
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe antitumor activity of 4,4′-bipyridinium amphiphiles†
Senlin Wang,
Hongshuai Wu,
Fanghui Chen,
Yu Zhang,
Yuchen Zhang and
Baiwang Sun *
*
School of Chemistry and Chemical Engineering, Southeast University, Nanjing 211189, PR China. E-mail: chmsunbw@seu.edu.cn; Fax: +86 25 52090614; Tel: +86 25 52090614
First published on 16th October 2019
Abstract
A series of 4,4′-bipyridinium amphiphiles were synthesized and their anticancer activities were further evaluated. MTT assay showed that the cytotoxicity first increased and then decreased with the growth of carbon chains (8–16 C) at both ends of bipyridyl. Specifically, compounds with saturated carbon chains consisting of 13 carbons at both ends of bipyridyl displayed the best cell inhibitory activity with IC50 values in the low-micromolar range, which were even superior to that of cisplatin, against all the tested human cancer cells and cisplatin-resistant A549 cancer cells in vitro. In addition, compound 6 could evidently arrest the G2/M phase of the cell cycle in a dose-dependent manner. Moreover, this study demonstrates the potent performance of compound 6 in cell growth inhibition and apoptosis induction via a conceivable approach of membrane damage.
1. Introduction
Cancer, as a serious disease, urgently needs to be eliminated due to its high mortality despite the considerable efforts that have been devoted to understanding cancer biology and developing available therapeutic options over the past decades.1–4 Among the current strategies for the treatment of cancer, chemotherapy still remains a significant approach for overcoming cancer. Although some impressive advances have been achieved to improve the current status of the poor control of cancer metastasis, some drawbacks, such as severe drug resistance, and extremely strong drug-induced toxic side effects5–7 exist. Also, the efficacy of existing drugs is rather limited. Obviously, the need for discovering and developing novel and effective therapeutic agents with high targeting, low toxicity and no drug resistance is imminent.Amphiphiles, from simple soaps and benzalkonium chloride to more complex antimicrobial peptides and dendrimers, have a privileged platform for antimicrobials.8 Additionally, a new class of cationic bioactive peptide-based therapies have become a research hotspot in the exploitation of antitumor drugs.9 Particularly, many natural cationic antimicrobial peptides (CAPs) can kill tumor cells and damage tumor cell membranes, like bacterial cell membranes, which have a negative charge.10,11 Therefore, the cell cytotoxicity of such polypeptides can be attributed to the electrostatic and hydrophobic interactions of cationic amphiphilic polypeptides and cell membranes.12–14 It has been widely reported that a cationic peptide mainly destroys the cell membrane and avoids the drug resistance mechanism of cancer cells.15 However, a variety of CAPs having cancer-selective toxicity are peptide-based drugs that are difficult to be developed because of poor pharmacokinetics, potential systemic toxicity and high cost of manufacturing.16–18 Nevertheless, a small molecular cationic antibacterial peptide possessing a simple structure and synthesis approach has been demonstrated to produce a strong antibacterial activity and a remarkable membrane cleavage effect.19
Based on this idea, it was expected to develop a simple and an effective method to exploit cell membrane rupture cationic amphiphilic compounds and lay a foundation for the development of high-efficiency and low-toxic anti-cancer drugs. In spite of their prevalence and amphiphilic nature, the anticancer activity of these compounds remains largely unreported. Here, we prepared a series of 4,4′-bipyridinium amphiphiles to investigate the structure–activity relationships in the inhibition performance of cancer.
