DOI:
10.1039/C9RA05797H
(Paper)
RSC Adv., 2019,
9, 28241-28247
Synthesis of a 1,2-cis-indoxyl galactoside as a chromogenic glycosidase substrate†
Received
26th July 2019
, Accepted 3rd September 2019
First published on 9th September 2019
Abstract
A synthetically challenging 1,2-cis-indoxyl galactoside, X-α-galactoside, was first prepared in this study using a cyclic ketone indoxyl acceptor and a glycosyl trichloroacetimidate donor to produce an enol glycoside and a 4,6-O-di-tert-butylsilylene-protected galactosyl donor to complete the synthesis. The target compound shows enzyme activity in the presence of α-galactosidase.
Introduction
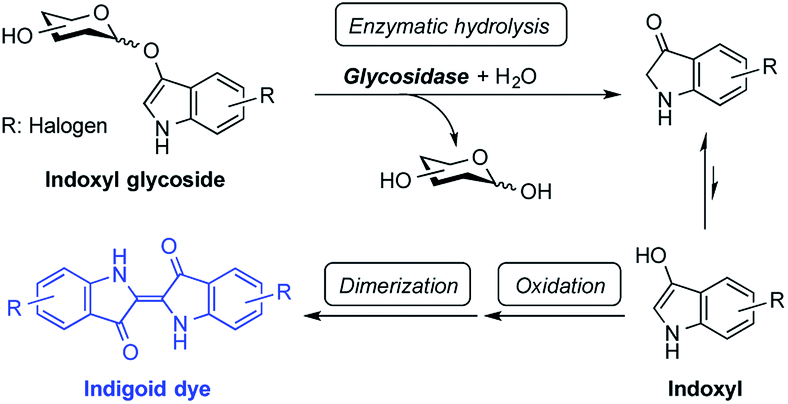
Indoxyl glycosides are chromogenic substrates that are useful in detecting glycosidase activity. These substrates have been widely used for this purpose in histochemical, biochemical, and bacteriological studies.1 In this process, the hydrolysis of an indoxyl substrate by glycosidase releases a sugar moiety and a free indoxyl, the liberated molecule of which then undergoes oxidation in the air followed by dimerization to generate an indigoid dye (Fig. 1). Indigoid dyes are insoluble in water, making the diffusion of the slightest amount of dye from enzymatic hydrolysis sites possible to determine the precise location of glycosidases.2 The color and physical properties (e.g., solubility and affinity for protein) of indigoid dyes depend on the nature of the halogen substituents located on the indole ring of the compound. For example, 5-bromo-4-chloro (greenish blue)-, 5-bromo-6-chloro (magenta)-, 5-chloro (red)-, and 5-iodo (purple)-substituted substrates are well known, and these different-colored compounds are used for tailored applications.1
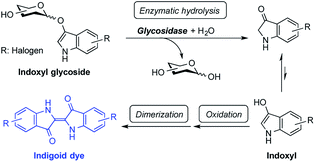 |
| | Fig. 1 Using indoxyl glycosides to detect glycosidase activity. | |
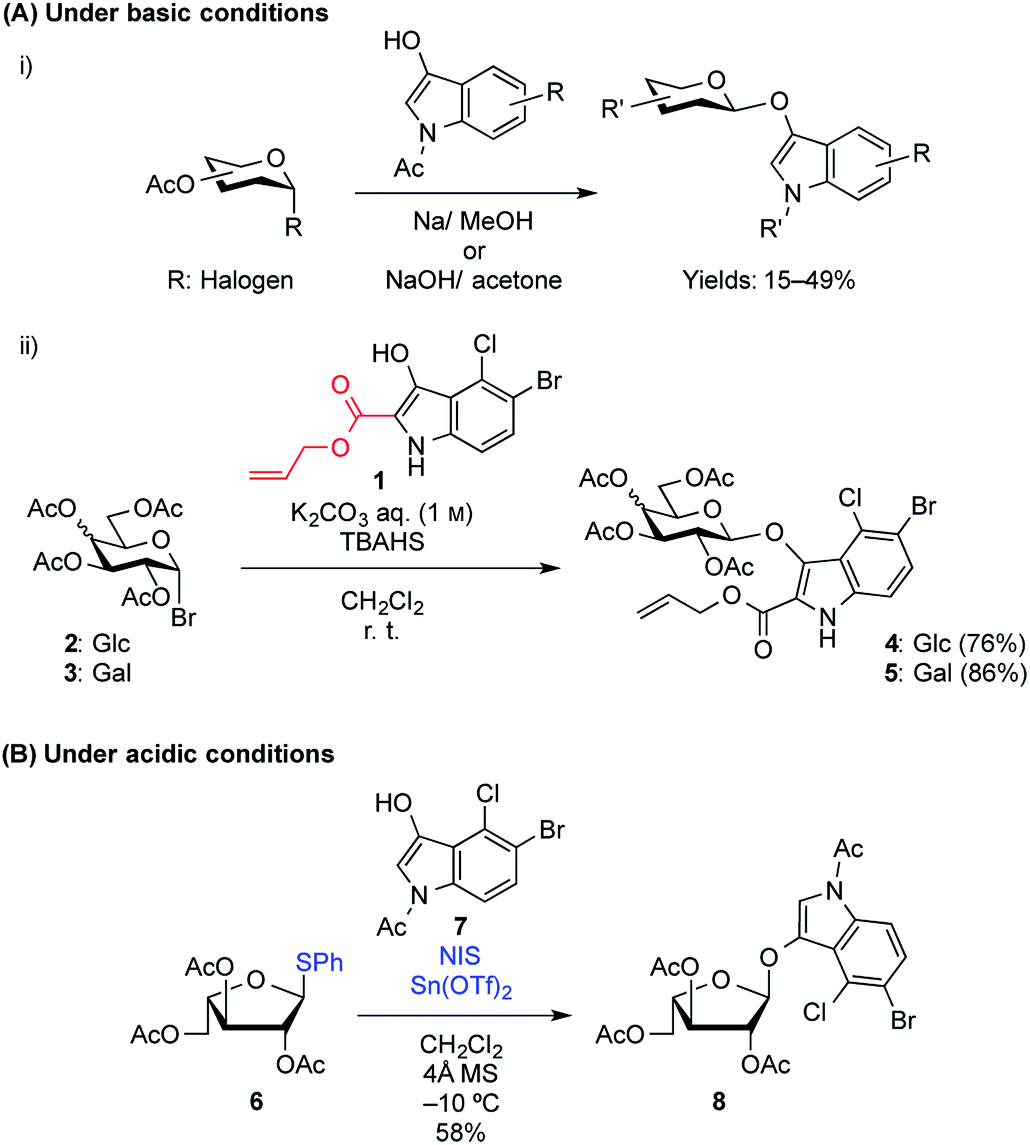
Many indoxyl glycosides are now commercially available, but halogen-substituted indoxyl glycosides are expensive because they are difficult to prepare. Their synthesis is difficult because of the low nucleophilicity of the indole hydroxy group, which suppresses the formation of the glycoside, in addition to side reactions that occur because of the indoxyl structure, leading to a decrease in the coupling yield. Furthermore, another synthetic challenge that exists for indoxyl glycosides is that they exist in the enol form and can be readily cleaved under normal glycosylation (acidic) conditions to release an aglycone in the keto form. Therefore, to date, reports on the synthesis of enol glycosides under acidic conditions have been extremely limited.3 SN2-like reaction between glycosyl halides and N-acetyl-indoxyl derivatives in the presence of NaOH in acetone or Na in MeOH is a commonly used synthetic approach used to produce indoxyl glycosides, which generally results in very low reaction yields of less than 50% (Fig. 2A-i).4 These basic conditions are required to generate the enolate form from the cyclic ketone version of the free indoxyl, which exists in keto–enol equilibrium in the reaction medium and almost none of the corresponding enol forms in nonpolar solvents.5 Unfortunately, these conditions result in low yields, in addition to the formation of various side products, including indigogenic compounds and acetyl migration and/or elimination products generated from the donor. In 2013, Thiem and coworkers synthesized indoxylic acid allyl ester 1 to improve the reactivity and instability of the indoxyl acceptor by blocking the reactive 2-position.6 In this study, they found that phase transfer glycosylation using 1 suppressed the formation of indigogenic side products, resulting in a drastic