
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAsymmetric amination of α,α-dialkyl substituted aldehydes catalyzed by a simple chiral primary amino acid and its application to the preparation of a S1P1 agonist†
Qiong Xiao a,
Yifan Tangb,
Ping Xie*a and
Dali Yin*a
a,
Yifan Tangb,
Ping Xie*a and
Dali Yin*a
aState Key Laboratory of Bioactive Substance and Function of Natural Medicines, Beijing Key Laboratory of Active Substance Discovery and Druggability Evaluation, Department of Medicinal Chemistry, Institute of Materia Medica, Peking Union Medical College, Chinese Academy of Medical Sciences, Beijing 100050, PR China. E-mail: yindali@imm.ac.cn
bBeijing Union Pharmaceutical Factory, PR China
First published on 18th October 2019
Abstract
The chiral catalytic amination of an α,α-dialkyl substituted aldehyde usually proceeds with low enantioselectivity. We selected naphthyl-L-alanine as the catalyst and observed improved enantioselectivity for the amination. Using this method, racemic α-methyl-α-benzyloxypropanal was aminated to give chiral serine derivatives in 74% ee, which was further increased to >99% ee after recrystallization. Moreover, we also successfully synthesized a chiral phosphonium salt 9 for the preparation of one α-substituted alaninol compound 14 as an S1P1 agonist in high overall yield.
Introduction
α,α-Disubstituted amino alcohols, aldehydes and acids are important chiral building blocks in organic synthesis. They are routinely found in a number of peptides,1–5 natural products6,7 and pharmaceuticals.8,9 Due to this importance, their synthesis has attracted sustained interest from the synthetic community. Existing methods for the asymmetric approach to scaffolds include classical Seebach's method,10,11 auxiliary Strecker synthesis,12 and a variety of asymmetric phase transfer catalysis reactions.13Recently, several methods have been reported describing the asymmetric Michael α-amination of achiral aldehydes via proline catalysis, resulting in the products being obtained in good yields and excellent enantioselectivities.14–17 However, these proline catalysts do not imbue high enantioselectivities in the amination of branched aldehydes. Wang et al. reported that 3-(1-naphthyl)-L-alanine (1d) successfully promoted the enantioselective α-amination of branched aldehydes with azadicarboxylates to give α-alkyl-α-aryl disubstituted aldehydes in up to 99% ee.18 However, low enantioselectivities only 4–28% ee were obtained with α-alkyl-α-alkyl disubstituted, potentially owing to poor stereo-differentiation between the two α-substituents.14 To some extent, the application of this kind of reaction is limited. In 2005, Barbas et al. reported higher stereoselectivities were possible utilizing proline derived tetrazole catalyst (1b) for the amination of α-alkyl-α-benzyl disubstituted aldehydes.19 In addition, no further progress about the asymmetric amination of α-alkyl-α-alkyl disubstituted aldehydes had been reported.
Results and discussion
Herein, we report the asymmetric Machel α-amination of α-methyl-α-protected hydroxymethyl aldehydes and their subsequent reduction and cyclisation to afford oxazolidinones in good ee. We initially chose 3-(benzyloxy)-2-methylpropanal and dibenzyl azodicarboxylate (DBAD) as a model substrate to determine to optimal reaction conditions. When L-proline (1a) (30 mol%) was used,14 the reaction was complete in 48 hours at room temperature and provided the amino aldehyde in 56% yield, however we obtained poor enantioselectivities (32% ee). To improve the enantioselectivity, we screened a number of catalysts (Fig. 1). For example, tetrazole catalyst (1b) (15 mol%) in CH3CN provided 42% ee with 68% yield (Table 1, entry 2).17 3-(1-Naphthyl)-L-alanine catalyst (1d) (15 mol%) in CH3CN gave the amino aldehyde in 70% yield with 46% ee (Table 1, entry 4).18 | ||
| Fig. 1 Chiral catalysts. | ||
|
||||
|---|---|---|---|---|
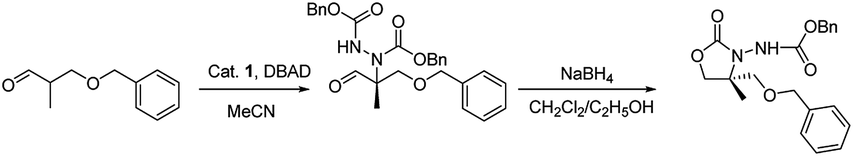
| Entry | Catalyst | Time (h) | Yieldb (%) | eec (%) |
| a All reactions were carried out with aldehyde (0.75 mmol), DBAD (0.5 mmol), catalyst (15 mol%) in THF solvent (4 mL) at rt under argon, subsequent reduction and cyclisation to the oxazolidinone.b Isolated yield.c Determined by HPLC with a Chiralpak-OD column.d With the opposite enantiomer. | ||||
| 1 | 1a | 48 | 56 | 32 |
| 2 | 1b | 12 | 68 | 42 |
| 3 | 1c | 24 | 45 | 34d |
| 4 | 1d | 24 | 70 | 46 |
| 5 | 1e | 24 | 53 | 44 |
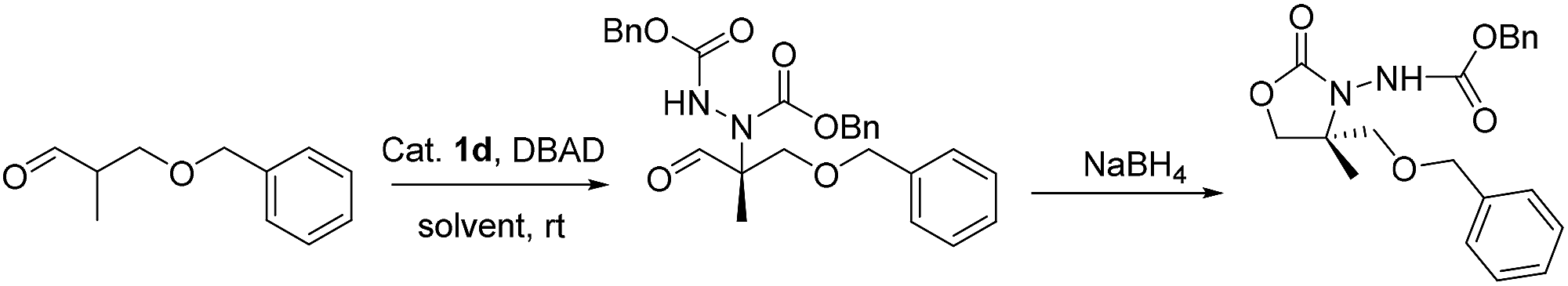
We then turned our attention to the effects of solvents on both yield and enantioselectivities (Table 2). Among them, dioxane, MeOH, MTBE and THF (entries 9, 10, 7 and 8) were all tolerated and produce the desired oxazolidinones in moderate to good enantioselectivities. Of particular note, THF delivered the highest enantioselectivity (69% ee) in synthetically useful yields.
|
||||
|---|---|---|---|---|
| Entry | Solvent | Time | Yieldb (%) | eec (%) |
| a Reaction conditions: the azodicarboxylate (1 equiv.) was added to the aldehyde (1.5 equiv.), with catalyst (15 mol%) in THF at rt for the stated period of time under argon. Reaction without isolation of intermediate.b Isolated yield.c Determined by chiral HPLC. | ||||
| 1 | n-Hexane | 24 | 52 | 48 |
| 2 | Toluene | 72 | 49 | 45 |
| 3 | CH2Cl2 | 72 | 41 | 30 |
| 4 | EtOAc | 24 | 67 | 54 |
| 5 | CH3OCH2CH2OCH3 | 36 | 47 | 40 |
| 7 | MTBE | 36 | 84 | 49 |
| 8 | THF | 36 | 81 | 69 |
| 9 | Dioxane | 36 | 76 | 57 |
| 10 | MeOH | 24 | 69 | 57 |
| 11 | Ethylene glycol | 24 | 42 | 57 |
Furthermore, when lowering temperature to 0 °C, we observed no improvement in enantioselectivity, however the reaction became notably more sluggish. Increasing catalyst loading up to 30 mol% did not improve either enantioselectivity or reaction time.
