DOI:
10.1039/C9RA05841A
(Paper)
RSC Adv., 2019,
9, 29659-29664
Facile synthesis of 3-substituted imidazo[1,2-a]pyridines through formimidamide chemistry†‡
Received
28th July 2019
, Accepted 3rd September 2019
First published on 19th September 2019
Abstract
A facile entry to 3-aryl/alkenyl/alkynyl substituted imidazo[1,2-a]pyridines (3a–p, 6a–d & 9a–9e) has been developed from readily available benzyl/allyl/propargyl halides and 2-amino pyridines as substrates via formimidamide chemistry that is devoid of caustic or expensive reagents, such as transition metal complexes. Quantum chemical calculations performed to understand the underlying mechanism of the transformation revealed a preference for intramolecular Mannich-type addition over pericyclic 1,5-electrocyclization for the systems reported herein that enable a Baldwin allowed 5-exo-trig cyclization instead of a formally anti-Baldwin 5-endo-trig process.
Introduction
Imidazo[1,2-a]pyridines, nitrogen containing fused bicyclic heterocycles, are considered to be privileged scaffolds in the field of medicinal chemistry, due to their ability to display a wide range of pharmacological activities, such as antifungal, antibacterial, antitumor, antipyretic, analgesic, antiulcer, anxiolytic, antineoplastic, cardiac stimulant and anti-osteoporotic activities.1–5 Zolpidem (hypnotic), miroprofen (analgesic), DS-1 (GABAA positive allosteric modulator), zolimidine (peptic ulcer), olprinone (cardiac stimulant) and minodronic acid (anti-osteoporotic) are some of the currently marketed imidazopyridine drugs (Fig. 1).
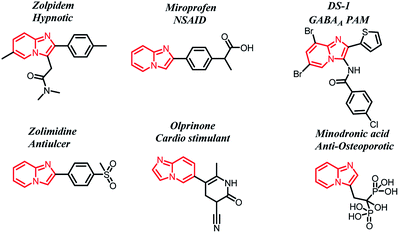 |
| | Fig. 1 Marketed imidazo[1,2-a]pyridine drugs. | |
Due to this promising potential to be a useful scaffold in drug design, continuous synthetic efforts are directed to access 2/3-substituted imidazo[1,2-a]pyridines in novel and facile ways. Several methods have been reported for the synthesis of substituted imidazopyridines, as shown in Scheme 1. For example, Jafarzadeh et al. have reported fluconazole-functionalized nanoparticle (Fe3O4@SiO2–Flu) catalyzed synthesis of 3-aryl or 3-amino-imidazo-[1,2-a]pyridines using standard α-halo ketones or aryl aldehydes and substituted isonitriles, respectively.6 Meshram et al. have reported DABCO mediated regioselective synthesis of 3-aryl imidazopyridines using standard α-halo ketones as substrates.7 Enolizable aldehydes have also been utilized as substrates for the 3-substituted imidazopyridine synthesis under oxidative conditions.8–10 Uma Maheshwari et al. have reported Et3N catalyzed oxidative amination and denitration reactions leading to 3-aryl imidazopyridines using substituted nitrostyrenes as substrates.11
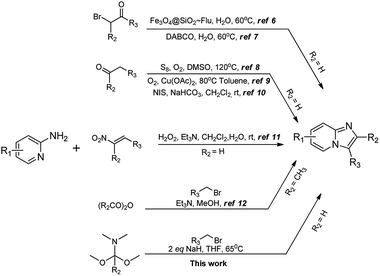 |
| | Scheme 1 Methods for the synthesis of 2/3-substituted imidazopyridines. | |
Kröhnke et al. reported synthesis of imidazopyridines using benzyl-2-iminopyridines and carboxylic anhydrides.12 All these methods have certain advantages, as well as disadvantages, such as formation of regioisomeric mixtures, harsh and demanding conditions, need for uncommon reagents or catalysts. Inspired by the lone example reported by Würthwein et al., involving base-mediated 6-electron cyclization of imino pyridinium salt to obtain 2,3-disubstituted imidazopyridine from benzyl bromide and benzaldehyde,13 and buoyed by our previous success in employing aminopyrimidine formimidamide chemistry,14 we became interested in extending and furthering the scope of the later functionality to obtain 3-substituted imidazopyridines. We reasoned that activating the simple halides through formation of corresponding formimidamide-pyridinium salts and thereby incorporating an electrophilic carbon onto 2-aminopyridine would obviate the need for α-halo ketones as the corresponding 1,2-bis-electrophiles.15–17 In addition to the undesirable characteristics and limitations of α-halo ketones, the regioselectivity of their cyclization with 2-aminopyridines is a major concern (Scheme 2). The results of our efforts towards the same are reported herein.