increase in the yields by up to 86% (Fig. 2A-ii).7 Recently, Wei et al. found that solid–liquid phase transfer catalysis (S–L PTC) glycosylation between acetobromo sugars and an indoxyl acceptor could be used to synthesize indoxyl glycosides, resulting in moderate glycosylation yields of 46–65%.8 However, the formation of indoxyl glycosides via a standard glycosylation reaction under acidic conditions has scarcely been reported, although there are limited reports on the synthesis of X-α-L-arabinofuranosides using the Koenigs–Knorr method9 or thioglycoside activation.10 In the latter, the authors used thiophenyl L-arabinofuranosyl donor 6 and indoxyl 7 in the presence of NIS-Sn(OTf)2 in CH2Cl2 to give 8 in a 58% yield (Fig. 2B). To date, various indoxyl glycosides, including β-Glc,11 β-Gal,11,12 β-GlcNAc,11a β-GlcA,13 α-L-Araf,9,10 β-LacNAc,7b and β-xylobioside,14 have been chemically synthesized, but they are all 1,2-trans-oriented glycosides. To the best of our knowledge, there have been no reports on the synthesis of 1,2-cis-indoxyl glycosides, which is due not only to the difficulty in preparing these compounds, but also to the lack of reliable methods available to control the anomeric stereoselectivity in 1,2-cis-glycoside formation. However, a large number of 1,2-cis-glycosides (e.g., α-Gal, α-Glc, and α-Fuc) and the corresponding glycosidases can be found in nature, where they play pivotal roles in various biological processes.15 Thus, developing a synthetic route for 1,2-cis-indoxyl glycosides is, therefore, of great importance.
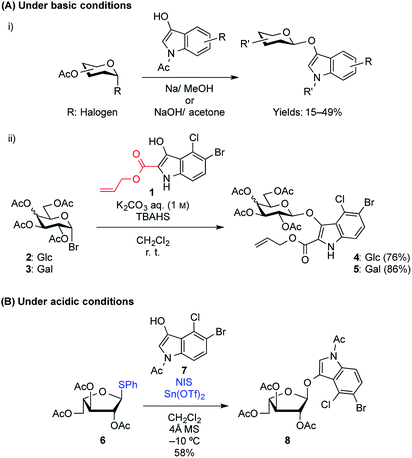 |
| | Fig. 2 Examples of the successful formation of 1,2-trans-indoxyl glycosides via (A) under basic conditions and (B) under acidic conditions. TBAHS: tetrabutylammonium hydrogen sulfate; NIS: N-iodosuccinimide. | |
Results and discussion
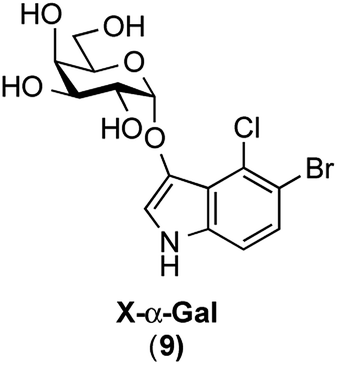
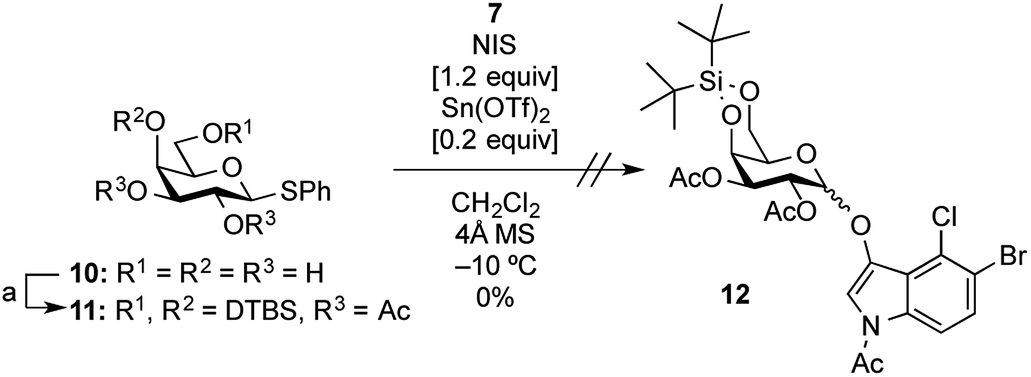
In this study, for the first time, the synthesis of X-α(1,2-cis-oriented)-galactoside (9, shown in Fig. 3, where X is the common abbreviation for 5-bromo-4-chloro-3-indoxyl) is reported. The first attempt to synthesize the target X-α-galactoside was made using thiophenyl 4,6-O-di-tert-butylsilylene- (DTBS-) protected galactosyl donor 11, readily prepared from known thiophenyl galactoside 10,16 and known indoxyl acceptor 7 (ref. 11b) to give 12. Based on our previous success in the synthesis of a large number of α-galactosides using DTBS-protected Gal donors, it was thought that this route would lead to a promising result.17 Furthermore, the abovementioned report on the synthesis of X-Araf using a thiophenyl arabinofuranosyl donor mediated by an NIS-Sn(OTf)2 system warranted further examination. However, the glycosylation reaction performed under the reported conditions did not proceed at all, and only the oxidative dimerization of 7 was observed, leading to a 91% recovery of the donor (Scheme 1). In this attempt, although 1.2 equiv. of the acceptor was initially used according to the preparation described in the literature,10 all of the acceptor was consumed in 1 ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) h, resulting in the precipitation of a blue dye. The use of 3.0 equiv. of the acceptor under the same conditions did not change the outcome of the reaction. These results suggest that the use of a thiophenyl pyranosyl donor was not feasible for the formation of X-glycosides, which might be due to the pyranoside donor being less reactive than its furanoside counterpart. In addition, in order to prevent the oxidative dimerization of the indoxyl acceptors, the use of thioglycosides, which usually have to be oxidatively activated, might be unsuitable in this study.
