
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePromotion of Pt/CeO2 catalyst by hydrogen treatment for low-temperature CO oxidation†
Asif Janab,
Jisu Shina,
Junsung Ahnac,
Sungeun Yang a,
Kyung Joong Yoona,
Ji-Won Sonab,
Hyoungchul Kima,
Jong-Ho Leeab and
Ho-Il Ji*ab
a,
Kyung Joong Yoona,
Ji-Won Sonab,
Hyoungchul Kima,
Jong-Ho Leeab and
Ho-Il Ji*ab
aCenter for Energy Materials Research, Korea Institute of Science and Technology (KIST), Seoul 02792, Republic of Korea. E-mail: hiji@kist.re.kr
bNanomaterials Science and Engineering, Korea University of Science and Technology (UST), KIST Campus, Seoul 02792, Republic of Korea
cDepartment of Materials Science & Engineering, Research Institute of Advanced Materials, Seoul National University, Seoul 08826, Republic of Korea
First published on 28th August 2019
Abstract
Low temperature CO oxidation reaction is known to be facilitated over platinum supported on a reducible cerium oxide. Pt species act as binding sites for reactant CO molecules, and oxygen vacancies on surface of cerium oxide atomically activate the reactant O2 molecules. However, the impacts of size of Pt species and concentration of oxygen vacancy at the surface of cerium oxide on the CO oxidation reaction have not been clearly distinguished, thereby various diverse approaches have been suggested to date. Here using the co-precipitation method we have prepared pure ceria support and infiltrated it with Pt solution to obtain 0.5 atomic% Pt supported on cerium oxide catalyst, and then systematically varied the size of Pt from single atom to ∼1.7 nm sized nanoparticles and oxygen vacancy concentration at surface of cerium oxide by controlling the heat-treatment conditions, which are temperature and oxygen partial pressure. It is found that Pt nanoparticles in range of 1–1.7 nm achieve 100% of CO oxidation reaction at ∼100 °C lower temperature compared to Pt single atom owing to the facile adsorption of CO but weaker binding strength between Pt and CO molecules, and the oxygen vacancy in the vicinity of Pt accelerates CO oxidation below 150 °C. Based on this understanding, we show that a simple hydrogen reduction at 550 °C for the single atom Pt supported on CeO2 catalyst induces the formation of highly dispersed Pt nanoparticles with size of 1.7 ± 0.2 nm and the higher concentration of surface oxygen vacancies simultaneously, enabling 100% conversion from CO to CO2 at 200 °C as well as 16% conversion even at 150 °C owing to the synergistic effects of Pt nanoparticles and oxygen vacancies.
1. Introduction
Carbon monoxide (CO) reaction is one of the most examined reactions in the field of heterogeneous catalysis. Its importance stems from its widespread applications in environmental and fundamental studies.1–5 CO is a colorless, odorless and poisonous gas produced by the incomplete combustion of various fuels such as coal, gasoline and natural gas. From an environmental perspective, since partially oxidized CO has been known to be harmful to living organisms, its elimination from the atmosphere is crucial for a healthy environment. From an industrial perspective, CO is known to be a catalyst poison, and thus its removal by oxidizing it to carbon dioxide (CO2) is imperative in the fields of fuel cells and automotive exhaust streams.6,7 In addition, CO is used as a probe molecule for understanding catalyst structure and reaction mechanism. Therefore, a better understanding of the CO oxidation reaction can help not only to evaluate catalysts itself more effectively, but also to design highly active catalysts.Owing to vast occurrence of CO oxidation reaction in various fields, numerous attempts have been made in the past to develop reliable catalysts for low temperature CO oxidation. Particularly, supported metal single atoms or nanoparticles (e.g., Pt, Pd, Ni) on reducible oxide supports (e.g., CeO2, TiO2, FeOx) have been widely investigated owing to the synergistic metal-support interactions8,9 and the active property of support itself in reactions involving redox steps.10 Pt among noble metals has been considered one of the best catalysts because of its stability and high turnover frequency at higher temperatures. Regarding the support materials, the most widely used reducible support is ceria (CeO2): the fascinating property of ceria which renders it useful for CO oxidation is its oxygen storage capacity (OSC), allowing ceria to release oxygen under reducing conditions and then reversibly take up oxygen under oxidizing conditions, thus making it more effective as compared to irreducible oxides (Al2O3, SiO2).11
For Pt supported on ceria (Pt/CeO2) catalysts, CO oxidation reaction has been extensively investigated with respect to the relevant parameters such as size and/or morphology of Pt nanoparticles,12,13 crystal orientation of CeO2 support,14,15 oxygen vacancy concentration at CeO2 surface,16 and lattice distortion caused by interaction of the Pt and CeO2.17 Indeed, a rate determining step in CO oxidation reaction has been reported as the reaction of adsorbed CO with oxygen species at the interface between Pt and CeO2, not the CO adsorption or the oxygen replenishment at the interface,13,18 and the corresponding slow reaction step can be affected by both (i) the desorption characteristic of CO on Pt and (ii) the reduction characteristic of O atom at the interface. However, in spite of continuous efforts, understanding on the corresponding parameters responsible for low temperature CO oxidation are still ambiguous, and in the worst case, contradict with each other. Regarding the size of Pt, recently single atom catalysis supported on ceria has drawn significant attention. It has been reported that the Pt in oxidized form (Pt2+) is strongly attached to the solid surface defects, and these highly dispersed under-coordinated Pt single atoms stationed on the surface of the ceria help maximize the atom efficiency, providing numerous adsorption sites for the reactant CO gas.19–21 On the contrary, there also exists a report showing that the oxidized Pt single atoms are not a suitable candidate for low temperature CO oxidation due to the fact that the binding strength between CO and Pt single atoms is very strong compared to the Pt nanoparticles, thereby lacking the catalytic activity.22 Thus in the field of heterogeneous catalysis, despite of significant work performed to understand the single atom catalysts, there exits discrepancy in the literature about the catalytic capabilities of the single atom catalysts, as such some reports show single atoms as the enhanced active centers;23–26 while, other reports show that the nanoparticles are the more active species in similar systems.27,28 In any cases, the common agreement is that the more the dispersed Pt sites, the more adsorption sites for CO, and hence the better CO adsorption properties. However, it alone is not a sufficient condition to facilitate the reaction at low temperature. How active the oxygen in vicinity of Pt reacting with the adsorbed CO at the interface between Pt and CeO2 is also considered to be one of the key parameters. It is well known that the O atoms at the metal/oxide interface exhibit a higher reactivity mainly owing to the metal-support interactions, i.e., weaker Ce–O bond at the interface.29,30 Furthermore, the higher density of surface oxygen vacancies in nano-sized CeO2 has been known to play an important role as catalyst support compared to micro-sized CeO2.31,32 However, while it has been commonly accepted that the oxygen vacancy at the surface of CeO2 effectively lowers the reaction temperature, the effect of oxygen vacancy concentration as only variable under fixed other parameters (e.g., morphology and size of Pt, size of CeO2) has not been clearly elucidated. The difficulty is arisen from that the heat-treatment in a reducing atmosphere, a typical way to increase the density of surface oxygen vacancy, simultaneously induces the increase of Pt size.33 Surprisingly, there exist several reports that heat-treatment above 800 °C in oxidizing atmosphere does not affect the size of Pt, even for the single atom catalysts;34 it implies that the subsequent oxygen treatment after hydrogen treatment can change only the density of surface oxygen vacancy with maintaining the size of Pt. Thus, the various heat-treatment conditions and their combinations can control the size of Pt and oxygen vacancy concentration at surface of cerium oxide independently, thereby distinguishing the effects of them.
