
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew synthesis of tetraoxaspirododecane-diamines and tetraoxazaspirobicycloalkanes†
Nataliya N. Makhmudiyarova *,
Kamil R. Shangaraev,
Lilya U. Dzhemileva*,
Tatyana V. Tyumkina,
Ekaterina S. Mescheryakova,
Vladimir A. D'yakonov,
Askhat G. Ibragimov and
Usein M. Dzhemilev
*,
Kamil R. Shangaraev,
Lilya U. Dzhemileva*,
Tatyana V. Tyumkina,
Ekaterina S. Mescheryakova,
Vladimir A. D'yakonov,
Askhat G. Ibragimov and
Usein M. Dzhemilev
Institute of Petrochemistry and Catalysis, Russian Academy of Sciences, 141 Prospekt Oktyabrya, 450075 Ufa, Russian Federation. E-mail: natali-mnn@mail.ru; dzhemilev@mail.ru
First published on 23rd September 2019
Abstract
An efficient method for the synthesis of new spiro-tetraoxadodecanediamines and tetraoxazaspirobicycloalkanes has been developed by reactions of primary arylamines with gem-dihydroperoxides and α,ω-dialdehydes (glyoxal, pentanedial) catalyzed by lanthanide catalysts. A potential pathway for formation of tetraoxaspirododecane-diamines and tetraoxazospirobicycloalkanes has been proposed that involves generation of intermediate tetraoxaspiroalkanediols under the reaction conditions. The structures of the crystalline products have been confirmed by XRD. It was shown that the synthesized tetraoxazaspirobicycloalkanes exhibit high cytotoxic activity against Jurkat, K562, and U937 tumor cultures and Fibroblasts.
Introduction
Discovery of the antimalarial activity of the natural peroxide compound artemisinin stimulated the development of synthetic routes leading to novel cyclic peroxides.1 According to the published data,2 tetraoxaspirocycloalkanes demonstrate, in vivo, high antimalarial activity comparable with that of artemisinin.3 Known methods to synthesize tetraoxaspirocycloalkanes include reactions of gem-dihydroperoxides with α,ω-dihaloalkanes in the presence of CsOH in DMF4 or the same in the presence of Ag2O in CH2Cl2.2,5 Additionally, tetraoxaspirocycloalkanes can be synthesized from unsaturated hydroperoxides6 or (alkenyldioxy)cyclododecyl hydroperoxides.7 However, known synthetic methods to obtain tetraoxaspirocycloalkanes are hindered by significant drawbacks, such as low yields of the target products2,4,5 and a multistage synthetic process.2,6 One of the effective methods for the synthesis of heteroatomic compounds of various structures in one stage with a good yield are catalytic multicomponent reactions.7–12Present communication concerns a new approach to the synthesis of tetraoxaspirocycloalkanes having amine substituents at α-positions relative to peroxide groups and of tetraoxazabicycloalkanes, via reactions of primary arylamines with gem-dihydroperoxides and α,ω-dialdehydes catalyzed by lanthanide complexes.
Presence of a heteroatom at α-position relative to the peroxide group in such compounds as artemisinin, artemether, DU-1301, OZ277,3 veruculogen13 and fumitremorgin,14 and in bicyclic15 and acyclic6 α-amino endoperoxides16 accounts for antimalarial and antimicrobial activities of these compounds. The data available on heteroatom-containing peroxides with high antimalarial activity1,13 suggest that tetraoxaspirocycloalkanediamines could be useful for the development of antimalarial agents.
Result and discusion
During preliminary experiments it has been shown that the reaction of 1,1-dihydroperoxycyclohexane 1 with an equimolar amount of glyoxal 2 and p-chloroaniline 3a in selected conditions (∼20 °C, THF, 6 h) catalyzed by 5 mol% of Sm(NO3)3·6H2O gives N,N′-bis(4-chlorophenyl)-7,8,11,12-tetraoxaspiro[5.6]dodecane-9,10-diamine 4a in 87% yield (Scheme 1). The Sm(NO3)3·6H2O catalyst has been selected due to its activity in the syntheses of pentaoxacanes,17 tetraoxazaspiroalkanes,18,19 and hexaoxazadispiroalkanes.20 In an absence of the catalyst, the aforesaid reaction proceeds with the formation, along with the target product 4a (10%), of N-(4-chlorophenyl)formamide (70%) and cyclohexanone (10%). Whether the reaction is conducted in presence of other lanthanide catalysts, the yield of N,N′-bis(4-chlorophenyl)-7,8,11,12-tetraoxaspiro[5.6]dodecane-9,10-diamine 4a decreases in the following order: La(NO3)3·6H2O (80%) > TbCl3·6H2O (73%) > Ho(NO3)3·5H2O (60%) > DyCl3·6H2O (51%) > NdCl3 ·6H2O (50%).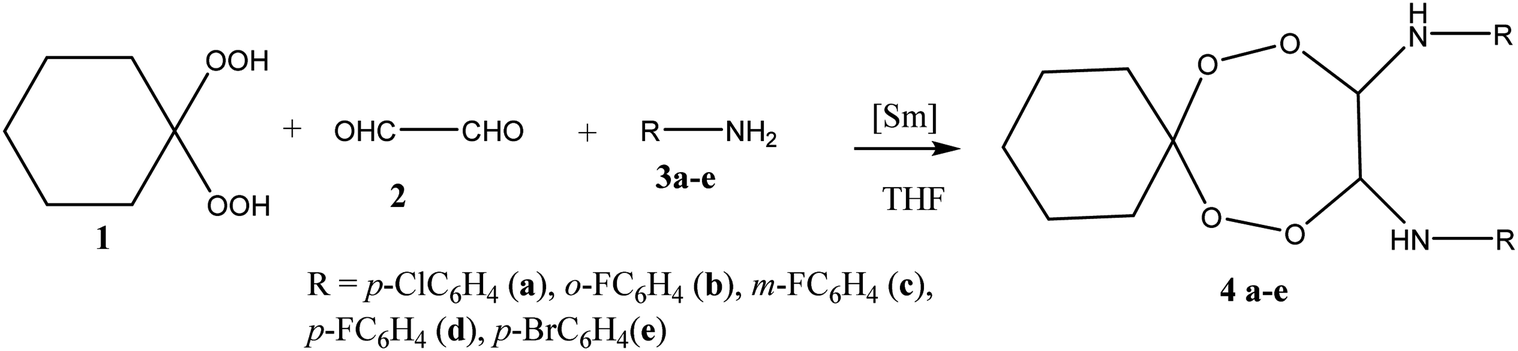 | ||
| Scheme 1 Synthesis of tetraoxaspirododecane-diamines. | ||
In selected conditions [5 mol% Sm(NO3)3·6H2O, 20 °C, 6 h], arylamines (m,p-fluoroanilines, p-bromoaniline) 3b–e enter the reaction with glyoxal 2 and 1,1-dihydroperoxycyclohexane 1 to result in formation of corresponding N,N′-bis(aryl)-7,8,11,12-tetraoxaspiro[5.6]dodecane-9,10-diamines 4b–d in 84–90% yields (Scheme 1). In the experiments, choice of the solvent has been stipulated by the fact that both the reactants and the target products are highly soluble in THF.
Structures of N,N′-bis(aryl)-7,8,11,12-tetraoxaspiro[5.6]dodecane-9,10-diamines 4a–e have been established using 1H and 13C NMR spectrometry methods, MALDI TOF/TOF mass spectrometry and X-ray diffraction (Fig. 1). In 1H NMR spectra, the signals for methine hydrogens atoms localized between the N and O atoms in the seven-membered rings resonate in a region between 4.60–4.75 ppm and emerge as a broadened singlet due to slow, in the NMR time scale, conformational flexibility of the ring, whereas methylene protons of the spiroalkane and alkane moieties occur as two multiplets in the regions between 1.40–1.70 ppm and 2.40–2.70 ppm. Aromatic protons resonate in a low-field region between 6.80–7.40 ppm. The mass spectrum of the heterocycles 4a–e displays the corresponding molecular ion peaks, accordingly.
