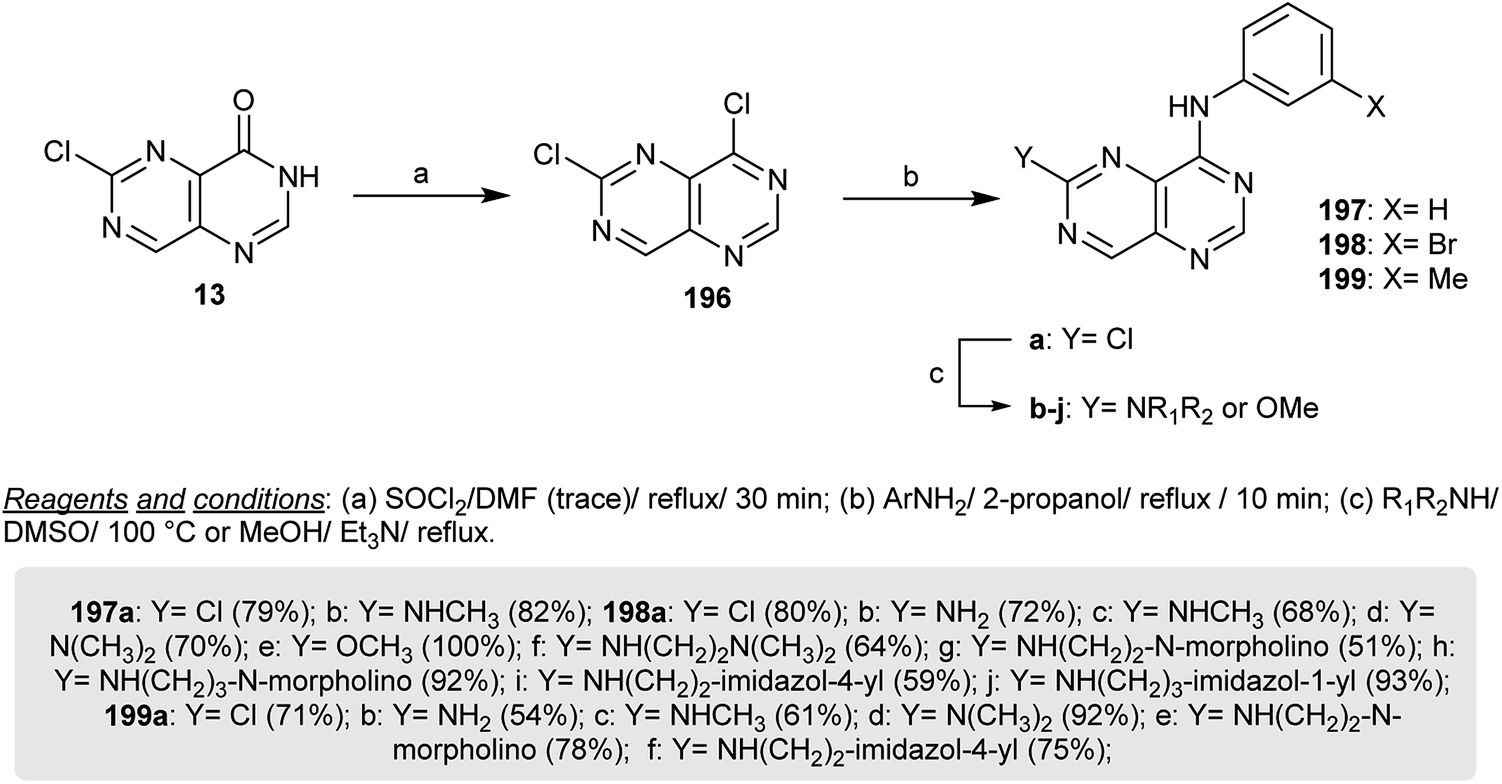
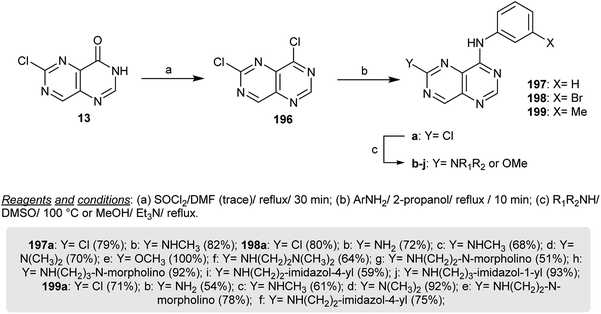
The chlorination of 6-chloropyrimido[5,4-d]pyrimidin-4(3H)-one (13) with thionyl chloride in refluxing DMF provides the dechlorinated product 196, which cannot be isolated and reacted directly with the respective aryl amines in refluxing isopropanol to yield the amination products 197a–199a in exceptional yields. Heating of 198a in methanol containing triethylamine is a simple route to prepare 198e, which has the possibility of reacting with nucleophilic amines. Reactions of secondary amines with 197a–199a by heating in DMSO yielded the respective diamines 197a, 197b, 198a–j and 199a–f by nucleophilic displacement of the chlorine atom at C6 (Scheme 51). The anticipated pyrimidopyrimidines 197–199 were investigated as inhibitors for EGFR autophosphorylation, in which the presence of amine substituents provides high solubility and weakly basic characteristics. Compound 198c has a higher potency for the inhibition of the autophosphorylation of EGFR in intact A431 cells.24
 |
| | Scheme 51 Synthesis of mono- and disubstituted-amines. | |
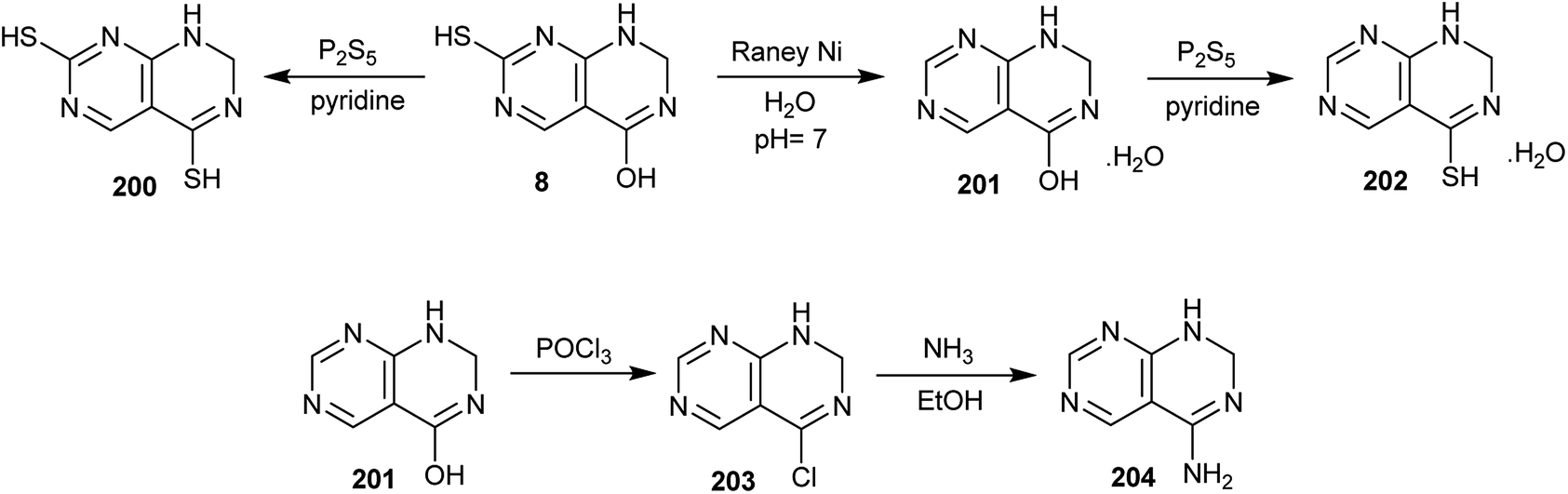
In addition, treatment of 7-mercapto-1,2-dihydropyrimido[4,5-d]pyrimidin-4-ol (8) with P2S5 in pyridine afforded the dimercapto analog 200. In addition, the reduction of 7-mercapto-1,2-dihydropyrimido[4,5-d]pyrimidin-4-ol (8) with RANEY® Ni in neutral medium gave 1,2-dihydropyrimido[4,5-d]pyrimidin-4-ol (201), which was treated with P2S5 in pyridine to afford the respective mercapto analog (202). Chlorination of compound 201 with phosphorus oxychloride yielded the desired 4-chloro-1,2-dihydropyrimido[4,5-d]pyrimidine (203), which was aminated by the reaction with ammonia in ethanol to afford 1,2-dihydropyrimido[4,5-d]pyrimidin-4-amine (204) (Scheme 52).53
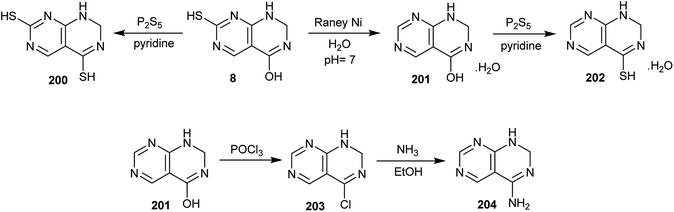 |
| | Scheme 52 Reactivity of 7-mercapto-1,2-dihydropyrimido[4,5-d]pyrimidin-4-ol. | |
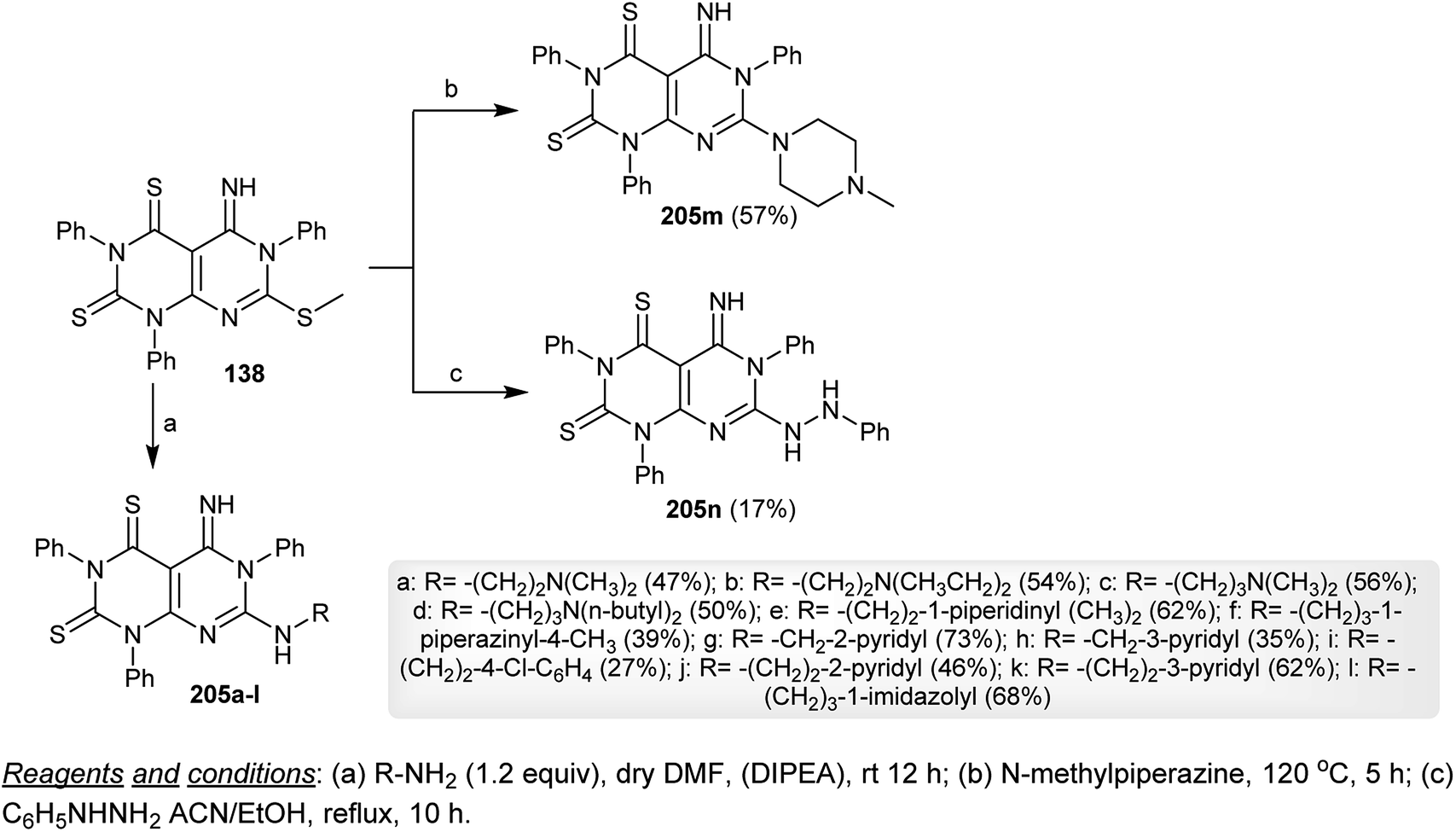