2. Experimental section
2.1. Materials
A549 (alveolar basal carcinoma), MCF-7 and MDA-MB-231 (breast adenocarcinoma), BEL-7402 (hepatoma), L929 (murine fibroblasts) and A549/CDDP (cisplatin-resistant lung cancer) human cell lines were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). 3-(4,5-Dimethyl-thinazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and 4,4′-bipyridine were obtained from Sigma-Aldrich. Annexin V-FITC apoptosis detection kit was purchased from Beyotime Biotechnology (Haimen, Jiangsu, China). Fetal bovine serum (FBS) was supplied by Shanghai Ponsure Biotechnology Co., Ltd. (Shanghai, China). RPMI-1640 medium was purchased from Gibco Laboratories (NY, USA). All other chemicals and solvents were commercially available and used without further purification.2.2. General procedure for the synthesis of 4,4′-bipyridinium amphiphiles
As shown in Fig. 1, 4,4′-bipyridinium amphiphiles were prepared according to a previously published paper8 with slight modifications. Briefly, the corresponding alkyl bromide was added to a solution of 4,4′-dipyridyl (0.39 g, 2.5 mmol) in acetonitrile (40 mL). The mixture was heated to reflux for 24 h, then cooled and filtered. The filtered solid was further washed with acetonitrile to obtain the pure product as a yellow powder.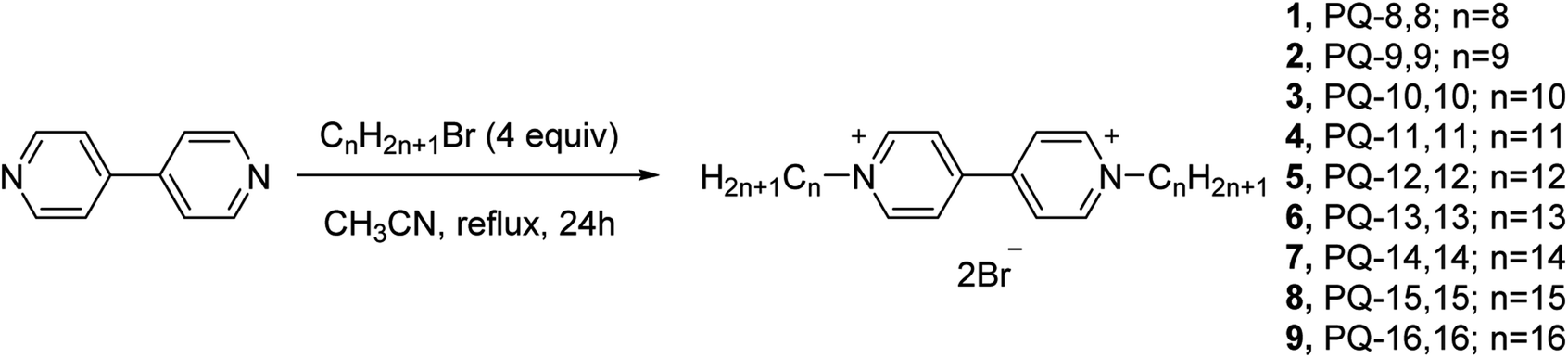 | ||
| Fig. 1 Synthesis route of 4,4′-bipyridinium amphiphiles. | ||
2.3. In vitro cytotoxicity
In this study, A549, MCF-7, BEL-7402, MDA-MB-231 cancer cells and mouse fibroblasts (L929) were cultured in the RPMI 1640 medium supplemented with 10% FBS and a 1% penicillin–streptomycin solution at 37 °C under 5% CO2. In brief, the cells were seeded in 96-well plates at a density of 7 × 103 per well. After 24 h of incubation, the cells were exposed to various compounds 1–10 for 24 h at 37 °C. 150 μL of the MTT solution (5.0 mg mL−1) was added to each well and incubated for 4 h at 37 °C. Subsequently, the medium was replaced with 150 μL DMSO to dissolve the resultant purple crystals. The plates were then shaken for 15 min. Finally, the absorption at 490 nm was measured by a microplate reader and the prism software was used for data analysis.2.4. Cell apoptosis
For the cell apoptosis, A549 cells were seeded in 6-well plates at a density of 1.0 × 105 cells per well and cultured for 24 h. The cells were treated with compound 6 (5 μM) for 24 h. The cells were harvested, and the Annexin V-FITC apoptosis detection kit was then used to detect and quantify the apoptosis by a flow cytometer.2.5. Cell cycle arrest
A549 cells were seeded in 6-well plates at a density of 1 × 105 cells per well and cultured for 24 h. The cells were treated with compound 6 (5, 10, 20 and 40 μM) for 24 h. Then, the cells were collected and washed with cold PBS. After centrifugation, the cells were fixed by the dropwise addition of 70% ethanol overnight at 4 °C. For staining, cells were centrifuged and resuspended in cold PBS and treated with propidium iodide (PI) for 30 min in the dark. The samples were recorded by a flow cytometer and the MFLT32 software was used for data analysis.2.6. Confocal cell imaging
Confocal laser scanning microscopy (CLSM) was used to qualitatively detect the cell interactive behaviors with compound 6. A549 cells were seeded in the Φ 20 mm confocal laser dish at a density of 1 × 105 cells per dish and cultured for 24 h. The cells were subsequently incubated with compound 6 for 1 h. The cells cultured with the medium alone were used as a blank control. The medium was removed, and the cells were washed twice with cold PBS. Subsequently, a final concentration of 50 μg mL−1 PI and a final concentration of 5 μg mL−1 DAPI were added and then incubated for 20 min at 37 °C. The cells were washed twice with cold PBS and the fluorescence images were immediately obtained via CLSM.3. Results and discussion
3.1. The synthesis and characterization of 4,4′-bipyridinium amphiphiles
As shown in Fig. 1, 4,4′-bipyridinium amphiphiles were prepared according to a previously published paper.8 The obtained compounds were fully characterized by 1H and 13C NMR spectroscopy. Mass spectrometry data of the corresponding compounds are also presented in Fig. S1–S9.† Chemical purity was analyzed by HPLC (Fig. S10–S12 in ESI†). All the samples were determined to possess >95% of purity. HPLC also provided information about the increase of hydrophobicity from compounds 1 to 9. The compounds showed a systematically increasing retention on a C18 reverse-phase stationary phase due to the increasing length of the chain. The lipophilicity of the compounds were evaluated by the calculatation of Clog![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values (Table S1†).20 As expected, the ClogP values increased from 1 (−1.25) to 9 (7.22).