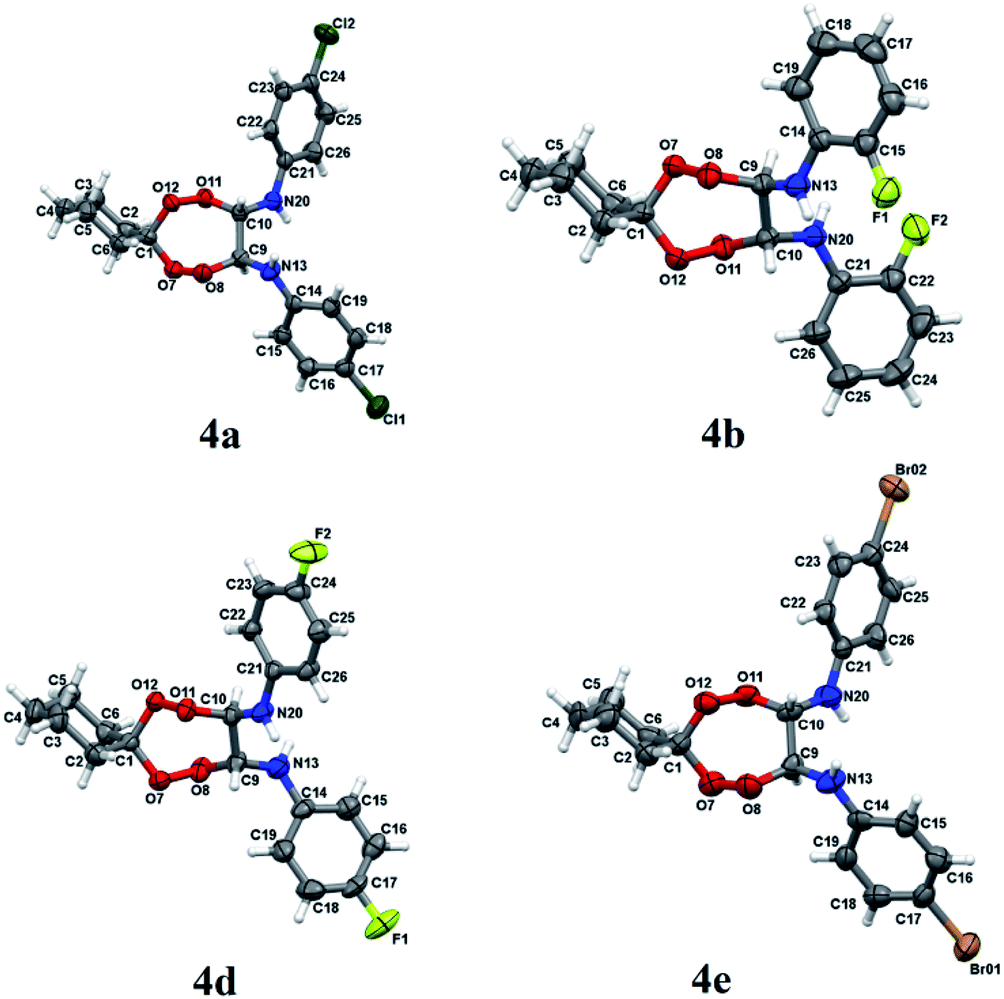 | ||
| Fig. 1 Molecular structure of peroxides 4a, b, d, e. The atoms are depicted as thermal ellipsoids (p = 50%). | ||
Crystals for the compounds 4a, b, d, e (Fig. 1) have been obtained from a solvent mixture of hexane and Et2O in 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, at room temperature. In the corresponding structures, a spiro-conjugated tetraoxepane ring adopts a twist boat conformation, similarly to the tetraoxepane derivative described in the literature.6 Chiral centers at atoms C9 and C10 adopt S configuration in the compounds 4a, 4d, and 4e and R configuration in the compound 4b. N-aryl substituents are anti-oriented relative to each other, whereupon the torsion angle N20–C10–C9–N13 constitutes 73.1(3), −79.4(2), 68.7(4) and 74.3(8) for the compounds 4a, 4b, 4d, and 4e, respectively. The cyclohexane moiety in all compounds 4a, b, d, e adopts a chair conformation. In all molecules, the nitrogen atoms adopt a planar configuration (wherein the sum of angles at the nitrogen atom is ∼360°), due to conjugation between the π-system of the aromatic substituent and an unshared pair of electrons at the nitrogen atom. The lengths of peroxide bonds are provided within a range of 1.458 to 1.468 Å.
:1 ratio, at room temperature. In the corresponding structures, a spiro-conjugated tetraoxepane ring adopts a twist boat conformation, similarly to the tetraoxepane derivative described in the literature.6 Chiral centers at atoms C9 and C10 adopt S configuration in the compounds 4a, 4d, and 4e and R configuration in the compound 4b. N-aryl substituents are anti-oriented relative to each other, whereupon the torsion angle N20–C10–C9–N13 constitutes 73.1(3), −79.4(2), 68.7(4) and 74.3(8) for the compounds 4a, 4b, 4d, and 4e, respectively. The cyclohexane moiety in all compounds 4a, b, d, e adopts a chair conformation. In all molecules, the nitrogen atoms adopt a planar configuration (wherein the sum of angles at the nitrogen atom is ∼360°), due to conjugation between the π-system of the aromatic substituent and an unshared pair of electrons at the nitrogen atom. The lengths of peroxide bonds are provided within a range of 1.458 to 1.468 Å.
In order to expand the scope of applicability of the method developed hereby, we have conducted a reaction of pentane-1,5-dial 5 with gem-bis-hydroperoxides and primary amines. In conditions determined for the cyclocondensation of glyoxal 2 (5 mol% Sm(NO3)3·6H2O, 20 °C, 6 h), the pentane-1,5-dial 5 enters the reaction with gem-bis-hydroperoxides and primary amines to give tetraoxazaspirobicycloalkanes (Scheme 3). In the reaction, the 1,1-dihydroperoxycycloalkane compounds based on cyclohexane 1, cyclopentane 6, 4-methylcyclohexane 7, cyclooctane 8, dodecane 9 and adamantane 10 have been utilized as gem-bis-hydroperoxides, whereas arylamines 3a–g have been utilized as the primary amines. The results obtainable in selected cyclocondensation conditions [pentane-1,5-dial:gem-bis-hydroperoxide:arylamine:Sm(NO3)3·6H2O = 1:1:1:0.05 (mol/mol); THF; 20 °C] indicate that the method thus developed is an efficient tool for the selective synthesis of bicyclic tetraoxazaspirobicycloalkanes 11–16 (71–83%) (Scheme 2). N-arylpiperidines 17a–g21 were isolated as by-products of the reaction with a yield not exceeding 25%.