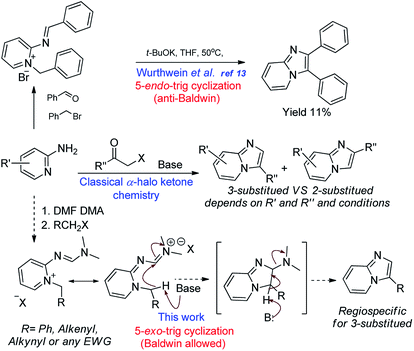 |
| | Scheme 2 Proposed chemistry for synthesis of 3-substituted imidazopyridines through formimidamide pyridinium salt to obviate the need for α-halo ketones and 5-endo-trig cyclization. | |
Results and discussions
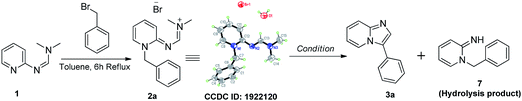
Our efforts commenced with the synthesis of N,N-dimethyl-N'-(pyridin-2-yl)formimidamide (1) accessed through reaction between 2-aminopyridine and N,N-dimethylformamide dimethyl acetal (DMF-DMA) under reflux in toluene (see Table 1).14 We initially attempted to access imidazopyridine 3a by exposing compound 1 and benzyl bromide together to elevated temperatures (toluene/115 °C in the pressure tube), but we could only isolate the quaternary ammonium salt (2a). We have reasoned that the cyclization of 2a may require the help of an acidic or basic promoter, as the attempted cyclization of 2a even at highly elevated temperature (i.e., 180 °C/DMSO) also failed to access compound 3a. Our efforts in acid catalysis to activate the imine of formimidamide in compound 2a using catalytic sulfuric acid or a super acid [i.e., triflic acid (TfOH)] were also unsuccessful, indicating the need for a base to activate the benzylic carbon attached to the quaternary nitrogen of compound 2a as a nucleophilic center. In the case of α-halo ketones as N-alkylating electrophiles, simple heating is enough to drive the cyclization as enolization and, thereby, introduction of nucleophilicity at the carbon attached to the quaternary nitrogen is facilitated by two flanking electron-withdrawing groups. In the case of benzyl halides as substrates though, the methylene carbon attached to the quaternary nitrogen is not acidic enough to drive the thermal cyclization thus demanding a base promoter. We have initially used sodium hydroxide as a base and water as a reaction medium, which failed to provide compound 3a, but yielded rather hydrolysed product 7 (Table 1) via hydrolysis of the formimidamide group. The formation of such compounds accounted for the loss of the desired product in subsequent experiments, as any remaining H2O in the reaction mixture resulted unavoidably in hydrolysis products. So, we then resorted to non-nucleophilic bases and water-free reaction conditions to suppress the same. The use of DABCO or DBU resulted in failure to generate the cyclised product 3a though traces of hydrolysis products were still observed, along with unreacted starting material and degradation products, as the above two bases are hygroscopic and had collected H2O upon storage. The use of potassium t-butoxide, however, was successful in providing compound 3a in 16% yield which was encouraging to intensify our efforts to find a better condition. The later attempts of using one molar equivalent of sodium hydride (60% w/w in mineral oil) yielded 23% of the desired product upon reflux in THF for 0.5 h. Gratifyingly, doubling the amount of sodium hydride increased the product yield to 44%, while further attempts of increasing molar excess of NaH over the cyclization precursor had no effect on product yield. This observation suggests that proton abstraction by the base is the rate limiting and, therefore, yield-controlling step in this process. With the above conditions in hand, we have explored the scope of this reaction, using various arylmethyl halides as substrates (Scheme 3) to access compounds 3a–3p. A closer look at the observed yields reveals that electron-withdrawing groups on the meta position of the benzyl substituent tend to increase the product recovery, especially for doubly substituted variants (c.f., 3m, 3n, 3e, 3k, 3f, 3h, and 3g).