The present study aims to comprehensively investigate the effects of Pt size and oxygen vacancy concentration in Pt/CeO2 catalyst using heat treatments under systematically controlled conditions, and consequentially suggest the optimal catalysts for low temperature CO oxidation reaction. In aspects of materials cost and simplicity of synthesis process towards actual applications, co-precipitation method and minimum amount of the Pt (0.5 atomic%) are employed. From the results of the catalytic activity of catalysts with different size of Pt and oxygen vacancy concentration at CeO2 surface, and from the characteristics of catalysts analyzed using complementary techniques (GC, XRD, H2-TPR, Raman, XPS, STEM, DRIFT, and X-ray absorption spectroscopy), the effects of both key parameters are discussed.
2. Results and discussion
The oxygen reduction behavior of pure CeO2 and as-prepared Pt/CeO2 catalysts were investigated using hydrogen temperature programmed reduction (H2-TPR) method as shown in Fig. 1. The H2-TPR profile of pure ceria exhibits a two-step reduction with peaks at 540 °C and 820 °C, which correspond to reduction of surface oxygen and bulk oxygen of ceria, respectively.35 Since the bulk oxygen is strongly bonded with Ce in the fluorite structure of CeO2, it reacts with H2 at higher temperature compared to the under-coordinated surface oxygen of CeO2. On the other hand, Pt/CeO2 which consists of highly dispersed Pt2+ single ions shows three peaks at 219 °C, 493 °C and 820 °C, respectively. The highly dispersed Pt species on CeO2 are responsible for the both reduction peaks obtained at temperatures of 219 °C and 493 °C. It has been reported that the reactant H2 gas is adsorbed on the Pt and then the Pt spills over the adsorbed H2 to the surface of CeO2, thus facilitating O2 reduction.10,30,36 In addition, Acerbi et al., suggested that the electronic interactions between the precious metal and the reducible support play a dominant role in the promotional effect of redox behavior.37 In any case, the oxygen atoms can be removed from the metal/oxide interface with much lower energy cost29 and the reduction at 219 °C can be ascribed to oxygen reduction of Pt–O–Ce, since the corresponding reaction has been established to take place in the temperature range of 40–250 °C and the bulk Pt oxides such as PtO and PtO2 species are reduced below room temperature because of weak interaction with the ceria.38,39 Furthermore, the position of the peak associated with ceria surface oxygen reduction is shifted to a lower temperature as compared to pure ceria, which is due to the fact that the hydrogen which is spilled over from Pt facilitates the reduction of O2 not only in close vicinity of Pt but also at surface of ceria. Moreover, the peak associated with the reduction of the bulk oxygen in ceria is not influenced by Pt. | ||
| Fig. 1 H2 temperature-programmed reduction curves of CeO2 and Pt/CeO2 catalysts. | ||
A typical approach to control the size of Pt on the support is changing the total amount of Pt in fabrication step, i.e., the higher amount of Pt, the bigger Pt.16,18 However, in that case the quantitative comparison on the catalytic activity is complicated owing to the fact that the density of active sites is a function of not only the Pt size but also the total amount of Pt. In this study, therefore, the heat-treatment temperature in H2 environment was varied from 200 to 550 °C for Pt/CeO2 with fixed amount of Pt as 0.5 atomic%. It is already known that the hydrogen treatment results in the formation of highly dispersed Pt or Pd nanoparticles on the surface of ceria.40,41 Ceria nanoparticles were basically of diameter in a range of 5–15 nm (Fig. 2a and b), and the size was invariant with temperature of H2 treatment (Fig. 2e–g). The Pt in the as-prepared Pt/CeO2 after calcination at 650 °C in air condition (Fig. 2c and d) remained as single atoms, indicating that strong Pt–O–Ce bond yields a sinter-resistant thermal stability.9,36 However, it also indicates that the heat-treatment in the reduced atmosphere (in this study, 4% H2 in Ar balance) would induce the loss of Pt–O–Ce bond, thereby the increase of Pt size can be readily achieved as the temperature increases in the reduced atmosphere. Based on H2-TPR result (Fig. 1), the temperatures of H2 heat-treatment above 200 °C were selected in which oxygen reduction in Pt–O–Ce bonds takes place. As temperature increased, Pt size also increased. The particle size distribution (PSD) for the reduced samples at 200 °C (R2-Pt/CeO2), 300 °C (R3-Pt/CeO2), and 550 °C (R5-Pt/CeO2), and for the re-oxidized at 500 °C (O5-R5-Pt/CeO2) were calculated by taking the average of Pt particle size from several STEM images; the Pt particles in all four samples have mean particle sizes of 0.8 ± 0.1 nm, 1.2 ± 0.2 nm, 1.7 ± 0.2 nm, and 1.6 ± 0.3 nm, respectively (Fig. S1†). STEM micrograph of the Pt/CeO2 composite (Fig. 2c and d) shows that the Pt is in the form of well dispersed single atoms adhered to the ceria nanoparticles and no Pt nanoparticle is found. For R2-Pt/CeO2, Pt nanoparticles of 0.8 nm in size are formed, but at the same time some of Pt still remains in the form of single atoms (Fig. 2e and S2†). For R3-Pt/CeO2 and R5-Pt/CeO2, no single atoms of Pt are observed (Fig. 2f and g) owing to all Pt–O–Ce bonds were broken above 300 °C as shown in H2-TPR result. The X-ray diffraction patterns of pure CeO2, Pt/CeO2, and R5-Pt/CeO2 belong to the ceria fluorite structure without any secondary phases (Fig. 3). The peaks corresponding to Pt were not detected, confirming the high dispersion of Pt in all samples. Furthermore, to elucidate the bulk properties of the CeO2, Pt/CeO2, and R5-Pt/CeO2 the calculated lattice parameters were identical to the value of 0.542 nm, indicating that (i) the size of ceria particles and the oxygen vacancy concentration in bulk of all three systems are same42 and (ii) Pt is not doped in Ce site.43