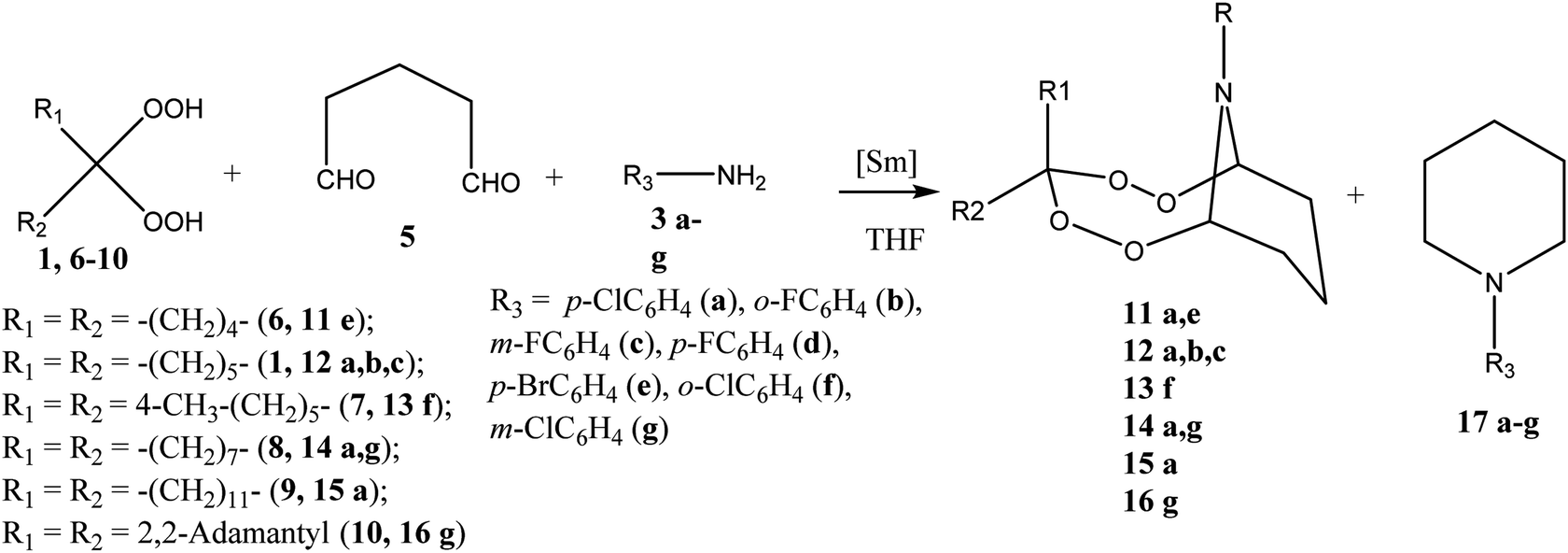 | ||
| Scheme 2 Synthesis of tetraoxazaspirobicycloalkanes. | ||
Crystals for the compound 12b (Fig. 2) have been obtained from a solvent mixture of hexane and Et2O in 10:1 ratio, at room temperature.
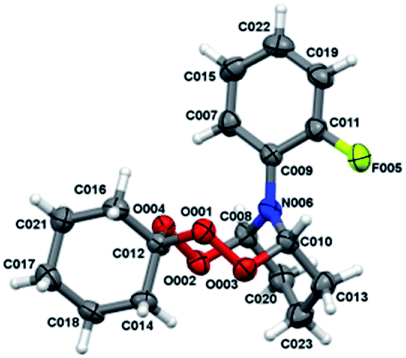 | ||
| Fig. 2 Molecular structure of peroxide 12b. The atoms are depicted as thermal ellipsoids (p = 50%). | ||
According to the X-ray diffraction data, a tetraoxazocane ring adopts a boat chair conformation, whereas cyclohexane and pyran rings adopt the chair conformation. Bond lengths of the peroxide bonds O001–O003 and O002–O004 constitute 1.4692 (14) and 1.4612 (14) Å, respectively. The nitrogen atom N006 in the compound 12b adopts a planar conformation similarly to that occurring in the compounds 4a, b, d, e (with the sum of angles at the nitrogen, ∑N006 = 359.7°).
As can be viewed from the figure, in a crystal phase the diperoxide moiety in the bicyclic structure adopts the chair conformation, while in solution a multicomponent conformational equilibrium exists, which is typical of both triperoxide20 and azadiperoxide18,19 compounds. Thus, in the 13C NMR spectra of the synthesized compounds 11–16, three signals with similar chemical shifts can be observed in a region between 85.92–86.87 ppm for each said compound, instead of individual signals characteristic of the bridgehead tertiary carbon atoms. At the same time, the methine protons also exhibit signals split into components with different intensity in the matching region 5.36–5.83 ppm of the 1H NMR spectra. By the way of an example, for the compound 12a, the integrated intensity ratio for signals at 5.83 ppm, 5.68 ppm, and 5.37 ppm constitutes 1:8:1, which correlates with the carbon signals at 85.92 ppm, 86.87 ppm, and 86.75 ppm, respectively, according to the data obtained by heteronuclear 2D HSQC spectroscopy. In order to assign signals in the NMR spectra and by using quantum chemical method B3LYP/6-31G(d,p), six stable conformers have been identified on a potential energy surface of the bicyclic tetraoxazaspiroalkane molecule 12a having the spirohexane substituent in the chair conformation.22 Three the most energetically favorable conformers are shown in Fig. 3 as preferred candidates for the structures observed in the NMR spectra.
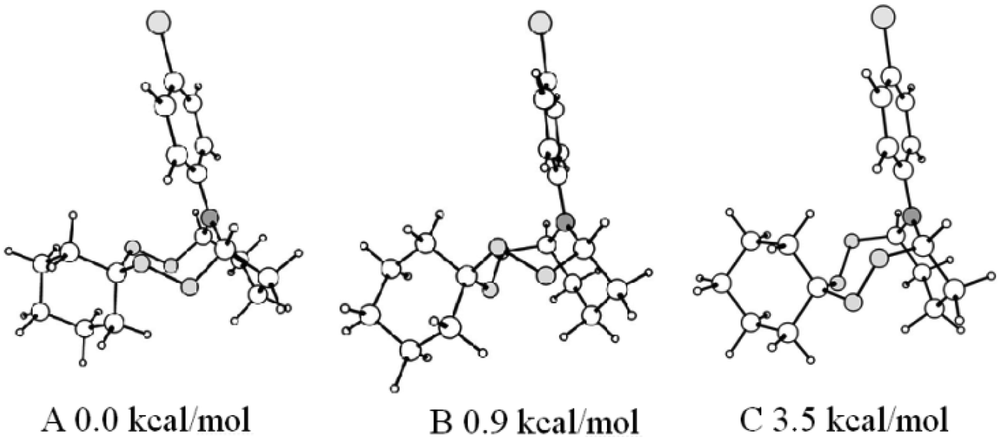 | ||
| Fig. 3 Optimized structures of the lowest energy conformers of tetraoxazaspirobicycloalkanes. | ||
According to the calculated data, the global minimum corresponds to a conformer A that occurs in the crystal phase. Slightly higher in energy conformational states of the spiroaminodiperoxide moiety are the twist chair (B) and the chair (C). For the conformation B, a conformation B′ of similar energy may exist, due to a lack of symmetry upon rigid fixation of rotational position of the N-substituent and the spiro moiety. Symmetry violation is also possible in an event of ortho- or meta-substitutions in the aromatic ring, which can cause doubling of the observed set of signals. Additionally, any changes in the bicyclic cage lead to a sharp increase in energy up to 28 kcal mol−1 and higher (ESI,† conformers D and E); therefore, these changes are unlikely.
Presumably, the scheme of formation of spiro-tetraoxepanes includes the initial formation of tetraoxaspirocycloalkanediols, which then undergo condensation with primary amines to give the target products. This assumption was verified by conducting the synthesis of tetraoxaspirocycloalkanediols 18–20 by the reaction of 1,1-dihydroperoxycycloalkanes 1, 6, and 7 with glyoxal 2 in the presence of 5 mol% of the Sm(NO3)3·6H2O catalyst (Scheme 3). Without a catalyst, the yield of diols 17–19 did not exceed 10%.
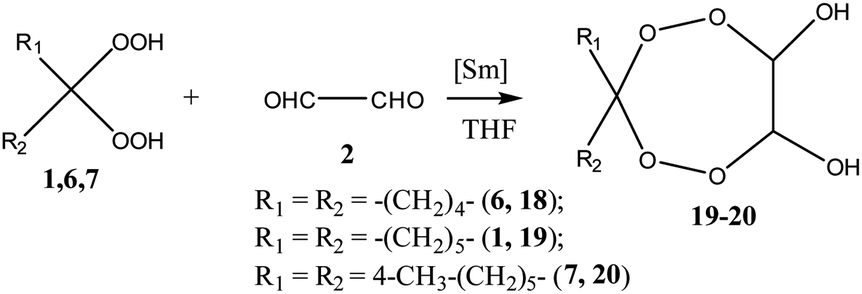 | ||
| Scheme 3 Synthesis of tetraoxaspirocycloalkanediols. | ||
According to X-ray diffraction data for compounds 19 (Fig. 4), in the crystalline state, the tetraoxepane and cyclohexane moieties exist in the chair conformation. Like in structure 4b, the chiral centers at the C005 and C008 carbon atoms have the R configuration.