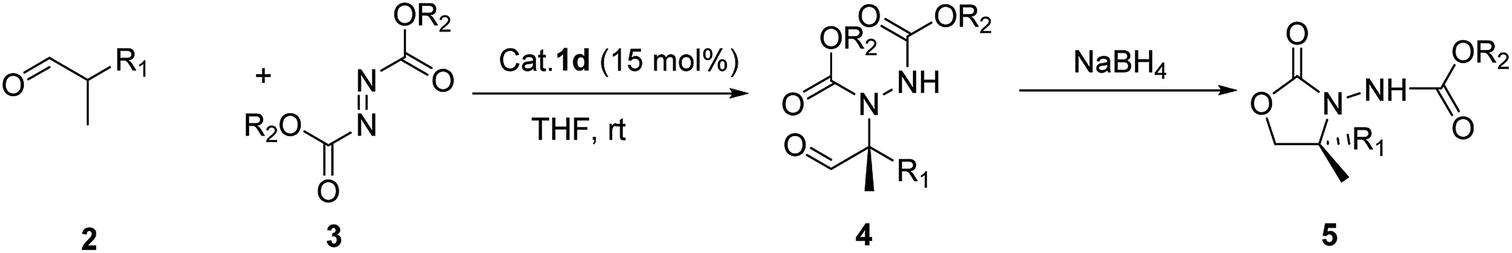
With these optimized conditions in hand, we probed the substrate scope of the reaction (Table 3). In general, various oxazolidinones 5 were obtained in moderate to good yields (54–89%) and enantioselectivities (24–73% ee). The reactions showed poor enantioselectivities for α-methyl-α-ethyl and α-methyl-α-carbethoxy disubstituted aldehydes, but not for α-methyl-α-protected hydroxymethyl substituted aldehydes with aromatic ring. The results also showed that electron-withdrawing groups were more successful than electron-donating groups. Moreover, p-F and p-CF3 substituents both showed similar enantioselectivities. We then investigated differing azodicarboxylates and observed that di-p-chlorobenzyl azodicarboxylate (DCAD) provided the desired products in excellent yields (90%) and good enantioselectivities (up to ee 74%) while lower enantioselectivities were obtained with diethyl azadicarboxylate (DEAD) or diisopropyl azadicarboxylate (DIAD). We also observed good enantioselective control with catalysts bearing naphthalene rings. This may be due to the π–π interaction between the aromatic ring of substrate and naphthalene ring limiting the conformation of the intermediate, thus improving the level of stereo-differentiation between the two α-substituents. Additionally, when the azo reagent contained an aromatic ring this π–π interaction may be further enhanced, resulting in the observed improvement of stereoselectivity.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Product | Time (h) | Yieldb (%) | eec (%) | eee (%) |
| a Reaction conditions: the azodicarboxylate (1 equiv.) was added to the aldehyde (1.5 equiv.), with catalyst (15 mol%) in THF at rt for the stated period of time under argon. Reaction performed without isolating the intermediate.b Isolated yield.c Isolated by silica gel column chromatography.d Determined by chiral HPLC.e ee determined by chiral HPLC after recrystallization. Absolute configuration of 5-R to determined be (R) on CD spectrum. | |||||||
| 1 | BnOCH2 | Et | 5a-Et | 38 | 79 | 57 | — |
| 2 | BnOCH2 | Bn | 5a-Bn | 36 | 81 | 69 | — |
| 3 | BnOCH2 | p-ClBn | 4a-p-ClBn | 48 | 80 | 71d | 97% |
| 4 | BnOCH2 | p-ClBn | 5a-p-ClBn | 1 | 94 | 72 | >99% |
| 5 | p-CH3BnOCH2 | Bn | 5b-Bn | 38 | 54 | 48 | — |
| 6 | p-CH3BnOCH2 | p-ClBn | 5b-p-ClBn | 36 | 56 | 54 | — |
| 7 | 3,4-DiMeOBnOCH2 | Et | 5c-Et | 38 | 78 | 38 | — |
| 8 | 3,4-DiMeOBnOCH2 | Bn | 5c-Bn | 36 | 67 | 45 | — |
| 9 | p-FBnOCH2 | Et | 5d-Et | 37 | 75 | 56 | — |
| 10 | p-FBnOCH2 | Bn | 5d-Bn | 36 | 80 | 68 | — |
| 11 | p-FBnOCH2 | p-ClBn | 5d-p-ClBn | 48 | 89 | 70 | — |
| 12 | p-ClBnOCH2 | Et | 5e-Et | 39 | 70 | 57 | — |
| 13 | p-ClBnOCH2 | Bn | 5e-Bn | 36 | 63 | 59 | — |
| 14 | p-BrBnOCH2 | Bn | 5f-Bn | 38 | 79 | 59 | — |
| 15 | p-BrBnOCH2 | p-ClBn | 5f-p-ClBn | 48 | 75 | 52 | — |
| 16 | p-CNBnOCH2 | Et | 5g-Et | 28 | 81 | 57 | — |
| 17 | p-CNBnOCH2 | Bn | 5g-Bn | 24 | 71 | 65 | — |
| 18 | p-CNBnOCH2 | p-ClBn | 5g-p-ClBn | 48 | 73 | 62 | — |
| 19 | p-CF3BnOCH2 | Et | 5h-Et | 39 | 76 | 56 | — |
| 20 | p-CF3BnOCH2 | Bn | 5h-Bn | 36 | 89 | 67 | — |
| 21 | p-CF3BnOCH2 | p-ClBn | 4h-p-ClBn | 48 | 90 | 74d | 97% |
| 22 | p-CF3BnOCH2 | p-ClBn | 5h-p-ClBn | 1 | 95 | 73 | 98% |
| 23 | p-NO2BnOCH2 | Bn | 5i-Bn | 48 | 77 | 55 | |
| 24 | THPOCH2 | Bn | 5j-Bn | 48 | 70 | 57 | |
| 25 | TrtOCH2 | Bn | 5k-Bn | 48 | — | — | |
| 26 | Et | Bn | 5l-Bn | 48 | 78 | 37 | |
| 27 | CO2Et | Bn | 4m-Bn | 48 | 76 | 24 | |
Upon recrystallization from 90% ethanol, the aldehyde 4a-p-ClBn was obtained in 97% ee (60% yield) and 4h-p-ClBn was obtained in 97% ee (65% yield), which was subsequently converted to oxazolidinone 5a-p-ClBn in >99% ee and 5h-p-ClBn was obtained in 98% ee respectively. The absolute configuration of 5-R was determined to be (R) on CD spectrum. Under ambient pressure, hydrogenation using 10% Pd/C in methanol/acetic acid, the benzyloxycarbonyl group was removed. Cleavage of the hydrazine moiety, 7 was accomplished by treating with NaNO2 (ref. 14) (Scheme 1). Alcohol 7 was treated with p-TsCl in pyridine, and the resulting tosylate was successively converted to iodide 8 with NaI in acetone under a reflux condition.20 8 with triphenylphosphine in DMF provided the desired phosphonium salt 9 in moderate yield as a stable white solid.20
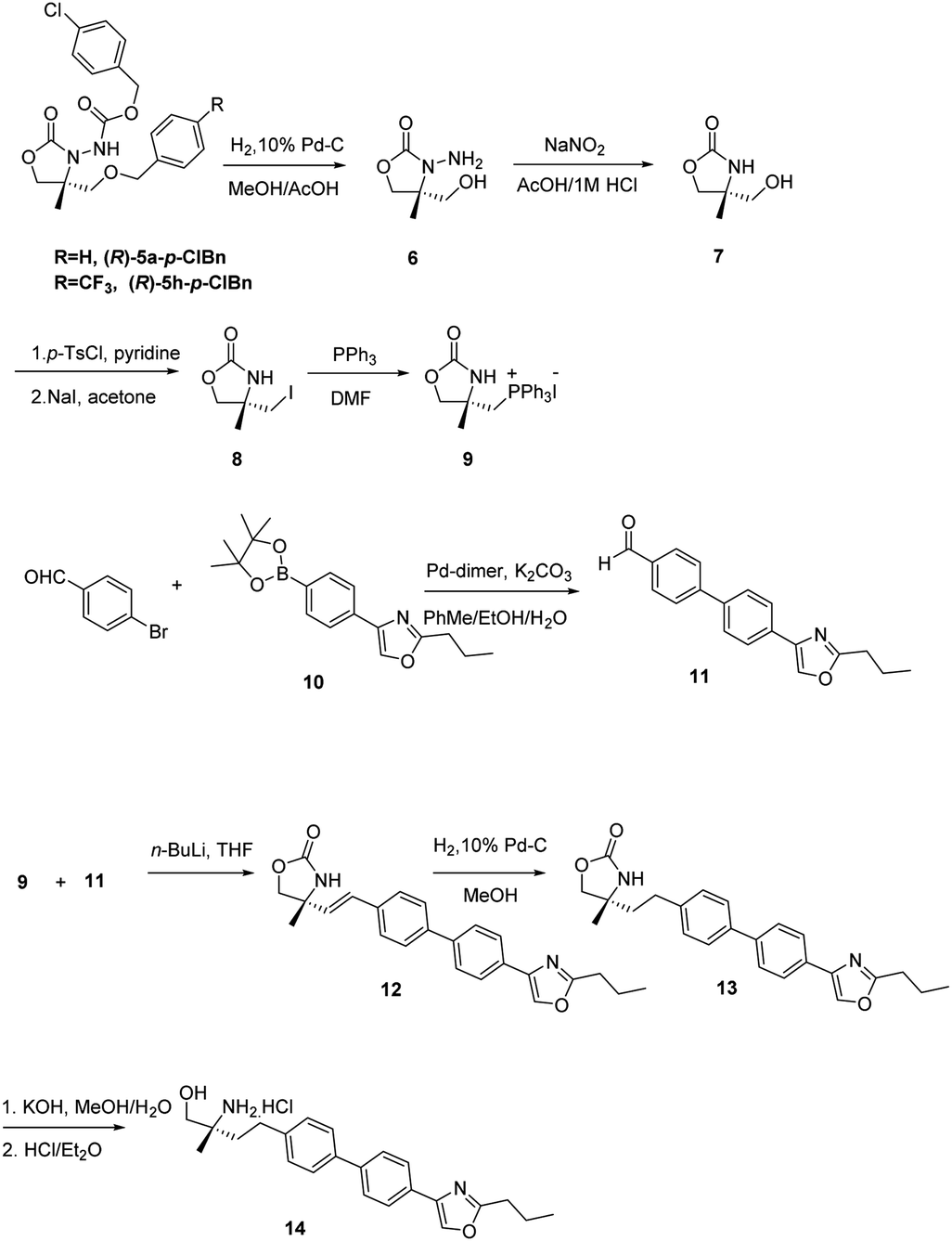 | ||
| Scheme 1 Synthesis of the α-substituted alaninol compound as S1P1 agonist. | ||