P values (Table S1†).20 As expected, the ClogP values increased from 1 (−1.25) to 9 (7.22).
3.2. In vitro cytotoxicity
To determine the effects of synthesized 4,4′-bipyridinium amphiphiles on cell proliferation and to gain the primary structure–activity relationship, the cytotoxic activity of compounds 1–9 were evaluated by standard MTT assays on four human cancer cell lines (A549, MCF-7, BEL-7402 and MDA-MB-231) and non-malignant cell line L929. Cisplatin was used as the positive control. The corresponding IC50 values (concentration required to reduce the viability to 50%) obtained after the treatment with different agents are listed in Table 1. The increase in the activity and then the decrease in the homo-sequence is a well-known phenomenon in medicinal chemistry.20 The lipophilicity directly contributed to the cytotoxicity of these complexes. In vitro evaluation results revealed that the IC50 values of compounds 1–9 decreases first and then increases gradually as the calculated ClogP values increase from compound 1 (−1.25) to compound 9 (7.22). It was much significant to observe that compounds 4–7, the 4,4′-bipyridinium amphiphiles with saturated carbon chains consisting of 11–14 carbons at both ends of bipyridyl, possessed a higher cytotoxicity against all test cancer cell lines than cisplatin as the calculated ClogP values increased from 1.93 of compound 1 to 5.10 of compound 7. In particular, compound 6 showed the best growth inhibitory activity against A549 (IC50: 8.26 μM), MCF-7 (IC50: 5.96 μM), BEL-7402 (IC50: 5.56 μM) and MDA-MB-231 (IC50: 7.06 μM) cells as the calculated ClogP value was 4.04, which is very suitable for the lipophilicity of the drug. The cytotoxicity of compounds against normal L929 cells was evaluated to investigate the selectivity of compounds for cancer cells and normal cells. As displayed in Table 1, the IC50 values of compounds 1–9 against L929 cells showed a certain cell killing activity, but lower IC50 values against all test cancer cell lines indicated that the compounds had a certain selectivity for cancer cells. This may be because the surface of cancer cells has a greater electronegativity than the normal cells.10,11