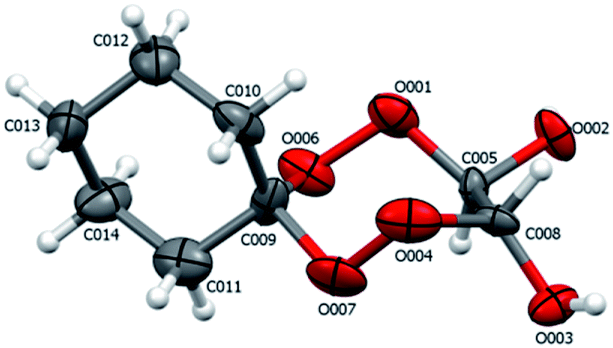 | ||
| Fig. 4 Structure of compound 19 according to X-ray diffraction. The atoms are depicted as thermal ellipsoids (p = 50%). | ||
Cytotoxicity of azaperoxide based compounds is well known,3,13,14,19,20b,23 so screened the representative compounds for their cytotoxicity activity against Jurkat, K562, U937 Fibroblasts cell lines and results are summarized in Table 1.
| C-d | Jurkat (IC50, μM) | K562 (IC50, μM) | U937 (IC50, μM) | Fibroblasts (IC50, μM) |
|---|---|---|---|---|
| 11e | 13.51 ± 0.72 | 12.38 ± 0.67 | 16.44 ± 1.43 | 127.63 ± 2.59 |
| 11a | 63.29 ± 2.25 | 45.13 ± 1.36 | 42.19 ± 1.24 | 231.42 ± 3.88 |
| 12c | 17.39 ± 1.87 | 12.35 ± 1.18 | 11.49 ± 0.84 | 145.19 ± 3.47 |
| 14g | 272.57 ± 5.68 | 45.87 ± 1.73 | 40.87 ± 1.58 | 368.68 ± 4.53 |
| 12b | 15.48 ± 0.31 | 15.09 ± 0.18 | 14.83 ± 0.24 | 129.42 ± 2.37 |
| 15a | 118.23 ± 2.74 | 61.34 ± 2.08 | 59.17 ± 1.79 | 389.23 ± 4.69 |
| 4b | 52.91 ± 1.41 | 28.37 ± 1.34 | 26.45 ± 1.17 | 249.37 ± 3.91 |
| 4a | >500 | 106.24 ± 3.95 | 141.28 ± 4.33 | >500 |
It was found that the synthesized spiro-tetraoxadodecanediamines 4a, f and tetraoxazaspirobicycloalkanes 11a, e, 12b, c, 14g, 15a exhibit a cytotoxic effect on all selected tumor cell lines in a wide range from 11.49 to >500 μM. The most potent cytotoxic activity was shown by peroxides 11e, 12c and 12b synthesized by the reaction of 1,1-dihydroperoxycyclopentane 6 or 1,1-dihydroperoxycyclohexane 1 and fluorine(bromine)arylamines. The replacement of bromine or fluorine atoms with chlorine atom in the phenyl substituent of the studied peroxides 11a, 14g and 15a leads to a significant decrease in their cytotoxicity, with a pronounced selective effect on myelocytic (K562) and monocytic (U937) cell cultures, in comparison with the cytotoxicity of the studied compounds to lymphocytes of the Jurkat line. At the same time, spiro-tetraoxadodecanediamines with two fluoroaromatic substituents 4b or chloroaromatic 4a fragments showed less cytotoxicity compared to tetraoxazaspirobicycloalkanes 11a, e, 12b, c, 14g, 15a.
The synthesized compounds have a selectivity index (SI) with respect to all tumor cells from 4 to 10 (SI = IC50 Fibroblasts/IC50 cancer cells).
Conclusions
Hence, a versatile method has been developed for the synthesis of new spiro-tetraoxepanediamines and tetraoxazaspirobicycloalkanes by the reactions of primary arylamines with gem-dihydroperoxides and α,ω-dialdehydes in presence of lanthanide catalysts. The method thus developed markedly expands structural diversity of nitrogen-containing cyclic diperoxide derivatives and, in most cases, allows synthesizing these compounds with higher yields (up to 95%) and selectivity. In addition, it was shown that the synthesized spiro-tetraoxepanediamines and tetraoxazaspirobicycloalkanes exhibit high cytotoxic activity against Jurkat, K562, U937 tumor cultures and Fibroblasts.Experimental section
General remarks
All reactions were performed at room temperature in air in round-bottom flasks equipped with a magnetic stir bar. The NMR spectra were recorded on a Bruker Avance 500 spectrometer at 500.17 MHz for 1H and 125.78 MHz for 13C according to standard Bruker procedures. CDCl3 was used as the solvent, and tetramethylsilane, as the internal standard. The mixing time for the NOESY experiments was 0.3 s. Mass spectra were recorded on a Bruker Autoflex III MALDI TOF/TOF instrument with α-cyano-4-hydroxycinnamic acid as a matrix. Samples were prepared by the dried droplet method. The C, H, and N were quantified by a Carlo Erba 1108 analyzer. The oxygen content was determined on a Carlo Erba 1108 analyzer. The progress of reactions was monitored by TLC on Sorbfil (PTSKh-AF-A) plates, with a 5:1 hexane:EtOAc mixture as the eluent and visualization with I2 vapor. For column chromatography, silica gel MACHEREY-NAGEL (0.063–0.2 mm) was used.
All calculations were carried out using a program Gaussian 09. Geometric parameter optimization, vibrational frequency analysis, and calculation of entropy and thermodynamic corrections to the total energy of the compounds were carried out on the B3LYP functional18 using the 6-31G(d,p) basis set. No limitation was imposed on the changes in the geometric parameters of the subsystems studied. Thermodynamic parameters were determined at 298 K. The minima were confirmed through the calculation of the force constant (Hessian) matrix and the analysis of the resulting frequencies. All minima were verified to have no negative frequencies. Visualization of quantum chemical data was carried out with the programs ChemCraft.24
The X-ray diffraction measurements for compounds 4a, 4b, 4d, 4e, 12b, 19 were performed on an XCalibur Gemini Eos automated four-circle diffractometer (graphite monochromator, MoKα radiation, λ = 0.71073 Å, ω-scan mode, 2θmax = 62°) at ambient temperature (293–298 K). Collected data were processed using the program CrysAlisPro.25 Structures determinations were carried out with the OLEX2 program.26 The structures were solved by direct methods and refined by the full-matrix least-squares method in the anisotropic approximation for non-hydrogen atoms. All hydrogen atoms are generated using the proper HFIX command and refined isotropically using the riding model. The calculations were performed using the SHELX program package.27 The molecular plots were drawn using mercury.28
The synthesis of the gem-dihydroperoxides 1, 6–10 was as reported in the literature.29 THF was freshly distilled over LiAlH4. Glyoxal was used as aqueous solution (40%).