Then we applied the chiral phosphonium salt 9 to the synthesis of biological active compound as S1P1 agonist 14. These types of compounds possessing a chiral 2-methyl-2-aminoethanol have shown promise in recent years as the immunosuppressant.21,22 This compound is an analogue of SYL930, an immunosuppressant we have been reported before.23 SYL930 is currently in phase I clinical stage. The synthesis of 14 started from the aldehyde 11 in only a three step manipulation.24 Aldehyde 11 was synthesized in good yield from 4-bromobenzaldehyde and dinary pinacol borate ester 10 via Suzuki reaction with Pd-dimer (dibromobis(tri-tert-butylphosphine)dipalladium) as the catalyst.24 The Wittig reaction of 9 with 11 in dry THF at −78 °C for 3 h furnished the alkenes 12 in good yield. Subsequently reducing with 10% Pd/C in MeOH for 1 h afforded compound 13 in virtually quantitative yield after a flash-filtration. Finally, hydrolysis of the oxazolidinone part and then acidification with 1 M HCl in Et2O produced the chiral α-substituted alaninol compound 14.
Conclusions
In this study, we presented an efficient asymmetric amination of branched racemic aldehydes catalyzed by the commercially available amino acid (3-(1-naphthyl)-L-alanine). Under the optimized conditions, we obtained α-methyl-α-protected hydroxymethyl substituted aldehydes in high ee. Importantly, we developed an efficient catalytic method for synthesizing the Wittig reagent involving a chiral 2-methyl-2-aminoethanol structure that could be applied to other syntheses. Further, a new S1P1 agonist 14 has been obtained by this method in high overall yield.Experimental
General procedure for the synthesis of 4,4-disubstituted 3-alkoxycarbonylamino-oxazolidin-2-ones (5-R) by one pot method
Catalyst 1d (15 mol% in respect to the azodicarboxylate) was added to a suspension of aldehydes (2, 1.5 eq. in respect to the azodicarboxylate) and azodicarboxylate (3) in THF. The mixture stirred at rt under argon until the colour of the azodicarboxylate had disappeared. NaBH4 (3 eq. in respect to the azodicarboxylate) was added in portions at room temperature. The reaction mixture was stirred for 1 h, and then it was quenched by adding 1 M HCl aq. until the mixture reached pH 7, and it was extracted with CH2Cl2. The combined organic phases were dried over Na2SO4, and the solvent was evaporated under reduced pressure. The resulting crude was purified by flash chromatography on silica gel eluted with light petroleum ether–ethyl acetate mixture (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) to afford products 5-R as oil or solid.
:1 v/v) to afford products 5-R as oil or solid.
3-Ethyloxycarbonylamino-4-methyl-4-benzyloxy-oxazolidin-2-one (5a-Et)
White solid, yield 79%; mp 50–55 °C; 1H NMR (400 MHz, CDCl3) δ 1.24 (m, 6H, 2CH3), 3.27 (d, 1H, J = 8.0 Hz, CH), 3.47 (d, 1H, J = 12.0 Hz, CH), 4.05 (d, 1H, J = 12.0 Hz, CH), 4.14 (q, 2H, J = 4.0 Hz, CH2), 4.33 (d, 1H, J = 8.0 Hz, CH), 4.51 (s, 2H, CH2), 6.33 (bs, 1H, NH), 7.27–7.38 (m, 5H, Har); 13C NMR (100 MHz, CDCl3) δ 14.3, 19.8, 29.7, 61.2, 62.5, 71.4, 71.8, 73.2, 127.8, 128.1, 128.7, 137.4, 156.3, 156.7; HRMS calcd for C15H21N2O5 [M + H]+ 309.1445, found 309.1442; HPLC (Daicel Chiralpak OD-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 16.37 min (major), tR = 20.63 min (minor), 57% ee.
3-Benzyloxycarbonylamino-4-methyl-4-benzyloxy-oxazolidin-2-one (5a-Bn)
Oil, yield 81%; 1H NMR (400 MHz, CDCl3) δ 1.26 (s, 3H, CH3), 3.27 (d, 1H, J = 12.0 Hz, CH), 3.44 (d, 1H, J = 12.0 Hz, CH), 4.06 (d, 1H, J = 12.0 Hz, CH), 4.32 (d, 1H, J = 12.0 Hz, CH), 4.46 (s, 2H, CH2), 5.14 (s, 2H, CH2), 6.38 (bs, 1H, NH), 7.24–7.35 (m, 10H, Har); 13C NMR (100 MHz, CDCl3) δ 19.8, 61.2, 68.1, 71.4, 71.7, 73.2, 127.8, 128.2, 128.3, 128.5, 128.6, 128.7, 135.3, 127.3, 156.1, 156.7; HRMS calcd for C20H23N2O5 [M + H]+ 371.1602, found 371.1591; HPLC (Daicel Chiralpak OD, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 17.15 min (major), tR = 25.28 min (minor) 69% ee.
3-(4-Chloro)benzyloxycarbonylamino-4-methyl-4-benzyloxy-oxazolidin-2-one (5a-p-ClBn)
Catalyst 1d (64 mg, 15 mol% in respect to the azodicarboxylate) was added to a suspension of 3-(benzyloxy)-2-methylpropanal (1.15 g, 6.46 mmol) and di-p-chlorobenzyl azodicarboxylate (1.58 g, 4.31 mmol) in THF (40 mL). The mixture stirred at rt under argon until the colour of the azodicarboxylate had disappeared and quenched by the addition H2O, then extracted three times with Et2O (50 ml ×3). The combined organic layers were dried over Na2SO4, filtered, and concentrated. The resulting crude was purified by flash chromatography on silica gel, eluting with light petroleum ether–ethyl acetate mixture (4:1 v/v) to afford 4a-p-ClBn (1.87 g) as solid in 80% yield with 71% ee. Recrystallization from 90% ethanol, the aldehyde 4a-p-ClBn (930 mg) was obtained in 97% ee (50% yield). [α]20D 9.72 (c 0.29, CHCl3). 1H NMR (400 MHz, CDCl3) δ 1.34 (s, 3H, CH3), 3.60–3.77 (m, 2H, CH2), 4.42 (s, 2H, CH2), 5.01–5.15 (m, 4H, 2CH2), 6.70 (s, 1H, NH), 7.16–7.30 (m, 13H, Har), 9.55 (s, 1H, CHO); HRMS calcd for C27H27N2O6Cl2 [M + H]+ 545.12407, found 545.12390; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 85:15, flow rate 1.0 mL min−1, λ = 213 nm): tR = 21.62 min (major), tR = 23.77 min (minor).