Nucleophilic substitution reactions of dihydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dithiones 138 with various alkyl-/cycloalkyl-/arylamines in DMF containing a catalytic amount of DIPEA yielded the respective amines 205a–l in moderate to good yields. The same reaction of 138 with N-methylpiperazine was accomplished by heating at 120 °C and the reaction of 138 with phenylhydrazine was processed in a refluxing mixture of acetonitrile and ethanol to afford the respective dihydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dithiones 205m and 205n, respectively, through amination step reactions by nucleophilic attack of the amines to the methylmercaptan moiety (Scheme 53).74
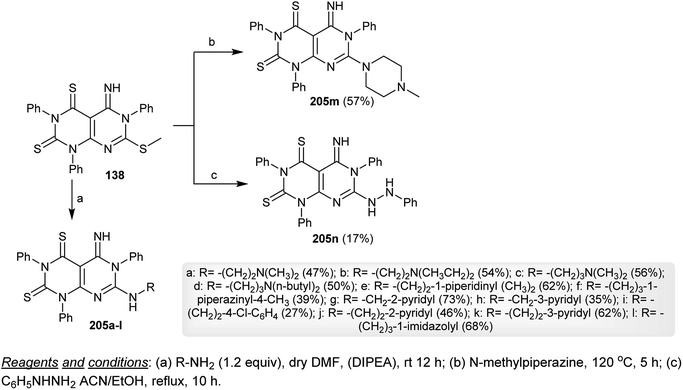 |
| | Scheme 53 Synthesis of 7-substituted-5-imino-1,3,6-triphenyl-5,6-dihydropyrimido[4,5-d]pyrimidine-2,4-(1H,3H)-dithiones. | |
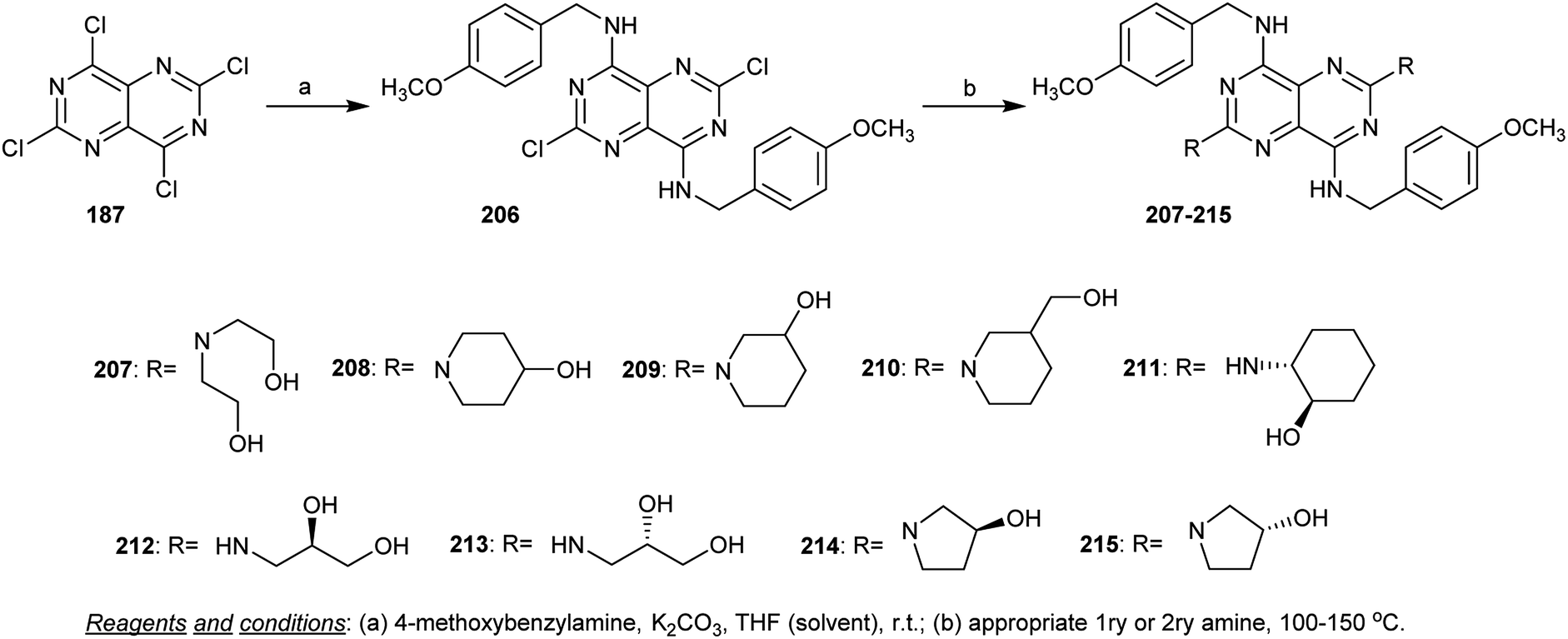
Treatment of perchloropyrimido[5,4-d]pyrimidine (187) with 4-methoxybenzylamine in THF containing potassium carbonate provided the diamines 206 through the nucleophilic displacement of chlorine atoms at C4 and C8. Heating the diamine 206 with the appropriate primary and secondary amines yielded the analogs pyrimidopyrimidines 207–215 (Scheme 54). The previous synthetic method is similar to those reported by Curtin et al.94 The compounds were assessed in vitro as inhibitors for cellular proliferation and motility prompted by h-prune in breast cancer, and high potency was found for compounds 214 and 215.8
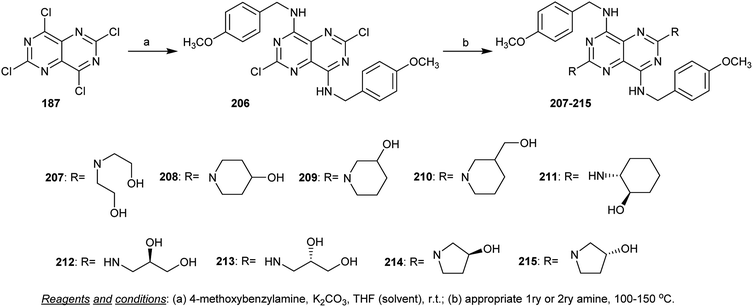 |
| | Scheme 54 Synthesis of tetra-substituted-amines. | |
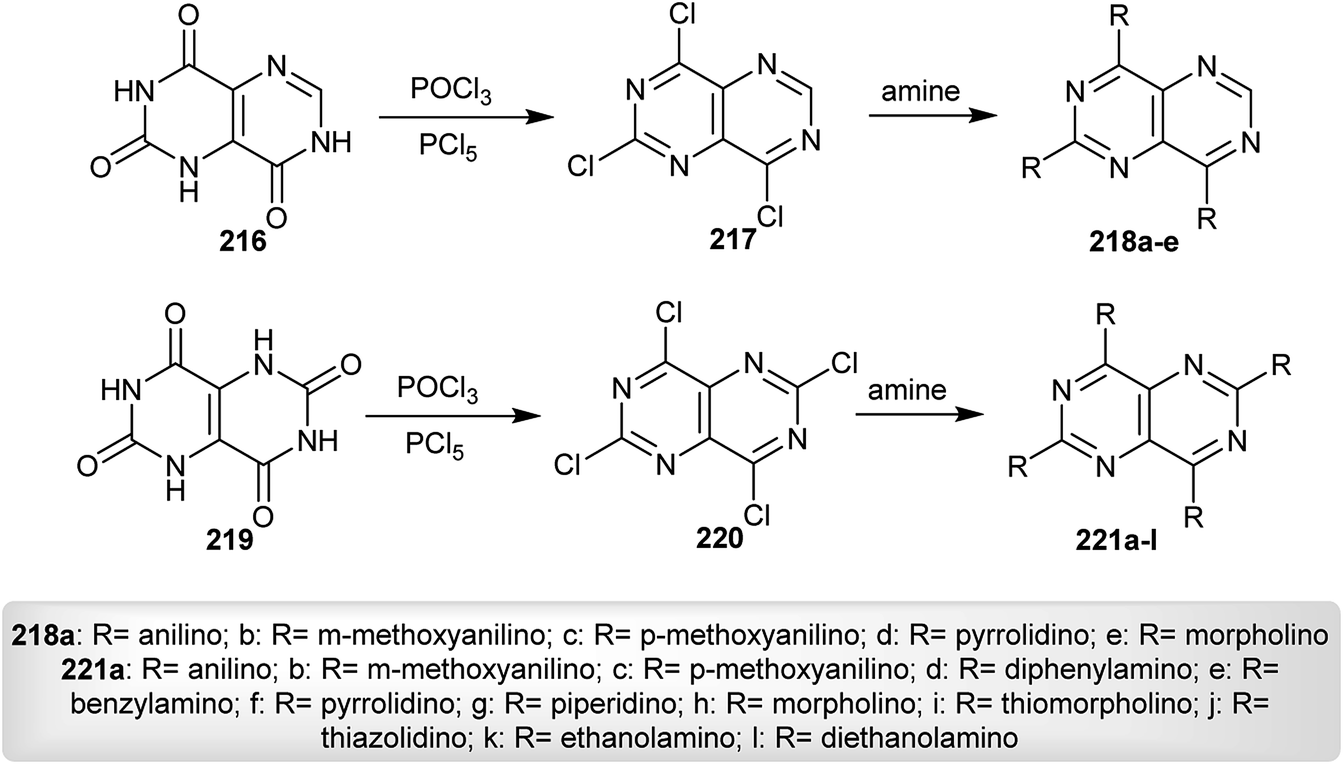
Nakashima and Akiyama95 prepared a series of tri- and tetra-substituted-amines of pyrimido[5,4-d]pyrimidines 218 and 221 by chlorination of 1,7-dihydropyrimido[5,4-d]pyrimidine-2,4,8(3H)-trione (216) and 1,5-dihydropyrimido[5,4-d]pyrimidine-2,4,6,8(3H,7H)-tetraone (219), each with phosphorus oxychloride or phosphorus pentasulfide followed by a nucleophilic substitution reaction of the respective halogenated products 217 and 220 with various amines (Scheme 55). The investigated pyrimidopyrimidines 218 and 221 were applied as fluorescence reagents in dioxane in a peroxyoxalate–chemiluminescence reaction, wherein the compounds with 3-methoxy-aniline substituents have the highest intensity.