h, resulting in the precipitation of a blue dye. The use of 3.0 equiv. of the acceptor under the same conditions did not change the outcome of the reaction. These results suggest that the use of a thiophenyl pyranosyl donor was not feasible for the formation of X-glycosides, which might be due to the pyranoside donor being less reactive than its furanoside counterpart. In addition, in order to prevent the oxidative dimerization of the indoxyl acceptors, the use of thioglycosides, which usually have to be oxidatively activated, might be unsuitable in this study.
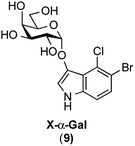 |
| | Fig. 3 X-α-galactoside, the target molecule in this study. X = 5-bromo-4-chloro-3-indoxyl. | |
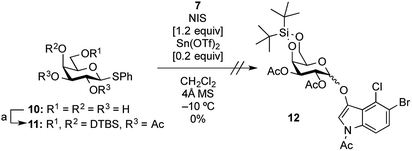 |
| | Scheme 1 Glycosylation of indoxyl 7 using DTBS-protected Gal thioglycoside donor 11. Reagents and conditions: (a) DTBS(OTf)2, Py, r.t.; Ac2O, r.t., 97% (2 steps in one-pot fashion). | |
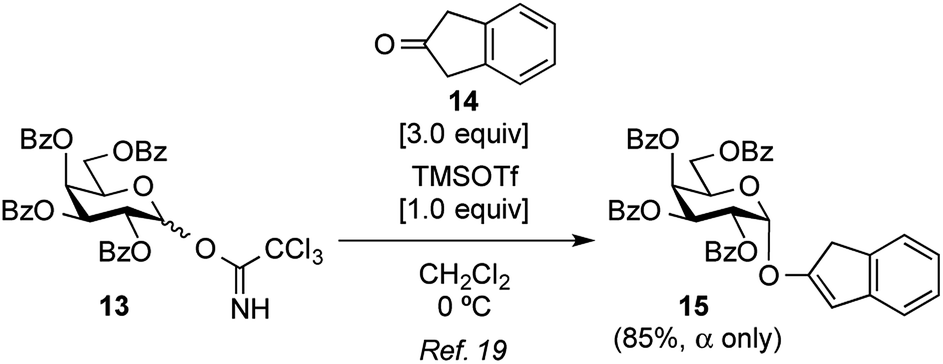
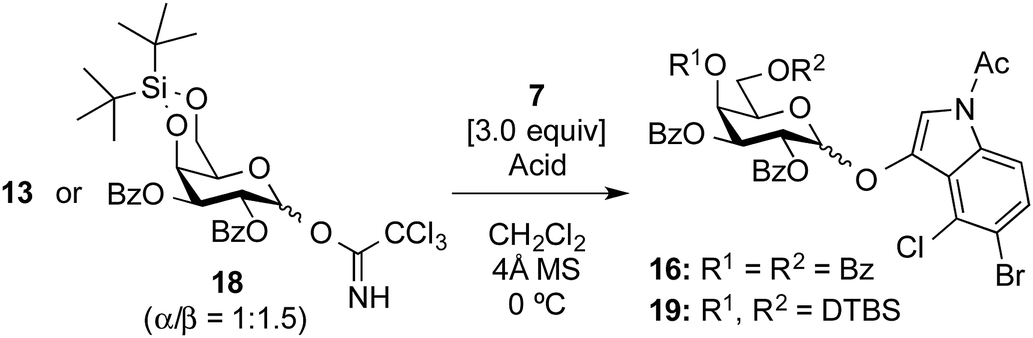
Attention was then turned to the acid activation of a trichloroacetimidate donor.18 Ding and coworkers reported the efficient direct α-glycosylation of a cyclic ketone using a glycosyl trichloroacetimidate donor, in which the coupling of 2,3,4,6-tetra-O-benzoyl-galactosyl trichloroacetimidate donor 13 (ref. 19) and cyclic ketone 14 in the presence of TMSOTf (1.0 equiv.) in CH2Cl2 at 0 °C provided the corresponding α-enol glycoside 15 as the sole product in an 85% yield (Scheme 2).20 The high α-stereo-selectivity observed was a result of the acid-catalyzed in situ anomerization of the kinetically formed β-isomer to the thermodynamic α-isomer. These glycosylation conditions were, thus, used in the present study (Table 1, entry 1). As a result, the reaction between 13 and 7 quantitatively proceeded to give the desired X-galactoside 16 contaminated with the dimerized dye product of 7, which was hard to remove by column chromatography. This result led to the first preparation of X-pyranosides under acidic conditions. However, the stereoselectivity in this reaction was very poor, with an α/β ratio of 1:1.5, favoring the unwanted β-isomer. The conversion of the β-isomer into the α-isomer was then attempted via in situ anomerization, in which the isolated β-isomer and 7 were stirred together under the above glycosylation conditions for 20 h. However, the formation of the α-isomer was not observed, with the reaction only resulting in the degradation of the β-isomer to the corresponding lactol and 7. Despite its poor stereoselectivity, the successful formation of an X-glycoside using a trichloroacetimidate donor prompted us to use DTBS-protected galactosyl donors again to improve the α-selectivity. Using 4,6-O-DTBS-protected galactosyl trichloroacetimidate donor 18, readily prepared from 10 via the corresponding thiophenyl glycoside 17, the glycosylation of 7 in the presence of TMSOTf (1.0 equiv.) was investigated (Table 1, entry 2). As anticipated, the reaction afforded the desired X-galactoside 19 in an almost quantitative yield with excellent α-selectivity (α/β ratio = >20:1). The installation of the DTBS group to endow the product with low polarity made the removal of the dye from the reaction mixture easy, but separation of the stereoisomers was not possible at this stage. However, the structure of 19α as the major product was confirmed by 1H NMR spectrum of the crude mixture, where the J1,2 value of 3.5 Hz for H-1 of the Gal residue (δ 5.94, doublet) and the presence of H-2 of the indole ring (δ 7.16, singlet) indicated the α-configuration of X-galactoside. In entry 3 of Table 1, it can be seen that reducing the amount of TMSOTf to 0.1 equiv. led to a sluggish reaction as well as a decrease in the coupling yield to 84%. Next, the acid catalyst was changed to triflic acid (Table 1, entry 4), but the outcome of the glycosylation was almost the same as that using TMSOTf. Subsequently, tin(II) triflate, which was used in the abovementioned thioglycoside activation, gave a result similar to that of TMSOTf (Table 1, entry 5). The results of both experiments, shown in entries 4 and 5 in Table 1, suggest that the nature of the acid species is not important in this reaction. Consequently, the use of a trichloroacetimidate donor was found to be of great importance in the synthesis of indoxyl glycosides.