NaBH4 (190 mg, 5.0 mmol) was added to a solution of 4a-p-ClBn (900 mg, 1.65 mmol) in CH2Cl2/C2H5OH (4 mL). The reaction mixture was stirred for 1 h, and then it was quenched by adding 1 M HCl aq. until the mixture reached pH 7, and it was extracted with CH2Cl2. The combined organic phases were dried over Na2SO4, and the solvent was evaporated under reduced pressure. The resulting crude was purified by flash chromatography on silica gel eluted with light petroleum ether–ethyl acetate mixture (4:1 v/v) to afford 5a-p-ClBn (650 mg) in 94% yield with >99% ee. As oil; [α]20D −12.3 (c 0.13, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.24 (s, 3H, CH3), 3.26 (d, 1H, J = 8.0 Hz, CH), 3.43 (d, 1H, J = 12.0 Hz, CH), 4.05 (d, 1H, J = 8.0 Hz, CH), 4.32 (d, 1H, J = 8.0 Hz, CH), 4.49 (s, 2H, CH2), 5.06 (s, 2H, CH2), 6.49 (bs, 1H, NH), 7.24–7.36 (m, 10H, Har);13C NMR (100 MHz, CDCl3) δ 19.8, 61.2, 67.2, 71.4, 71.7, 73.2, 127.8, 128.2, 128.5, 128.7, 128.8, 129.6, 134.0, 134.3, 137.3, 156.0, 156.7; HRMS calcd for C20H22N2O5Cl [M + H]+ 405.1212, found 405.1204; HPLC (Daicel Chiralpak OD-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 213 nm): tR = 27.77 min (major), tR = 35.0 min (minor).
3-(4-Chloro)benzyloxycarbonylamino-4-methyl-4-(4-methyl) benzyloxy-oxazolidin-2-one (5b-Bn)
Oil, yield 54%; 1H NMR (400 MHz, CDCl3) δ 1.24 (s, 3H, CH3), 2.29 (s, 3H, CH3), 3.24 (d, 1H, J = 12.0 Hz, CH), 3.40 (d, 1H, J = 12.0 Hz, CH), 3.99 (d, 1H, J = 8.0 Hz, CH), 4.31 (d, 1H, J = 8.0 Hz, CH), 4.40–4.48 (m, 2H, CH2), 5.12 (s, 2H, CH2), 6.39 (bs, 1H, NH), 7.13–7.18 (m, 4H, Har), 7.31–7.37 (m, 5H, Har); 13C NMR (100 MHz, CDCl3) δ 19.8, 21.2, 61.2, 68.0, 71.3, 71.4, 73.0, 73.1, 128.1, 128.2, 128.5, 128.6, 129.2, 129.4, 134.3, 135.4, 138.0, 156.2, 156.7; HRMS calcd for C21H25N2O5 [M + H]+ 385.1758, found 385.1738; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 22.42 min (major), tR = 25.10 min (minor), 48% ee.
3-(4-Chloro)benzyloxycarbonylamino-4-methyl-4-(4-methyl) benzyloxy-oxazolidin-2-one (5b-p-ClBn)
Oil, yield 56%; 1H NMR (400 MHz, CDCl3) δ 1.23 (s, 3H, CH3), 2.30 (s, 3H, CH3), 3.24 (d, 1H, J = 12.0 Hz, CH), 3.39 (d, 1H, J = 12.0 Hz, CH), 4.04 (d, 1H, J = 8.0 Hz, CH), 4.31 (d, 1H, J = 12.0 Hz, CH), 4.39–4.49 (m, 2H, CH2), 5.08 (s, 2H, CH2), 6.22 (bs, 1H, NH), 7.13–7.16 (m, 4H, Har), 7.26 (d, 2H, J = 8.0 Hz, Har), 7.32 (d, 2H, J = 8.0 Hz, Har); 13C NMR (100 MHz, CDCl3) δ 19.8, 21.2, 61.2, 67.2, 71.2, 71.4, 73.0, 128.1, 128.8, 129.4, 129.6, 133.9, 134.2, 134.4, 138.1, 156.0, 156.7; HRMS calcd for C21H24N2O5Cl [M + H]+ 419.1368, found 419.1359; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 23.38 min (major), tR = 26.35 min (minor), 54% ee.
3-Benzyloxycarbonylamino-4-methyl-4-(3,4-dimethoxy) benzyloxy-oxazolidin-2-one (5c-Et)
Oil, yield 78%; 1H NMR (400 MHz, CDCl3) δ 1.20 (m, 6H, 2 CH3), 3.21 (d, 1H, J = 8.0 Hz, CH), 3.40 (d, 1H, J = 8.0 Hz, CH), 3.82 (s, 3H, CH3), 3.84 (s, 3H, CH3), 4.01 (d, 1H, J = 8.0 Hz, CH), 4.08–4.13 (m, 2H, CH2), 4.28 (d, 1H, J = 8.0 Hz, CH), 4.37–4.44 (m, 2H, CH2), 6.45 (bs, 1H, NH), 6.76–6.80 (m, 3H, Har); 13C NMR (100 MHz, CDCl3) δ 14.3, 14.5, 19.7, 55.8, 55.9, 61.2, 62.4, 71.3, 71.4, 72.9, 110.9, 111.0, 120.3, 130.0, 148.9, 149.3, 156.3, 156.9; HRMS calcd for C17H24N2O7Na [M + Na]+ 391.1476, found 391.1471; HPLC (Daicel Chiralpak OD-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 21.88 min (major), tR = 27.05 min (minor), 38% ee.
3-Benzyloxycarbonylamino-4-methyl-4-(3,4-dimethoxy)benzyloxy-oxazolidin-2-one (5c-Bn)
Oil, yield 67%; 1H NMR (400 MHz, CDCl3) δ 1.22 (s, 3H, CH3), 3.22 (d, 1H, J = 8.0 Hz, CH2), 3.40 (d, 1H, J = 12.0 Hz, CH2), 3.80 (s, 3H, CH3), 3.84 (s, 3H, CH3), 4.03 (d, 1H, J = 8.0 Hz, CH), 4.30 (d, 1H, J = 8.0 Hz, CH), 4.41 (m, 2H, CH2), 5.11 (s, 2H, CH2), 6.347 (bs, 1H, NH), 6.79–6.82 (m, 3H, Har), 7.25–7.35 (m, 5H, Har); 13C NMR (100 MHz, CDCl3) δ 19.7, 55.9, 61.3, 68.0, 71.3, 71.4, 72.9, 111.0, 120.3, 128.2, 128.4, 128.6, 129.9, 135.4, 148.9, 149.3, 156.2, 156.9; HRMS calcd for C22H26N2O7Na [M + H]+ 453.1632, found 453.1638; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 85:15, flow rate 1.0 mL min−1, λ = 254 nm): tR = 26.31 min (major), tR = 32.42 min (minor), 45% ee.
3-Ethyloxycarbonylamino-4-methyl-4-(4-trifluoro)benzyloxy-oxazolidin-2-one (5d-Et)
White solid, yield 75%; 1H NMR (400 MHz, CDCl3) δ 1.21–1.26 (m, 6H, 2 CH3), 3.26 (d, 1H, J = 12.0 Hz, CH), 3.46 (d, 1H, J = 8.0 Hz, CH), 4.04 (d, 1H, J = 8.0 Hz, CH), 4.13 (m, 2H, CH2), 4.30 (d, 1H, J = 12.0 Hz, CH), 4.43–4.50 (m, 2H, CH2), 6.61 (bs, 1H, NH), 6.70–7.04 (m, 2H, Har), 7.22–7.26 (m, 2H, Har); 13C NMR (100 MHz, CDCl3) δ 14.3, 19.7, 61.2, 62.5, 71.4, 71.9, 72.5, 115.4, 115.6, 129.4, 129.5, 133.2, 156.4, 156.8, 161.3, 163.7; HRMS calcd for C15H20N2O5F [M + H]+ 327.1351, found 327.1341; HPLC (Daicel Chiralpak AS-H, hexane/isopropanol = 80:20, flow rate 1.0 mL min−1, λ = 254 nm): tR = 51.09 min (major), tR = 76.63 min (minor), 56% ee.
3-Benzyloxycarbonylamino-4-methyl-4-(4-fluoro)benzyloxy-oxazolidin-2-one (5d-Bn)
Oil, yield 80%; 1H NMR (400 MHz, CDCl3) δ 1.33 (s, 3H, CH3), 3.32 (d, 1H, J = 8.0 Hz, CH), 3.51 (d, 1H, J = 8.0 Hz, CH), 4.12 (d, 1H, J = 4.0 Hz, CH), 4.37 (d, 1H, J = 8.0 Hz, CH), 4.51 (s, 2H, CH2), 5.21 (s, 2H, CH2), 6.42 (bs, 1H, NH), 7.08 (t, J = 4.0 Hz, 2H, Har), 7.30 (d, 2H, J = 8.0 Hz, Har), 7.40 (bs, 5H, Har); 13C NMR (100 MHz, CDCl3) δ 19.8, 60.4, 68.1, 71.4, 71.9, 72.5, 115.7, 128.2, 128.5, 128.6, 129.4, 129.5, 132.5, 133.2, 135.2, 156.2, 156.7; HRMS calcd for C20H22N2O5F [M + H]+ 389.1507, found 389.1502; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 22.44 min (major), tR = 24.32 min (minor), 68% ee.