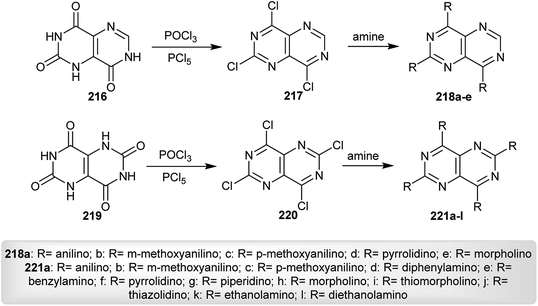 |
| | Scheme 55 Synthesis of tri- and tetra-substituted-amines of pyrimido[5,4-d]pyrimidines. | |
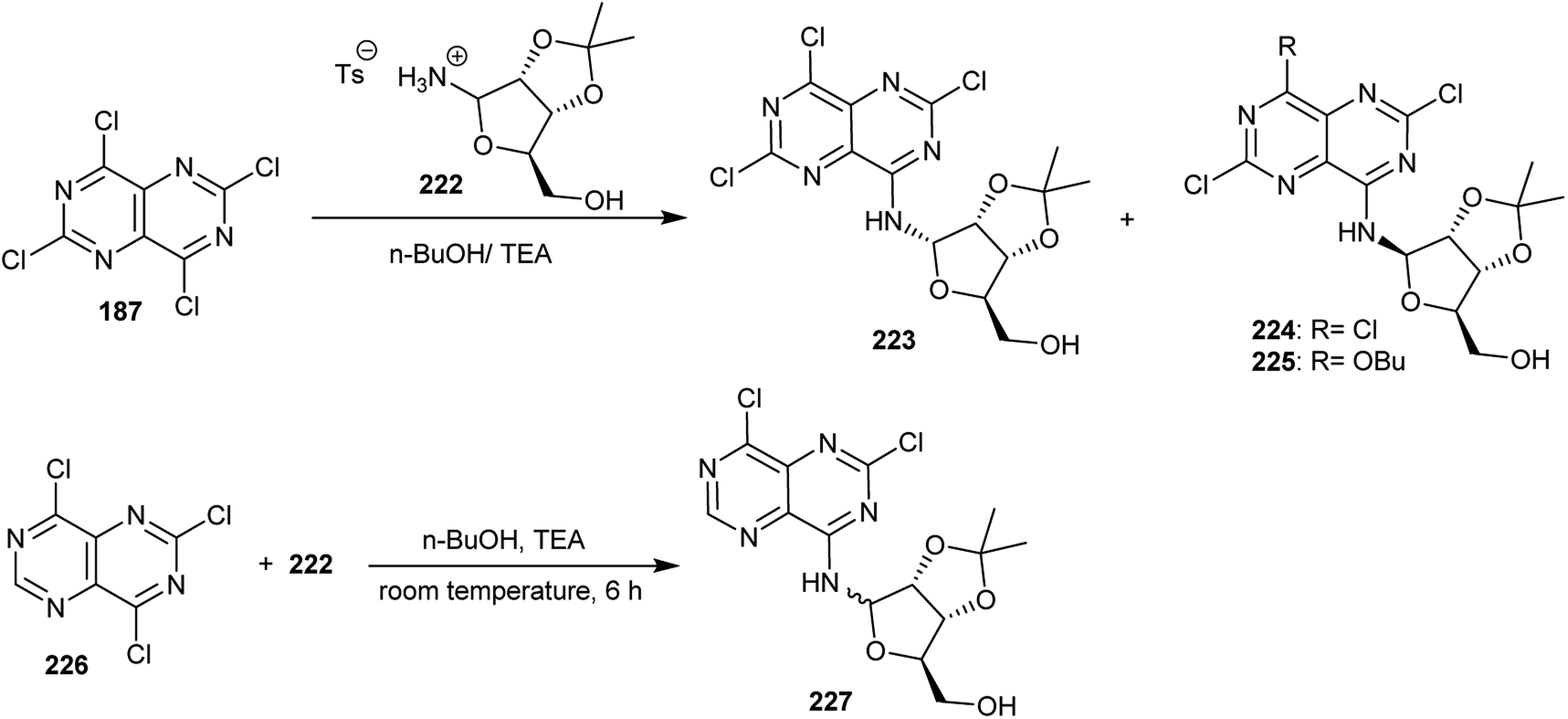
Treatment of perchloropyrimido[5,4-d]pyrimidine (187) with free 2,3-O-isopropylidine-D-ribofuranosyl-amine, which was formed in situ from its stable salt 222 by the addition of triethylamine in n-butanol at room temperature, yielded a mixture of two nucleosides 224 and its α-anomer 223 in a ratio of 1:0.77. At ambient temperature, the product 225 was also obtained after 16 h from the same reaction, providing the possibility of a chlorine atom at the C4 position of compound 224 to be displaced by the nucleophile. Similarly, an anomeric mixture of pyrimidopyrimidines 227 was obtained by treatment of 226 with 222 in n-butanol containing triethylamine at room temperature after 6 h (Scheme 56).20
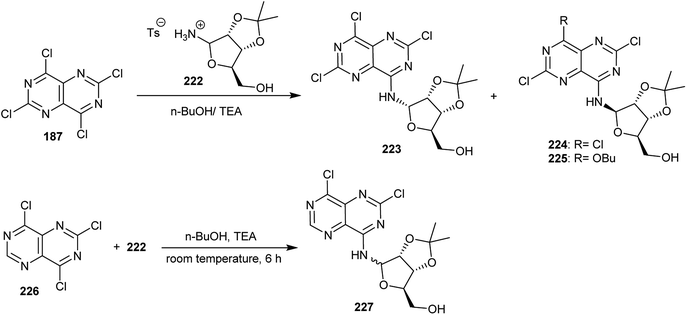 |
| | Scheme 56 Synthesis of nucleosides incorporated with a pyrimido[5,4-d]pyrimidine core. | |
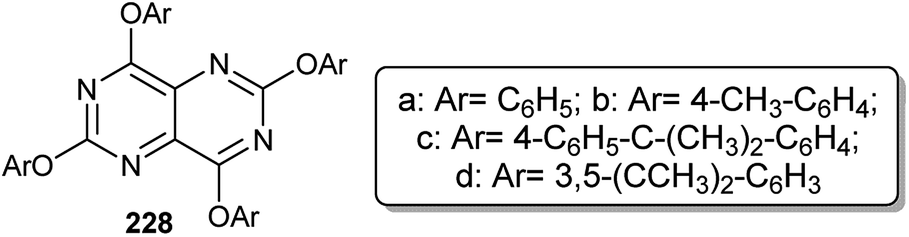
Gall and coworkers96 reported a synthetic technique for the preparation of 2,4,6,8-tetrakis(3,5-di-tert-butyl-phenoxy)pyrimido[5,4-d]pyrimidine (228d) in a yield of 28.5% from the reaction of 2,4,6,8-tetrachloropyrimido[5,4-d]pyrimidine (187) with four equivalents of 3,5-di-t-butylphenol in THF containing sodium hydride. The structure of 228d was established using X-ray crystallography. The relative pyrimidopyrimidines 228a–c were obtained in 67, 33, and 72% yields, respectively (Fig. 2).
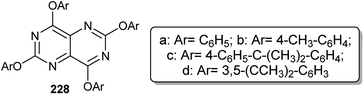 |
| | Fig. 2 The structure of 2,4,6,8-tetrakis(substituted-phenoxy)pyrimido[5,4-d]pyrimidines. | |
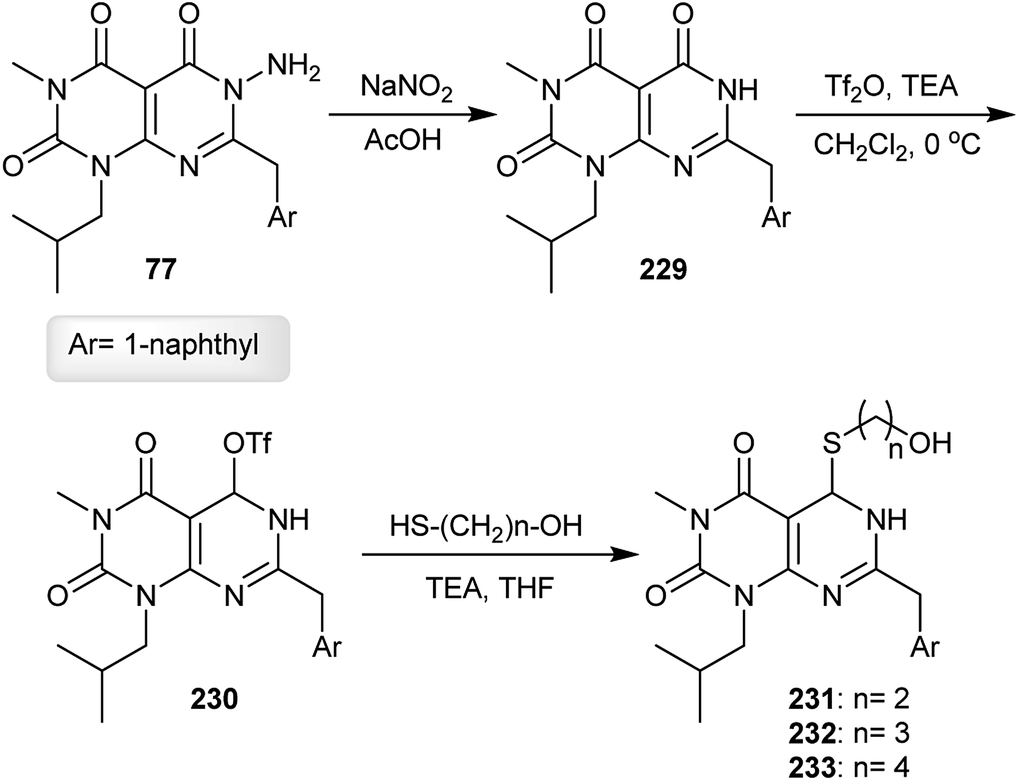
The respective pyrimido[4,5-d]pyrimidine 229 was obtained in a 38% yield under diazotization conditions of the analogs 77 with sodium nitrite in acetic acid through the cleavage of the N–N bond. Treatment of 229 with triflic anhydride in the presence of triethylamine gave the corresponding triflate 230 in a 64% yield. The nucleophilic substitution reactions of 230 with mercaptoalcohols yielded the thioethers 231–233 in 67–77% yields (Scheme 57).62
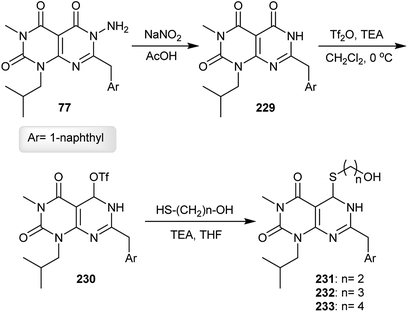 |
| | Scheme 57 Reactivity of the ring substituents. | |
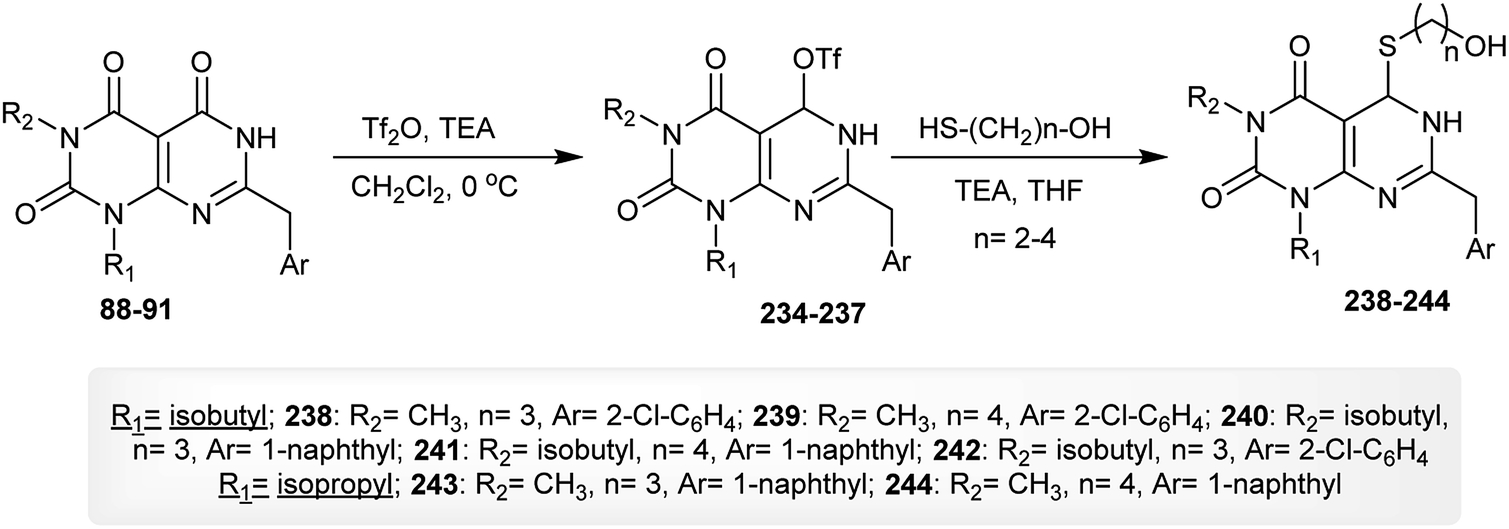
As stated in the previous reaction sequence, dihydropyrimido[4,5-d]pyrimidine-triones 88–91 reacted with triflic anhydride in dichloromethane containing triethylamine to give the anticipated triflates 234–237 in 52–83% yields. The reactions of 234–237 with mercaptoalcohols in THF/TEA afforded the dihydropyrimido[4,5-d]pyrimidine-diones 238–244 in 45–82% yields through a nucleophilic substitution step (Scheme 58).62
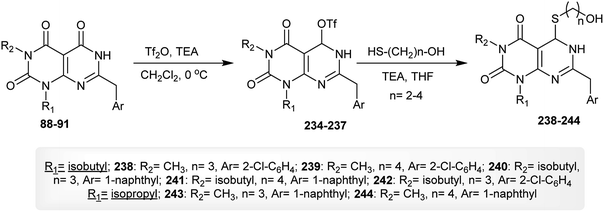 |
| | Scheme 58 Synthesis of 1,3,5,7-tetra-substituted-5,6-dihydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-diones. | |