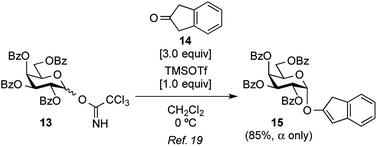 |
| | Scheme 2 The successful conditions for the stereoselective glycosylation of cyclic ketone derivative 14 using Gal trichloroacetimidate donor 13 reported by Liu et al. | |
Table 1 Reaction scheme and conditions used for the glycosylation of 7 using Gal trichloroacetimidate donors 13 or 18
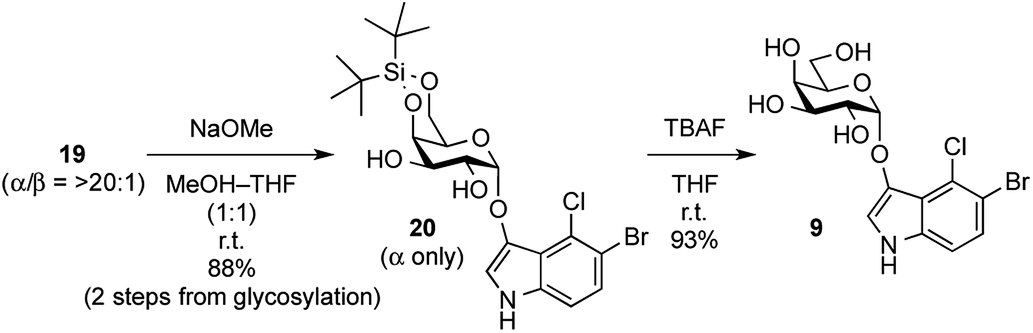
In order to complete the synthesis of X-α-galactoside, its protecting groups, including acyl groups and the DTBS group, were removed (Scheme 3). Before acyl group deprotection, it was thought that the 1,2-cis-enol glycosides would be alkali-sensitive because the 1,2-cis-configuration could lead to β-elimination at the anomeric carbon, producing the enolate form of the indoxyl and the 1,2-ene derivative of the sugar. Fortunately, these fears proved to be unfounded and the acyl groups in 19 (a mixture of α- and β-isomers) were cleaved under Zemplén conditions, affording 20 as a single isomer in good yield without any degradation of the enol glycoside. Subsequently, clean removal of the DTBS group by TBAF in THF furnished the target X-α-Gal 9 in excellent yield. During the deprotection process, it should be noted that the protected X-glycoside derivatives 19 and 20 were not very stable during even short-term storage in the freezer and underwent degradation to the corresponding lactol and indigoid dye. Therefore, after the glycosylation of the indoxyl, global deprotection should be performed as soon as possible.
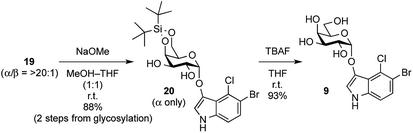 |
| | Scheme 3 Global deprotection of fully protected X-α-Gal. | |
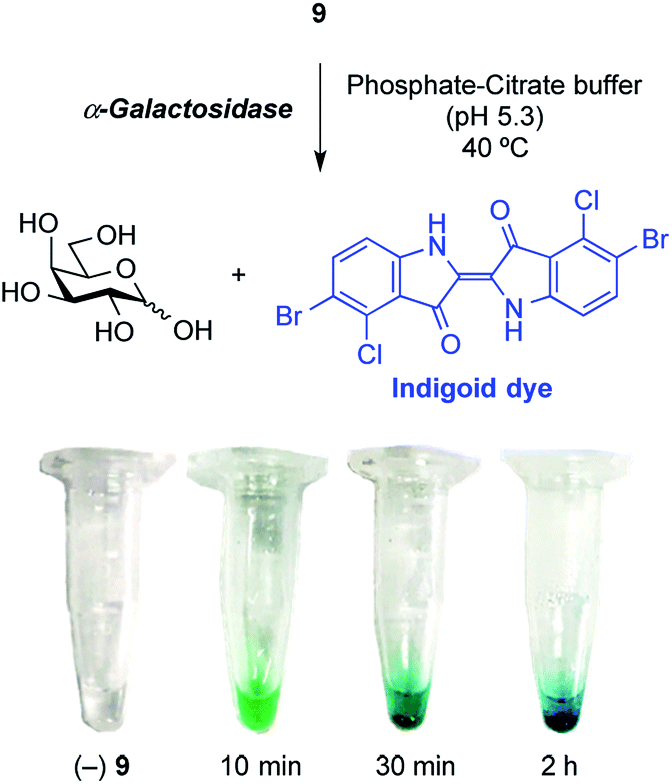
Finally, enzymatic hydrolysis of the synthesized X-α-Gal 9 using an α-galactosidase (from green coffee bean) was carried out (Fig. 4). The glycosidase activity in a phosphate-citrate buffer (pH 5.3) at 40 °C was monitored in Eppendorf tubes for 10 min, 30 min, and 2 h. A greenish-blue color evolved over 10 min, after which the precipitation of dye particles was observed after 30 min and 2 h. In the tube, the substrate was hydrolyzed by glycosidase, and the liberated indoxyl was gradually oxidized and then dimerized to form a water-insoluble indigoid dye, providing a clear indication of enzyme activity. This result demonstrated that the synthesized X-α-Gal was applicable to the detection of α-galactosidase activity.