Table 1 Optimization of reaction conditions for the synthesis of 3a
|
| Entry |
Solvent |
Reagent/catalyst |
Equivalents |
Temperature/reaction time |
% isolated yield of 3a |
% yield of hydrolysis product 7 |
| 1 |
DMSO |
— |
— |
180 °C/6 h |
Not observed |
Trace |
| 2 |
THF |
H2SO4 |
1 |
65 °C/12 h |
Not observed |
— |
| 3 |
THF |
Triflic acid |
1 |
65 °C/12 h |
Not observed |
— |
| 4 |
H2O |
NaOH |
1 |
RT/30 min |
Not observed |
41% |
| 5 |
THF |
NaOH |
1 |
RT/1 h |
Not observed |
35% |
| 6 |
THF |
DABCO |
1 |
65 °C/12 h |
Not observed |
Trace |
| 7 |
THF |
DBU |
1 |
65 °C/12 h |
Not observed |
Trace |
| 8 |
THF |
KOtBu |
1 |
65 °C/2 h |
16% |
Trace |
| 9 |
DCM |
KOtBu |
1 |
40 °C/2 h |
14% |
Trace |
| 10 |
THF |
NaH |
1 |
65 °C/0.5 h |
23% |
Trace |
| 11 |
THF |
NaH |
2 |
65 °C/0.5 h |
44% |
Trace |
| 12 |
DCM |
NaH |
2 |
40 °C/0.5 h |
22% |
Trace |
| 13 |
DMF |
NaH |
2 |
65 °C/0.5 h |
20% |
Trace |
| 14 |
Dioxane |
NaH |
2 |
65 °C/0.5 h |
11% |
Trace |
| 15 |
DMA |
NaH |
2 |
65 °C/0.5 h |
21% |
Trace |
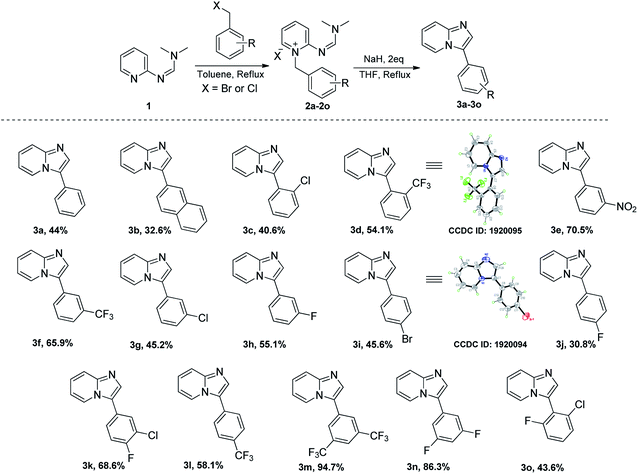 |
| | Scheme 3 Synthesis of substituted 3-aryl imidazo[1,2-a]pyridines – substrate scope. | |
This yield trend follows closely the Hammett meta-effect constants,18 which correlate with both electron-withdrawing strength of substituents and acidity at the benzylic carbon, confirming the central role that the base plays in this reaction. That is, once the pyridinium ylide is formed, the cyclization and subsequent aromatization through β-elimination of dimethylamine occurs with few (if any) interferences from unwanted side processes. In fact, the reaction can be followed visually since the orange colour of the ylide solution dissipates quite rapidly. It is also quite notable that in the above reactions, no hydrolysis product was detected.