3-(4-Chloro)benzyloxycarbonylamino-4-methyl-4-(4-fluoro)benzyloxy-oxazolidin-2-one (5d-p-ClBn)
Oil, yield 89%; 1H NMR (400 MHz, CDCl3) δ 1.24 (s, 3H, CH3), 3.24 (d, 1H, J = 8.0 Hz, CH), 3.43 (d, 1H, J = 12.0 Hz, CH), 4.04 (d, 1H, J = 8.0 Hz, CH), 4.30 (d, 1H, J = 8.0 Hz, CH), 4.40–4.48 (m, 2H, CH2), 5.07 (s, 2H, CH2), 6.84 (bs, 1H, NH), 7.00 (t, 2H, J = 8.0 Hz, Har), 7.21–7.25 (m, 2H, Har), 7.30 (d, 2H, J = 12.0 Hz, Har); 13C NMR (100 MHz, CDCl3) δ 19.7, 61.2, 67.3, 71.4, 71.8, 72.5, 115.4, 115.7, 128.8, 129.0, 129.4, 129.5, 133.1, 133.2, 134.0, 134.2, 156.1, 156.8, 161.3, 163.7; HRMS calcd for C20H21N2O5ClF [M + H]+ 423.1118, found 423.1104; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 36.19 min (major), tR = 45.33 min (minor), 70% ee.
3-Ethyloxycarbonylamino-4-methyl-4-(4-chloro)benzyloxy-oxazolidin-2-one (5e-Et)
White solid, yield 78%, mp 80–85 °C; 1H NMR (400 MHz, CDCl3) δ 1.25 (m, 6H, 2CH3), 3.27 (d, 1H, J = 8.0 Hz, CH), 3.48 (d, 1H, J = 12.0 Hz, CH), 4.06 (d, 1H, J = 8.0 Hz, CH), 4.17 (q, 2H, J = 16 Hz, 8 Hz, CH2), 4.32 (d, 1H, J = 8.0 Hz, CH), 4.47–4.48 (m, 2H, CH2), 6.39 (bs, 1H, NH), 6.76–6.80 (m, 4H, Har); 13C NMR (100 MHz, CDCl3) δ 14.3, 19.8, 61.2, 62.6, 71.4, 72.1, 72.5, 128.8, 129.0, 133.9, 135.9, 156.3, 156.7; HRMS calcd for C15H20N2O5Cl [M + H]+ 343.1055, found 343.1048; HPLC (Daicel Chiralpak OJ-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 32.27 min (major), tR = 37.97 min (minor), 57% ee.
3-Benzyloxycarbonylamino-4-methyl-4-(4-chloro)benzyloxy-oxazolidin-2-one (5e-Bn)
White solid, yield 63%; mp 75–80 °C; 1H NMR (400 MHz, CDCl3) δ 1.26 (s, 3H, CH3), 3.25 (d, 1H, J = 8.0 Hz, CH), 3.45 (d, 1H, J = 8.0 Hz, CH), 4.06 (d, 1H, J = 8.0 Hz, CH), 4.31 (d, 1H, J = 8.0 Hz, CH), 4.41–4.49 (m, 2H, CH2), 5.13 (s, 2H, CH2), 6.60 (bs, 1H, NH), 7.19 (d, 2H, J = 8.0 Hz, Har), 7.29–7.37 (m, 7H, Har); 13C NMR (100 MHz, CDCl3) δ 19.8, 60.4, 61.2, 68.1, 71.4, 71.7, 72.1, 72.5, 127.8, 128.2, 128.5, 128.7, 128.8, 129.0, 133.9, 135.3, 135.9, 156.2, 156.7; HRMS calcd for C20H22N2O5Cl [M + H]+ 405.1212, found 405.1201; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 16.12 min (major), tR = 17.62 min (minor), 59% ee.
3-Benzyloxycarbonylamino-4-methyl-4-(4-bromo)benzyloxy-oxazolidin-2-one (5f-Bn)
White solid, yield 79%; mp 80–84 °C; 1H NMR (400 MHz, CDCl3) δ 1.33 (s, 3H, CH3), 3.32 (d, 1H, J = 8.0 Hz, CH), 3.52 (d, 1H, J = 8.0 Hz, CH), 4.12 (d, 1H, J = 8.0 Hz, CH), 4.37 (d, 1H, J = 8.0 Hz, CH), 4.49–4.50 (m, 2H, CH2), 5.20 (s, 2H, CH2), 6.49 (bs, 1H, NH), 7.19 (d, 2H, J = 4.0 Hz, Har), 7.40 (m, 5H, Har), 7.52 (d, 2H, J = 8.0 Hz, Har); 13C NMR (100 MHz, CDCl3) δ 19.7, 61.2, 68.1, 71.4, 72.2, 72.5, 121.9, 128.2, 128.5, 128.7, 129.3, 131.7, 135.4, 136.5, 156.3, 156.7; HRMS calcd for C20H22N2O5Br [M + H]+ 449.0707, found 449.0710; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 85:15, flow rate 1.0 mL min−1, λ = 254 nm): tR = 19.46 min (major), tR = 21.85 min (minor), 59% ee.
4-Chlorobenzyl(4-(((4-bromobenzyl)oxy)methyl)-4-methyl-2-oxooxazolidin-3-yl)carbamate (5f-p-ClBn)
Oil, yield 75%; 1H NMR (400 MHz, CDCl3) δ 1.27 (s, 3H, CH3), 3.27 (d, 1H, J = 10 Hz, CH), 3.45 (d, 1H, J = 10 Hz, CH), 4.08 (d, 1H, J = 8.0 Hz, CH), 4.32 (d, 1H, J = 8.0 Hz, CH), 4.45 (s, 2H, CH2), 5.11 (s, 2H, CH2), 6.27 (bs, 1H, NH), 7.14 (d, 2H, J = 8.0 Hz, Har), 7.27 (d, 2H, J = 8.0 Hz, Har), 7.33 (d, 2H, J = 8.0 Hz, Har), 7.47 (d, 2H, J = 8.0 Hz, Har); 13C NMR (100 MHz, CDCl3) δ 19.7, 61.2, 68.1, 71.4, 72.2, 72.5, 121.9, 128.2, 128.5, 128.7, 129.3, 131.7, 135.4, 136.5, 156.3, 156.7; HRMS calcd for C20H21N2O5ClBr [M + H]+ 483.0317, found 483.0315; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 85:15, flow rate 1.0 mL min−1, λ = 254 nm): tR = 22.65 min (major), tR = 27.78 min (minor), 59% ee.
3-Ethyloxycarbonylamino-4-methyl-4-(4-cyano)benzyloxy-oxazolidin-2-one (5g-Et)
Oil, yield 81%; 1H NMR (400 MHz, CDCl3) δ 1.22 (m, 6H, 2 CH3), 3.33 (d, 1H, J = 8.0 Hz, CH), 3.55 (d, 1H, J = 8.0 Hz, CH), 4.06 (d, 1H, J = 4.0 Hz, CH), 4.11–4.16 (m, 2H, CH2), 4.34 (d, 1H, J = 12.0 Hz, CH), 4.51–4.62 (m, 2H, CH2), 6.892 (bs, 1H, NH), 7.36 (d, 2H, J = 8.0 Hz, Har), 7.58 (d, 2H, J = 8.0 Hz, Har); 13C NMR (100 MHz, CDCl3) δ 14.3, 19.6, 61.2, 62.5, 70.8, 71.3, 72.3, 72.8, 111.4, 118.8, 127.6, 130.3, 132.3, 132.4, 143.1, 156.5, 156.7; HRMS calcd for C16H20N3O5 [M + H]+ 334.1398, found 334.1416; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 43.74 min (minor), tR = 48.41 min (major), 57% ee.