| Compounds | Cytotoxicity in different cell lines (IC50, μM) | ||||||
|---|---|---|---|---|---|---|---|
| A549 | MCF-7 | BEL-7402 | MDA-MB-231 | L929 | A549/CDDP | RFa | |
| a RF (resistant factor) is defined as IC50 in A549/CDDP/IC50 in A549. Mean values based on three independent experiments, and the results of the representative experiments are shown. | |||||||
| 4,4′-dipyridyl | >1000 | >1000 | >1000 | >1000 | >1000 | >1000 | — |
| 1 | 141.4 ± 5.66 | 161 ± 5.98 | 86.1 ± 3.42 | 163.15 ± 7.68 | 240.2 ± 3.68 | 158.8 ± 6.98 | 1.13 |
| 2 | 27.96 ± 1.23 | 31.34 ± 1.38 | 18.98 ± 0.86 | 30.32 ± 1.12 | 54.52 ± 1.21 | 34.21 ± 1.45 | 1.22 |
| 3 | 16.17 ± 0.8 | 14.87 ± 0.68 | 18.2 ± 0.76 | 16.72 ± 0.76 | 39.72 ± 0.63 | 22.58 ± 1.1 | 1.4 |
| 4 | 13.13 ± 0.5 | 10.76 ± 0.46 | 12.25 ± 0.6 | 11.53 ± 0.51 | 25.63 ± 0.48 | 15.74 ± 0.72 | 1.2 |
| 5 | 9.5 ± 0.42 | 7.31 ± 0.33 | 9.51 ± 0.43 | 9.36 ± 0.45 | 17.95 ± 0.78 | 13.88 ± 0.69 | 1.46 |
| 6 | 8.26 ± 0.38 | 5.96 ± 0.3 | 5.56 ± 0.2 | 7.06 ± 0.35 | 13.24 ± 0.53 | 8.44 ± 0.42 | 1.02 |
| 7 | 10.38 ± 0.5 | 14.1 ± 0.7 | 6.536 ± 0.31 | 7.09 ± 0.33 | 19.65 ± 0.41 | 11.34 ± 0.54 | 1.09 |
| 8 | 13.88 ± 0.67 | 19.64 ± 0.97 | 12.22 ± 0.54 | 9.61 ± 0.42 | 30.59 ± 0.36 | 16.07 ± 0.75 | 1.16 |
| 9 | 20.45 ± 0.96 | 21.82 ± 1.08 | 27.54 ± 1.11 | 32.32 ± 1.45 | 59.97 ± 1.25 | 28.58 ± 1.32 | 1.4 |
| Cisplatin | 15.35 ± 0.68 | 20.16 ± 0.89 | 15.69 ± 0.73 | 22.16 ± 1.08 | 18.96 ± 1.16 | 141.83 ± 6.9 | 9.24 |
Drug resistance is an important therapeutic obstacle, which can confine the treatment efficacy of cisplatin for some human cancer cells. Thus, we further evaluated the cytotoxicity of compound 6 against A549/CDDP (cisplatin resistant cancer cells). As shown in Table 1, the IC50 values of compounds 1–9 against