To get more insight about the nature of Pt as a function of H2-treatment temperature, XPS of Pt/CeO2, R2-, R3-, R5-Pt/CeO2, and R5-O5-Pt/CeO2 catalysts in the Pt 4f region were carried out to identify and quantify the respective Pt species present in each catalyst. As shown in Fig. 4, the XPS results were de-convoluted into two characteristics at 72.2–72.4 and 75.5–75.7 corresponding to Pt2+, and 70.7–71 and 74.1–74.3 eV corresponding to Pt0, respectively.44,45 For Pt/CeO2, all of the Pt on the surface of CeO2 is in the form of Pt2+ (Fig. 4a). The reduction of the catalysts in the hydrogen atmosphere at various temperatures results in transition of Pt2+ single ions to Pt0 nanoparticles; the higher the temperature of H2-treatment, the higher [Pt0]/([Pt2+] + [Pt0]), and the re-oxidation of the R5-Pt/CeO2 catalyst results in re-formation of oxidized Pt species as denoted in Fig. 4. This finding was further backed-up by X-ray absorption near edge structure (XANES) analysis presented in the Fig S3.† First, clear trend of decreasing white line intensity, peak intensity around 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 567 eV, with reduction treatment was observed. White line intensity of Pt L3 edge reflects the vacancy in the 5d orbital of Pt, thus average oxidation state of Pt can be estimated.46 In addition, the decrease of intensity represents the decrease of oxidation state of Pt.47 This result well agreed with XPS analysis, where dominant Pt2+ from as prepared Pt/CeO2 gradually decreased and became Pt0 after reduction treatment at 200–550 °C. This clearly explains that the increase in size of the Pt nanoparticles shown by the STEM images: as more Pt2+ single ions are reduced to Pt0, accompanying with being agglomerated and/or sintered to form larger nanoparticles.
567 eV, with reduction treatment was observed. White line intensity of Pt L3 edge reflects the vacancy in the 5d orbital of Pt, thus average oxidation state of Pt can be estimated.46 In addition, the decrease of intensity represents the decrease of oxidation state of Pt.47 This result well agreed with XPS analysis, where dominant Pt2+ from as prepared Pt/CeO2 gradually decreased and became Pt0 after reduction treatment at 200–550 °C. This clearly explains that the increase in size of the Pt nanoparticles shown by the STEM images: as more Pt2+ single ions are reduced to Pt0, accompanying with being agglomerated and/or sintered to form larger nanoparticles.
 | ||
| Fig. 2 STEM images of pure CeO2 and Pt/CeO2 variants: (a and b) pure ceria; (c and d) Pt/CeO2 showing Pt single atoms adsorbed on ceria particles; (e) R2-Pt/CeO2; (f) R3-Pt/CeO2; (g) R5-Pt/CeO2; (h) O5-R5-Pt/CeO2. | ||
 | ||
| Fig. 3 X-ray diffraction patterns of CeO2, Pt/CeO2 and R5-Pt/CeO2. | ||
 | ||
| Fig. 4 Pt 4f XPS of (a) Pt/CeO2; (b) R2-Pt/CeO2; (c) R3-Pt/CeO2; (d) R5-Pt/CeO2; (e) R5-O5-Pt/CeO2. The contributions from Pt2+ and Pt0 species are de-convoluted and marked on the figures. | ||
As summarized in the introduction, the catalytic activity of Pt/CeO2 for low-temperature CO oxidation reaction is highly probable to be correlated with both (i) the size of metal catalyst12,19,48 and (ii) the oxygen vacancy (or Ce3+) at the surface of ceria in vicinity of Pt, not within the lattice of ceria.16,49,50 Whereas Pt supported on non-reducible oxides follows a Langmuir–Hinshelwood mechanism, Pt on reducible oxides such as CeO2 is known to follow a Mars–Van Krevelen mechanism:51 for Pt/CeO2, the dissociative adsorption of O2 on ceria and the CO adsorption on Pt preferentially occur, thereafter the conversion of CO to CO2 takes place. Therefore, how strong the binding strength between Pt and CO molecule is and how active the surface oxygen in vicinity of Pt is would be decisive factors on CO oxidation reaction. Particularly, the oxygen vacancies at surface can play a role as adsorption sites of oxygen molecules from the atmosphere and/or available sites for taking the diffused lattice oxygen from sub-surface, thereby affecting the activity of surface oxygen. In such point of view, since H2 treatment not only increases the size of Pt but also induces the formation of surface oxygen vacancies, the re-oxidized sample at 500 °C after the reduction at 550 °C was additionally prepared (O5-R5-Pt/CeO2) which has similar Pt size (∼1.6 nm; Fig. 2h) but less oxygen vacancies at ceria surface (will be discussed below regarding the oxygen vacancy). To verify that in quantitative manner, XPS analysis was carried out. From XPS results of Pt/CeO2 (Fig. 5), R2-, R3-, R5-, and O5-R5-Pt/CeO2 (Fig. S4†) catalysts, the Ce 3d core levels of CeO2 were investigated. Cerium is known to have two oxidation states of Ce(III) and Ce(IV), with Ce(IV) being more stable in presence of air. As it can be seen from the Pt/CeO2 spectra that there are 10 de-convoluted Gaussian peak assignments, the peaks at 881.8, 888.0, 894.6, 897.8, 900, 906, 912, and 916 eV are characteristic of Ce4+, whereas the peaks at 884.5 and 902 eV are characteristic of Ce3+.52–55 In order to identify the oxygen vacancy concentration at the surface in Pt/CeO2 based on the correlation of  the concentration of Ce3+ ion, i.e.,
the concentration of Ce3+ ion, i.e., 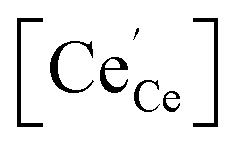 in Kröger–Vink notation, was calculated from the ratio between fitted peak areas of Ce3+ and sum of Ce3+ and Ce4+, i.e. [Ce3+]/([Ce3+] + [Ce4+]). The concentration of Ce3+ ions in Pt/CeO2 comes out to be 13.3%. Similarly, [Ce3+] in R2-, R3-, R5-, O5-R5-Pt/CeO2 were calculated as summarized in Table 1. Although the depth information of XPS is known to be ∼10 nm, and thus the exact oxygen vacancy concentration at the very top surface layer cannot be directly calculated from [Ce3+] in XPS result, the tendency of that the higher temperature of H2 treatment results in the higher [Ce3+] at the surface of ceria, meaning the higher oxygen vacancy concentration, was verified. Particularly, for O5-R5-Pt/CeO2, the oxygen vacancy concentration was significantly reduced. It needs to be noted that the temperatures of H2-treatment below and above 300 °C are expected to induce the different nature of oxygen vacancies as schematically shown in Fig. 6: for the former (T ≤ 300 °C), only those at the sites of Pt single atom would be generated; for the latter (300 °C ≤ T ≤ ∼600 °C), those only at the surface of ceria including the sites in the vicinity of Pt nanoparticle, not in the lattice, would be generated.