3.2.3. Alkylation.
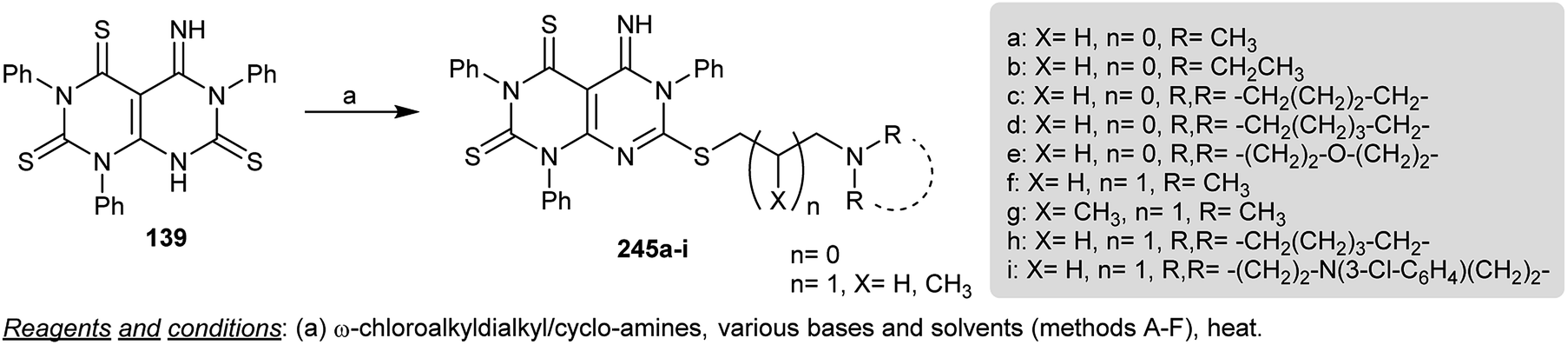
Regio-alkylation of dihydropyrimido[4,5-d]pyrimidine-trithione 139 was accomplished by treatment with a stirred solution of 40% Triton (dried from methanol) in DMF followed by subsequent addition of o-chloroalkyl- and cycloalkyl amines to give the anticipated S-alkyl derivatives 245a–i. The alkylation process occurred at the sulfur atom attached to the C7 of the pyrimidopyrimidine ring (Scheme 59). The compounds of the series 245 were assessed as antiproliferative agents, in which compounds 245b, 245d, 245f, 245h, 245i, and 245j revealed potent activities against the tested leukemia and prostate cell lines.75
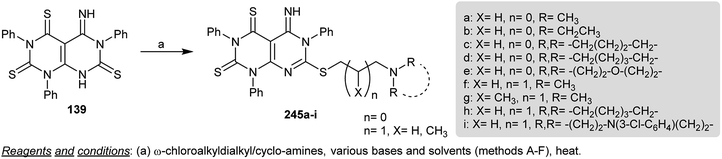 |
| | Scheme 59 Synthesis of 7-(alkylthio)-5-imino-1,3,6-triphenyl-5,6-dihydropyrimido[4,5-d]pyrimidine-2,4-(1H,3H)-dithiones. | |
3.2.4. Oxidation and substitution reactions.
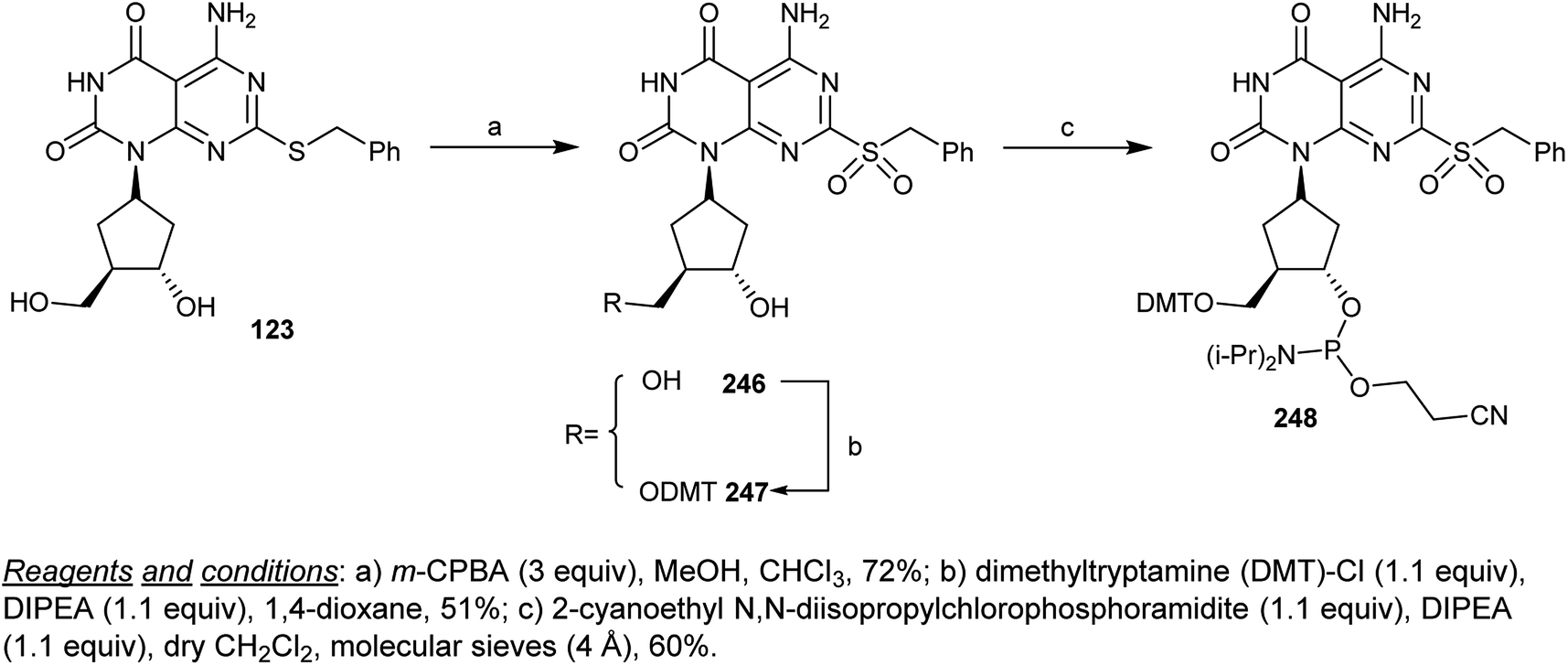
Oxidation of cyclopentyl-pyrimido[4,5-d]pyrimidine-dione 123 with m-chloroperoxybenzoic acid (m-CPBA) yielded the sulfone 246 in a 72% yield. Protection of the hydroxymethyl group was achieved by reaction of 246 with dimethyltryptamine (DMT)-Cl in a 1,4-dioxane/DIPEA system to yield the protected nucleotide 247, which was converted into the respective phosphoramidite 248 at a yield of 60% by reaction with 2-cyanoethyl N,N-diisopropylchlorophosphoramidite in dichloromethane containing DIPEA through the nucleophilic displacement of the chlorine atom of the desired chlorophosphoramidite (Scheme 60).72
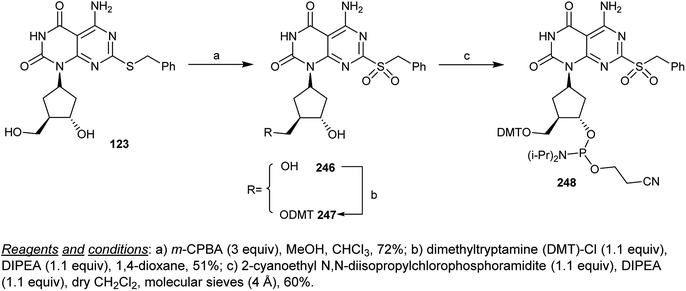 |
| | Scheme 60 Reactivity of cyclopentyl-pyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione. | |