A549/CDDP cells showed the same pattern as other cells, first decreasing and then increasing gradually. Interestingly, compound 6 was also found to exhibit obvious anticancer activities against A549/CDDP cells. The IC50 values of compound 6 against A549/CDDP cells did not obviously change (IC50: 8.44 μM). On the contrary, the corresponding IC50 values of cisplatin against A549/CDDP increased to 141.83 μM. It was gratifying to observe that compounds 1–9 had much lower resistance factors (1.02–1.46) compared to cisplatin (9.24).
The results indicated that the hydrophobic chains at both ends of bipyridyl had a great effect on the cytotoxicity of 4,4′-bipyridinium amphiphiles. As the carbon chains (8–16 C) at both ends of bipyridyl grew, the cytotoxicity increased first and then decreased, which also demonstrated that the compounds with good cytotoxicity had calculated ClogP values of 1.93 to 5.10. The compound with saturated carbon chains consisting of 13 carbons at both ends of bipyridyl displayed the best antitumor activity against all four of the tested human cancer cells and cisplatin resistant A549 cancer cells. Therefore, compound 6 with a superior cytotoxicity was chosen for further study.
3.3. Cell apoptosis
Compound 6 was found to exhibit a broad spectrum and the best antitumor activity against all the tested five human cancer cells in vitro. In view of this, compound 6 was chosen to further evaluate the mechanism of action. The induced apoptosis performance of compound 6 on A549 cells was performed with FITC and PI dual-staining method (Q1, Q2, Q3 and Q4 represent four different cell states: necrotic cells, late apoptotic or necrotic cells, apoptotic cells and living cells, respectively). The percentages of apoptotic cells were determined by flow cytometry. A549 cells were treated with 5 μM of compound 6 for 24 h. As shown in Fig. 2, compared to the 0.62% apoptosis induced by the medium alone (control group), the cells incubated with compound 6 showed a remarkable apoptosis ratio (21.59%). This result clearly informs that compound 6 could effectively induce apoptosis in A549 cells at the indicated concentration, which is consistent with the results of MTT.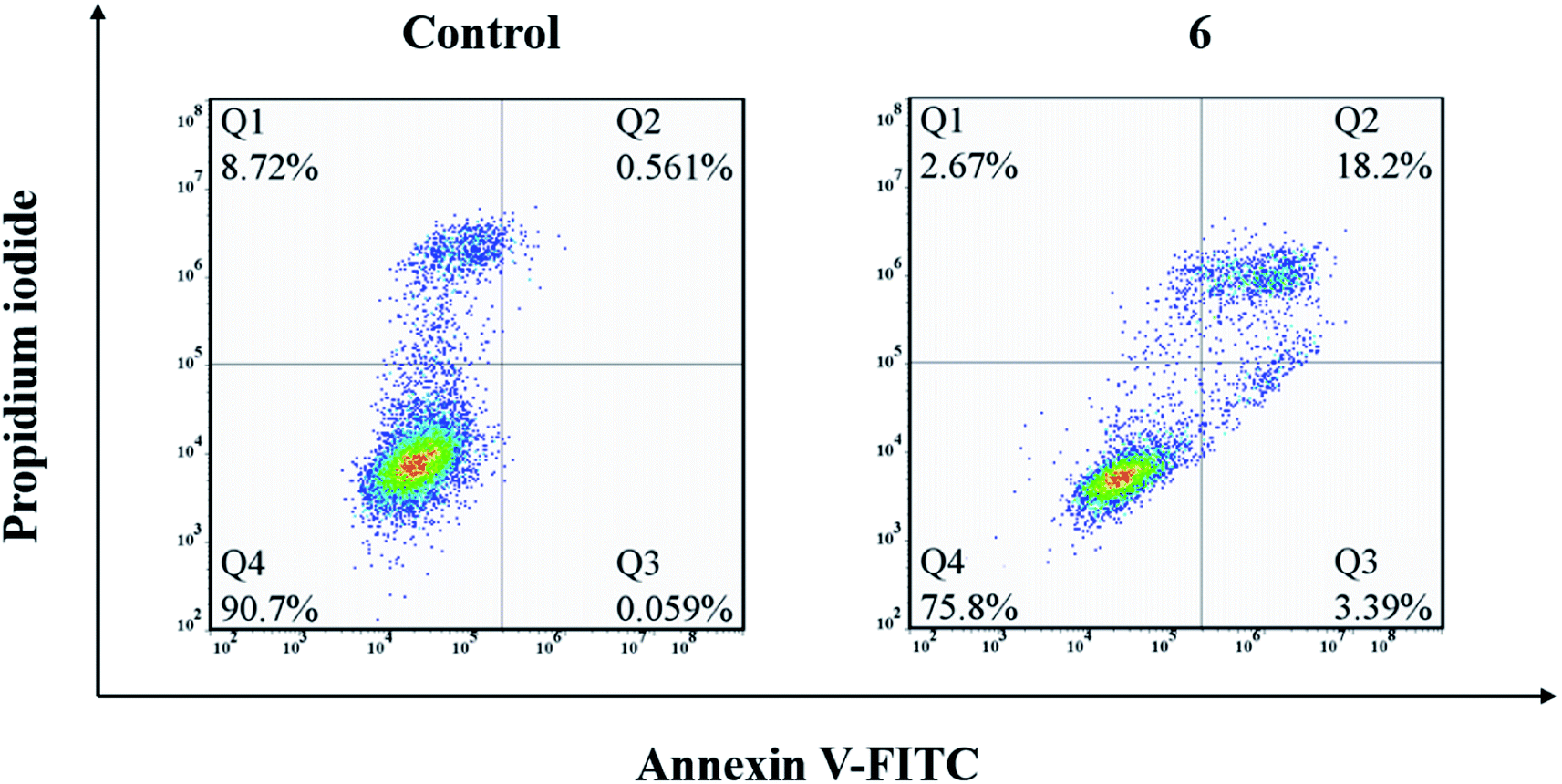 | ||