in Kröger–Vink notation, was calculated from the ratio between fitted peak areas of Ce3+ and sum of Ce3+ and Ce4+, i.e. [Ce3+]/([Ce3+] + [Ce4+]). The concentration of Ce3+ ions in Pt/CeO2 comes out to be 13.3%. Similarly, [Ce3+] in R2-, R3-, R5-, O5-R5-Pt/CeO2 were calculated as summarized in Table 1. Although the depth information of XPS is known to be ∼10 nm, and thus the exact oxygen vacancy concentration at the very top surface layer cannot be directly calculated from [Ce3+] in XPS result, the tendency of that the higher temperature of H2 treatment results in the higher [Ce3+] at the surface of ceria, meaning the higher oxygen vacancy concentration, was verified. Particularly, for O5-R5-Pt/CeO2, the oxygen vacancy concentration was significantly reduced. It needs to be noted that the temperatures of H2-treatment below and above 300 °C are expected to induce the different nature of oxygen vacancies as schematically shown in Fig. 6: for the former (T ≤ 300 °C), only those at the sites of Pt single atom would be generated; for the latter (300 °C ≤ T ≤ ∼600 °C), those only at the surface of ceria including the sites in the vicinity of Pt nanoparticle, not in the lattice, would be generated.
 | ||
| Fig. 5 Ce 3d XP spectra of Pt/CeO2. | ||
| Concentration | Pt/CeO2 | R2-Pt/CeO2 | R3-Pt/CeO2 | R5-Pt/CeO2 | O5-R5-Pt/CeO2 |
|---|---|---|---|---|---|
| Ce3+ (%) | 13.3 | 14.5 | 17.6 | 18.1 | 10.6 |
| Ce4+ (%) | 86.7 | 85.5 | 82.4 | 81.9 | 89.4 |
| Pt0/Pt2+ | 0 | 0.1 | 0.7 | 3.1 | 0 |
 | ||
| Fig. 6 Schematic representing (top) Pt single atoms decorated on the ceria particles (Pt/CeO2); (middle) formation of oxygen vacancies and simultaneous reduction of Pt2+ single atoms to Pt0 nanoparticles (R2-Pt/CeO2 & R3-Pt/CeO2); (bottom) enhancement of oxygen vacancies in vicinity of Pt nanoparticles and growth of Pt nanoparticles. | ||
The catalytic activities of pure CeO2, Pt/CeO2, R2-, R3-, R5-, and O5-R5-Pt/CeO2 in CO oxidation reaction were analyzed as a function of temperature using gas chromatography under continuous flow of feed gas mixtures of 3 mole% CO and 20 mole% O2 in argon balance (Fig. 7). As anticipated, all Pt supported on CeO2 catalysts exhibit better catalytic performance than pure CeO2 and R5-CeO2 certainly owing to the interaction between Pt and CeO2 associated with highly reducible O atom in vicinity of Pt as well as the better adsorption of CO on Pt compared to CeO2 free surface. As H2 reduction temperature increases (R2-, R3-, R5-Pt/CeO2), the CO conversion curves shift to lower temperature. In addition, after re-oxidation of R5-Pt/CeO2 at 500 °C, i.e., O5-R5-Pt/CeO2, while the temperature showing 100% conversion is maintained at 200 °C, the catalytic activity below 150 °C is suppressed in comparison with R5-Pt/CeO2. It should be noted here that the oxygen vacancies in the vicinity of the Pt nanoparticles and oxygen vacancies on the surface of ceria away from the Pt nanoparticles contribute to different extents to the CO oxidation activity of the catalysts. As mentioned before, the oxygen vacancies in vicinity of Pt nanoparticles are in the front line for CO oxidation, whereas oxygen vacancies away from Pt nanoparticles play a supporting role for augmenting the CO oxidation. To prove that the oxygen vacancies in the reduced samples lie in the vicinity of Pt nanoparticles, extended X-ray absorption fine structure (EXAFS) was conducted as presented in Fig. S5a,† providing the information about Pt–O and Pt–Pt coordination numbers. Strongest interaction at 1.6 Å was evident from all Pt/CeO2 samples, originated from the oxygen atoms at ∼2.0 Å distance. From the Fig. S5b,† we can see that the Pt–O coordination number decreases as the reduction temperature increases, supporting our argument that the reduction treatment removes oxygen coordinated with Pt, probably of Pt–O–Ce, making oxygen vacancies available at the vicinity of Pt. The remaining peaks over 2 Å radial distance were much smaller, indicating small coordination number of Pt–Pt and/or broad distance distribution reflecting disordered nature of Pt/CeO2 samples. To provide further insight, we heat-treated pure ceria (without Pt) in H2 atmosphere at 550 °C (R5-CeO2), where the surface oxygen vacancies can be generated based on H2-TPR results of pure ceria (Fig. 1), The catalytic activity of R5-CeO2 is far better than pure ceria, and we can attribute this enhanced catalytic activity to the surface oxygen vacancies. Based on this, we speculate that the oxygen vacancies on surface of the ceria help ameliorate the catalytic activity for CO oxidation by providing adsorption sites for the reactant O2 molecule: dissociating O2 to form atomic O, and subsequently being diffused to the Pt and ceria interface by the oxygen vacancy channels. The catalytic activities of five Pt/CeO2 catalysts are probable to be resulted from the convolution of both effects of Pt size and oxygen vacancy at CeO2 surface.