Cell culturing
Human cancer cell line HeLa was obtained from the HPA Culture Collections (UK). Cells (Jurkat, K562, U937, Fibroblasts) were purchased from Russian Cell Culture Collection (Institute of Cytology of the Russian Academy of Sciences) and cultured according to standard protocols and sterile technique. The cell lines were shown to be free of viral contamination and mycoplasma. Cells were maintained in RPMI 1640 (Jurkat, K562, U937, Fibroblast) (Gibco) supplemented with 4 μM glutamine, 10% FBS (Sigma) and 100 units per ml penicillin–streptomycin (Sigma). All types of cells were grown in an atmosphere of 5% CO2 at 37 °C. The cells were subcultured at 2–3 days intervals. Cells were then seeded in 24 well plates at 5 × 104 cells per well and incubated overnight. Jurkat, K562, U937, Fibroblast cells were subcultured at 2 day intervals with a seeding density of 1 × 105 cells per 24 well plates in RPMI with 10% FBS.Cytotoxicity assay
Viability (live/dead) assessment was performed by staining cells with 7-AAD (7-aminoactinomycin D) (Biolegend). After treatment cells were harvested, washed 1–2 times with phosphate-buffered saline (PBS) and centrifuged at 400g for 5 min. Cell pellets were resuspended in 200 μL of flow cytometry staining buffer (PBS without Ca2+ and Mg2+, 2.5% FBS) and stained with 5 μL of 7-AAD staining solution for 15 min at room temperature in the dark. Samples were acquired on NovoCyte TM 2000 Flow Cytometry System (ACEA) equipped with 488 nm argon laser. Detection of 7-AAD emission was collected through a 675/30 nm filter in the FL4 channel.Cyclocondensation reactions of primary arylamines with gem-dihydroperoxides and α,ω-dialdehydes (glyoxal, pentanedial) catalyzed by Sm(NO3)3·6H2O
General procedure: a Schlenk vessel mounted on a magnetic stirrer was charged at ∼20 °C with tetrahydrofuran (5 ml), α,ω-dialdehydes (glyoxal, pentanedial) (10 mmol), and specified gem-dihydroperoxides (10 mmol).29 Then Sm(NO3)2·6H2O (0.062 g, 5 mol% relative to 1,1′-peroxybis(1-hydroperoxycycloalkane)) was added. The reaction mixture was stirred at ∼20 °C for 1 h, after which primary arylamines (20 mmol) was added, and the reaction mixture was stirred at ∼20 °C for 6 h more. After completion of the reaction H2O (5 ml) and CH2Cl2 (5 ml) were added. The organic layer was separated, dried (anhydrous MgSO4) and concentrated to isolate products stable during storage at room temperature. Products of the reaction were purified by column chromatography on SiO2 using 10:1 PE:Et2O as the eluent. The progress of reactions was monitored by TLC, with a 5:1 hexane:EtOAc mixture as the eluent, visualization was performed with I2 vapor.
N9,N10-bis(4-Chlorophenyl)-7,8,11,12-tetraoxaspiro[5.6]dodecane-9,10-diamine 4a
White crystals; 0.37 g (87% yield), Rf 0.77 (PE/Et2O = 10/1), mp 120–122 °C. 1H NMR (400 MHz, CDCl3, 25 °C): δ = 1.28–1.48 (m, 2H, CH2), 1.69 (br.s, 4H, 2CH2), 1.77–1.89 (m, 4H, 2CH2), 5.57 (br.s, 2H, 2CH), 7.13–7.15 (m, 4H, CH), 7.18–7.21 (m, 4H, CH). 13C NMR (100 MHz, CDCl3, 25 °C): δ = 22.6 (conformer A), 22.8 (conformers B+C), 25.1, 31.2 (conformer A), 31.4 (conformers B+C), 85.1, 115.8, 116.3, 124.7, 128.6, 145.1. MALDI TOF/TOF, m/z: 424 [M − H]+. Anal. calcd for C20H22Cl2N2O4: C, 56.48; H, 5.21; N, 6.59%. Found: C, 56.46; H, 5.19; N, 6.57%.N9,N10-bis(2-Fluorophenyl)-7,8,11,12-tetraoxaspiro[5.6]dodecane-9,10-diamine 4b
White crystals; 0.33 g (85% yield), Rf 0.75 (PE/Et2O = 10/1), mp 134–136 °C. 1H NMR (400 MHz, CDCl3, 25 °C): δ = 1.28–1.36 (m, 2H, CH2), 1.43–1.45 (m, 4H, 2CH2), 1.60–1.61 (m, 4H, 2CH2), 5.70 (br.s, 2H, 2CH), 6.87–6.89 (m, 2H, CH), 7.02–7.10 (m, 4H, CH), 7.18–7.24 (m, 2H, CH). 13C NMR (100 MHz, CDCl3, 25 °C): δ = 22.6, 25.1, 29.7, 89.7, 111.5, 114.5, 115.2 (J = 19), 120.3 (J = 17), 124.7, 142.5, 164.1 (J = 192). MALDI TOF/TOF, m/z: 391 [M − H]+. Anal. calcd for C20H22F2N2O4: C, 61.22; H, 5.65; N, 7.14%. Found: C, 61.20; H, 5.63; N, 7.11%.N9,N10-bis(3-Fluorophenyl)-7,8,11,12-tetraoxaspiro[5.6]dodecane-9,10-diamine 4c
White crystals; 0.34 g (85% yield), Rf 0.78 (PE/Et2O = 10/1), mp 138–140 °C. 1H NMR (400 MHz, CDCl3, 25 °C): δ = 1.30–1.46 (m, 2H, CH2), 1.61 (br.s, 4H, 2CH2), 1.77–1.84 (m, 4H, 2CH2), 5.62 (br.s, 2H, 2CH), 6.58–6.68 (m, 6H, CH), 7.18–7.25 (m, 2H, CH). 13C NMR (100 MHz, CDCl3, 25 °C): δ = 22.6 (conformer A), 22.7 (conformers B+C), 25.0 (conformer A), 25.2 (conformers B+C), 31.2 (conformer A), 31.6 (conformers B+C), 88.2, 102.5 (J = 7),111.1, 112.1, 107.2 (J = 17), 130.7 (J = 8), 145.6, 163.8 (J = 195). MALDI TOF/TOF, m/z: 391 [M − H]+. Anal. calcd for C20H22F2N2O4: C, 61.22; H, 5.65; N, 7.14%. Found: C, 61.19; H, 5.63; N, 7.12%.N9,N10-bis(4-Fluorophenyl)-7,8,11,12-tetraoxaspiro[5.6]dodecane-9,10-diamine 4d
White crystals; 0.35 g (88% yield), Rf 0.74 (PE/Et2O = 10/1), mp 128–130 °C. 1H NMR (400 MHz, CDCl3, 25 °C): δ = 1.28–1.45 (m, 2H, CH2), 1.61 (br.s, 4H, 2CH2), 1.77–1.89 (m, 4H, 2CH2), 5.57 (br.s, 2H, 2CH), 6.80–6.88 (m, 4H, CH), 6.96–7.01 (m, 4H, CH). 13C NMR (100 MHz, CDCl3, 25 °C): δ = 22.6, 25.1, 31.2 (conformer A), 31.4 (conformers B+C), 89.4, 111.9, 116.0 (J = 18), 121.6, 139.9, 157.7 (J = 190). MALDI TOF/TOF, m/z: 391 [M − H]+. Anal. calcd for C20H22F2N2O4: C, 61.22; H, 5.65; N, 7.14%. Found: C, 61.20; H, 5.62; N, 7.12%.N9,N10-bis(4-Bromophenyl)-7,8,11,12-tetraoxaspiro[5.6]dodecane-9,10-diamine 4e