3.2.5. Miscellaneous reactions.
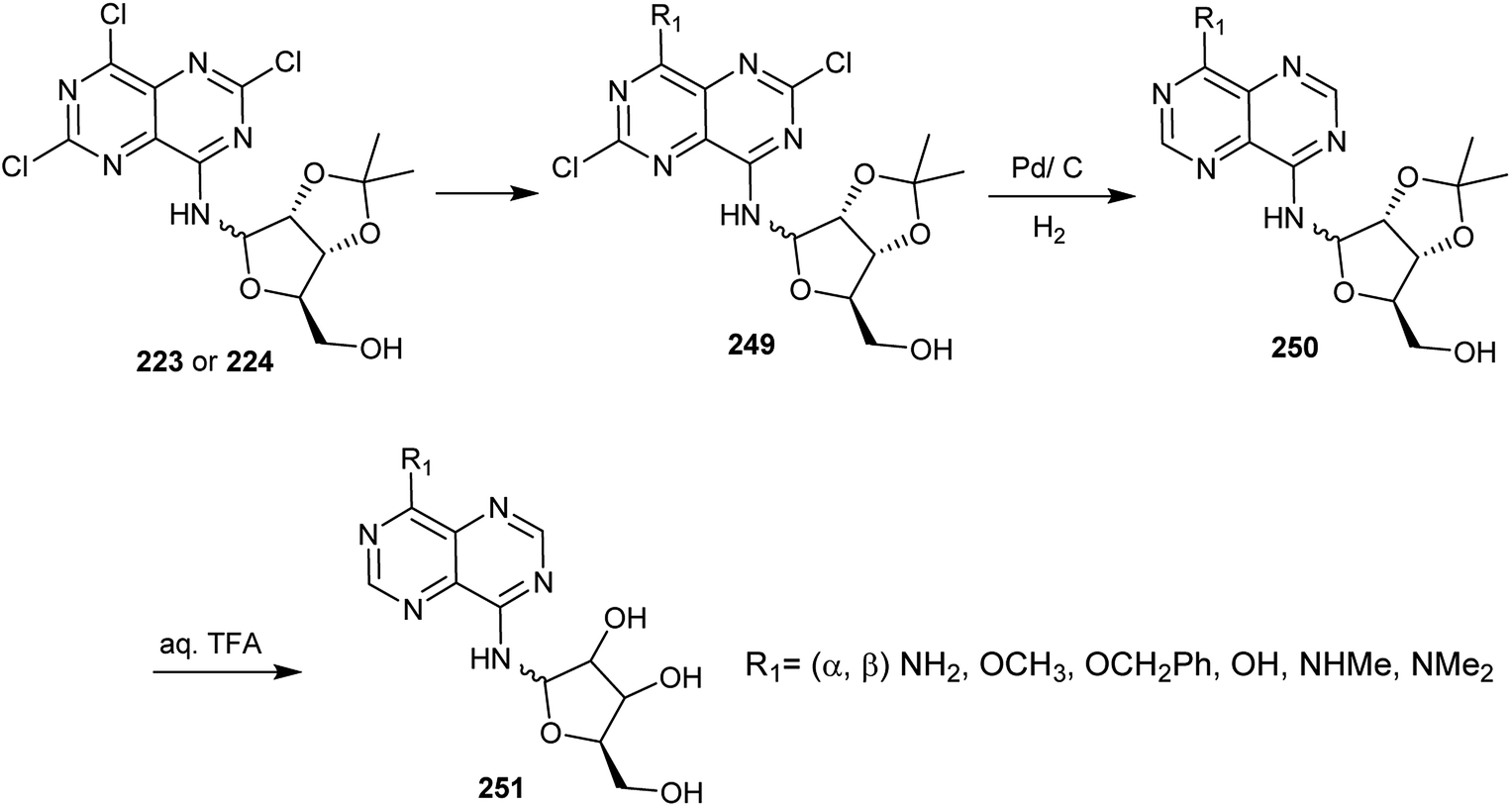
Treatment of compounds 223 and 224, each with ammonia in ethanol at 0 °C, sodium methoxide solution in methanol, benzyl alcohol in the presence of TEA at rt (>60% yield), methyl amine or dimethyl amine gave the respective amines and their α-anomer 249 (R1 = (α, β) NH2, OCH3, OCH2Ph, OH, NHMe, NMe2). Catalytic hydrogenation of 249 using Pd/C yielded the desired dehalogenation pyrimidopyrimidines and their α-anomer 250. Subsequent treatment of 250 with an aqueous solution of trifluoroacetic acid (90%) at room temperature afforded pyrimidopyrimidines 251 through isopropylidene ring cleavage in high yields (Scheme 61).20
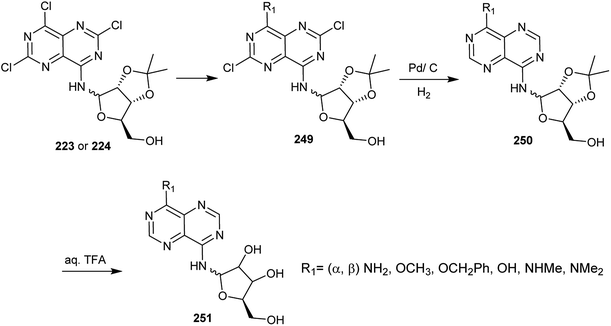 |
| | Scheme 61 Reactivity of chlorine atoms attached to the ring carbon atoms. | |
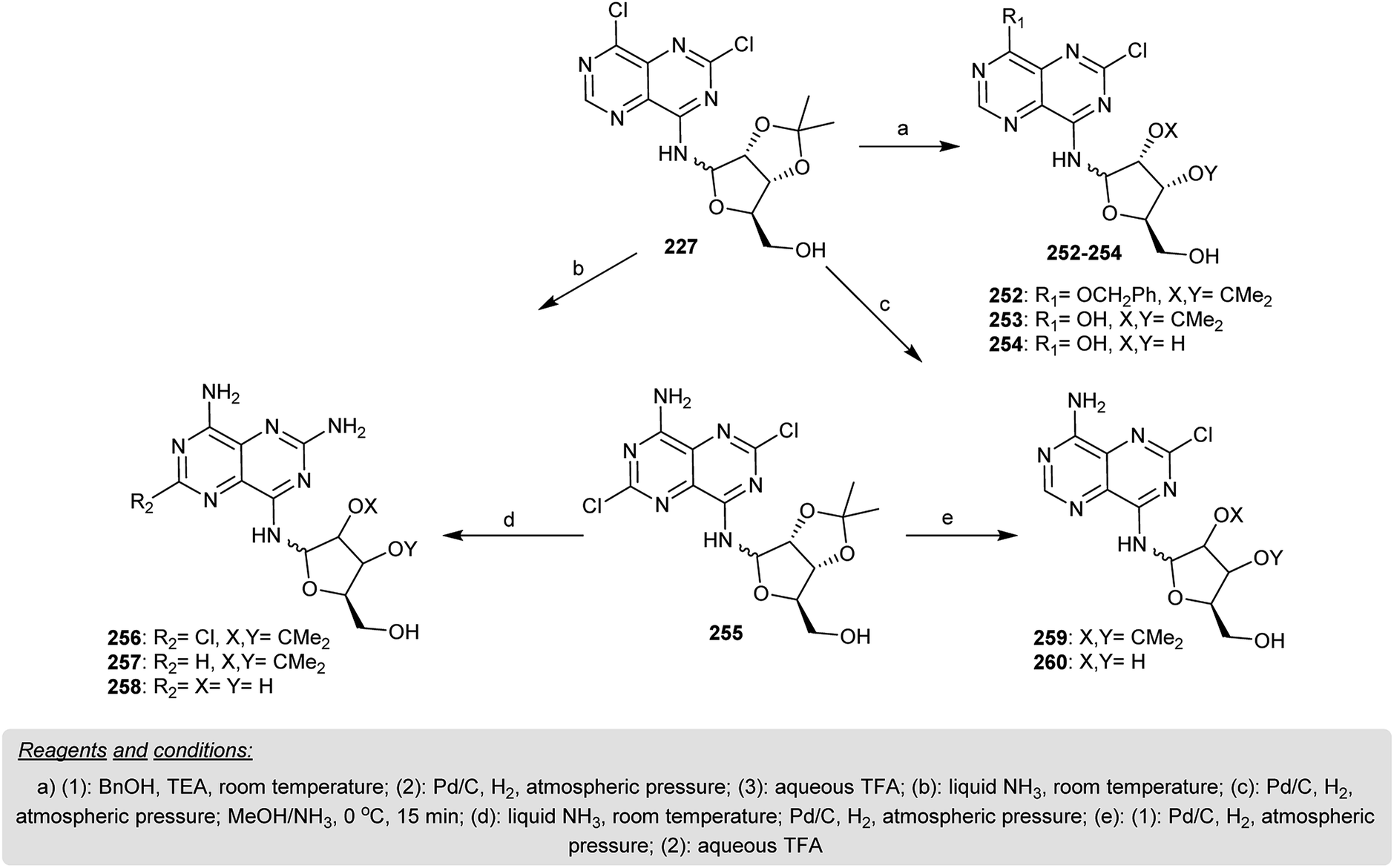
Treatment of 227 with benzyl alcohol in the presence of triethylamine gave 252. In addition, the catalytic reduction of 30 with Pd/C yielded 253, which after hydrolysis with trifluoroacetic acid afforded 254 in a yield of 58%. Treatment of 255 with liquid ammonia for six days gave a mixture of isomers identified as β-anomer 256 (59% yield) and α-anomer 256 (15% yield). Catalytic hydrogenation of 256 afforded the respective 34. Amonolysis of 29 with liquid ammonia after six days furnished another synthetic method for 257. Deisopropylidenation of 257 yielded the corresponding pyrimidopyrimidine 258, which was isolated as a salt of TFA. Pyrimidopyrimidine 259 was obtained in a yield of 65% by hydrogenation of 235 with the removal of the chlorine atom at C2. Successive deisopropylidenation of 259 yielded the respective pyrimidopyrimidine 260 in a yield of 67% (Scheme 62).20
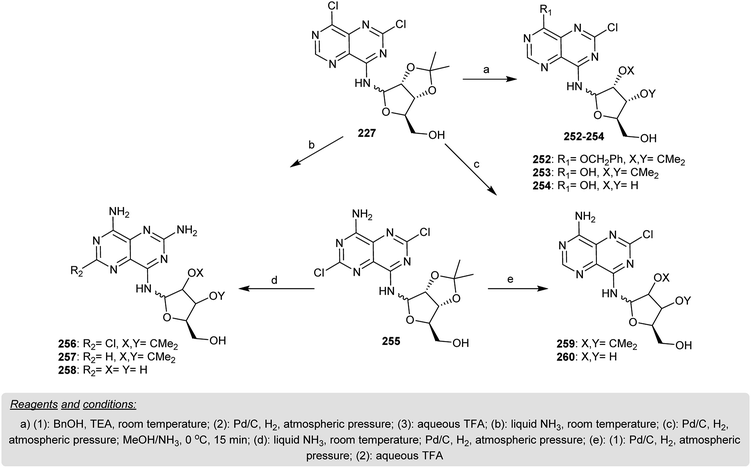 |
| | Scheme 62 Synthesis of the pyrimido[5,4-d]pyrimidine skeleton incorporating nucleosides. | |
Under reflux conditions, the electrophilic substitution of the proton at N3 of 46a–d in the reaction with alkyl halides in pyridine yielded the corresponding N-alkyl derivatives 261a–f in good yields. On the other hand, refluxing of thiones 46a–d with hydrazine hydrate yielded the respective hydrazines 262a–d, which were condensed with benzaldehydes in acetic acid to give Schiff bases 263a–d, respectively (Scheme 63).56
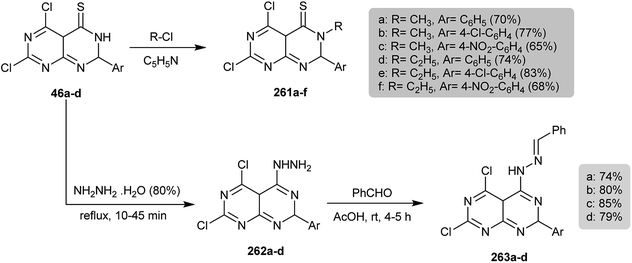 |
| | Scheme 63 Synthesis of N-alkyl- and hydrazine-substituents. | |
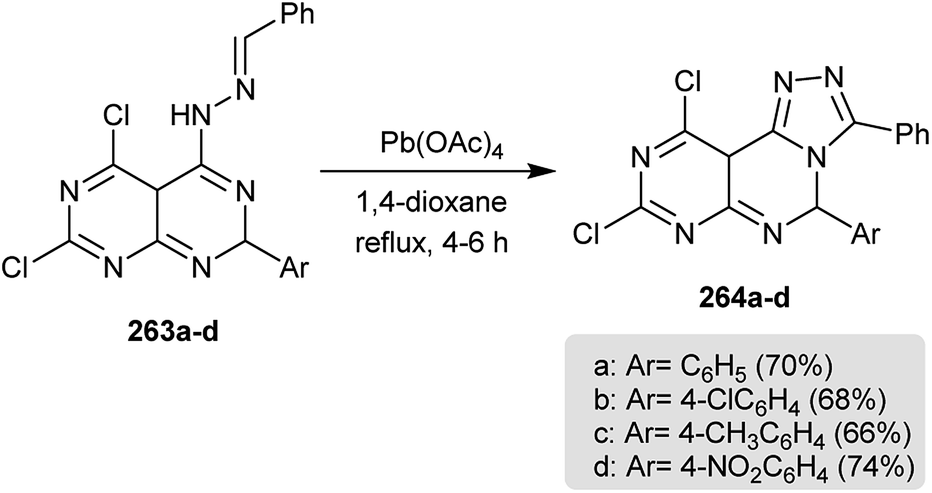
Intramolecular cyclization of 263a–d in refluxing 1,4-dioxane containing catalytic palladium acetate gave the desired dihydropyrimido[1,2,4]triazolo-pyrimidines 264a–d (Scheme 64). The transformation mechanism to products 6 was reported through proton, 1,5-hydrogen abstraction and subsequent cyclization.56
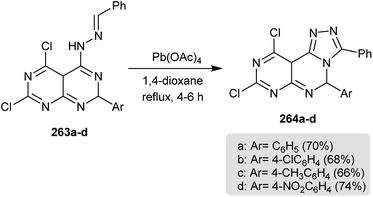 |
| | Scheme 64 Synthesis of 3-aryl-5,10a-dihydropyrimido[5,4-e][1,2,4]triazolo[4,3-c]pyrimidines. | |