 | ||
| Fig. 7 Light-off curves of CO oxidation over pure and reduced CeO2, Pt/CeO2, R2-, R3-, R5-, and O5-R5-Pt/CeO2. | ||
To distinguish the effects of Pt size and surface oxygen vacancy, CO adsorption/desorption properties on Pt with respect to Pt size were investigated using diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy in two different conditions: (i) CO adsorption followed by He flushing and heating, (ii) CO adsorption followed by O2 flushing and heating. DRIFT was performed for Pt/CeO2 and the variant which showed highest catalytic activity, i.e. R5-Pt/CeO2. For the former condition (He flushing), the CO desorption characteristic with respect to Pt size can be evaluated regardless of oxygen vacancy concentration, because there is no oxygen source. For the latter condition (O2 flushing), based on the result of He flushing, it is possible to observe how oxygen vacancy accelerates CO oxidation. The IR spectra of adsorbed CO on Pt/CeO2 and R5-Pt/CeO2 is shown in Fig. 8. The DRIFT desorption spectra for the Pt/CeO2 (He flushing) reveals two sets of CO adsorption bands centered at 2176 cm−1 and a broad peak with a shoulder at 2097 cm−1 (Fig. 8a). The peak at 2176 cm−1 is because of CO gas in the atmosphere of the DRIFT chamber and the broad peak with shoulder at 2097 cm−1 is associated with the CO adsorbed on Pt single atoms, which can be confirmed from the XPS and STEM data. As the flow of CO gas is stopped and He gas is flown, no red shift was observed in the peaks, thus proving that all of Pt species are in the form of single atoms. The desorption spectra of the R5-Pt/CeO2 also contains two peaks at 2176 cm−1 and 2076 cm−1, which are characteristics of CO in the atmosphere of DRIFT chamber and CO on Pt nanoparticles, respectively (Fig. 8b). In contrast, red shift during desorption process was observed. It means that dipole–dipole coupling between CO molecules took place, and thus Pt nanoparticles were present as the adsorption sites in the system.56,57 The DRIFT oxidation spectra for the Pt/CeO2 (O2 flushing) shows that the peak associated with the CO adsorbed on Pt single atoms (Pt2+) still existed even at 100 °C (Fig. 8c), whereas the peak associated with CO adsorbed on Pt nanoparticles (Pt0) in R5-Pt/CeO2 disappeared (Fig. 8d), indicating that all of CO was readily converted to CO2. This result suggests that the stronger binding between Pt single atoms and CO molecules results in ∼100 °C higher temperature for 100% CO conversion in Pt/CeO2 compared to R3-, R5-, O5-R5-Pt/CeO2 which consist of Pt nanoparticles. Moreover, from the result that the peak corresponding to the adsorbed CO in R5-Pt/CeO2 significantly decreased at 50 °C (Fig. 8d) whereas such decrease was not observed in He flushing (Fig. 8b), it can be concluded that high surface oxygen vacancy concentration of R5-Pt/CeO2 is also responsible for CO conversion. The higher concentration of surface oxygen vacancies on the R5-Pt/CeO2 catalyst activate the reactant O2 molecules and result in the formation of the superoxide O2− and peroxide O22− species. Li et al. reported that these active O2 species can easily be identified over reduced pure ceria (without Pt), but for the Pt/CeO2 system these active O2 species being mobile and labile migrate onto Pt where they are converted to atomic oxygen species (O, O−) in a process called reverse spillover of oxygen, thus making their detection really difficult.58 Our results comply with their findings; we analyzed the DRIFT oxidation spectra for Pt/CeO2 and R5-Pt/CeO2 in the range of 800–1200 cm−1, and any peaks associated with the active O2 species at 883 cm−1 and 1126 cm−1 were not detected (Fig. S6†), implying that the active atomic oxygen species, not the diatomic oxygen species, are involved in R5-Pt/CeO2 catalyst for the CO oxidation reaction.
 | ||
| Fig. 8 CO desorption DRIFTS for (a) Pt/CeO2; (b) R5-Pt/CeO2. CO oxidation DRIFTS for (c) Pt/CeO2; (d) R5-Pt/CeO2. | ||
If it is assumed that the reduction characteristic of O atoms is not dependent on Pt size but only correlated with the surface oxygen vacancy, the effect of Pt size can be evaluated from the comparison between Pt/CeO2 (single atom Pt; low  ) and O5-R5-Pt/CeO2 (∼1.6 nm sized nanoparticle Pt; low
) and O5-R5-Pt/CeO2 (∼1.6 nm sized nanoparticle Pt; low 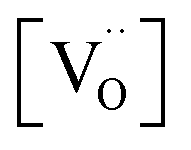 ) catalysts. Even though the XPS results for Ce 3d showed that O5-R5-Pt/CeO2 had less Ce3+ (i.e., less oxygen vacancies) than Pt/CeO2 (Table 1), O5-R5-Pt/CeO2 exhibited 100% CO conversion at 200 °C, whereas Pt/CeO2 was activated above 250 °C and 100% conversion was achieved at 300 °C (Fig. 7 or S7†). It implies that the reduction of O atom at the interface is rather fast enough at temperatures above 200 °C even in catalysts with not-sufficient surface oxygen vacancies, and the dominant rate determining is the hardly desorbed CO on Pt single atoms. On the other hand, CO oxidation reaction below 200 °C seems to be correlated with oxygen vacancies. In comparison between R5-Pt/CeO2 (1.7 ± 0.2 nm sized nanoparticle Pt; high
) catalysts. Even though the XPS results for Ce 3d showed that O5-R5-Pt/CeO2 had less Ce3+ (i.e., less oxygen vacancies) than Pt/CeO2 (Table 1), O5-R5-Pt/CeO2 exhibited 100% CO conversion at 200 °C, whereas Pt/CeO2 was activated above 250 °C and 100% conversion was achieved at 300 °C (Fig. 7 or S7†). It implies that the reduction of O atom at the interface is rather fast enough at temperatures above 200 °C even in catalysts with not-sufficient surface oxygen vacancies, and the dominant rate determining is the hardly desorbed CO on Pt single atoms. On the other hand, CO oxidation reaction below 200 °C seems to be correlated with oxygen vacancies. In comparison between R5-Pt/CeO2 (1.7 ± 0.2 nm sized nanoparticle Pt; high  ) and O5-R5-Pt/CeO2 (1.6 ± 0.3 nm sized nanoparticle Pt; low
) and O5-R5-Pt/CeO2 (1.6 ± 0.3 nm sized nanoparticle Pt; low  ), R5-Pt/CeO2 was active showing 16% CO conversion at 150 °C whereas O5-R5-Pt/CeO2 was catalytically inactive at that temperature (Fig. 7 or S7†). To sum, the best catalyst for CO oxidation is the one which has uniformly dispersed fine Pt nanoparticles over CeO2, and these nanoparticles must be accompanied with the oxygen vacancies in vicinity of them to be active below 200 °C.