We have also explored the scopes of substituted amino pyridines as substrates (Scheme 4). While the listed yields are unoptimized, the relatively poor performance of trifluoromethyl-substituted 2-aminopyridine suggests that the nucleophilicity of the pyridine nitrogen is important for the overall reaction success. Notably, the alkylation step could be optimized further via solvent optimization and/or catalyst use. Finally, we have also explored the scope of allyl and propargyl bromides to access the corresponding 3-vinyl and 3-ethynyl imidazopyridines, as they provide attractive functional handles for further manipulation in organic synthesis.19,20 Such compounds are generally accessed through palladium-mediated coupling reactions.21–23 Using our formimidamide chemistry under basic conditions, we were able to access compounds 9a–9e through transition metal-free formamidine-pyridinium route (Scheme 5), albeit in relatively low yield (unoptimized) due to, most likely, reduced acidity of the cyclization precursors.
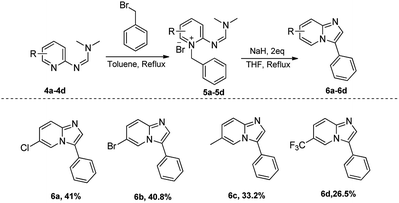 |
| | Scheme 4 Substrate scope of substituted 2-aminopyridines. | |
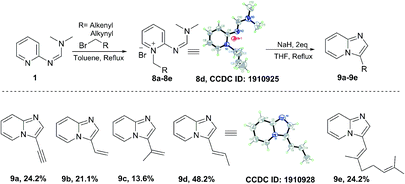 |
| | Scheme 5 Substrate scope of alkenyl/alkynyl bromides. | |
The X-ray crystallography data on some of the pyridinium salts (2a & 2o, see ESI†) revealed that they are hygroscopic and absorb moisture upon storage, which could negatively impact the cyclization reaction as it requires anhydrous conditions to supress the hydrolysis product. We then thought of condensing the formation of imine, pyridinium salt, and the cyclization of the corresponding ylide to a one-pot solvent-free procedure to avoid the isolation of potentially hygroscopic pyridinium salts. Thus, we have attempted this procedure to access compound 3a (Scheme 6) and successfully synthesized it in 58% yield, which is greater than the yield from the route shown in Scheme 3. We have also witnessed the absence of the hydrolysed product 7 in the one-pot synthesis. We are currently focusing on further optimization of this procedure and extending this to diverse substrates.
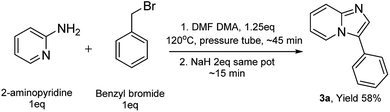 |
| | Scheme 6 One-pot synthesis of compound 3a. | |
Reaction mechanism & computational studies
Previous studies by Würthwein et al. on a cyclization reaction of similar imino pyridinium salts, which lack the dimethylamino group, suggested that these precursors undergo an apparent base-initiated anti-Baldwin24 (5-endo-trig) cyclization to provide imidazo[1,2-a]pyridines in low yields, upon 4 h exposure to KOt-Bu (2 eq.) in heated THF.13 Quantum-chemical calculations and subsequent analysis of reaction coordinates have suggested that this sluggish anti-Baldwin reaction proceeds via a pericyclic 1,5-electrocyclization process, rather than an intramolecular addition. The superior yields of imidazopyridines and broader scope of our system prompted us to investigate the mechanism and the role of the dimethylamino substituent in the key cyclization step. The positioning of counter anions in the crystal structures of pyridinium salts revealed that the dimethyl amino substituent participates actively in the delocalization of positive charge throughout the trinitrogen conjugated system (2a, 2o & 8d, see ESI†), with a significant resonance contribution from the dimethylimino form. Thus, the formation of 3a could arise from either 1,5-electrocyclization, as in the Würthwein et al. system, or Mannich-type intramolecular nucleophilic addition, which can now proceed via Baldwin-allowed 5-exo-trig ring closure, as shown in Scheme 7.