| Fig. 2 Apoptosis of A549 cells induced by compound 6 (5 μM). | ||
3.4. Cell cycle arrest
To further investigate the cell cycle arrest induced by compound 6, the cell cycle distribution of A549 cells treated with compound 6 was detected by flow cytometry under the PI staining. As shown in Fig. 3a and b, compound 6 could effectively arrest the cell cycle at the G2/M phase in a concentration-dependent manner compared to the control. Meanwhile, the percentage of untreated A549 cells in the G0/G1 phase was at 56.42% with only 15.33% in the G2/M phase. After the treatment with compound 6 at 10 μM, the percentage of the G2/M phase only increased to 17.76%, while in the cells treated with compound 6 at 20 and 40 μM, the percentages of the G2/M phase increased to 30.70% and 33.25%, respectively. These results show that compound 6 evidently arrested the G2/M phase of the cell cycle in a dose-dependent manner.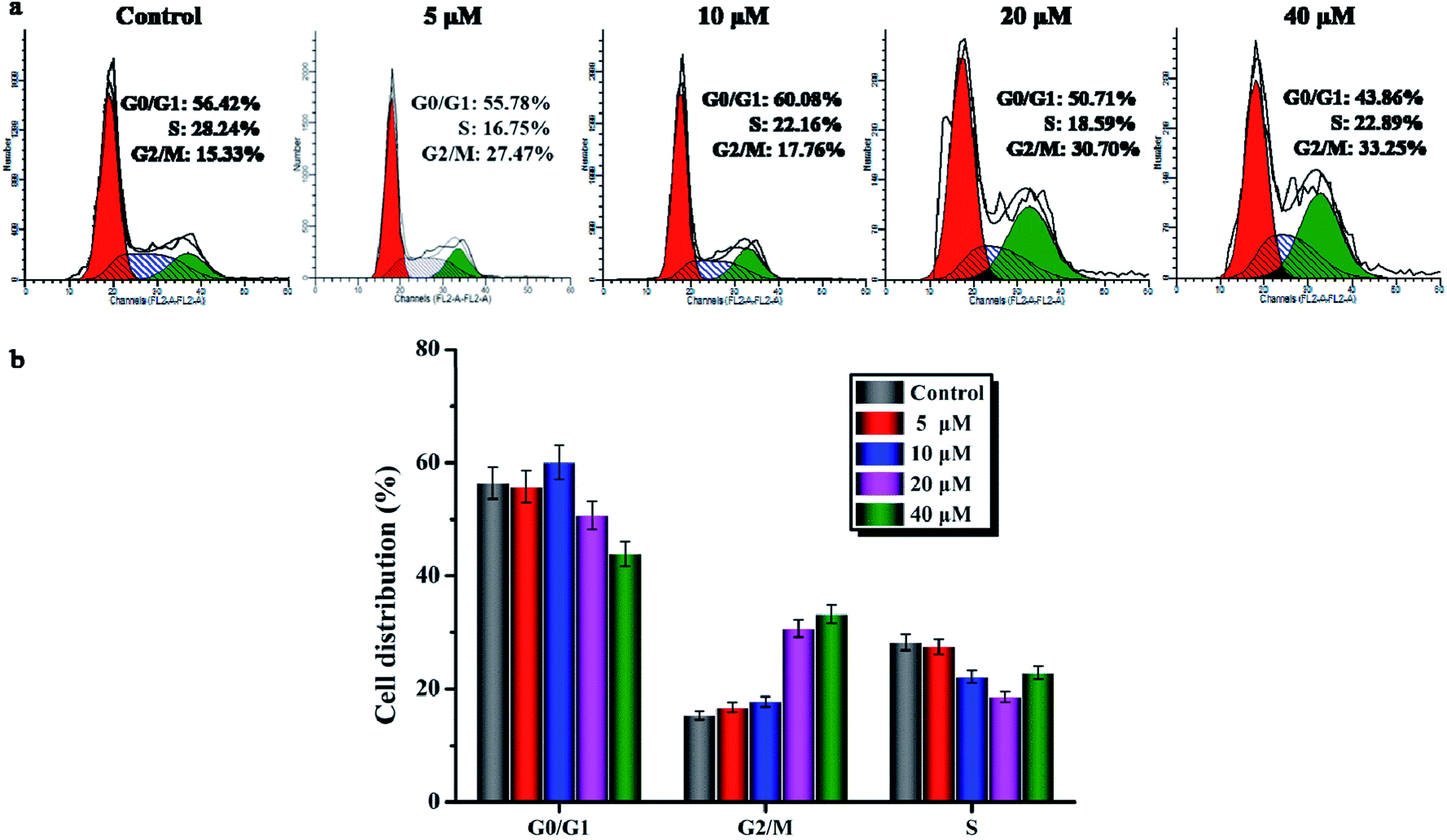 | ||
| Fig. 3 A549 cell cycle arrest after the treatment with different concentrations of 6 (5, 10, 20, 40 μM) for 24 h. (a) Cell cycle analysis examined by flow cytometry. (b) Bar chart showing cell distributions in various cell cycles. | ||
3.5. Confocal cell imaging
After incubation with compound 6 for 1 h, A549 cells exhibited very different morphological characteristics. As shown in Fig. 2, in the control group, their morphology stayed intact. On the contrary, the cells incubated with compound 6 exhibited some membrane leakage and dispersing of many membrane fragments in the culture medium could be observed. The difference in the cell membrane integrity after drug treatments could be clearly demonstrated by confocal imaging using nuclear