), R5-Pt/CeO2 was active showing 16% CO conversion at 150 °C whereas O5-R5-Pt/CeO2 was catalytically inactive at that temperature (Fig. 7 or S7†). To sum, the best catalyst for CO oxidation is the one which has uniformly dispersed fine Pt nanoparticles over CeO2, and these nanoparticles must be accompanied with the oxygen vacancies in vicinity of them to be active below 200 °C.
It is further noteworthy that Pt/CeO2 catalysts in this study were synthesized via simplistic co-precipitation method and contained only 0.5 atomic% Pt. Besides, it was shown that the simple H2 reduction treatment at 550 °C of Pt/CeO2 catalyst can significantly enhance the activity for CO oxidation reaction which is attributed to the facilitated CO desorption on Pt nanoparticles and the higher oxygen vacancy concentration at CeO2 surface. Although further study will be required to elucidate the underlying mechanism on how oxygen vacancies at surface of CeO2 enhances the reduction characteristic of oxygen species, it is expected that the findings in this study possess significant implications for the design and preparation of highly active catalysts for low temperature CO oxidation.
3. Conclusion
It has been known that Pt/CeO2 catalysts obey Mars–Van Krevelen mechanism and the rate determining step is the reaction of adsorbed CO with oxygen species at the interface between Pt and CeO2. Since the corresponding step can be affected by key parameters which are the desorption characteristic of CO on Pt and the reduction characteristic of O atom at the interface, this work investigates their effects on CO oxidation reaction. Pt/CeO2 catalysts with the minimum amount of Pt (0.5 atomic%) is prepared using a simple co-precipitation method, and the size of Pt and the oxygen vacancy concentration at CeO2 surface are systematically controlled by heat-treatment at various temperatures in H2 or air environments. It is found that the bonding strength between the Pt single atoms and CO is very strong, thus making CO oxidation difficult, whereas Pt nanoparticles of 1–1.7 nm uniformly dispersed over CeO2 are more effective owing to the weaker binding strength between them and CO. At temperatures above 200 °C, the reduction of O atom at the interface is rather fast enough even in catalysts with not-sufficient surface oxygen vacancies, and the dominant rate determining is the hardly desorbed CO on Pt single atoms. On the other hand, at temperatures below 200 °C, higher oxygen vacancies at CeO2 surface effectively enhance the activity of catalysts. Our findings prove that the best catalyst for CO oxidation is the one which has uniformly dispersed fine Pt nanoparticles over CeO2, accompanying with the oxygen vacancies in vicinity of them, and it is demonstrated that a simple hydrogen reduction at 550 °C for the single atom Pt supported on CeO2 catalyst induces the formation of highly dispersed Pt nanoparticles with size of ∼1.7 nm and the higher concentration of surface oxygen vacancies simultaneously, enabling 100% conversion from CO to CO2 at 200 °C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Korea Institute of Energy Technology Evaluation and Planning (KETEP), a granted financial resource from the Ministry of Trade, Industry & Energy, Republic of Korea (No. 20173010032140), and the institutional research program of the Korea Institute of Science and Technology.References
- E. W. McFarland and H. Metiu, Catalysis by Doped Oxides, Chem. Rev., 2013, 113(6), 4391–4427 CrossRef CAS PubMed.
- R. V. Gulyaev, E. M. Slavinskaya, S. A. Novopashin, D. V. Smovzh, A. V. Zaikovskii, D. Y. Osadchii, O. A. Bulavchenko, S. V. Korenev and A. I. Boronin, Highly active PdCeOx composite catalysts for low-temperature CO oxidation, prepared by plasma-arc synthesis, Appl. Catal., B, 2014, 147, 132–143 CrossRef CAS.
- A. K. Santra and D. W. Goodman, Catalytic oxidation of CO by platinum group metals: from ultrahigh vacuum to elevated pressures, Electrochim. Acta, 2002, 47(22–23), 3595–3609 CrossRef CAS.
- M. A. Newton, D. Ferri, G. Smolentsev, V. Marchionni and M. Nachtegaal, Room-temperature carbon monoxide oxidation by oxygen over Pt/Al2O3 mediated by reactive platinum carbonates, Nat. Commun., 2015, 6, 8675 CrossRef CAS PubMed.
- L. Q. Liu, F. Zhou, L. G. Wang, X. J. Qi, F. Shi and Y. Q. Deng, Low-temperature CO oxidation over supported Pt, Pd catalysts: Particular role of FeOx support for oxygen supply during reactions, J. Catal., 2010, 274(1), 1–10 CrossRef CAS.
- X. Cheng, Z. Shi, N. Glass, L. Zhang, J. J. Zhang, D. T. Song, Z. S. Liu, H. J. Wang and J. Shen, A review of PEM hydrogen fuel cell contamination: Impacts, mechanisms, and mitigation, J. Power Sources, 2007, 165(2), 739–756 CrossRef CAS.
- H. S. Gandhi, G. W. Graham and R. W. McCabe, Automotive exhaust catalysis, J. Catal., 2003, 216(1–2), 433–442 CrossRef CAS.
- Y. Zhou, Z. Y. Wang and C. J. Liu, Perspective on CO oxidation over Pd-based catalysts, Catal. Sci. Technol., 2015, 5(1), 69–81 RSC.
- J. Jones, H. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga and X. I. P. Hernández, Thermally stable single-atom platinum-on-ceria catalysts via atom trapping, Science, 2016, 353(6295), 150–154 CrossRef CAS PubMed.
- S. Alayoglu, K. An, G. Melaet, S. Chen, F. Bernardi, L. W. Wang, A. E. Lindeman, N. Musselwhite, J. Guo, Z. Liu, M. A. Marcus and G. A. Somorjai, Pt-Mediated Reversible Reduction and Expansion of CeO2 in Pt Nanoparticle/Mesoporous CeO2 Catalyst: In Situ X-ray Spectroscopy and Diffraction Studies under Redox (H2 and O2) Atmospheres, J. Phys. Chem. C, 2013, 117(50), 26608–26616 CrossRef CAS.