3-Benzyloxycarbonylamino-4-methyl-4-(4-cyano)benzyloxy-oxazolidin-2-one (5g-Bn)
Oil, yield 71%; 1H NMR (400 MHz, CDCl3) δ 1.27 (s, 3H, CH3), 3.31 (d, 1H, J = 8.0 Hz, CH), 3.52 (d, 1H, J = 8.0 Hz, CH), 4.05 (d, 1H, J = 8.0 Hz, CH), 4.33 (d, 1H, J = 8.0 Hz, CH), 4.48–4.58 (m, 2H, CH2), 5.10 (s, 2H, CH2), 7.09 (bs, 1H, NH), 7.25–7.35 (m, 7H, Har), 7.55 (d, 2H, J = 8.0 Hz, Har); 13C NMR (100 MHz, CDCl3) δ 19.7, 61.2, 68.1, 71.4, 72.3, 72.9, 111.5, 118.7, 127.5, 128.2, 128.5, 128.7, 132.4, 135.3, 143.0, 156.3, 156.7; HRMS calcd for C21H22N3O5 [M + H]+ 396.1554, found 396.1573; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 65.86 min (major), tR = 69.48 min (minor), 65% ee.
3-(4-Chloro)benzyloxycarbonylamino-4-methyl-4-(4-cyano)benzyloxy-oxazolidin-2-one (5g-p-ClBn)
Oil, yield 73%; 1H NMR (400 MHz, CDCl3) δ 1.28 (s, 3H, CH3), 3.31 (d, 1H, J = 8.0 Hz, CH), 3.50 (d, 1H, J = 12.0 Hz, CH), 4.08 (d, 1H, J = 8.0 Hz, CH), 4.34 (d, 1H, J = 8.0 Hz, CH), 4.51–4.60 (m, 2H, CH2), 5.09 (s, 2H, CH2), 6.62 (bs, 1H, NH), 7.25 (d, 2H, J = 4.0 Hz, Har), 7.31 (d, 2H, J = 8.0 Hz, Har), 7.37 (d, 2H, J = 8.0 Hz, Har), 7.59 (d, 2H, J = 8.0 Hz, Har); 13C NMR (100 MHz, CDCl3) δ 19.7, 61.2, 67.3, 71.4, 72.5, 120.0, 122.7, 125.4, 125.5, 125.6, 125.7, 127.5, 128.9, 133.8, 134.5, 141.4, 156.1, 156.7; HRMS calcd for C21H21N3O5Cl [M + H]+ 430.1164, found 430.1159; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 85:15, flow rate 1.0 mL min−1, λ = 254 nm): tR = 50.46 min (major), tR = 57.77 min (minor), 62% ee.
3-Ethyloxycarbonylamino-4-methyl-4-(4-trifluoro)benzyloxy-oxazolidin-2-one (5h-Et)
White solid, yield 76%; 1H NMR (400 MHz, CDCl3) δ 1.23 (t, 3H, J = 8.0 Hz, CH3), 1.29 (s, 3H, CH3), 3.31 (d, 1H, J = 12.0 Hz, CH), 3.52 (d, 1H, J = 12.0 Hz, CH), 4.06 (d, 1H, J = 8.0 Hz, CH), 4.14 (q, 2H, J = 12.0 Hz, 4 Hz, CH2), 4.34 (d, 1H, J = 8.0 Hz, CH), 4.52–4.62 (m, 2H, CH2), 6.78 (bs, 1H, NH), 7.38 (d, 2H, J = 8.0 Hz, Har), 7.58 (d, 2H, J = 8.0 Hz, Har); 13C NMR (100 MHz, CDCl3) δ 14.3, 19.6, 61.2, 62.5, 71.3, 72.4, 72.5, 125.5, 127.5, 141.7, 156.4, 156.8; HRMS calcd for C16H20N2O5F3 [M + H]+ 377.1319, found 377.1315; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 13.40 min (minor), tR = 15.24 min (major), 56% ee.
3-Benzyloxycarbonylamino-4-methyl-4-(4-trifluoro)benzyloxy-oxazolidin-2-one (5h-Bn)
Oil, yield 89%; 1H NMR (400 MHz, CDCl3) δ 1.27 (s, 3H, CH3), 3.30 (d, 1H, J = 12.0 Hz, CH), 3.51 (d, 1H, J = 8.0 Hz, CH), 4.05 (d, 1H, J = 8.0 Hz, CH), 4.33 (d, 1H, J = 8.0 Hz, CH), 4.49–4.59 (m, 2H, CH2), 5.12 (s, 2H, CH2), 6.87 (bs, 1H, NH), 7.25–7.38 (m, 7H, Har), 7.58 (d, 2H, J = 4.0 Hz, Har); 13C NMR (100 MHz, CDCl3) δ 19.7, 61.2, 68.1, 71.4, 72.5, 72.6, 125.5, 125.6, 127.4, 128.2, 128.5, 128.6, 135.3, 141.6, 156.3, 156.8; HRMS calcd for C21H22N2O5F3 [M + H]+ 439.1475, found 439.1470; HPLC (Daicel Chiralpak AS-H, hexane/isopropanol = 80:20, flow rate 1.0 mL min−1, λ = 254 nm): tR = 29.18 min (major), tR = 61.72 min (minor), 67% ee.
3-(4-Chloro)benzyloxycarbonylamino-4-methyl-4-(4-trifluoro)benzyloxy-oxazolidin-2-one (5h-p-ClBn)
Catalyst 1d (191 mg, 15 mol% in respect to the azodicarboxylate) was added to a suspension of 2-methyl-3-((4-(trifluoromethyl)benzyl)oxy)propanal (2.2 g, 8.93 mmol) and di-p-chlorobenzyl azodicarboxylate (2.17 g, 5.93 mmol) in THF (50 mL). The mixture stirred at rt under argon until the colour of the azodicarboxylate had disappeared and quenched by the addition H2O, then extracted three times with Et2O (50 mL ×3). The combined organic layers were dried over Na2SO4, filtered, and concentrated. The resulting crude was purified by flash chromatography on silica gel, eluting with light petroleum ether–ethyl acetate mixture (4:1 v/v) to afford 4h-p-ClBn (3.27 g) as solid in 90% yield with 74% ee. Mp 145–149 °C; 1H NMR (400 MHz, CDCl3) δ 1.32 (s, 3H, CH3), 3.71–3.87 (m, 2H, CH2), 4.54 (s, 2H, CH2), 5.10–5.20 (m, 4H, 2CH2), 6.71 (s, 1H, NH), 7.22–7.38 (m, 10H, Har), 7.62 (d, 2H, J = 4.0 Hz, Har), 9.62 (s, 1H, CHO); HRMS calcd for C28H26N2O6Cl2F3 [M + H]+ 613.1115, found 613.1110; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 85:15, flow rate 1.0 mL min−1, λ = 254 nm): tR = 18.00 min (major), tR = 20.29 min (minor).
Upon recrystallization from 90% ethanol, the aldehyde 4h-p-ClBn (2.1 g) was obtained in 98% ee (65% yield). After reduction and cyclization with NaBH4 (380 mg, 10 mmol), 5h-p-ClBn (1.53 g) was obtained in 95% yield with 98% ee. [α]20D −17.84 (c 0.7, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.34 (s, 3H, CH3), 3.36 (d, 1H, J = 8.0 Hz, CH), 3.56 (d, 1H, J = 8.0 Hz, CH), 4.12 (d, 1H, J = 4.0 Hz, CH), 4.38 (d, 1H, J = 4.0 Hz, CH), 4.56–4.46 (m, 2H, CH2), 5.18 (s, 2H, CH2), 6.84 (bs, 1H, NH), 7.38–7.43 (m, 6H, Har), 7.63 (d, 2H, J = 4.0 Hz, Har); 13C NMR (100 MHz, CDCl3) δ 19.7, 61.2, 67.3, 71.4, 72.5, 125.4, 122.7, 125.5, 125.6, 127.5, 128.8, 129.6, 133.8, 134.4, 141.4, 156.1, 156.7; HRMS calcd for C21H21N2O5ClF3 [M + H]+ 473.1084, found 473.1086; HPLC (Daicel Chiralpak AS-H, hexane/isopropanol = 70:30, flow rate 1.0 mL min−1, λ = 254 nm): tR = 34.57 min (major), tR = 56.49 min (minor).