White crystals; 0.43 g (90% yield), Rf 0.72 (PE/Et2O = 10/1), mp 122–124 °C. 1H NMR (400 MHz, CDCl3, 25 °C): δ = 1.28–1.48 (m, 2H, CH2), 1.63 (br.s, 4H, 2CH2), 1.77–1.85 (m, 4H, 2CH2), 5.56 (br.s, 2H, 2CH), 6.73–6.75 (m, 4H, CH), 7.34–7.35 (m, 4H, CH). 13C NMR (100 MHz, CDCl3, 25 °C): δ = 22.6, 24.9, 30.7 (conformer A), 31.3 (conformers B+C), 89.3, 106.4, 112.2, 116.0, 121.5, 132.2. MALDI TOF/TOF, m/z: 513 [M − H]+. Anal. calcd for C20H22Br2N2O4: C, 46.72; H, 4.31; N, 5.45%. Found: C, 46.70; H, 4.29; N, 5.43%.11-(4-Chlorophenyl)-2,3,5,6-tetraoxa-11-azaspiro[bicyclo[5.3.1]undecane-4,1′-cyclopentane] 11a
Brown oil; 0.24 g (75% yield), Rf 0.79 (PE/Et2O = 10/1). 1H NMR (500.17 MHz, CDCl3, 25 °C): δ = 1.54–1.59 (m, 1H, CHa, conformers B+C), 2.40–2.44 (m, 1H, CHb, conformers B+C), 2.13–2.19 (m, 2H, CH2, conformer A), 1.53–2.10 (m, 4H, 2CH2), 1.70–1.97 (m, 4H, 2CH2), 2.45–2.35 and 2.13–2.20 and 1.76–1.82 and 1.50–1.60 (m, 4H, 2CH2), 5.35–5.36 (m, 2H, 2CH, conformer B), 5.65 (s, 2H, 2CH, conformer A), 5.77 (s, 2H, 2CH, conformer C), 6.77–6.87 (m, 2H, CH), 7.13–7.20 (m, 2H, CH). 13C NMR (125.78 MHz, CDCl3, 25 °C): δ = 14.2 (conformer A), 16.0 (conformers B+C), 23.2 (conformer A), 24.3 (conformers B+C), 24.6 (conformer A), 26.4 (conformer A), 27.1 (conformers B+C), 33.5 (conformers B+C), 33.7 (conformer A), 34.0 (conformers B+C), 34.2 (conformer A), 88.2 (conformer B), 88.9 (conformer A), 89.1 (conformer C), 113.4, 114.2 (conformers B+C), 120.7 (conformer A), 128.0, 129.2 (conformer A), 140.2 (conformer B+C), 159.7. MALDI TOF/TOF, m/z: 324 [M − H]+. Anal. calcd for C16H20ClNO4: C, 58.99; H, 6.19; N, 4.30%. Found: C, 58.97; H, 6.17; N, 4.27%.11-(4-bromophenyl)-2,3,5,6-tetraoxa-11-azaspiro[bicyclo[5.3.1]undecane-4,1′-cyclopentane] 11e
Brown solid; 0.26 g (71% yield), Rf 0.77 (PE/Et2O = 10/1), mp 110–112 °C. 1H NMR (500.17 MHz, CDCl3, 25 °C): δ = 1.57–1.60 (m, 1H, CHa, conformers B+C), 2.43–2.47 (m, 1H, CHb, conformers B+C), 2.13–2.20 (m, 2H, CH2, conformer A), 1.57–2.11 (m, 4H, 2CH2), 1.70–1.97 (m, 4H, 2CH2), 2.47–2.39 and 2.13–2.20 and 1.74–1.80 and 1.52–1.60 (m, 4H, 2CH2), 5.27–5.28 (m, 2H, 2CH, conformer B), 5.67 (s, 2H, 2CH, conformer A), 5.81 (s, 2H, 2CH, conformer C), 7.06–7.10 (m, 2H, CH), 7.35–7.39 (m, 2H, CH). 13C NMR (125.78 MHz, CDCl3, 25 °C): δ = 14.3 (conformer A), 16.0 (conformers B+C), 23.3 (conformer A), 24.6 (conformers B+C), 24.9 (conformer A), 26.4 (conformer A), 27.1 (conformers B+C), 32.4 (conformers B+C), 33.5 (conformer A), 33.7 (conformers B+C), 34.6 (conformer A), 86.5 (conformer B), 87.1 (conformer A), 87.4 (conformer C), 113.1, 119.7 (conformers B+C), 120.50 (conformer A), 124.5, 131.6 (conformer A), 132.4 (conformer B+C), 149.2. MALDI TOF/TOF, m/z: 369 [M − H]+. Anal. calcd for C16H20BrNO4: C, 51.91; H, 5.45; N, 3.78%. Found: C, 51.89; H, 5.43; N, 3.76%.11-(4-Chlorophenyl)-2,3,5,6-tetraoxa-11-azaspiro[bicyclo[5.3.1]undecane-4,1′-cyclohexane] 12a
Orange oil; 0.27 g (80% yield), Rf 0.75 (PE/Et2O = 10/1). 1H NMR (500.17 MHz, CDCl3, 25 °C): δ = 1.57–1.62 (m, 1H, CHa, conformers B+C), 2.11–2.20 (m, 1H, CHb, conformers B+C), 2.39–2.46 (m, 2H, CH2, conformer A), 1.44–1.62 (m, 4H, 2CH2), 1.44–1.50 (m, 4H, 2CH2), 1.78–1.87 and 1.97–2.00 (m, 4H, 2CH2), 1.31–1.34 and 2.20–2.24 (m, 4H, 2CH2), 5.37–5.38 (m, 2H, 2CH, conformer B), 5.68 (s, 2H, 2CH, conformer A), 5.84 (s, 2H, 2CH, conformer C), 7.05–7.19 (m, 1H, CH), 7.21–7.22 (m, 1H, CH), 6.89–6.95 (m, 1H, CH), 7.17–7.19 (m, 1H, CH). 13C NMR (125.78 MHz, CDCl3, 25 °C): δ = 14.6 (conformer A), 16.2 (conformers B+C), 22.0 (conformer A), 22.5 (conformers B+C), 22.7 (conformer A), 22.9 (conformer A), 25.3 (conformers B+C), 25.4 (conformers B+C), 26.4 (conformer A), 26.9 (conformers B+C), 28.7 (conformer A), 29.7 (conformers B+C), 30.7 (conformer B+C), 31.2 (conformer A), 85.9 (conformer B), 86.8 (conformer C), 86.9 (conformer A), 108.8 (conformers B+C), 109.5 (conformers A), 116.2 (conformers B+C), 116.8 (conformer A), 118.4 (conformers B+C), 119.0 (conformers A), 120.6 (conformers A), 121.3 (conformers B+C), 129.7 (conformers A), 129.9 (conformers B+C), 134.4, 151.2. MALDI TOF/TOF, m/z: 338 [M − H]+. Anal. calcd for C17H22ClNO4: C, 60.09; H, 6.53 N, 4.12%. Found: C, 60.07; H, 6.51 N, 4.10%.11-(2-Fluorophenyl)-2,3,5,6-tetraoxa-11-azaspiro[bicyclo[5.3.1]undecane-4,1′-cyclohexane] 12b