3.2.6. Synthesis of polymers bearing alkylamino groups.
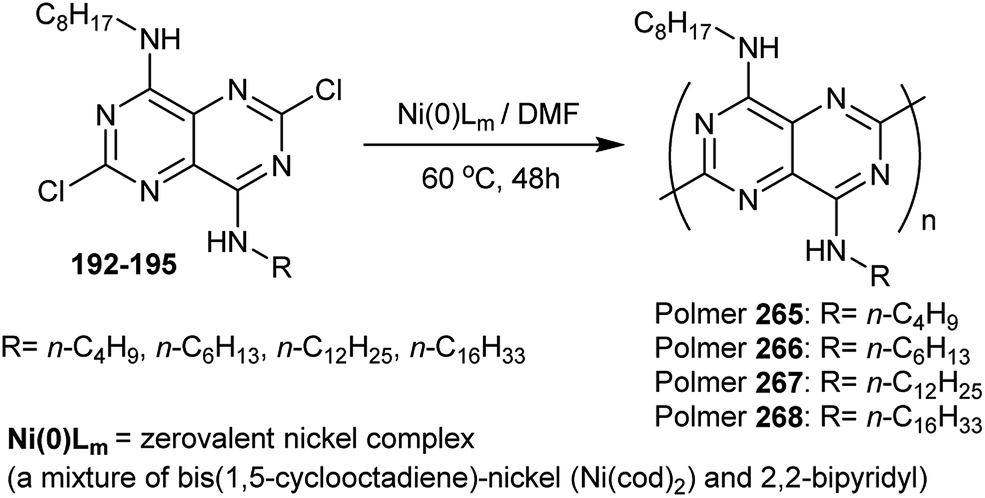
A series of conjugated polymers of pyrimidopyrimidines 265–268 were prepared in high yields by heating the corresponding pyrimidopyrimidines with a nickel(0) complex in DMF. The procedure involved dehalogenation polycondensation of the diamines 192–195 at C2 and C6 of the monomers. The polymers displayed photo-luminescence in the solution and in the film. In the solid-state, the polymers have an inclination to form a stacked structure (Scheme 65).93
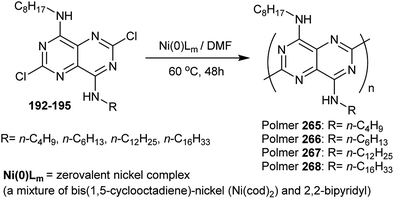 |
| | Scheme 65 Synthesis of polymers bearing alkylamino groups. | |
3.2.7. Synthesis of ribose–supramolecular complex.
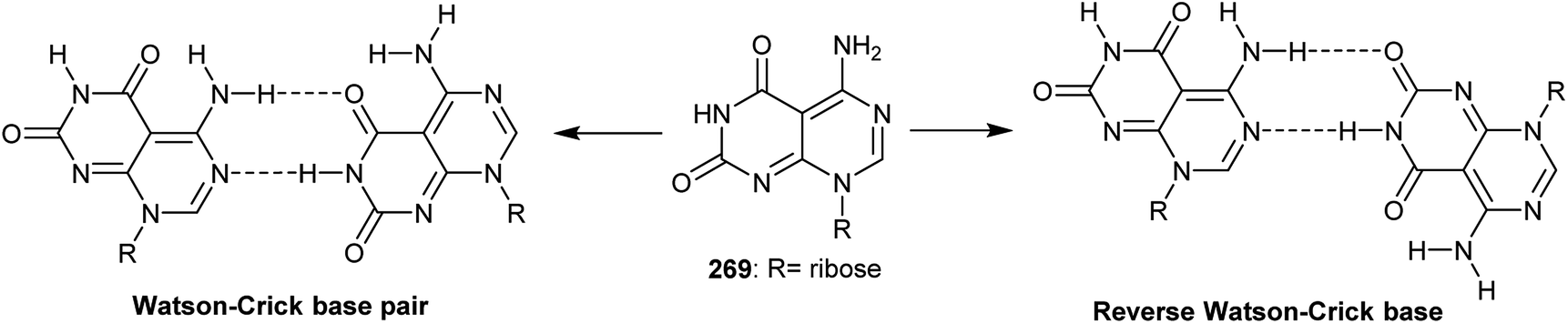
Ribose bearing 5-amino-pyrimido[4,5-d]pyrimidine-2,4(3H,8H)-dione (269) is considered as a structurally biological active molecule and has received attention from different industrial fields. Zhao et al.97 studied the mechanism of the formation of a flower-shaped supramolecular complex structure of a nucleoside derived pyrimido[4,5-d]pyrimidine using X-ray crystallography, NMR spectroscopy, dynamic light scattering, and electron microscopy scanning. The structure has two possible conformers, a Watson–Crick base pair and a reverse Watson–Crick base (Fig. 3).
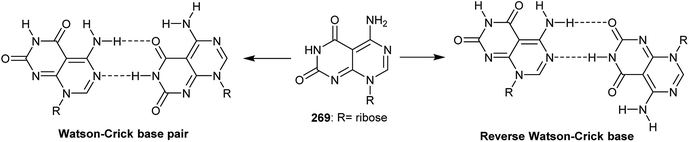 |
| | Fig. 3 The structures of the two probable monomeric conformers as base pair motifs. | |
4. Conclusions
The present study reviews and summarizes the research into the chemistry of pyrimido[4,5-d]pyrimidine and pyrimido[5,4-d]pyrimidine analogs published to date. The diverse biological efficacy of pyrimidopyrimidine compounds has been extensively tested on a large scale. This type of compound can be obtained via several synthetic routes using different techniques, for example: (a) the reactions of β-haloester with thiourea, or heating β-aminoester with formamide or phenyl isocyanate, or a multistep-synthesis starting from β-aminoester or α,β-unsaturated esters; (b) preparation from 4-amino-2,6-dichloropyrimidine and 2,6-diaminopyrimidin-4(3H)-one; (c) multicomponent reactions of barbituric and thiobarbituric acids with aryl aldehydes and urea, aryl isothiocyanates, thiourea or arylamines; (d) via a definite route, in which pyrimidopyrimidines were also synthesized from the reaction of 1,5-diketones with hydrazine hydrate; (e) preparation from multicomponent reactions of acyclic reagents such as ureas with ketones (or aldehydes) and aryl aldehydes or 2-(ethoxymethylene)malononitrile; and (f) other techniques that have been applied for the synthesis of tricyclic and tetracyclic systems or the transformation of another ring.
On the other hand, the substituted compounds are reactive with respect to nucleophilic, electrophilic, substitution and condensation reactions and provide the possibility of obtaining a number of compounds incorporating a pyrimidopyrimidine nucleus with the aim of obtaining novel antibiotics. Therefore, improvement of the method used for the synthesis of pyrimido[4,5-d]pyrimidines and pyrimido[5,4-d]pyrimidines would be particularly useful for the preparation of bicyclic pyrimidines, which could be useful in several areas of chemistry, for example, chelating derivatives, bases, and drug design.
5. Literature overview
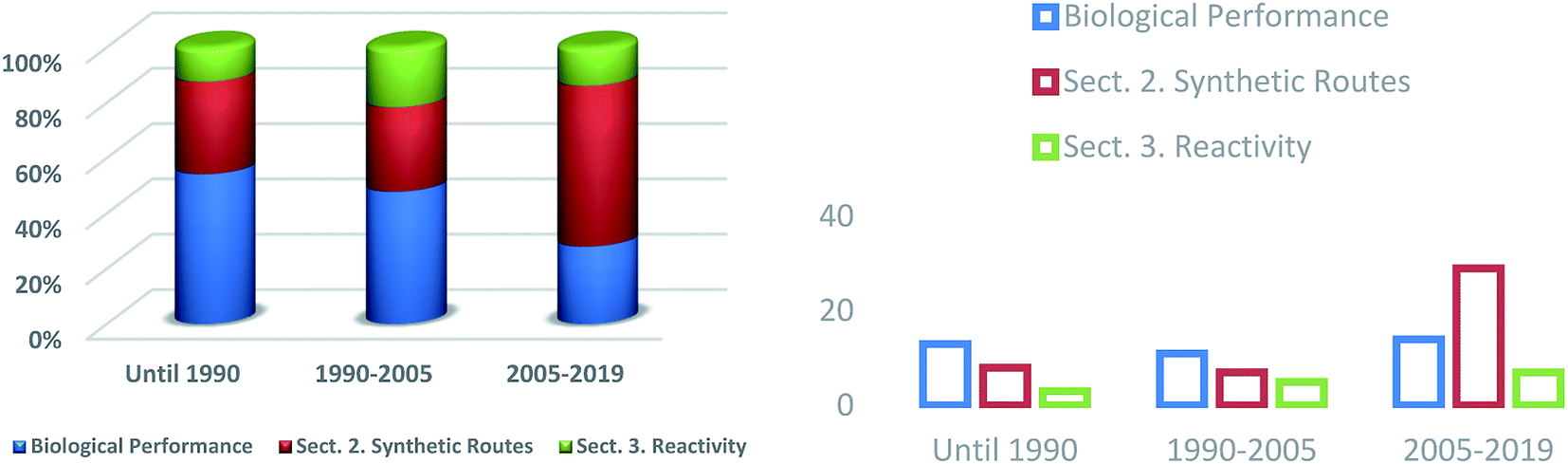
Numerous investigations have been performed and the frequency with which published articles from different fields have been published has shown continual growth(Fig. 4). Synthetic strategies (Section 2) used to access heterocycles with a pyrimido[4,5-d]pyrimidine and pyrimido[5,4-d]pyrimidine skeleton have been considerably extended and there has been a significant increase in interest from researchers for the synthesis of this class of compounds in recent years (2005–2019). Modified methods of preparation have reached a remarkable level of versatility and efficiency. In view of the synthetic methods, it has been found that they have increased considerably in recent years owing to the high biological diversity of this class of compounds. During the period before 1990, the biological importance of the compounds and the synthetic procedures produced great interest, as well as the reactivity of substituents, which has increased over time.