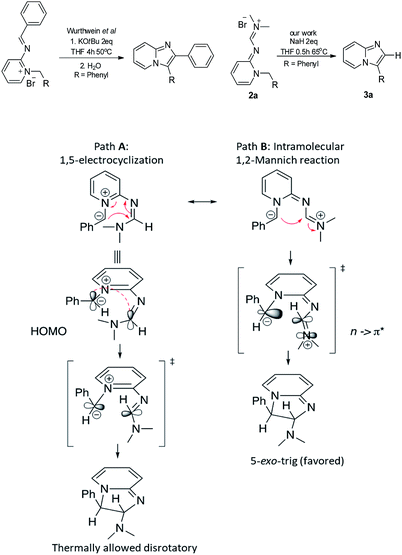 |
| | Scheme 7 Plausible mechanistic pathways for the formation of 3a from compound 2a. | |
Geometry optimization and assessment of pKa of benzylpyridinium salt 2a (pKa ≈ 27 in DMSO), using quantum-chemical approach implemented within program Jaguar (Schrödinger, LLC), rationalized both the need for a relatively strong base, such as KOtBu (pKa of conjugate acid ≈ 29 in DMSO)25 and NaH (pKa of conjugate acid ≥ 34),26 to initiate this reaction and the observed dependence of the yield on acidity of the ylide.
Search for minima and transition states (TS) for the cyclization reaction, their geometry optimizations and validation of stationary points via vibrational frequencies indicated that the lowest energy conformations of pyridinium ylide (Ra and Sa) have to undergo significant reorganization via bond rotations to bring into proximity and align the reactive centers: nucleophilic carbon of the ylide and distal electrophilic carbon of the formimidamide group (Scheme 8). The activation barriers for achieving the TS geometries (6.0 and 7.6 kcal mol−1 for those leading to trans and cis products, respectively) were found to be significantly lower than those reported by Würthwein et al. for the dimethylamino-free imino pyridinium system (10.5 kcal mol−1) at the same level of Density Functional Theory (B3LYP/6-31+G*). The less sterically congested TS leading to the trans product is expected to favour its formation in preference to cis. However, this could not be established experimentally due to facile elimination of dimethylamine with concomitant aromatization of the resulting bicycle under the reaction conditions.
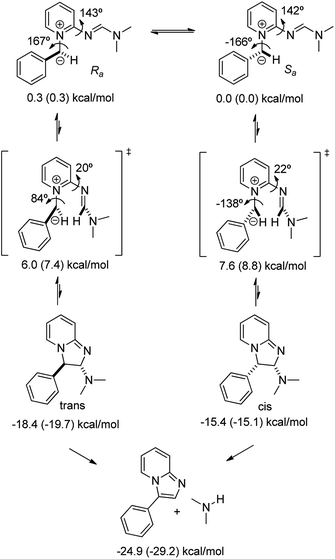 |
| | Scheme 8 Minima and transition states for cyclization of ylide derived from compound 2a to yield compound 3a. | |
The significantly reduced activation barrier resulting in improved imidazopyridine yield within the formimidamide series over the imino one raises the question of mechanistic differences in the two highly related cyclization processes. Further analysis of the frontier molecular orbitals of the formimidamide TS revealed that the highest molecular orbital coefficients at the reactive centers (ylide carbon anion and distal formimidamide carbon) reside on the highest occupied molecular orbital (HOMO) and the second lowest occupied molecular orbital (LUMO+1; LUMO is centered on pyridinium ring), respectively (Fig. 2). Notably, none of the frontier molecular orbitals extend through the 5-atom/6-electron system that would be expected for a pericyclic process (path A in Scheme 7). Both HOMO and LUMO+1, on the other hand, are fully consistent with path B mechanism, whereby the carbanion, reacts with dimethyliminium, both stabilized by extensive conjugation with adjacent groups. Thus, the dimethylamine substituent, which participates in delocalization of positive charge, as seen in the crystal structures of formimidamides (see above Scheme 5), appears to play a key role in the reaction described herein. Notably, this group converts the formally anti-Baldwin 5-endo-trig cyclization, forced to undergo a thermal pericyclic process, into a formally allowed 5-exo-trig cyclization, which yields a practical and versatile synthetic procedure for a medicinally important class of aromatic heterobicycles.