Brown crystals; 0.26 g (83% yield), Rf 0.77 (PE/Et2O = 10/1), mp 80–82 °C. 1H NMR (500.17 MHz, CDCl3, 25 °C): δ = 1.56–1.60 (m, 1H, CHa), 2.14–2.24 (m, 1H, CHb), 1.60–1.66 (m, 2H, CH2), 1.54–1.59 (m, 2H, CH2), 1.46–1.50 (m, 4H, 2CH2), 1.87–2.02 (m, 4H, 2CH2), 2.28–2.30 (m, 2H, CH2), 5.15 (d, 2H, J = 10 Hz, 2CH, conformer B), 5.34 (br.s, 2H, 2CH, conformer A), 5.44–5.45 (m, 2H, 2CH, conformer C), 6.93–7.12 (m, 3H, CH), 7.84–7.87 (m, 1H, CH). 13C NMR (125.78 MHz, CDCl3, 25 °C): δ = 14.9 (conformer A), 16.4 (conformers B+C), 22.0 (conformer A), 22.6 (conformers B+C), 22.9 (conformer B+C), 22.9 (conformer A), 25.4 (conformers B+C), 25.5 (conformers B+C), 26.1 (conformer A), 30.8 (conformers B+C), 31.6 (conformer A), 88.9 (conformer B), 89.3 (conformer A), 89.5 (conformer C), 108.7 (conformers B+C), 115.9 (J = 17, conformers B+C), 116.1 (J = 17 Hz, conformers A), 123.6 (J = 6), 124.4, 124.8, 137.9 (J = 6), 156.3 (J = 193). MALDI TOF/TOF, m/z: 322 [M − H]+. Anal. calcd for C17H22FNO4: C, 63.14; H, 6.86; N, 4.33%. Found: C, 63.12; H, 6.84; N, 4.30%.11-(3-Fluorophenyl)-2,3,5,6-tetraoxa-11-azaspiro[bicyclo[5.3.1]undecane-4,1′-cyclohexane] 12c
Brown oil; 0.24 g (75% yield), Rf 0.81 (PE/Et2O = 10/1). 1H NMR (500.17 MHz, CDCl3, 25 °C): δ = 1.31–1.46 (m, 4H, CH2, 2CHa), 1.48–1.62 (m, 5H, 2CH2, CHa), 1.72–1.99 (m, 4H, 2CH2), 2.14–2.19 (m, 1H, CHb), 2.22–2.25 (m, 2H, CHb), 5.37–5.39 (m, 2H, 2CH, conformer C), 5.69 (br.s, 2H, 2CH, conformer A), 5.85–5.86 (m, 2H, 2CH, conformer B), 6.59–6.68 (m, 1H, CH), 6.89–6.98 (m, 2H, CH), 7.15–7.25 (m, 1H, CH). 13C NMR (125.78 MHz, CDCl3, 25 °C): δ = 14.2 (conformer B+C), 14.6 (conformer A), 21.0 (conformers B+C), 22.0 (conformer A), 22.5 (conformer B), 22.7 (conformer A), 22.8 (conformer C), 25.0 (conformer C), 25.3 (conformer B), 25.4 (conformer A), 26.4 (conformer A), 26.9 (conformer C), 27.0 (conformer B), 85.6 (conformer B), 86.7 (conformer C), 86.9 (conformer A), 104.8 (J = 17, conformer C), 105.3 (J = 20, conformer B), 105.9 (J = 20, conformer A), 107.2 (J = 17 conformer A), 107.8 (J = 17, conformers B+C), 108.8 (conformers B+C), 109.5 (conformer A), 113.3 (conformers B+C), 114.0 (conformer A), 129.7 (J = 8, conformers A), 129.9 (J = 7, conformer B), 130.4 (J = 8, conformer C), 151.3 (J = 8, conformers B+C), 151.8 (J = 8, conformer A), 163.3 (J = 194). MALDI TOF/TOF, m/z: 322 [M − H]+. Anal. calcd for C17H22FNO4: C, 63.14; H, 6.86; N, 4.33%. Found: C, 63.11; H, 6.85; N, 4.32%.11-(2-Chlorophenyl)-4′-methyl-2,3,5,6-tetraoxa-11-azaspiro[bicyclo[5.3.1]undecane-4,1′-cyclohexane] 13f
Orange oil; 0.27 g (78% yield), Rf 0.76 (PE/Et2O = 10/1). 1H NMR (500.17 MHz, CDCl3, 25 °C): δ = 0.97 (d, 3H, J = 10 Hz, CH3, conformer A), 1.04 (d, 3H, J = 10 Hz, CH3, conformer B+C), 1.58–1.59 and 2.21–2.28 (m, 2H, CH2), 1.89–1.92 and 2.14–2.19 (m, 4H, 2CH2), 1.59–1.61 and 3.09–3.11 (m, 4H, CH2), 1.23–1.73 (m, 4H, 2CH2), 1.98–2.03 (m, 1H, CH), 5.03–5.52 (m, 2H, 2CH), 6.69–6.78 (m, 2H, 2CH), 6.97–7.40 (m, 2H, 2CH). 13C NMR (125.78 MHz, CDCl3, 25 °C): δ = 14.2 (conformer C), 15.0 (conformers A), 16.6 (conformer B), 21.6 (conformer A), 21.0 (conformers B+C), 25.9 and 26.0 and 26.7 (conformers A+B+C), 30.9 and 31.2 (conformer B+C), 31.0 (conformers A), 31.4 (conformers B+C), 31.5 (conformer A), 31.7 and 31.9 (conformers B+C), 31.7 (conformer A), 34.8, 89.7 (conformer A), 89.6 and 89.8 and 89.9 (conformer A+B+C), 109.6, 113.7 (conformers B+C), 115.9 (conformers A), 119.0 (conformer A), 119.3 and 119.4 (conformers B+C), 127.4 and 127.6 and 127.8 (conformers B+C), 127.6 (conformers A), 129.2 (conformers B+C), 129.4 (conformers A), 130.2 (conformers B+C), 130.5 (conformer A), 143.0 (conformer B+C), 147.1 (conformer A), MALDI TOF/TOF, m/z: 322 [M − H]+. Anal. calcd for C18H24ClNO4: C, 61.10; H, 6.84; N, 3.96%. Found: C, 61.08; H, 6.82; N, 3.94%.11-(2-Chlorophenyl)-2,3,5,6-tetraoxa-11-azaspiro[bicyclo[5.3.1]undecane-4,1′-cyclooctane] 14a
Orange oil; 0.26 g (70% yield), Rf 0.77 (PE/Et2O = 10/1). 1H NMR (500.17 MHz, CDCl3, 25 °C): δ = 1.56–1.59 (m, 1H, CHa), 2.14–2.15 (m, 1H, CHb), 1.63–1.67 and 1.51–1.53 (m, 4H, 2CH2), 2.38–2.40 (m, 2H, 2CH2), 2.17–2.24 and 1.95–2.00 and 1.77–1.87 (m, 6H, 3CH2), 1.42–1.44 and 1.63–1.67 and 1.80–1.85 (m, 4H, 2CH2), 5.67 (s, 2H, 2CH, conformer A), 5.80 (s, 2H, 2CH, conformer B+C), 7.01–7.03 (m, 2H, 2CH, conformer A), 7.10–7.15 (m, 2H, 2CH, conformer B+C), 6.96–6.97 (m, 2H, 2CH, conformer B+C), 7.19–7.27 (m, 2H, 2CH, conformer A). 13C NMR (125.78 MHz, CDCl3, 25 °C): δ = 14.6 (conformer A), 16.3 (conformer B+C), 22.0 (conformers B+C), 22.3 (conformer A), 22.1, 26.4 (conformer A), 26.6 (conformer B+C), 31.2 (conformer A), 31.4 (conformer B+C), 86.0 (conformer B+C), 86.9 (conformer A), 111.1, 119.0 (conformer A), 119.8 (conformer B+C), 124.5, 128.7 (conformers A), 129.0 (conformers B+C), 144.8. MALDI TOF/TOF, m/z: 366 [M − H]+. Anal. calcd for C19H26ClNO4: C, 62.04; H, 7.12; N, 3.81%. Found: C, 62.02; H, 7.10; N, 3.79%.11-(2-Chlorophenyl)-2,3,5,6-tetraoxa-11-azaspiro[bicyclo[5.3.1]undecane-4,1′-cyclooctane] 14g