3-Benzyloxycarbonylamino-4-methyl-4-(4-nitro)benzyloxy-oxazolidin-2-one (5i-Bn)
Oil, yield 77%; 1H NMR (400 MHz, CDCl3) δ 1.30 (s, 3H, CH3), 3.35 (d, 1H, J = 12.0 Hz, CH), 3.56 (d, 1H, J = 12.0 Hz, CH), 4.10 (d, 1H, J = 4.0 Hz, CH), 4.36 (d, 1H, J = 8.0 Hz, CH), 4.54–4.64 (m, 2H, CH2), 5.13 (s, 2H, CH2), 6.82 (bs, 1H, NH), 7.25–7.35 (m, 5H, Har), 7.41 (d, 2H, J = 8.0 Hz, Har), 8.15 (d, 2H, J = 12.0 Hz, Har); 13C NMR (100 MHz, CDCl3) δ 18.4, 19.8, 29.7, 30.9, 61.2, 68.2, 71.4, 72.1, 73.0, 76.7, 77.1, 77.2, 77.4, 123.8, 127.7, 128.2, 128.6, 128.7, 136.2, 144.8, 147.6, 164.2, 164.6, 207.2; HRMS calcd for C20H22N3O7 [M + H]+ 416.1452, found 416.1435; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 72.96 min (major), tR = 76.57 min (minor), 55% ee.
4-Chlorobenzyl((4R)-4-methyl-2-oxo-4-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)oxazolidin-3-yl)carbamate (5j-Bn)
Oil, yield 70%; 1H NMR (400 MHz, CDCl3): δ 1.26 (s, 3H, CH3), 1.41–1.56 (m, 4H, 2 CH2), 1.60–1.74 (m, 4H, CH2), 3.34–3.84 (m, 4H, CH2), 4.05–4.11 (m, 1H, CH), 4.33–4.38 (m, 1H, CH), 4.43–4.55 (m, 1H, CH), 5.14 (s, 2H, CH2), 6.93 (bs, 1H, NH), 7.26–7.33 (m, 5H, Har); HRMS calcd for C18H24N2O6Na [M + Na]+ 387.1527, found 387.1508; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 26.0 min (major), tR = 34.5 min (minor), 60% ee.
Benzyl(4-ethyl-4-methyl-2-oxooxazolidin-3-yl)carbamate (5l-Bn)
Oil, yield 78%; 1H-NMR (600 MHz, CDCl3): δ 0.86–0.96 (m, 3H, CH3), 1.24–1.33 (s, 3H, CH3), 1.55–1.70 (m, 2H, CH2), 4.06 (d, J = 6.0 Hz, 1H, CH), 4.20 (d, J = 6.0 Hz, 1H, CH), 5.19 (s, 2H, CH2), 6.53 (bs, 1H, NH), 7.26–7.38 (m, 5H, Har); 13C NMR (150 MHz, CDCl3) δ 7.7, 22.1, 29.8, 53.4, 61.8, 68.2, 72.1, 128.3, 128.5, 128.6, 135.3, 156.0, 156.3; HRMS calcd for C14H18N2O4Na [M + Na]+ 301.1159, found 301.1154; HPLC (Daicel Chiralpak AD-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 21.53 min (major), tR = 23.23 min (minor), 37% ee.
Dibenzyl 1-(1-ethoxy-2-methyl-1,3-dioxopropan-2-yl)hydrazine-1,2-dicarboxylate (4m-Bn)
Oil, yield 76%; 1H-NMR (300 MHz, CDCl3): δ 1.26 (t, J = 6.0 Hz, 3H, CH3), 1.56 (s, 3H, CH3), 4.18–4.22 (m, 2H, CH2), 5.16 (s, 4H, 2 CH2), 6.60 (brs, 1H, NH), 7.25–7.32 (m, 10H, Har), 9.60 (s, 1H, CHO); HRMS calcd for C22H25N2O7 [M + H]+ 429.1653, found 429.1656; HPLC (Daicel Chiralpak OD-H, hexane/isopropanol = 90:10, flow rate 1.0 mL min−1, λ = 254 nm): tR = 20.65 min (major), tR = 22.78 min (minor), 24% ee.
(R)-3-Amino-4-(hydroxymethyl)-4-methyloxazolidin-2-one (6)
To a solution of 5h-p-ClBn (670 mg, 1.42 mmol) in 8 ml of methanol and acetic acid (4 mL). 360 mg of 10% palladium on charcoal was added. The mixture hydrogenated at ambient pressure for 12 h and filtered. The filtrate was evaporated to dryness under reduced pressure. Column chromatography on silica gel (dichloromethane/methanol, 20:1 to 10:1) delivered 152 mg (1.03 mmol, 73%) of a colourless solid. Mp 113–115 °C; [α]20D −3.86 (c 0.9, CH3OH); 1H NMR (400 MHz, CDCl3) δ 1.20 (s, 3H, CH3), 3.31 (dd, 1H, J = 12.0 Hz, 1 Hz, CH), 3.55 (bs, 3H, NH2 and OH), 3.76–3.79 (m, 1H, CH), 3.96 (d, 1H, J = 8.0 Hz, CH), 4.40 (d, 1H, J = 8.0 Hz, CH); HRMS calcd for C5H11N2O3 [M + H]+ 147.0762, found 147.0764.
(R)-4-(Hydroxymethyl)-4-methyloxazolidin-2-one (7)
146 mg (1 mmol) of NaNO2 was added dropwise to a solution of 45.0 mg (0.234 mmol) of 6 in 18 ml of acetic acid and 6 ml of 1 M HCl. The mixture was refluxed for 1 h. The solvent was evaporated to dryness under reduced pressure. Column chromatography on silica gel (dichloromethane/methanol, 20:1 to 10:1) delivered 79 mg (0.6 mmol, 60%) of a white solid. [α]20D −8.8 (c 0.5, CH3OH); 1H NMR (400 MHz, CDCl3) δ 1.34 (s, 3H, CH3), 3.55 (dd, 1H, J = 12.0 Hz, 4 Hz, CH2), 4.04 (d, 1H, J = 8.0 Hz, CH), 4.33 (d, 1H, J = 8.0 Hz, CH), 5.59 (bs, 1H, NH); 13C NMR (150 MHz, CDCl3) δ 22.6, 58.9, 67.5, 72.8, 159.4; HRMS calcd for C5H10NO3 [M + H]+ 132.0654, found 132.0655.
4′-(2-Propyloxazol-4-yl)-[1,1′-biphenyl]-4-carbaldehyde (11)
Catalyst Pd-dimer (2.5 mg, 1 mol% in respect to 4-bromobenzaldehyde) was added to a suspension of 4-bromobenzaldehyde (101 mg, 0.55 mmol), K2CO3 (207 mg, 1.5 mmol) and 10 (157 mg, 0.5 mmol) in toluene:EtOH:H2O = 1:1:1 (v/v/v). The mixture was refluxed for 4 h. Then the solvent was removed under vacuum. The crude material was extracted with Et2O and washed with brine. The organic phase was dried (Na2SO4) and the solvent was evaporated under reduced pressure. The crude product was chromatographed (silica gel, light petroleum ether/ethyl acetate = 20:1) to afford the aldehyde (116 mg, 80%) as a white solid. Mp 100 °C; 1H NMR (400 MHz, CDCl3) δ 1.04 (t, 3H, J = 8.0 Hz, CH3), 1.84–1.89 (m, 2H, CH2), 2.83 (m, 2H, CH2), 7.69 (m, 2H, Har), 7.78–7.85 (m, 4H, Har), 7.89 (s, 1H, Har), 7.95–8.01 (m, 4H, Har), 10.06 (s, 1H, CHO); HRMS calcd for C19H18NO2 [M + H]+ 292.1332, found 292.1335.
(R,E)-4-Methyl-4-(2-(4'-(2-propyloxazol-4-yl)-[1,1′-biphenyl]-4-yl)vinyl)oxazolidin-2-one (12)
To a suspension of the phosphonium salt (235 mg, 0.48 mmol) in THF was added n-butyllithium (2.5 M in hexane, 0.37 mL, 0.937 mmol) at −78 °C and then the solution was stirred for 30 min at the same temperature. After the addition of benzaldehyde (70 mg, 0.24 mmol) at −78 °C, the reaction mixture was warmed to ambient temperature and stirred for 3 h. After quenching with saturated aq. NH4Cl, the resulting biphasic mixture was extracted with AcOEt. The combined organic layer was washed with water and brine, dried over Na2SO4, filtered, and evaporated. Purification by silica gel column chromatography (hexane:AcOEt = 4:1 to 1:1) provided 12 (131 mg, 73%) as a white solid. Mp 235 °C; [α]20D −17.8 (c 0.1, CH3OH); 1H NMR (400 MHz, CDCl3) δ 1.03 (t, 3H, J = 8.0 Hz, CH3), 1.81–1.90 (m, 2H, CH2), 2.82 (t, 2H, J = 8.0 Hz, CH2), 4.17–4.21 (m, 1H, CH), 4.58–4.65 (m, 2H, CH2), 5.12 (s, 1H, NH), 6.16–6.22 (m, 1H, CH), 6.66 (d, 1H, J = 16.0 Hz, CH), 7.46 (d, 2H, J = 8.0 Hz, Har), 7.59–7.65 (m, 4H, Har), 7.80 (d, 2H, J = 8.0 Hz, Har), 7.87 (s, 1H, Har); 13C NMR (100 MHz, CDCl3) δ 13.7, 20.7, 30.2, 56.2, 70.2, 125.9, 126.3, 127.2, 127.3, 130.6, 133.2, 133.7, 134.3, 139.6, 140.1, 140.9, 158.9, 165.5; HRMS calcd for C24H25N2O3 [M + H]+ 389.1860, found 389.1882.