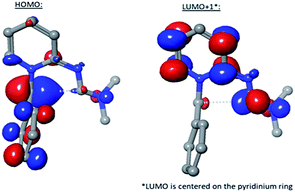 |
| | Fig. 2 FMO's of lowest energy transition state. | |
Conclusions
We have successfully developed a facile entry to 3-aryl/alkenyl/alkynyl substituted imidazo[1,2-a]pyridines using respective benzyl/allyl/propargyl halides and 2-amino pyridines as substrates (3a–p & 6a–d) via formimidamide chemistry that is devoid of expensive or caustic reagents or transition metals. Anhydrous conditions are necessary for suppressing major side reaction and for efficient synthesis of 3-substituted imidazopyridines. We have also developed a one-pot procedure, which furnished the corresponding heterobicycle in a better yield, and are currently focussing on further optimization and scope extension. Quantum chemical analysis of the reaction mechanism revealed that dimethyliminium group, acting as an exo substituent in the cyclization reaction, is beneficial in reducing the activation energy by permitting a formally allowed Mannich-type reaction over Baldwin-forbidden 1,5-electrocylization. We have also been able to access both vinyl and ethynyl substituted imidazopyridines (9a–e) that can, in turn, be used as starting points for further synthetic elaboration of fused heterocycles through the convenient functional handles. We have successfully expanded the repertoire of halides and 2-aminopyridines as substrates in this chemical transformation to access the imidazopyridine which otherwise was restricted to α-halo ketones. Currently, we are focusing on optimizing reaction conditions and on extending the scope of this formimidinium way of transforming a formally anti-Baldwin 5-endo-trig cyclization into a formally allowed 5-exo-trig cyclization to obtain diversely substituted fused imidazosystems through, and the results will be reported in due course.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work has been supported by LSMF, and OTCV Phase-I funds of UMass System and instrumentation grants from the NSF (Grants CHE-1229339 and CHE-1429086 for funds to purchase of the 400 MHz NMR spectrometer and X-ray diffractometer, respectively, used in this research).
References
- I. B. Sayeed, V. Lakshma Nayak, M. A. Shareef, N. K. Chouhan and A. Kamal, MedChemComm, 2017, 8, 1000 RSC.
- H. Lee, S. J. Kim, K. J. Jung, M. K. Son, H. H. Yan, S. Hong and S. S. Hong, Oncol. Rep., 2013, 30, 863 CrossRef CAS PubMed.
- T. O'Malley, T. Alling, J. V. Early, H. A. Wescott, A. Kumar, G. C. Moraski, M. J. Miller, T. Masquelin, P. A. Hipskind and T. Parish, Antimicrob. Agents Chemother., 2018, 62, e02439 CrossRef PubMed.
- L. Almirante, L. Polo, A. Mugnaini, E. Provinciali, P. Rugarli, A. Biancotti, A. Gamba and W. Murmann, J. Med. Chem., 1965, 8, 305 CrossRef CAS PubMed.
- L. Dymińska, Bioorg. Med. Chem., 2015, 23, 6087 CrossRef PubMed.
- M. Jafarzadeh, E. Soleimani, H. Sepahvand and R. Adnan, RSC Adv., 2015, 5, 42744 RSC.
- V. M. Bangade, B. C. Reddy, P. B. Thakur, B. Madhu Babu and H. M. Meshram, Tetrahedron Lett., 2013, 54, 4767 CrossRef CAS.
- J. Tan, P. Ni, H. Huang and G.-J. Deng, Org. Biomol. Chem., 2018, 16, 4227 RSC.
- S. K. Rasheed, D. N. Rao and P. Das, Asian J. Org. Chem., 2016, 5, 1213 CrossRef.
- S. K. Lee and J. K. Park, J. Org. Chem., 2015, 80, 3723 CrossRef CAS PubMed.
- E. S. Devi, A. Alanthadka, S. Nagarajan, V. Sridharan and C. U. Maheswari, Tetrahedron Lett., 2018, 59, 3485 CrossRef CAS.
- F. Kröhnke and B. Kickhofen, Chem. Ber., 1955, 88, 1103 CrossRef.
- B. Neue, R. Fröhlich, E.-U. Würthwein and Z. Naturforsch, Z. Naturforsch., B: J. Chem. Sci., 2012, 67, 295 CAS.