dyes. DAPI can stain all living and dead cell nuclei, while PI can only stain dead cells or cells with large changes in membrane permeability. As depicted in Fig. 4, compound 6 treated cells showed a co-staining feature of PI (red) and DAPI (blue) after 1 h. However, the cell nuclei of the control group were stained only by DAPI and the membranes were stained by PI. The results indicate that the treatment of cells with compound 6 could result in large changes in membrane permeability.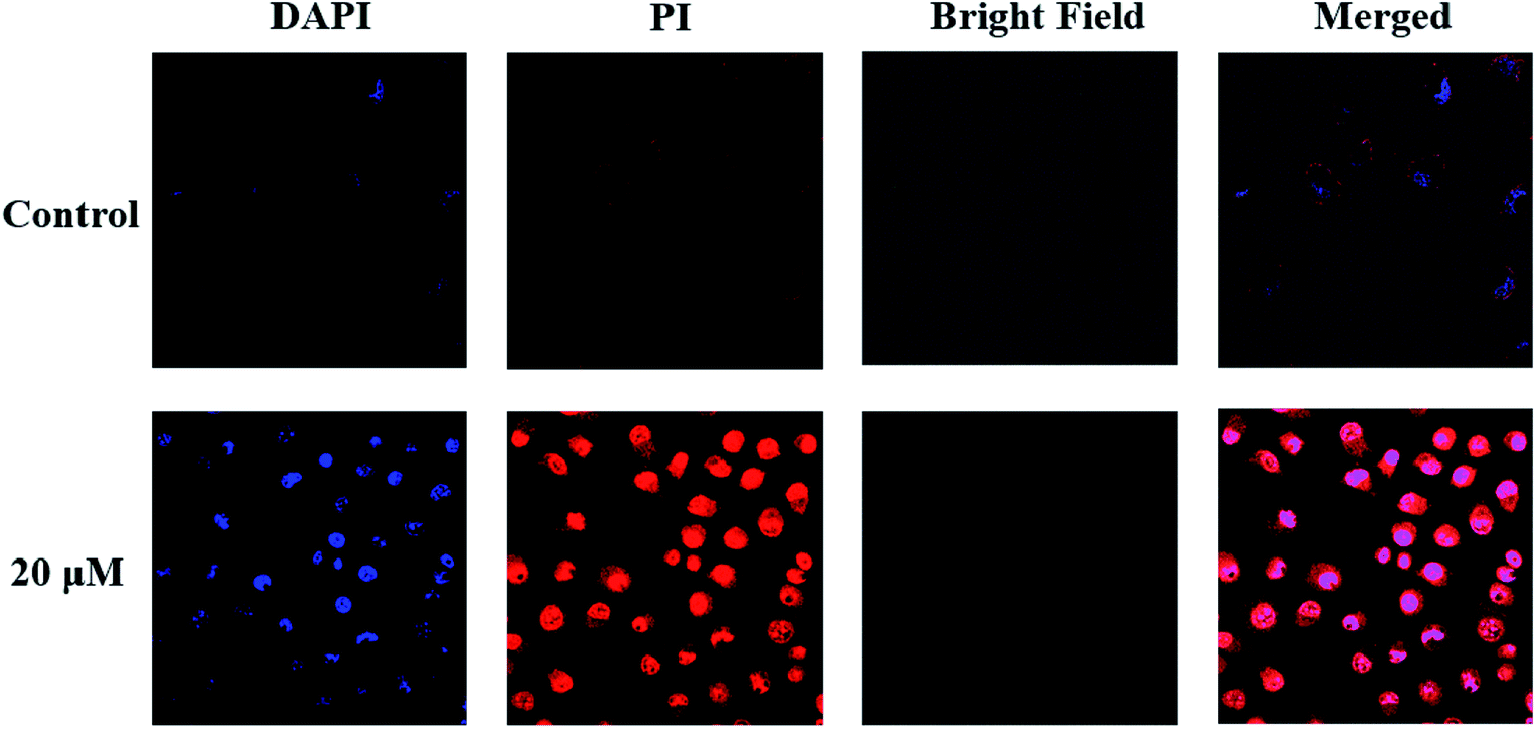 | ||
| Fig. 4 The interactive behaviors of A549 cells after incubation with compound 6 (20 μM) for 1 h. | ||
4. Conclusions
In summary, a series of 4,4′-bipyridinium amphiphiles were synthesized and their antitumor effects in vitro were further investigated. The results indicated that the hydrophobic chains at both ends of bipyridyl had a great effect on the cytotoxicity of 4,4′-bipyridinium amphiphiles. As the carbon chains (8–16 C) at both ends of bipyridyl grow, the cytotoxicity first increases and then decreases. Compounds with saturated carbon chains consisting of 13 carbons at both ends of bipyridyl displayed the best cell growth inhibitory activity with IC50 values in the low-micromolar range against all four of the tested human cancer cells and the cisplatin resistant A549 cancer cells in vitro. In addition, compound 6 evidently arrested the G2/M phase of the cell cycle in a dose-dependent manner. Finally, our study demonstrated that the potent activity in the cell growth inhibition and apoptosis induction of compound 6 may be related to membrane damage.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (Grant No. 21628101 and 21371031), the International S&T Cooperation Program of China (No. 2015DFG42240) and the Priority Academic Program Development (PAPD) of Jiangsu Higher Education Institutions.References
- Y. Huang, Q. Feng, Q. Yan, X. Hao and Y. Chen, Mini-Rev. Med. Chem., 2015, 15, 73–81 CrossRef CAS PubMed.