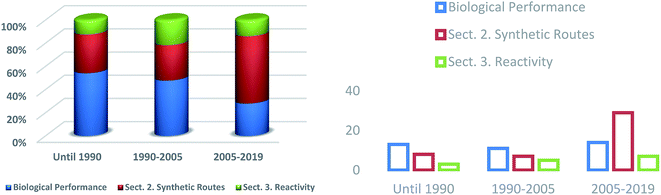 |
| | Fig. 4 A comparison of the different areas of research in the last decade. | |
6. Future perspectives
The synthesis of pyrimido[4,5-d]pyrimidines was first reported by Taylor et al.18 from the reaction of 4-aminopyrimidine-5-carboxamides with 4-amino-5-cyanopyrimidines. Since then, many types of research have been published for the synthesis of compounds incorporating a pyrimidopyrimidine core and their subsequent application in the medicinal field, as these compounds have a diverse and remarkable biological significance. The interest of researchers should be focused on the evaluation of these compounds in different areas of medicinal chemistry, for example, ATPase activity enzymes that catalyze the decomposition of ATP”, human CaR antagonism, hepatic metabolism, antitumor activity, fungicidal activity, antimycobacterial activity, and tyrosine kinase inhibitors, which are applications of the analogs of pyrido[4,3-d]pyrimidines.49a
Conflicts of interest
The authors declare no conflicts of interest.
Abbreviations
| HBV-DNA | Hepatitis B virus DNA |
| KDR | Kinase insert domain receptor (a type III receptor tyrosine kinase) |
| EGFR | Epidermal growth factor receptor |
| FGFRs | Fibroblast growth factor receptors |
| PDGFR | Platelet-derived growth factor receptors |
| TFAA | Trifluoroacetic anhydride |
| DMF |
N,N-Dimethylformamide |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofuran |
| DMU |
L-(+)-Tartaric acid-dimethyl urea |
| CDI | 1,1′-Carbonyldiimidazole |
| NIS |
N-Iodosuccinimide |
|
B. subtilis
|
Bacillus subtilis (bacteria) |
|
S. aureus
|
Staphylococcus aureus
|
|
E. coli
|
Escherichia coli
|
|
A. niger
|
Aspergillus niger
|
|
C. albicans
|
Candida albicans
|
|
P. diminuta
|
Pseudomonas diminuta
|
| ppm | Parts per million |
|
P. aeruginosa
|
Pseudomonas aeruginosa
|
| LiHMDS | Lithium bis(trimethylsilyl)amide |
|
p-TSA |
p-Toluenesulfonic acid |
| NHC-PPIm | N-heterocyclic carbene 1,3-dipropylimidazole-2-ylidene |
| DMF-DMA |
N,N-Dimethylformamide dimethyl acetal |
| EC50 | Half maximal effective concentration |
| DMSO | Dimethyl sulfoxide |
| IC | Inhibitory concentration |
| A431 cells | Human epidermoid carcinoma cell line |
| P2S5 | Phosphorus pentasulfide |
| DIPEA |
N,N-Diisopropylethylamine |
References
- H. Gastpar, Acta Med. Scand., Suppl., 1972, 525, 269–271 Search PubMed.
- R. D. Bunag, C. R. Douglas, S. Imai and R. M. Berne, Circ. Res., 1964, 15, 83–88 CrossRef CAS.
- P. R. Emmons, M. G. J. Harrison, A. J. Honour and J. R. A. Mitchell, Nature, 1965, 208, 255–257 CrossRef CAS.
- L. A. Harker and R. A. Kadatz, Thromb. Res., 1983, 4, 39–46 CrossRef CAS.
- H. J. Kaiser, D. Stiimpfig and J. Flammer, Int. Ophthalmol., 1996, 19, 355–358 CrossRef CAS PubMed.
- R. Eliasson and S. Bygdeman, Scand. J. Clin. Lab. Invest., 1969, 24, 145–151 CrossRef CAS.
- D. D. Ivy, J. P. Kinsella, J. W. Ziegler and S. H. Abman, J. Thorac. Cardiovasc. Surg., 1998, 115(4), 875–882 CrossRef CAS.
- A. Virgilio, D. Spano, V. Esposito, V. Di Dato, G. Citarella, N. Marino, V. Maffia, D. De Martino, P. De Antonellis, A. Galeone and M. Zollo, Eur. J. Med. Chem., 2012, 57, 41–50 CrossRef CAS.
- J. P. De la Cruz, T. Carrasco, G. Ortega and F. S. De la Cuesta, Lipids, 1992, 27(3), 192–194 CrossRef CAS.
- M. G. Gebauer, C. Mc Kinlay and J. E. Gready, Eur. J. Med. Chem., 2003, 38(7–8), 719–728 CrossRef CAS PubMed.
- Y. Fang, J. Xu, Z. Li, Z. Yang, L. Xiong, Y. Jin, Q. Wang, S. Xie, W. Zhu and S. Chang, Bioorg. Med. Chem., 2018, 26(14), 4080–4087 CrossRef CAS.
- M. Simcox, B. Higgins, L. McDermott, T. Nevins, K. Kolinsky, M. Smith, H. Yang, J. Li, Y. Chen and K. Luk, Eur. J. Cancer Suppl., 2004, 2(8), 59 CrossRef.
- H. C. Barlow, K. J. Bowman, N. J. Curtin, A. H. Calvert, B. T. Golding, B. Huang, P. J. Loughlin, D. R. Newell, P. G. Smith and R. J. Griffin, Bioorg. Med. Chem. Lett., 2000, 10(6), 585–589 CrossRef CAS.
- A. H. Bacelar, M. A. Carvalho and M. F. Proença, Eur. J. Med. Chem., 2010, 45(7), 3234–3239 CrossRef CAS.
- S. J. Haggarty, T. U. Mayer, D. T. Miyamoto, R. Fathi, R. W. King, T. J. Mitchison and S. L. Schreiber, Chem. Biol., 2000, 7(4), 275–286 CrossRef CAS.
- G. J. Grover, S. Dzwonczyk, D. M. McMullen, D. E. Normandin, C. S. Parham, P. G. Sleph and S. Moreland, J. Cardiovasc. Pharmacol., 1995, 26(2), 289–294 CrossRef CAS.
- M. W. Martin, J. Newcomb, J. J. Nunes, C. Boucher, L. Chai, L. F. Epstein, T. Faust, S. Flores, P. Gallant, A. Gore, Y. Gu, F. Hsieh, X. Huang, J. L. Kim, S. Middleton, K. Morgenstern, A. Oliveirados-Santos, V. F. Patel, D. Powers, P. Rose, Y. Tudor, S. M. Turci, A. A. Welcher, D. Zack, H. L. Zhao, L. Zhu, X. T. Zhu, C. Ghiron, M. Ermann and D. Johnston, J. Med. Chem., 2008, 51(6), 1637–1648 CrossRef CAS.
- E. C. Taylor, R. J. Knopf, R. F. Meyer, A. Holmes and M. L. Hoefle, J. Am. Chem. Soc., 1960, 82(21), 5711–5718 CrossRef CAS.
-
N. Kitamura, and A. Onishi, Eur. Pat., 163599, 1984Chem. Abstr, 1984, 104, 186439.
- Y. S. Sanghvi, S. B. Larson, S. S. Matsumoto, L. D. Nord, D. F. Smee, R. C. Willis, T. L. Avery, R. K. Robins and G. R. Revankar, J. Med. Chem., 1989, 32(3), 629–637 CrossRef CAS PubMed.
- R. B. Tenser, A. Gaydos and K. A. Hay, Antimicrob. Agents Chemother., 2001, 45(12), 3657–3659 CrossRef CAS.
-
(a) R. Niess and R. K. Robins, J. Heterocycl. Chem., 1970, 7, 243–244 CrossRef CAS;
(b) T. Hirano, K. Kuroda, H. Kodama, M. Kataoka and Y. Hayakawa, Lett. Org. Chem., 2007, 4, 530–534 CrossRef CAS;
(c) H. Bredereck, G. Simchen and M. Kraemer, Angew. Chem., Int. Ed. Engl., 1969, 8, 383–384 CrossRef CAS;
(d) J. L. Bernier, J. P. Henichart, V. Warin and F. Baert, J. Pharm. Sci., 1980, 11, 1343–1345 CrossRef.
- V. J. Ram, A. Goel, S. Sarkhel and P. R. Maulik, Bioorg. Med. Chem., 2002, 10(5), 1275–1280 CrossRef CAS.
- G. W. Rewcastle, A. J. Bridges, D. W. Fry, J. R. Rubin and W. A. Denny, J. Med. Chem., 1997, 40(12), 1820–1826 CrossRef CAS.
- D. W. Fry, M. A. Becker and R. L. Switzer, Mol. Pharmacol., 1995, 47(4), 810–815 CAS.
- M. Dabiri, H. Arvin-Nezhad, H. R. Khavasi and A. Bazgir, Tetrahedron, 2007, 63, 1770–1774 CrossRef CAS.
- M. Liu, J. Li, S. Chen, D. Huang, H. Chai, Q. Zhang and D. Shi, RSC Adv., 2014, 4, 35629–35634 RSC.
- J. Xiang, C. Geng, L. Yi, Q. Dang and X. Bai, Mol. Diversity, 2011, 15(4), 839–847 CrossRef CAS.
- S. Rostamizadeh, M. Nojavan, R. Aryan and M. Azad, Catal. Lett., 2014, 144, 1772–1783 CrossRef CAS.
- S. Das, A. J. Thakur, T. Medhi and B. Das, RSC Adv., 2013, 3, 3407–3413, 10.1039/c3ra22089c.
- F. M. Abd El Latif, M. A. Barsy, A. M. Arefa and K. U. Sadek, Green Chem., 2002, 4, 196–198 RSC.
- J. Liu, M. Lei and L. Hu, Green Chem., 2012, 14, 2534–2539 RSC.
- F. Shi, R. Jia, X. Zhang, S. Tu, S. Yan, Y. Zhang, B. Jiang, J. Zhang and C. Yao, Synthesis, 2007, 18, 2782–2790 Search PubMed.
- D. Prajapati, K. J. Borah and M. Gohain, Synlett, 2007, 4, 0595–0598 CrossRef.
- S. B. Katiyar, A. Kumar and P. M. S. Chauhan, Synth.
Commun., 2006, 36(20), 2963–2973 CrossRef CAS.
- S. Majumder, P. Borah and P. J. Bhuyan, Tetrahedron Lett., 2014, 55(6), 1168–1170 CrossRef CAS.
- P. Sharma, N. Rane and V. K. Gurram, Bioorg. Med. Chem. Lett., 2004, 14, 4185–4190 CrossRef CAS.
- A. Mobinikhaledi, T. Mosleh and N. Foroughifar, Res. Chem. Intermed., 2015, 41(5), 2985–2990 CrossRef CAS.
- R. A. Kadatz, Arzneimittelforschung, 1959, 9, 39–45 CAS.
- R. L. Magar, P. B. Thorat, P. B. Thorat, V. V. Thorat, B. R. Patil and R. P. Pawar, Chin. Chem. Lett., 2013, 24(12), 1070–1074 CrossRef CAS.
- D. Prajapati, M. Gohain and A. J. Thakur, Bioorg. Med. Chem. Lett., 2006, 16(13), 3537–3540 CrossRef CAS.
- A. Echavarren, A. Galan, J. M. Lehn and J. de Mendoza, J. Am. Chem. Soc., 1989, 111(13), 4994–4995 CrossRef CAS.
- H. Kurzmeier and F. P. Schmidtehen, J. Org. Chem., 1990, 55(12), 3749–3755 CrossRef CAS.
- H. A. Burch, L. E. Benjamin, H. E. Russell and R. Freedman, J. Med. Chem., 1974, 17(4), 451–453 CrossRef CAS.
- P. Sharma, A. Kumar, N. Rane and V. K. Gurram, Tetrahedron, 2005, 61, 4237–4248 CrossRef CAS.
- K. Saravanan, H. C. Barlow, M. Barton, A. H. Calvert, B. T. Golding, D. R. Newell, J. S. Northen, N. J. Curtin, H. D. Thomas and R. J. Griffin, J. Med. Chem., 2011, 54, 1847–1859 CrossRef CAS.