- S. Rasapalli, V. Kumbam, A. N. Dhawane, J. A. Golen, C. J. Lovely and A. L. Rheingold, Org. Biomol. Chem., 2013, 11, 4133 RSC.
- H. Jiang, X. Dong, X. Jin, D. Zhu, R. Yin, R. Yu, S. Wan, L. Zhang and T. Jiang, Chem.–Asian J., 2018, 13, 2009 CrossRef CAS PubMed.
- F. Tufail, S. Singh, M. Saquib, J. Tiwari, J. Singh and J. Singh, ChemistrySelect, 2017, 2, 6082 CrossRef CAS.
- D.-J. ZhuI, J.-X. ChenI, M.-C. LiuI, J.-C. Ding and H.-Y. WuI, J. Braz. Chem. Soc., 2009, 20, 482 Search PubMed.
- L. P. Hammett, J. Am. Chem. Soc., 1937, 59, 96 CrossRef CAS.
- Y. Li Y, M. Shen, Z. Zhang, J. Luo, X. Pan, X. Lu, H. Long, D. Wen, F. Zhang, F. Leng, Y. Li, Z. Tu, X. Ren and K. Ding, J. Med. Chem., 2012, 55, 10033 CrossRef PubMed.
- B. Desai, K. Dixon, E. Farrant, Q. Feng, K. R. Gibson, W. P. van Hoorn, J. Mills, T. Morgan, D. M. Parry, M. K. Ramjee, C. N. Selway, G. J. Tarver, G. Whitlock and A. G. Wright, J. Med. Chem., 2013, 56, 3033 CrossRef CAS PubMed.
- J. Koubachi, S. El Kazzouli, S. Berteina-Raboin, A. Mouaddib and G. Guillaumet, Synthesis, 2008, 16, 2537 Search PubMed.
- C. A. Metcalf, R. Sundaramoorthi, Y. Wang, D. Zou, R. M. Thomas, X. Zhu, L. Cai, D. Wen, S. Liu, J. Romero, J. Qi, I. Chen, G. Banda, S. P. Lentini, S. Das, Q. Xu, J. Keats, F. Wang, S. Wardwell, Y. Ning, J. T. Snodgrass, M. I. Broudy, K. Russian, T. Zhou, L. Commodore, N. I. Narasimhan, Q. K. Mohemmad, J. Iuliucci, V. M. Rivera, D. C. Dalgarno, T. K. Sawyer, T. Clackson and W. C. Shakespeare, J. Med. Chem., 2010, 53, 4701 CrossRef PubMed.
- M. Najjar, C. Suebsuwong, S. S. Ray, R. J. Thapa, J. L. Maki, S. Nogusa, S. Shah, D. Saleh, P. J. Gough, J. Bertin, J. Yuan, S. Balachandran, G. D. Cuny and A. Degtere, Cell Rep., 2015, 10, 1850 CrossRef CAS PubMed.
- J. E. Baldwin, J. Chem. Soc., Chem. Commun., 1976, 734 RSC.
- W. N. Olmstead, Z. Margolin and F. G. Bordwell, J. Org. Chem., 1980, 45, 3295 CrossRef CAS.
- C. A. Kelly and D. R. Rosseinsky, Phys. Chem. Chem. Phys., 2001, 3, 2086 RSC.
Footnotes |
| † Dedicated to Prof. Alan P. Marchand in recognition of his contributions to Physical Organic Chemistry on the occasion of 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Complete experimental procedures are provided, including copies of 1H and 13C NMR spectra of all new compounds and HRMS analysis. X-ray data for 2a, 3d, 3i, 8d and 9d is also provided. CCDC 1920094–1920096, 1922120, 1910925 and 1910928. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra05841a |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Vamshikrishna Reddy Sammetaa,
Yanchang Huanga,
James A. Golena and
Sergey N. Savinov
*a,
Vamshikrishna Reddy Sammetaa,
Yanchang Huanga,
James A. Golena and
Sergey N. Savinov