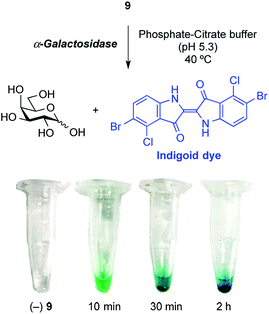 |
| | Fig. 4 Monitoring of α-galactosidase activity using the synthesized X-α-Gal compound. | |
Conclusions
This was the first successful preparation of a synthetically challenging 1,2-cis-oriented indoxyl galactoside, the reaction steps of which proceeded in a facile and high-yielding manner. The formation of the α-galactosidic linkage was achieved using a 4,6-O-DTBS-protected galactosyl donor with excellent stereoselectivity. Importantly, the use of a trichloroacetimidoyl leaving group was found to be effective in the formation of an enol glycoside. Upon exposure to α-galactosidase, the solution of the synthesized X-α-Gal in a buffer underwent a color change as the substrate was hydrolyzed by the enzyme, oxidized by the air, and then dimerized, showing that the target X-α-Gal exhibits enzyme activity. The successful synthesis of a 1,2-cis-indoxyl galactoside in this study can be expanded upon to allow access to a variety of indoxyl glycosides. We are currently underway with the efficient synthesis of other 1,2-cis-indoxyl glycosides including X-α-D-glucoside and X-α-L-fucoside. The results will be reported in the near future.
Experimental
Synthetic procedures
General methods. All reactions were performed under a positive argon pressure, unless otherwise stated. All chemicals were obtained from commercial suppliers and used without further purification, unless otherwise stated. Molecular sieves were purchased from Wako Chemicals Inc. and dried at 300 °C for 2 h in a muffle furnace before use. TLC analysis including high performance TLC (HPTLC) was conducted on Merck TLC (silica gel 60F254 on glass plate). Compounds were detected either by exposure to UV light (2536 Å) or by soaking in a solution of 10% H2SO4 in ethanol followed by heating. Silica gel (80 mesh and 300 mesh) manufactured by Fuji Silysia Chemical Ltd. was employed for flash column chromatography. The quantity of silica gel was usually estimated as 100- to 150-fold the weight of the sample to be charged. Solvent systems in chromatography are specified in v/v. Evaporation and concentration were performed in vacuo. High-resolution mass spectrometry (ESI-TOF MS) was performed on a Bruker micrOTOF instrument. Optical rotations were measured using a “Horiba SEPA-300” high-sensitivity polarimeter. The 1H NMR and 13C NMR spectra were recorded using Bruker Biospin AVANCE III 500 spectrometer. Chemical shifts in 1H NMR spectra are expressed in ppm (δ) relative to the Me4Si signal, adjusted to δ 0.00 ppm. Data are presented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, dd = doublet of doublet, td = triplet of doublet, m = multiplet and/or multiple resonance), integration, coupling constant in Hertz (Hz), and position of the corresponding proton.
Enzyme assay. α-Galactosidase from green coffee bean (pH optimum 5.3 and no data for temperature optimum) was purchased from Sigma-Aldrich. To a suspension of the enzyme (ca. 25 U) in 3.2 M ammonium sulfate (pH 6.0) (100 μL) was added 1.0 mL of phosphate-citrate buffer prepared at pH 5.3, giving the enzyme solution. The synthetic X-α-Gal (1.0 mg) was added to the enzyme solution (275 μL) and the enzyme activity at 40 °C was monitored in 10 min, 30 min, and 2 h.
1-Acetyl-5-bromo-4-chloroindol-3-ol (7). Known indoxyl acceptor 7 was prepared from commercially available 1-acetyl-5-bromo-4-chloro-3-indolyl acetate according to the literature.11b To 95% sulfuric acid (4.65 mL) with vigorously stirring was added portionwise 1-acetyl-5-bromo-4-chloro-3-indolyl acetate (1.00 g, 3.04 mmol) at 0 °C. After stirring for 2 min at 0 °C, the completion of the reaction was confirmed by TLC (1:1 n-hexane/EtOAc). The reaction mixture was poured on crushed ice in a beaker and stirred quickly by glass rod. The precipitate formed was filtered off and washed with generous quantities of 1% solution of sodium acetate followed by water. The resulting residue was exposed to high vacuum, giving 7 (856 mg, 98%) as yellow solid. Prepared 7 should be stored in a temperature of less than −60 °C until use. 1H NMR (500 MHz, CDCl3): δ 8.42 (d, 1H, 3Jortho = 8.5 Hz, Ar), 7.82 (d, 1H, Ar), 4.36 (s, 2H, CH2), 2.33 (s, 3H, Ac); 13C NMR (125 MHz, CDCl3): δ 190.7, 168.1, 154.1, 140.9, 131.8, 122.8, 119.0, 117.8, 56.4, 24.3. HRMS (ESI-TOF): m/z calcd for C8H5BrClNO: 267.9135 [M + Na]+; found: 267.9135.
Phenyl 2,3-di-O-acetyl-4,6-O-di-tert-butylsilylene-1-thio-β-D-galactopyranoside (11). To a solution of known phenyl 1-thio-β-D-galactopyranoside 10 (1.00 g, 3.67 mmol) in pyridine (36.7 mL) was added di-tert-butylsilyl bis(trifluoromethanesulfonate) (1.43 mL, 4.41 mmol) at 0 °C. After stirring for 45 min at rt, the completion of the reaction was confirmed by TLC (5:1 n-hexane/EtOAc). Subsequently, acetic anhydride (3.45 mL, 36.7 mmol) was added to the reaction mixture at 0 °C. After stirring for 1 h at rt, the completion of the reaction was confirmed by TLC (5:1 n-hexane/EtOAc). The reaction was quenched by the addition of dry methanol at 0 °C. Solvents were removed by co-evaporation with toluene, and then the residue was diluted with CHCl3, washed with 2 M HCl, H2O, satd aq NaHCO3, and brine. The organic layer was subsequently dried over Na2SO4, filtered, and concentrated. The resulting residue was purified by silica gel column chromatography (5:1 n-hexane/EtOAc) to give 11 (1.76 g, 97%) as white foamy material. [α]D +33.8 (c 1.0 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.50–7.25 (m, 5H, Ph), 5.47 (t, 1H, J1,2 = J2,3 = 10.0 Hz, H-2), 4.80 (dd, 1H, J3,4 = 3.0 Hz, H-3), 4.73 (d, 1H, H-1), 4.69 (d, 1H, H-4), 4.25 (dd, 1H, J5,6a = 1.5 Hz, Jgem = 13.0 Hz, H-6a), 4.22 (dd, 1H, J5,6b = 2.0 Hz, H-6a), 3.49 (s, 1H, H-5), 2.10 (s, 3H, Ac), 2.08 (s, 3H, Ac), 1.11 (s, 9H, tBu), 1.01 (s, 9H, tBu); 13C NMR (125 MHz, CDCl3): δ 170.6, 169.5, 134.0, 132.0, 128.9, 127.7, 87.2, 77.3, 74.9, 74.8, 70.1, 67.4, 67.0, 27.5, 27.5, 23.2, 20.9, 20.8, 20.6. HRMS (ESI-TOF): m/z calcd for C24H36O7SSi: 519.1843 [M + Na]+; found: 519.1844.