- E. Teerasak, P. Thongararm, S. Roytrakul, L. Meesuk and P. Chumnanpuen, Comput. Struct. Biotechnol. J., 2016, 14, 49–57 CrossRef.
- H. P. Varbanov, F. Kuttler, D. Banfi, G. Turcatti and P. J. Dyson, PLoS One, 2017, 12, e0171052 CrossRef.
- H. Wu, C. You, J. Jiao, F. Chen, B. Sun and X. Zhu, Nanotechnology, 2019, 30, 035601 CrossRef CAS.
- S. Elhady, A. Al-Abd, A. El-Halawany, A. Alahdal, H. Hassanean and S. Ahmed, Mar. Drugs, 2016, 14, 130 CrossRef.
- N. L.-X. Syn, W.-P. Yong, B.-C. Goh and S.-C. Lee, Expert Opin. Drug Metab. Toxicol., 2016, 12, 911–922 CrossRef CAS.
- H. Wu, C. You, F. Chen, J. Jiao, Z. Gao, P. An, B. Sun and R. Chen, Mater. Sci. Eng. C, 2019, 103, 109738 CrossRef CAS.
- M. C. Grenier, R. W. Davis, K. L. Wilson-Henjum, J. E. LaDow, J. W. Black, K. L. Caran, K. Seifert and K. P. Minbiole, Bioorg. Med. Chem. Lett., 2012, 22, 4055–4058 CrossRef CAS.
- M. L. Leite, N. B. da Cunha and F. F. Costa, Pharmacol. Ther., 2018, 183, 160–176 CrossRef CAS.
- D. Gaspar, A. S. Veiga and M. A. Castanho, Front. Microbiol., 2013, 4, 294 Search PubMed.
- J. S. Mader and D. W. Hoskin, Expert Opin. Invest. Drugs, 2006, 15, 933–946 CrossRef CAS.
- H.-M. Zhou, D.-C. Li, Y.-Y. Wang, H. Zhu, Y.-Q. Su and Y. Mao, Fish Shellfish Immunol., 2018, 81, 368–373 CrossRef CAS.
- Y. Wan, C. Ma, M. Zhou, X. Xi, L. Li, D. Wu, L. Wang, C. Lin, J. Lopez and T. Chen, Toxins, 2015, 7, 5182–5193 CrossRef CAS.
- R. Roudi, N. L. Syn and M. Roudbary, Front. Immunol., 2017, 8, 1320 CrossRef.
- Q. Wu, Z. Yang, Y. Nie, Y. Shi and D. Fan, Cancer Lett., 2014, 347, 159–166 CrossRef CAS PubMed.
- C. Dos Santos, S. Hamadat, K. Le Saux, C. Newton, M. Mazouni, L. Zargarian, M. Miro-Padovani, P. Zadigue, J. Delbé and Y. Hamma-Kourbali, PLoS One, 2017, 12, e0182926 CrossRef PubMed.
- S. Maijaroen, N. Jangpromma, J. Daduang and S. Klaynongsruang, Environ. Toxicol. Pharmacol., 2018, 62, 164–176 CrossRef CAS.
- X. Liu, R. Cao, S. Wang, J. Jia and H. Fei, J. Med. Chem., 2016, 59, 5238–5247 CrossRef CAS PubMed.
- A. A. Baxter, F. T. Lay, I. K. Poon, M. Kvansakul and M. D. Hulett, Cell. Mol. Life Sci., 2017, 74, 3809–3825 CrossRef CAS.
- H. Wang, B. Huwaimel, K. Verma, J. Miller, T. M. Germain, N. Kinarivala, D. Pappas, P. S. Brookes and P. C. Trippier, ChemMedChem, 2017, 12, 1033–1044 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06172j |
| This journal is © The Royal Society of Chemistry 2019 |