- H.-Z. Yang, M.-Y. Pan, D.-W. Jiang and Y. He, Org. Biomol. Chem., 2011, 9, 1516–1522 RSC.
-
(a) P. T. Patil, P. P. Warekar, K. T. Patil, D. K. Jamale, G. B. Kolekar and P. V. Anbhule, Res. Chem. Intermed., 2017, 43, 4103–4114, DOI:10.1007/s11164-017-2868-9;
(b) T. J. Delia, J. Org. Chem., 1984, 49, 2065–2067 CrossRef CAS;
(c) Y. Hao, J. Lyu, R. Qu, Y. Tong, D. Sun, F. Feng, L. Tong, T. Yang, Z. Zhao, L. Zhu, J. Ding, Y. Xu, H. Xie and H. Li, J. Med. Chem., 2018, 61, 5609–5622 CrossRef CAS.
-
(a) K. M. Elattar and B. D. Mert, RSC Adv., 2016, 6, 71827–71851 RSC;
(b) A. A. Fadda, S. A. El-Hadidy and K. M. Elattar, Synth. Commun., 2015, 45(24), 2765–2801 CrossRef CAS;
(c) A. A. Fadda, A. El-Mekabaty and K. M. Elattar, Synth. Commun., 2013, 43(20), 2685–2719 CrossRef CAS.
-
(a) K. M. Elattar, I. Youssef and A. A. Fadda, Synth. Commun., 2016, 46, 719–744 CrossRef CAS;
(b) K. M. Elattar, R. Rabie and M. M. Hammouda, Synth. Commun., 2016, 46, 1477–1498 CrossRef CAS.
-
(a) K. M. Elattar, B. D. Mert, M. A. Abozeid and A. El-Mekabaty, RSC Adv., 2016, 6, 37286–37307 RSC;
(b) A. A. Fadda and K. M. Elattar, Synth. Commun., 2016, 46, 1–30 CrossRef CAS;
(c) K. M. Elattar and A. A. Fadda, Synth. Commun., 2016, 46, 1567–1594 CrossRef CAS;
(d) K. M. Elattar, R. Rabie and M. M. Hammouda, Monatsh. Chem., 2017, 148, 601–627 CrossRef CAS;
(e) A. A. Fadda and K. M. Elattar, J. Heterocycl. Chem., 2014, 51(6), 1697–1704 CrossRef CAS.
- N. H. Ouf, A. E.-G. E. Amr and A. A. Fayed, Monatsh. Chem., 2008, 139, 281–287, DOI:10.1007/s00706-007-0781-4.
- M. Dymicky and W. Caldwell, J. Org. Chem., 1962, 27(12), 4211–4215 CrossRef CAS.
- L. A. McDermott, M. Simcox, B. Higgins, T. Nevins, K. Kolinsky, M. Smith, H. Yang, J. K. Li, Y. Chen, J. Ke, N. Mallalieu, T. Egan, S. Kolis, A. Railkar, L. Gerber and K.-C. Luk, Bioorg. Med. Chem., 2005, 13, 4835–4841 CrossRef CAS.
- X. Huang, N. Zorn, A. Palani and R. Aslanian, Tetrahedron Lett., 2012, 53, 7154–7158 CrossRef CAS.
- P. Sharma, A. Kumar and N. Rane, Heteroat. Chem., 2006, 17(4), 245–253 CrossRef CAS.
- R. Gupta, A. Jain, R. Joshi and M. Jain, Bull. Korean Chem. Soc., 2011, 32(3), 899–904, DOI:10.5012/bkcs.2011.32.3.899.
- M. A. A. Mohamed, N. F. H. Mahmoud and A. M. M. El-Saghier, Chem. J., 2012, 02(2), 64–68 CAS.
- P. Gupta, S. Gupta, A. Sachar, D. Kour, J. Singh and R. L. Sharma, J. Heterocycl. Chem., 2010, 47, 324–333 CrossRef CAS.
- M. R. Bhosle, L. D. Khillare, J. R. Mali, A. P. Sarkate, D. K. Lokwani and S. V. Tiwari, New J. Chem., 2018, 42, 18621–18632, 10.1039/c8nj04622k.
- W. S. Hamama, M. A. Ismail, H. A. Al-Saman and H. H. Zoorob, J. Adv. Res., 2013, 4, 115–121 CrossRef CAS.
- H. Wang, C. Wang and T. D. Bannister, Tetrahedron Lett., 2015, 56, 1949–1952 CrossRef CAS PubMed.
- F. Shirini, M. S. N. Langarudi, N. Daneshvar, N. Jamasbi and M. Irankhah-Khanghah, J. Mol. Struct., 2018, 1161, 366–382, DOI:10.1016/j.molstruc.2018.02.069.
- M. Kidwai, K. Singhal and S. Kukreja, One-pot green synthesis for pyrimido[4,5-d]pyrimidine derivatives, Z. Naturforsch., B: J. Chem. Sci., 2007, 62, 732–736 CAS.
- A. H. Kategaonkar, S. A. Sadaphal, K. F. Shelke, B. B. Shingate and M. S. Shingare, Microwave assisted synthesis of pyrimido[4,5-d]pyrimidine derivatives in dry media, Ukr. Bioorg. Acta, 2009, 1, 3–7 Search PubMed.
- S. V. Shinde, W. N. Jadhav and N. N. Karade, Three component solvent-free synthesis and fungicidal activity of substituted pyrimido [4,5-d]pyrimidine-2-(1H)-one, Orient. J. Chem., 2010, 26, 307–317 CAS.
- L. Suresh, P. S. V. Kumar, Y. Poornachandra, C. G. Kumar and G. V. P. Chandramouli, Bioorg. Med. Chem. Lett., 2017, 27, 1451–1457, DOI:10.1016/j.bmcl.2017.01.087.
- V. F. Sedova and O. P. Shkurko, Chem. Heterocycl. Compd., 2004, 40(2), 194–202 CrossRef CAS.
- P. B. Thorat, S. V. Goswami, R. L. Magar, S. S. Pendalwar and S. R. Bhusare, Chem. Biol.
Interface, 2014, 4(3), 200–205 Search PubMed.
- Y. Ohtsuka, J. Org. Chem., 1978, 43(16), 3231–3234 CrossRef CAS.
- S. Gore, S. Baskaran and B. Koenig, Adv. Synth. Catal., 2012, 354, 2368–2372, DOI:10.1002/adsc.201200257.
- E. Largy, W. Liu, A. Hasan and D. M. Perrin, Chem.–Eur. J., 2014, 20, 1495–1499, DOI:10.1002/chem.201303867.
- A. C. Krueger, D. L. Madigan, D. W. Beno, D. A. Betebenner, R. Carrick, B. E. Green, W. He, D. Liu, C. J. Maring, K. F. McDaniel, H. Mo, A. Molla, C. E. Motter, T. J. Pilot-Matias, M. D. Tufano and D. J. Kempf, Bioorg. Med. Chem. Lett., 2012, 22, 2212–2215 CrossRef.
- A. Spallarossa, C. Rotolo, C. Sissi, G. Marson, M. L. Greco, A. Ranise, P. La Colla, B. Busonera and R. Loddo, Bioorg. Med. Chem., 2013, 21, 6328–6336 CrossRef CAS.
- A. Ranise, A. Spallarossa, S. Schenone, O. Bruno, F. Bondavalli, A. Pani, M. E. Marongiu, V. Mascia, P. La Colla and R. Loddo, Bioorg. Med. Chem., 2003, 11, 2575–2589 CrossRef CAS.
- V. A. Dorokhov, V. A. Voronkova, A. V. Komkov and A. S. Shashkov, Russ. Chem. Bull., Int. Ed., 2011, 60(11), 2320–2324 CrossRef CAS.
- S. V. Hese, R. D. Kamble, P. P. Mogle, S. S. Kamble, M. J. Hebade, A. N. Ambhore, S. N. Kadam, R. N. Gacche and B. S. Dawane, J. Chem. Pharm. Res., 2015, 7(7), 784–790 CAS.
- A. A. Arutyunyan, Russ. J. Org. Chem., 2014, 50(2), 257–262 CrossRef CAS.
- A. M. S. Youssef, A. M. Fouda and R. M. Faty, Chem. Cent. J., 2018, 12, 50–63, DOI:10.1186/s13065-018-0419-0.
- A. Rocha, A. H. Bacelar, J. Fernandes, M. F. Proenca and M. A. Carvalho, Synlett, 2014, 25(3), 343–348, DOI:10.1055/s-0033-1340344.
- P. C. Srivastava, G. R. Revankar, R. K. Robins and R. J. Rousseau, J. Med. Chem., 1981, 24, 393–398 CrossRef CAS.
- T. E. Mabry, C. D. Jones, T. S. Chou, J. M. Colacino, G. B. Grindey, J. F. Worzalla and H. L. Pearce, Nucleosides Nucleotides, 1994, 13, 1125–1133 CrossRef CAS.
- J. S. Northen, F. T. Boyle, W. Clegg, N. J. Curtin, A. J. Edwards, R. J. Griffin and B. T. Golding, J. Chem. Soc., Perkin Trans. 1, 2002, 1, 108–115 Search PubMed.
- M. El-Araby, R. S. Pottorf and M. R. Player, Comb. Chem. High Throughput Screening, 2004, 7, 413–421 CrossRef CAS.
- G. W. Rewcastle, W. A. Denny and D. H. Showalter, Curr. Org. Chem., 2000, 4, 679–706 CrossRef CAS.
- H. M. Berman, R. J. Rousseau, R. W. Mancuso, G. P. Kreishman and R. K. Robins, Tetrahedron Lett., 1973, 33, 3099–3101 CrossRef.
- J. D. Westover, G. R. Revankar, R. K. Robins, R. D. Madsen, J. R. Orgden, J. A. North, R. W. Mancuso, R. J. Rousseau and E. L. Stephen, J. Med. Chem., 1981, 24, 941–946 CrossRef CAS.
- T. S. Rao and G. R. Revankar, Nucleosides Nucleotides, 1995, 14, 1601–1612 CrossRef CAS.
- M. A. Carvalho, S. Esperança, T. Esteves and M. F. Proença, Eur. J. Org. Chem., 2007, 2007, 1324–1331 CrossRef.
-
A. Takamizawa, and H. Sato, US Pat., 3,787,408, 1974.
- A. Sheykhi-Estalkhjani, N. O. Mahmoodi, A. Yahyazadeh, M. P. Nadamani and H. T. Nahzomi, Tetrahedron, 2019, 75, 749–756 CrossRef CAS.
- P. Sharma, N. Rane and P. Pandey, Arch. Pharm. Chem. Life Sci., 2006, 339, 572–578 CrossRef CAS.
- H. Fukumoto, S. Takatsuki, B.-L. Lee and T. Yamamoto, Synth. Met., 2009, 159, 900–904 CrossRef CAS.
- N. J. Curtin, H. C. Barlow, K. J. Bowman, A. H. Calvert, R. Davison, B. T. Golding, B. Huang, P. J. Loughlin, D. R. Newell, P. G. Smith and R. J. Griffin, J. Med. Chem., 2004, 47, 4905–4922 CrossRef CAS PubMed.
- K. Nakashima and S. Akiyama, Dyes Pigm., 1990, 12, 21–26 CrossRef CAS.
- J. H. Gall, D. D. MacNicol, R. MacSween and C. S. Frampton, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2019, 75, 383–387, DOI:10.1107/S2056989019002470.
- H. Zhao, X. Guo, S. He, X. Zeng, X. Zhou, C. Zhang, J. Hu, X. Wu, Z. Xing, L. Chu, Y. He and Q. Chen, Nat. Commun., 2014, 5, 3108–3118, DOI:10.1038/ncomms4108.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Khaled M.
Elattar
b and
Khaled M.
Elattar