(R)-4-Methyl-4-(2-(4′-(2-propyloxazol-4-yl)-[1,1′-biphenyl]-4-yl)ethyl)oxazolidin-2-one (13)
To a solution of 12 (120 mg, 0.32 mmol) in methanol was added 10% Pd/C (30 mg), and then the suspension was stirred for 2 h under a hydrogen atmosphere at ambient temperature. The reaction mixture was filtered and evaporated in vacuo, providing the product 13 (112 mg, 92%) as a white solid. Mp 185 °C; [α]20D 14.4 (c 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 1.04 (t, 3H, J = 8.0 Hz, CH3), 1.44 (s, 3H, CH3), 1.84–1.89 (m, 2H, CH2), 1.93–1.97 (m, 2H, CH2), 2.70–2.74 (m, 2H, CH2), 2.75–2.86 (m, 2H, CH2), 4.10 (d, 1H, J = 8.0 Hz, CH), 4.22 (d, 1H, J = 8.0 Hz, CH), 5.42 (bs, 1H, NH), 7.25 (d, 1H, J = 8.0 Hz, CH), 7.55 (d, 1H, J = 8.0 Hz, CH), 7.61 (d, 2H, J = 8.0 Hz, Har), 7.79 (m, 2H, J = 8.0 Hz, Har), 7.86 (s, 1H, Har); 13C NMR (100 MHz, CDCl3) δ 13.7, 20.6, 26.0, 30.0, 30.1, 42.2, 57.6, 75.6, 125.9, 127.2, 127.3, 128.7, 129.8, 133.2, 138.8, 139.8, 139.9, 140.3, 158.7, 165.6; HRMS calcd for C24H27N2O3 [M + H]+ 391.2016, found 391.2011.(R)-2-Amino-2-methyl-4-(4′-(2-propyloxazol-4-yl)-[1,1′-biphenyl]-4-yl)butan-1-ol hydrochloride (14)
Compound 13 (100 mg, 0.26 mmol) was diluted with methanol:H2O = 10:1 (v/v), then potassium hydroxide (146 mg, 2.6 mmol) was added, which was refluxed for 18 h. After cooling to room temperature, water was added to the reaction mixture and extracted with CH2Cl2. The combined organic layer was washed with water and brine, dried over Na2SO4, filtered, and evaporated. The crude product was chromatographed (silica gel, dichloromethane/methanol = 10:1), then added 1 M HCl in Et2O (2 mL) to afford the product 14 (82 mg, 80%) as a white solid. Mp 214 °C; [α]20D −1.61 (c 0.2, CH3OH); 1H NMR (400 MHz, CDCl3) δ 1.01 (t, 3H, J = 8.0 Hz, CH3), 1.34 (s, 3H, CH3), 1.81–1.92 (m, 2H, CH2), 2.68–2.72 (m, 2H, CH2), 2.83–2.86 (m, 2H, CH2), 3.52–3.57 (m, 2H, CH2), 3.62–3.65 (m, 2H, CH2), 7.31 (d, 2H, J = 8.0 Hz, Har), 7.58 (d, 2H, J = 8.0 Hz, Har), 7.65 (d, 2H, J = 8.0 Hz, Har), 7.77 (d, 2H, J = 8.0 Hz, Har), 8.23 (s, 1H, Har); 13C NMR (100 MHz, CDCl3) δ 14.0, 20.4, 21.6, 30.2, 30.8, 38.7, 58.8, 66.3, 127.2, 128.2, 128.3, 130.0, 136.0, 139.9, 140.7, 141.9, 142.1, 167.8; HRMS calcd for C23H29N2O2 [M + H]+ 365.2224, found 365.2212.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Drug Innovation Major Project (No. 2018ZX09711001-005-012), National Key R&D Program of China (No. 2018YFC1706403) and CAMS Innovation Fund for Medical Sciences (No. 2016-I2M-2-002).Notes and references
- E. Katz, H. Schmitt, M. Aydin, W. A. König and G. Jung, Liebigs Ann. Chem., 1985, 365 CrossRef CAS.
- C. Auvin-Guette, S. Rebuffat, I. Vuidepot, M. Massias and B. Bodo, J. Chem. Soc., Perkin Trans. 1, 1993, 249 RSC.
- I. Augeven-Bour, S. Rebuffat, C. Auvin, C. Goulard, Y. Prigent and B. Bodo, J. Chem. Soc., Perkin Trans. 1, 1997, 1587 RSC.
- M. Ahrend, Angew. Chem., 1999, 111, 3047 (Angew. Chem., Int. Ed., 1999, 38, 2873) CrossRef.
- K. L. Reddy and K. B. Sharpless, J. Am. Chem. Soc., 1998, 120, 1207 CrossRef CAS.
- H. F. Wang, G. H. Ma, S. B. Yang, R. G. Han and P. F. Xu, Tetrahedron: Asymmetry, 2008, 19, 1630 CrossRef CAS.
- G. P. Miley, J. C. Rote, R. B. Silverman, N. L. Kelleher and R. J. Thomson, Org. Lett., 2018, 20, 2369 CrossRef CAS.
- T. Tsuji, K. Suzuki, T. Nakamura and T. Nishi, Tetrahedron, 2014, 70, 5234 CrossRef CAS.
- M. A. Jones, A. D. Hislop and J. S. Snaith, Org. Biomol. Chem., 2006, 4, 3769 RSC.
- D. Seebach, J. D. Aebi, M. Gander-Coquoz and R. Naef, Helv. Chim. Acta, 1987, 70, 1194 CrossRef CAS.
- M. Di Giacomo, V. Vinci, M. Serra and L. Colombo, Tetrahedron: Asymmetry, 2008, 19, 247 CrossRef CAS.
- P. Vachal and E. N. Jacobsen, Org. Lett., 2000, 2, 867 CrossRef CAS.
- K. Maruoka and T. Ooi, Chem. Rev., 2003, 103, 3013 CrossRef CAS.
- H. Vogt, S. Vanderheiden and S. Bräse, Chem. Commun., 2003, 19, 2448 RSC.
- N. Kumaragurubaran, K. Juhl, W. Zhuang, A. Bøgevig and K. A. Jørgensen, J. Am. Chem. Soc., 2002, 124, 6254 CrossRef CAS.
- C. E. Hartmann, T. Baumann, M. Bächle and S. Bräse, Tetrahedron: Asymmetry, 2010, 21, 1341 CrossRef CAS.
- J. Ferreira, S. C. Rees-Jones, V. Ramaotsoa and R. Hunter, Org. Biomol. Chem., 2016, 14, 1545 RSC.
- J. Y. Fu, Q. C. Yang, Q. L. Wang, J. N. Ming, F. Y. Wang, X. Y. Xu and L. X. Wang, J. Org. Chem., 2011, 76, 4661 CrossRef CAS PubMed.
- N. S. Chowdari and C. F. Barbas, Org. Lett., 2005, 7, 867 CrossRef CAS.
- T. Tsuji, K. Suzuki, T. Nakamura and T. Nishi, Tetrahedron, 2014, 70, 5234 CrossRef CAS.
- H. Deng, S. G. Bernier, E. Doyle, J. Lorusso, B. A. Morgan, W. F. Westlin and G. Evindar, ACS Med. Chem. Lett., 2013, 4, 942 CrossRef CAS.
- T. Nishi, S. Miyazaki, T. Takemoto, K. Suzuki, Y. Iio, K. Nakajima, T. Ohnuki, Y. Kawase, F. Nara, S. Inaba, T. Izumi, H. Yuita, K. Ohshima, H. Doi, R. Inoue, W. Tomisato, T. Kagari and T. Shimozato, ACS Med. Chem. Lett., 2011, 2, 368 CrossRef CAS.
- J. Jin, J. P. Hu, W. Q. Zhou, X. J. Wang, Q. Xiao, N. N. Xue, D. L. Yin and X. G. Chen, Biochem. Pharmacol., 2014, 90, 50 CrossRef CAS.
- S. Chen, et al., under review.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra06210f |
| This journal is © The Royal Society of Chemistry 2019 |