- K. Reed, A. Cormack, A. Kulkarni, M. Mayton, D. Sayle, F. Klaessig and B. Stadler, Exploring the properties and applications of nanoceria: is there still plenty of room at the bottom?, Environ. Sci.: Nano, 2014, 1(5), 390–405 RSC.
- L. Liu and A. Corma, Metal Catalysts for Heterogeneous Catalysis: From Single Atoms to Nanoclusters and Nanoparticles, Chem. Rev., 2018, 118(10), 4981–5079 CrossRef CAS PubMed.
- J. Ke, W. Zhu, Y. Jiang, R. Si, Y.-J. Wang, S.-C. Li, C. Jin, H. Liu, W.-G. Song, C.-H. Yan and Y.-W. Zhang, Strong Local Coordination Structure Effects on Subnanometer PtOx Clusters over CeO2 Nanowires Probed by Low-Temperature CO Oxidation, ACS Catal., 2015, 5(9), 5164–5173 CrossRef CAS.
- F. Lin, D. T. Hoang, C.-K. Tsung, W. Huang, S. H.-Y. Lo, J. B. Wood, H. Wang, J. Tang and P. Yang, Catalytic properties of Pt cluster-decorated CeO2 nanostructures, Nano Res., 2011, 4(1), 61–71 CrossRef CAS.
- Y. Gao, W. Wang, S. Chang and W. Huang, Morphology Effect of CeO2 Support in the Preparation, Metal–Support Interaction, and Catalytic Performance of Pt/CeO2 Catalysts, ChemCatChem, 2013, 5(12), 3610–3620 CrossRef CAS.
- H.-H. Liu, Y. Wang, A.-P. Jia, S.-Y. Wang, M.-F. Luo and J.-Q. Lu, Oxygen vacancy promoted CO oxidation over Pt/CeO2 catalysts: A reaction at Pt–CeO2 interface, Appl. Surf. Sci., 2014, 314, 725–734 CrossRef CAS.
- A. B. Kehoe, D. O. Scanlon and G. W. Watson, Role of Lattice Distortions in the Oxygen Storage Capacity of Divalently Doped CeO2, Chem. Mater., 2011, 23(20), 4464–4468 CrossRef CAS.
- M. Cargnello, V. V. Doan-Nguyen, T. R. Gordon, R. E. Diaz, E. A. Stach, R. J. Gorte, P. Fornasiero and C. B. Murray, Control of metal nanocrystal size reveals metal-support interface role for ceria catalysts, Science, 2013, 341(6147), 771–773 CrossRef CAS PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis, Acc. Chem. Res., 2013, 46(8), 1740–1748 CrossRef CAS PubMed.
- B. C. Gates, M. Flytzani-Stephanopoulos, D. A. Dixon and A. Katz, Atomically dispersed supported metal catalysts: perspectives and suggestions for future research, Catal. Sci. Technol., 2017, 7(19), 4259–4275 RSC.
- F. Dvořák, M. Farnesi Camellone, A. Tovt, N.-D. Tran, F. R. Negreiros, M. Vorokhta, T. Skála, I. Matolínová, J. Mysliveček, V. Matolín and S. Fabris, Creating single-atom Pt-ceria catalysts by surface step decoration, Nat. Commun., 2016, 7, 10801 CrossRef PubMed.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Identification of active sites in CO oxidation and water-gas shift over supported Pt catalysts, Science, 2015, 350(6257), 189 CrossRef CAS PubMed.
- M. Yang, S. Li, Y. Wang, J. A. Herron, Y. Xu, L. F. Allard, S. Lee, J. Huang, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2014, 346(6216), 1498–1501 CrossRef CAS PubMed.
- Y. Zhai, D. Pierre, R. Si, W. Deng, P. Ferrin, A. U. Nilekar, G. Peng, J. A. Herron, D. C. Bell, H. Saltsburg, M. Mavrikakis and M. Flytzani-Stephanopoulos, Science, 2010, 329(5999), 1633 CrossRef CAS PubMed.
- M. Flytzani-Stephanopoulos and B. C. Gates, Annu. Rev. Chem. Biomol. Eng., 2012, 3(1), 545–574 CrossRef CAS PubMed.
- M. Yang, J. Liu, S. Lee, B. Zugic, J. Huang, L. F. Allard and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2015, 137(10), 3470–3473 CrossRef CAS PubMed.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321(5894), 1331 CrossRef CAS PubMed.
- S. Liu, J. M. Tan, A. Gulec, L. A. Crosby, T. L. Drake, N. M. Schweitzer, M. Delferro, L. D. Marks, T. J. Marks and P. C. Stair, Organometallics, 2017, 36(4), 818–828 CrossRef CAS.
- A. Ruiz Puigdollers, P. Schlexer, S. Tosoni and G. Pacchioni, Increasing oxide reducibility: the role of metal/oxide interfaces in the formation of oxygen vacancies, ACS Catal., 2017, 7(10), 6493–6513 CrossRef CAS.
- H. C. Yao and Y. F. Y. Yao, Ceria in automotive exhaust catalysts: I. Oxygen storage, J. Catal., 1984, 86(2), 254–265 CrossRef CAS.
- C. Sun, H. Li and L. Chen, Nanostructured ceria-based materials: synthesis, properties, and applications, Energy Environ. Sci., 2012, 5(9), 8475–8505 RSC.
- T. Bunluesin, R. Gorte and G. Graham, Studies of the water-gas-shift reaction on ceria-supported Pt, Pd, and Rh: implications for oxygen-storage properties, Appl. Catal., B, 1998, 15(1–2), 107–114 CrossRef CAS.
- M. Kurnatowska, L. Kepinski and W. Mista, Appl. Catal., B, 2012, 117–118, 135–147 CrossRef CAS.
- X. I. Pereira-Hernández, A. DeLaRiva, V. Muravev, D. Kunwar, H. Xiong, B. Sudduth, M. Engelhard, L. Kovarik, E. J. M. Hensen, Y. Wang and A. K. Datye, Nat. Commun., 2019, 10(1), 1358 CrossRef PubMed.
- G. Gao, J.-W. Shi, C. Liu, G. Chen, Z. Fan and C. Niu, Mn/CeO2 catalysts for SCR of NOx with NH3: comparative study on the effect of supports on low-temperature catalytic activity, Appl. Surf. Sci., 2017, 411 Search PubMed.
- J. Lee, Y. Ryou, X. Chan, T. J. Kim and D. H. Kim, How Pt Interacts with CeO2 under the Reducing and Oxidizing Environments at Elevated Temperature: The Origin of Improved Thermal Stability of Pt/CeO2 Compared to CeO2, J. Phys. Chem. C, 2016, 120(45), 25870–25879 CrossRef CAS.