N-Acetyl-5-bromo-4-chloroindoxyl 2,3,4,6-tetra-O-benzoyl-D-galactopyranoside (16). A solution of 13 (200 mg, 0.271 mmol) in CH2Cl2 (1.0 mL) was transferred into a two-neck round bottom flask, in which pre-activated 4 Å molecular sieves (200 mg) were placed, using a cannula at 0 °C. To the suspension was transferred a solution of 7 (233 mg, 0.813 mmol) in CH2Cl2 (3.0 mL) using a cannula at 0 °C. Subsequently, TMSOTf (49.0 μL, 0.271 mmol) was added to the mixture at 0 °C. After stirring for 10 h at the same temperature, the completion of the reaction was confirmed by HPTLC (1:1 n-hexane/EtOAc). The reaction was quenched by the addition of satd aq NaHCO3 at 0 °C. The precipitate was filtered through Celite pad and the pad was washed with EtOAc. The combined filtrate and washings were washed with satd aq NaHCO3, 2 M HCl, and brine. The organic layer was subsequently dried over Na2SO4, filtered, and concentrated. The resulting residue was purified by silica gel column chromatography (7:1 n-hexane/EtOAc) to give 16 (a mixture of the stereoisomers, α/β = 1/1.5), 7, and the corresponding dye (426 mg as the mixture). 16β (contaminated with some compounds): 1H NMR (500 MHz, CDCl3): δ 8.23–7.27 (m, 22H, Ar), 7.25 (s, 1H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–N), 6.20 (dd, 1H, J1,2 = 7.5 Hz, J2,3 = 10.0 Hz, H-2), 6.08 (d, 1H, J3,4 = 3.5 Hz, H-4), 5.72 (dd, 1H, H-3), 5.36 (d, 1H, H-1), 4.76 (dd, 1H, J5,6a = 7.0 Hz, Jgem = 12.0 Hz, H-6a), 4.58 (dd, 1H, J5,6b = 4.5 Hz, H-6b), 4.54 (m, 1H, H-5), 2.24 (s, 3H, Ac); 13C NMR (125 MHz, CDCl3): δ 168.3, 166.2, 165.6, 165.6, 165.2, 139.7, 133.8, 133.6, 133.5, 133.4, 133.4, 130.5, 130.1, 129.8, 129.8, 129.8, 129.7, 129.3, 129.1, 128.8, 128.6, 128.6, 128.5, 128.4, 125.3, 122.5, 118.4, 116.1, 112.8, 101.2, 72.4, 71.4, 69.1, 68.2, 63.0, 23.4. HRMS (ESI-TOF): m/z calcd for C44H33BrClNO11: 888.0818 [M + Na]+; found: 888.0814.
CH–N), 6.20 (dd, 1H, J1,2 = 7.5 Hz, J2,3 = 10.0 Hz, H-2), 6.08 (d, 1H, J3,4 = 3.5 Hz, H-4), 5.72 (dd, 1H, H-3), 5.36 (d, 1H, H-1), 4.76 (dd, 1H, J5,6a = 7.0 Hz, Jgem = 12.0 Hz, H-6a), 4.58 (dd, 1H, J5,6b = 4.5 Hz, H-6b), 4.54 (m, 1H, H-5), 2.24 (s, 3H, Ac); 13C NMR (125 MHz, CDCl3): δ 168.3, 166.2, 165.6, 165.6, 165.2, 139.7, 133.8, 133.6, 133.5, 133.4, 133.4, 130.5, 130.1, 129.8, 129.8, 129.8, 129.7, 129.3, 129.1, 128.8, 128.6, 128.6, 128.5, 128.4, 125.3, 122.5, 118.4, 116.1, 112.8, 101.2, 72.4, 71.4, 69.1, 68.2, 63.0, 23.4. HRMS (ESI-TOF): m/z calcd for C44H33BrClNO11: 888.0818 [M + Na]+; found: 888.0814.
Phenyl 2,3-di-O-benzoyl-4,6-O-di-tert-butylsilylene-1-thio-β-D-galactopyranoside (17). To a solution of 10 (1.00 g, 3.67 mmol) in pyridine (18.4 mL) was added di-tert-butylsilyl bis(trifluoromethanesulfonate) (1.43 mL, 4.41 mmol) at 0 °C. After stirring for 1 h at rt, the completion of the reaction was confirmed by TLC (2:1 n-hexane/EtOAc). Subsequently, benzoyl chloride (3.41 mL, 29.4 mmol) and a catalytic amount of N,N-dimethyl-4-aminopyridine were added to the reaction mixture at 0 °C. After stirring for 1 h at rt, the completion of the reaction was confirmed by TLC (5:1 n-hexane/EtOAc). The reaction was quenched by the addition of dry methanol at 0 °C. Solvents were removed by co-evaporation with toluene, and then the residue was diluted with CHCl3, washed with 2 M HCl, H2O, satd aq NaHCO3, and brine. The organic layer was subsequently dried over Na2SO4, filtered, and concentrated. The resulting residue was purified by silica gel column chromatography (8:1 n-hexane/EtOAc) to give 17 (2.00 g, 88%) as white foamy material. [α]D +96.5 (c 1.0 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 8.14–7.21 (m, 15H, Ph), 5.93 (t, 1H, J1,2 = J2,3 = 10.0 Hz, H-2), 5.22 (dd, 1H, J3,4 = 3.0 Hz, H-3), 4.95 (d, 1H, H-1), 4.89 (d, 1H, H-4), 4.34 (d, 1H, Jgem = 12.0 Hz, H-6a), 4.30 (dd, 1H, J5,6b = 2.0 Hz, H-6b), 3.66 (s, 1H, H-5), 1.16 (s, 9H, tBu), 0.96 (s, 9H, tBu); 13C NMR (125 MHz, CDCl3): δ 166.1, 165.5, 133.8, 133.3, 133.2, 132.5, 129.8, 129.8, 129.6, 129.4, 128.9, 128.4, 128.4, 127.8, 87.5, 75.5, 75.0, 70.5, 68.1, 67.1, 27.5, 27.5, 23.3, 20.7. HRMS (ESI-TOF): m/z calcd for C34H40O7SSi: 643.2156 [M + Na]+; found: 643.2155.