- N. Acerbi, S. E. Tsang, G. Jones, S. Golunski and P. Collier, Rationalization of Interactions in Precious Metal/Ceria Catalysts Using the d-Band Center Model, Angew. Chem., 2013, 125(30), 7891–7895 CrossRef.
- T. Tanabe, Y. Nagai, T. Hirabayashi, N. Takagi, K. Dohmae, N. Takahashi, S. Matsumoto, H. Shinjoh, J. N. Kondo, J. C. Schouten and H. H. Brongersma, Low temperature CO pulse adsorption for the determination of Pt particle size in a Pt/cerium-based oxide catalyst, Appl. Catal., A, 2009, 370(1–2), 108–113 CrossRef CAS.
- J. F. Moulder and J. Chastain, Handbook of X-ray photoelectron spectroscopy: a reference book of standard spectra for identification and interpretation of XPS data, Physical Electronics, Eden Prairie Minnesota, 1995 Search PubMed.
- S. Gatla, D. Aubert, G. Agostini, O. Mathon, S. Pascarelli, T. Lunkenbein, M. G. Willinger and H. Kaper, Room-Temperature CO Oxidation Catalyst: Low-Temperature Metal–Support Interaction between Platinum Nanoparticles and Nanosized Ceria, ACS Catal., 2016, 6(9), 6151–6155 CrossRef CAS.
- M. Kurnatowska, L. Kepinski and W. Miśta, Structure evolution of nanocrystalline Ce1−xPdxO2−y mixed oxide in oxidizing and reducing atmosphere: Reduction-induced activity in low-temperature CO oxidation, Appl. Catal., B, 2012, 117–118, 135–147 CrossRef CAS.
- L. Wu, H. Wiesmann, A. Moodenbaugh, R. Klie, Y. Zhu, D. Welch and M. Suenaga, Oxidation state and lattice expansion of CeO2−x nanoparticles as a function of particle size, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69(12), 125415 CrossRef.
- S. J. Hong and A. V. Virkar, Lattice Parameters and Densities of Rare-Earth Oxide Doped Ceria Electrolytes, J. Am. Ceram. Soc., 1995, 78(2), 433–439 CrossRef CAS.
- J. Lee, Y. Ryou, J. Kim, X. Chan, T. J. Kim and D. H. Kim, Influence of the Defect Concentration of Ceria on the Pt Dispersion and the CO Oxidation Activity of Pt/CeO2, J. Phys. Chem. C, 2018, 122(9), 4972–4983 CrossRef CAS.
- N. Zanfoni, L. Avril, L. Imhoff, B. Domenichini and S. Bourgeois, Direct liquid injection chemical vapor deposition of platinum doped cerium oxide thin films, Thin Solid Films, 2015, 589, 246–251 CrossRef CAS.
- A. N. Mansour, J. W. Cook and D. E. Sayers, Quantitative technique for the determination of the number of unoccupied d-electron states in a platinum catalyst using the L2,3 X-ray absorption edge spectra, J. Phys. Chem., 1984, 88(11), 2330–2334 CrossRef CAS.
- S. Gatla, D. Aubert, V. Flaud, R. Grosjean, T. Lunkenbein, O. Mathon, S. Pascarelli and H. Kaper, Facile synthesis of high-surface area platinum-doped ceria for low temperature CO oxidation, Catal. Today, 2018, 105–112 Search PubMed.
- L. Liu, D. N. Zakharov, R. Arenal, P. Concepcion, E. A. Stach and A. Corma, Evolution and stabilization of subnanometric metal species in confined space by in situ TEM, Nat. Commun., 2018, 9(1), 574 CrossRef PubMed.
- R. Kopelent, J. A. van Bokhoven, J. Szlachetko, J. Edebeli, C. Paun, M. Nachtegaal and O. V. Safonova, Catalytically Active and Spectator Ce3+ in Ceria-Supported Metal Catalysts, Angew. Chem., Int. Ed., 2015, 54(30), 8728–8731 CrossRef CAS PubMed.
- X. Liu, K. Zhou, L. Wang, B. Wang and Y. Li, Oxygen Vacancy Clusters Promoting Reducibility and Activity of Ceria Nanorods, J. Am. Chem. Soc., 2009, 131(9), 3140–3141 CrossRef CAS PubMed.
- L. Nie, D. Mei, H. Xiong, B. Peng, Z. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Activation of surface lattice oxygen in single-atom Pt/CeO2 for low-temperature CO oxidation, Science, 2017, 358(6369), 1419 CrossRef CAS PubMed.
- F. Meng and L. Wang, Hydrothermal synthesis of monocrystalline CeO2 nanopoles and their room temperature ferromagnetism, Mater. Lett., 2013, 100, 86–88 CrossRef CAS.
- F. Meng, Z. Fan, C. Zhang, Y. Hu, T. Guan and A. Li, Morphology-controlled synthesis of CeO2 microstructures and their room temperature ferromagnetism, J. Mater. Sci. Technol., 2017, 33(5), 444–451 CrossRef.
- A. Younis, D. Chu and S. Li, Oxygen level: the dominant of resistive switching characteristics in cerium oxide thin films, J. Phys. D: Appl. Phys., 2012, 45(35), 355101 CrossRef.
- S. Liu, X. Wu, J. Tang, P. Cui, X. Jiang, C. Chang, W. Liu, Y. Gao, M. Li and D. Weng, An exploration of soot oxidation over CeO2-ZrO2 nanocubes: do more surface oxygen vacancies benefit the reaction?, Catal. Today, 2017, 281, 454–459 CrossRef CAS.
- A. D. Allian, K. Takanabe, K. L. Fujdala, X. Hao, T. J. Truex, J. Cai, C. Buda, M. Neurock and E. Iglesia, Chemisorption of CO and Mechanism of CO Oxidation on Supported Platinum Nanoclusters, J. Am. Chem. Soc., 2011, 133(12), 4498–4517 CrossRef CAS PubMed.
- A. Crossley and D. A. King, Infrared spectra for co isotopes chemisorbed on Pt “111”: evidence for strong absorbate coupling interactions, Surf. Sci., 1977, 68, 528–538 CrossRef CAS.
- C. Li, Y. Chen, W. Li and Q. Xin in Studies in Surface Science and Catalysis, ed. T. lnui, K. Fujimoto, T. Uchijima, M. Masai, Elsevier, 1993, vol. 77, pp. 217–222 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra05965b |
| This journal is © The Royal Society of Chemistry 2019 |