2,3-Di-O-benzoyl-4,6-O-di-tert-butylsilylene-D-galactopyranosyl trichloroacetimidate (18). To a solution of 17 (1.00 g, 1.61 mmol) in the mixed solvent (16.1 mL, 5:1 acetone–H2O) was added N-bromosuccinimide (1.43 g, 8.05 mmol) at 0 °C. After stirring for 15 min at rt, the completion of the reaction was confirmed by TLC (2:1 n-hexane/EtOAc). The reaction mixture was diluted with EtOAc, washed with satd aq Na2SO3 and brine. The organic layer was subsequently dried over Na2SO4, filtered, and concentrated. The resulting residue was purified by silica gel column chromatography (7:1 n-hexane/EtOAc) to give the lactol compound (746 mg, 88%). The lactol (600 mg, 1.14 mmol) was then dissolved in CH2Cl2 (11.4 mL). To the solution were added CCl3CN (1.14 mL, 11.4 mmol) and 1,8-diazabicyclo[5.4.0]undec-7-ene (136 μL, 0.912 mmol) at 0 °C. After stirring for 6 h at 0 °C as the reaction was monitored by TLC (1:1 n-hexane/EtOAc), the reaction mixture was concentrated. The resulting residue was purified by silica gel column chromatography (15:1 → 2:1 n-hexane/EtOAc) to give 18 (711 mg, 93%, α/β = 1/1.5) as white foamy material. 18α: 1H NMR (500 MHz, CDCl3): δ 8.56 (s, 1H, NH), 8.03–7.35 (m, 10H, Ph), 6.79 (d, 1H, J1,2 = 3.5 Hz, H-1), 6.04 (dd, 1H, J2,3 = 10.5 Hz, H-2), 5.69 (dd, 1H, J3,4 = 3.0 Hz, H-3), 5.01 (d, 1H, H-4), 4.34 (dd, 1H, J5,6a = 2.0 Hz, Jgem = 12.5 Hz, H-6a), 4.30 (dd, 1H, J5,6b = 1.5 Hz, H-6b), 4.17 (s, 1H, H-5), 1.15 (s, 9H, tBu), 0.98 (s, 9H, tBu); 13C NMR (125 MHz, CDCl3): δ 166.2, 165.7, 160.8, 133.4, 133.3, 129.9, 129.7, 129.6, 129.0, 128.4, 124.8, 94.6, 91.0, 70.9, 70.7, 69.8, 67.3, 66.6, 27.5, 27.2, 23.3, 20.8. HRMS (ESI-TOF): m/z calcd for C30H36Cl3NO7Si: 694.1168 [M + Na]+; found: 694.1172.
N-Acetyl-5-bromo-4-chloroindoxyl 2,3-di-O-benzoyl-4,6-O-di-tert-butylsilylene-D-galactopyranoside (19). A solution of 18 (200 mg, 0.298 mmol) in CH2Cl2 (1.0 mL) was transferred into a two-neck round bottom flask, in which pre-activated 4 Å molecular sieves (222 mg) were placed, using a cannula at 0 °C. To the suspension was transferred a solution of 7 (258 mg, 0.894 mmol) in CH2Cl2 (3.0 mL) using a cannula at 0 °C. Subsequently, TMSOTf (54.0 μL, 0.298 mmol) was added to the mixture at 0 °C. After stirring for 0.5 h at the same temperature, the completion of the reaction was confirmed by HPTLC (1:1 n-hexane/EtOAc). The reaction was quenched by the addition of satd aq NaHCO3 at 0 °C. The precipitate was filtered through Celite pad and the pad was washed with EtOAc. The combined filtrate and washings were washed with satd aq NaHCO3, 2 M HCl, and brine. The organic layer was subsequently dried over Na2SO4, filtered, and concentrated. The resulting residue was purified by silica gel column chromatography (9:1 n-hexane/EtOAc) to give 19 (229 mg, 96%, α/β = 20/1) as purple powder. 19α: 1H NMR (500 MHz, CDCl3): δ 8.23 (d, 1H, 3Jortho = 8.5 Hz, Ar), 8.05–7.36 (m, 11H, Ar), 7.16 (s, 1H, CH–N), 5.96 (dd, 1H, J1,2 = 3.5 Hz, J2,3 = 10.5 Hz, H-2), 5.94 (d, 1H, H-1), 5.90 (dd, 1H, J3,4 = 3.0 Hz, H-3), 5.03 (d, 1H, H-4), 4.35 (dd, 1H, J5,6a = 2.0 Hz, Jgem = 13.0 Hz, H-6a), 4.22 (dd, 1H, J5,6b = 1.5 Hz, H-6b), 4.10 (s, 1H, H-5), 2.56 (s, 3H, Ac), 1.19 (s, 9H, tBu), 1.00 (s, 9H, tBu); 13C NMR (125 MHz, CDCl3): δ 168.1, 166.2, 166.0, 140.2, 133.6, 133.5, 133.2, 130.6, 129.9, 129.7, 129.0, 128.5, 128.5, 128.4, 125.4, 122.3, 118.4, 116.2, 107.9, 97.5, 77.2, 70.9, 70.7, 68.3, 68.1, 66.8, 27.6, 27.4, 24.0, 23.3, 20.8. HRMS (ESI-TOF): m/z calcd for C38H41BrClNO9Si: 820.1315 [M + Na]+; found: 820.1315.
5-Bromo-4-chloroindoxyl 4,6-O-di-tert-butylsilylene-α-D-galactopyranoside (20). To a solution of 19 (210 mg, 0.256 mmol, α/β = 20/1) in the mixed solvent (2.6 mL, 1:1 MeOH–THF) was added sodium methoxide (1.0 M solution in MeOH, 77 μL, 76.8 μmol) at 0 °C. After stirring for 1.5 h at rt as the reaction was monitored by TLC (1:1 n-hexane/EtOAc), the reaction was neutralized with Muromac (H+) resin. The resin was filtered off and washed with MeOH. The filtrate and washings were concentrated. The resulting residue was purified by silica gel column chromatography (1:1 n-hexane/EtOAc) to give 20 (225 mg, 88%) as white powder. [α]D +106.7 (c 1.0 in CHCl3); 1H NMR (500 MHz, CDCl3): δ 7.87 (br s, 1H, NH), 7.37 (d, 1H, 3Jortho = 8.5 Hz, Ar), 7.10 (d, 1H, JCCH,NH = 3.0 Hz, CCH), 7.09 (d, 1H, Ar), 5.46 (d, 1H, J1,2 = 3.0 Hz, H-1), 4.56 (d, 1H, J3,4 = 1.5 Hz, H-4), 4.30 (dd, 1H, J5,6a = 2.0 Hz, Jgem = 12.5 Hz, H-6a), 4.17 (dd, 1H, J5,6b = 1.5 Hz, H-6b), 4.04–3.97 (m, 2H, H-2, H-3), 3.95 (s, 1H, H-5), 2.67 (m, 1H, OH), 2.45 (m, 1H, OH), 1.08 (s, 9H, tBu), 1.06 (s, 9H, tBu); 13C NMR (125 MHz, CDCl3): δ 136.7, 133.4, 127.2, 124.3, 118.2, 113.5, 111.4, 110.3, 100.7, 77.2, 73.2, 71.5, 70.1, 68.2, 66.8, 27.6, 27.2, 23.4, 20.7. HRMS (ESI-TOF): m/z calcd for C38H41BrClNO9Si: 570.0685 [M + Na]+; found: 570.0688.
5-Bromo-4-chloroindoxyl α-D-galactopyranoside (9). To a solution of 20 (225 mg, 0.395 mmol) in THF (3.9 mL) were added acetic acid (54 μL, 0.948 mmol) and tetrabutylammonium fluoride (1.0 M solution in THF, 948 μL, 0.948 mmol) at 0 °C. After stirring for 0.5 h at rt, the completion of the reaction was confirmed by TLC (5:1 CHCl3/MeOH). Solvents were removed by co-evaporation with toluene. The resulting residue was purified by silica gel column chromatography on Iatrobeads 6RS-8060 (10:1→ 5:1 CHCl3/MeOH) to give 9 (150 mg, 93%) as light sky blue powder. [α]D +117.2 (c 1.0 in CHCl3); 1H NMR (500 MHz, CD3OD): δ 7.28 (d, 1H, 3Jortho = 9.0 Hz, Ar), 7.25 (s, 1H, CCH), 7.14 (d, 1H, Ar), 5.38 (d, 1H, J1,2 = 3.5 Hz, H-1), 4.07 (t, 1H, J5,6a = J5,6b = 6.0 Hz, H-5), 4.05–4.00 (m, 2H, H-3, H-4), 3.97 (dd, 1H, J2,3 = 10.0 Hz, H-2), 3.73 (d, 2H, H-6a, H-6b); 13C NMR (125 MHz, CD3OD): δ 137.1, 135.1, 127.1, 125.0, 119.6, 114.1, 113.4, 112.7, 101.9, 73.4, 71.6, 71.0, 70.4, 62.7, 49.9. HRMS (ESI-TOF): m/z calcd for C14H15BrClNO6: 429.9663 [M + Na]+; found: 429.9668.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
This work was supported in part by JSPS KAKENHI Grant Number JP18K05455 (to A. I.). We thank Prof. Dr Thiem and Dr Böttcher (University of Hamburg) for technical support on the synthesis of indoxyl derivatives.
Notes and references
- J. A. Kiernan, Biotech. Histochem., 2007, 82, 73–103 CrossRef CAS PubMed.
- R. J. Barrnett and A. M. Seligman, Science, 1951, 114, 579–582 CrossRef CAS PubMed.
-
(a) S. Saito, S. Sumita, Y. Kanda and Y. Sasaki, Tetrahedron Lett., 1992, 33, 7381–7384 CrossRef CAS;
(b) S. Saito, S. Sumita, Y. Kanda and Y. Sasaki, Chem. Pharm. Bull., 1994, 42, 1016–1027 CrossRef CAS PubMed.
- For review see: S. Böttcher and J. Thiem, Trends Carbohydr. Res., 2014, 6, 1–10 Search PubMed and references therein.
- N-Acetyl-indoxyl acceptor is more likely present in the keto form in non-polar and aprotic solvents, which was confirmed in 1H NMR spectrum measured in this study (see ESI†).
- S. Böttcher, M. Hederos, E. Champion, G. Dékány and J. Thiem, Org. Lett., 2013, 15, 3766–3769 CrossRef PubMed.
-
(a) S. Böttcher and J. Thiem, Eur. J. Org. Chem., 2014, 564–574 CrossRef;
(b) S. Böttcher and J. Thiem, RSC Adv., 2014, 4, 10856–10861 RSC.
- X. Wei, Q. Wu, J. Zhang, Y. Zhang, W. Guo, M. Chen, Q. Gu, Z. Cai and M. Lu, Chem. Commun., 2017, 53, 103–106 RSC.
- W. Berlin and B. Sauer, Anal. Biochem., 1996, 243, 171–175 CrossRef CAS PubMed.
- L. Marmuse, M. Asther, E. Fabre, D. Navarro, L. Lesage-Meessen, M. Asther, M. O'Donohue, S. Fort and H. Driguez, Org. Biomol. Chem., 2008, 6, 1208–1214 RSC.
-
(a) F. B. Anderson and D. H. Leaback, Tetrahedron, 1961, 12, 236–239 CrossRef CAS;
(b) J. P. Horwitz, J. Chua, R. J. Curby, A. J. Tomson, M. A. Da Rooge, B. E. Fisher, J. Mauricio and I. Klundt, J. Med. Chem., 1964, 7, 574–575 CrossRef CAS PubMed.
- M. E. van Dort, K. C. Lee, C. A. Hamilton, A. Rehemtulla and B. D. Ross, Mol. Imaging, 2008, 7, 187–197 CrossRef CAS PubMed.
- K. Yoshida, N. Iino and I. Koga, Chem. Pharm. Bull., 1975, 32, 1759–1763 CrossRef PubMed.
- S. Kaneko, M. Kitaoka, A. Kuno and K. Hayashi, Biosci., Biotechnol., Biochem., 2000, 64, 741–745 CrossRef CAS PubMed.
- Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, A. G. Darvill, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor Laboratory Press, New York, 3rd edn, 2017 Search PubMed.
- M. Yde and C. K. De Bruyne, Carbohydr. Res., 1973, 26, 227–229 CrossRef CAS.
-
(a) A. Imamura, H. Ando, S. Korogi, G. Tanabe, O. Muraoka, H. Ishida and M. Kiso, Tetrahedron Lett., 2003, 44, 6725–6728 CrossRef CAS;
(b) A. Imamura, A. Kimura, H. Ando, H. Ishida and M. Kiso, Chem.: Eur. J., 2006, 12, 8862–8870 CrossRef CAS PubMed;
(c) A. Imamura, N. Matsuzawa, S. Sakai, T. Udagawa, S. Nakashima, H. Ando, H. Ishida and M. Kiso, J. Org. Chem., 2016, 81, 9086–9104 CrossRef CAS PubMed.
-
(a) R. R. Schmidt and J. Michel, Angew. Chem., Int. Ed. Engl., 1980, 19, 731–732 CrossRef;
(b) R. R. Schmidt, Angew. Chem., Int. Ed. Engl., 1986, 25, 212–235 CrossRef.
- S. Rio, J.-M. Beau and J.-C. Jacquinet, Carbohydr. Res., 1991, 219, 71–90 CrossRef CAS PubMed.
- X. Liu, S. Ren, Q. Gao, C. Hu, Y. Li and N. Ding, Org. Lett., 2018, 20, 5186–5189 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data for all new compounds. See DOI: 10.1039/c9ra05797h |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence bc,
Akihiro Imamura
bc,
Akihiro Imamura