Orange solid; 0.27 g (74% yield), Rf 0.74 (PE/Et2O = 10/1), mp 98–100 °C. 1H NMR (500.17 MHz, CDCl3, 25 °C): δ = 1.46–1.51 (m, 1H, CHa), 2.09–2.19 (m, 1H, CHb), 1.38–1.69 (m, 4H, 2CH2), 2.34–2.36 and 1.38–1.50 (m, 2H, 2CH2), 1.29–1.84 (m, 6H, 3CH2), 1.34–1.40 (m, 4H, 2CH2), 5.61 (s, 2H, 2CH, conformer A), 5.75 (s, 2H, 2CH, conformer B+C), 6.45–6.47 (m, 1H, CH), 6.58–6.59 (m, 1H, CH), 6.62–6.63 (m, 1H, CH), 6.95–7.19 (m, H, CH). 13C NMR (125.78 MHz, CDCl3, 25 °C): δ = 14.2 (conformer A), 16.1 (conformer B+C), 22.0 (conformers A), 22.3 (conformer B+C), 24.7 (conformer A), 25.1 (conformer B+C), 25.7, 31.0 (conformer B+C), 31.4 (conformers A), 85.8 (conformer B), 86.7 (conformer C), 86.7 (conformer A), 113.3, 112.5 (conformer A), 113.2 (conformer B+C), 114.7, 117.9 (conformers A), 118.2 (conformers B+C), 129.7 (conformers B+C), 130.3 (conformers A), 134.3 (conformers B+C), 134.6 (conformers A), 147.4 (conformers B+C), 148.1 (conformers A). MALDI TOF/TOF, m/z: 366 [M − H]+. Anal. calcd for C19H26ClNO4: C, 62.04; H, 7.12; N, 3.81%. Found: C, 62.01; H, 7.09; N, 3.79%.11-(2-Chlorophenyl)-2,3,5,6-tetraoxa-11-azaspiro[bicyclo[5.3.1]undecane-4,1′-cyclododecane] 15a
Orange crystal; 0.33 g (79% yield), Rf 0.78 (PE/Et2O = 10/1), mp 82–84 °C. 1H NMR (500.17 MHz, CDCl3, 25 °C): δ = 1.27–1.36 (m, 12H, 6CH2), 1.70–1.75 (m, 2H, CH2), 1.27–1.98 (m, 4H, 2CH2), 2.15–2.20 and 1.98–2.20 (m, 4H, 2CH2), 2.15–2.41 (m, 2H, CH2), 5.35–5.36 (m, 2H, 2CH, conformer B), 5.66 (s, 2H, 2CH, conformer A), 5.82 (s, 2H, 2CH, conformer C), 7.03–7.08 (m, 1H, CH), 7.15–7.21 (m, 2H, CH), 6.87–6.94 (m, 1H, CH). 13C NMR (125.78 MHz, CDCl3, 25 °C): δ = 14.2 (conformer A), 16.1 (conformer B+C), 22.0 (conformers A), 22.3 (conformer B+C), 24.7 (conformer A), 25.1 (conformer B+C), 25.7, 31.0 (conformer B+C), 31.4 (conformers A), 85.8 (conformer B), 86.7 (conformer C), 86.7 (conformer A), 113.3, 112.5 (conformer A), 113.2 (conformer B+C), 114.7, 117.9 (conformers A), 118.2 (conformers B+C), 129.7 (conformers B+C), 130.3 (conformers A), 134.3 (conformers B+C), 134.6 (conformers A), 147.4 (conformers B+C), 148.1 (conformers A). MALDI TOF/TOF, m/z: 366 [M − H]+. Anal. calcd for C23H34ClNO4: C, 65.16; H, 8.08; N, 3.30%. Found: C, 65.12; H, 8.06; N, 3.28%.11'-(3-Chlorophenyl)-2′,3′,5′,6′-tetraoxa-11′-azaspiro[adamantane-2,4′-bicyclo[5.3.1]undecane] 16g
Brown oil; 0.28 g (72% yield), Rf 0.80 (PE/Et2O = 10/1). 1H NMR (400 MHz, CDCl3, 25 °C): δ = 1.66–1.74 (m, 2H, CH2-Ad), 1.77 (m, 1H, CH-Ad, conformer A), 1.82–1.86 (m, 8H, CH2-Ad), 1.95–2.01 (m, 4H, CH2-Ad, conformer A), 2.04–2.06 (m, 4H, CH2-Ad), 2.10–2.17 (m, 4H, 2CH2), 2.19–2.28 (m, 2H, CH2), 2.31 (br.s, 2H, CH2-Ad), 2.39 (br.s, 1H, CH2-Ad, conformer A), 5.48–5.49 (m, 2H, 2CH, conformer B), 5.68 (s, 2H, 2CH, conformer A), 5.81 (s, 2H, 2CH, conformer C), 6.77–6.84 (m, 1H, CH), 6.89–6.95 (m, 1H, CH), 7.02–7.13 (m, 1H, CH), 7.15–7.23 (m, 1H, CH). 13C NMR (100 MHz, CDCl3, 25 °C): δ = 14.6 (conformer A), 16.3 (conformer B+C), 26.5, 27.1 (conformer B+C), 27.2 (conformer A), 30.3, 33.1 (conformer A), 33.6 (conformer B+C), 34.2 (conformers B+C), 34.4 (conformer A), 37.3 (conformer B+C), 37.4 (conformer A), 84.8 (conformer B), 86.1 (conformer C), 86.9 (conformer A), 110.9 (conformer B), 111.5 (conformer A), 112.6 (conformer C), 116.6 (conformers B+C), 116.9 (conformer A), 118.8 (conformers A), 119.1 (conformers B+C), 120.5 (conformer A), 121.4 (conformers B+C), 129.7, 151.3. MALDI TOF/TOF, m/z: 390 [M − H]+. Anal. calcd for C21H26ClNO4: C, 64.36; H, 6.69; N, 3.57%. Found: C, 64.33; H, 6.67; N, 3.55%.Cyclocondensation reactions of gem-dihydroperoxides with glyoxal catalyzed by Sm(NO3)3·6H2O
General procedure: a Schlenk vessel mounted on a magnetic stirrer was charged at ∼20 °C with tetrahydrofuran (5 ml), glyoxal (10 mmol), and specified gem-dihydroperoxides (10 mmol).29 Then Sm(NO3)2·6H2O (0.062 g, 5 mol% relative to 1,1′-peroxybis(1-hydroperoxycycloalkane)) was added. The reaction mixture was stirred at ∼20 °C for 1 h. After completion of the reaction H2O (5 ml) and CH2Cl2 (5 ml) were added. The organic layer was separated, dried (anhydrous MgSO4) and concentrated to isolate products stable during storage at room temperature. Products of the reaction were purified by column chromatography on SiO2 using 10:1 PE:Et2O as the eluent. The progress of reactions was monitored by TLC, with a 5:1 hexane:EtOAc mixture as the eluent, visualization was performed with I2 vapor.
6,7,10,11-Tetraoxaspiro[4.6]undecane-8,9-diol 18
White solid; 0.16 g (82% yield), Rf 0.79 (PE/Et2O = 10/1), mp = 96–98 °C. 1H NMR (500.17 MHz, CDCl3, 25 °C): δ = 1.72–1.80 (m, 4H, CH2) 1.90–2.04 (m, 4H, CH2), 5.15–5.22 (m, 2H, 2CH). 13C NMR (125.78 MHz, CDCl3, 25 °C): δ = 24.2, 24.3, 33.5, 33.7, 90.9, 91.1, 110.4. MALDI TOF/TOF, m/z: 191 [M − H]+. Anal. calcd for C7H12O6: C, 43.75; H, 6.29%. Found: C, 43.73; H, 6.27%.7,8,11,12-Tetraoxaspiro[5.6]dodecane-9,10-diol 19
White solid; 0.17 g (85% yield), Rf 0.76 (PE/Et2O = 10/1), mp = 54–56 °C. 1H NMR (500.17 MHz, CDCl3, 25 °C): δ = 1.46–1.47 (m, 4H, CH2), 1.59–1.64 (m, 2H, CH2), 1.88–1.90 (m, 4H, CH2) 5.27–5.33 (m, 2H, 2CH). 13C NMR (125.78 MHz, CDCl3, 25 °C): δ = 22.5, 22.6, 25.2, 25.4, 29.8, 30.7, 90.5, 91.0, 113.5. MALDI TOF/TOF, m/z: 205 [M − H]+. Anal. calcd for C8H14O6: C, 46.60; H, 6.84%. Found: C, 46.58; H, 6.81%.3-Methyl-7,8,11,12-tetraoxaspiro[5.6]dodecane-9,10-diol 20
White solid; 0.18 g (85% yield), Rf 0.76 (PE/Et2O = 10/1), mp = 68–70 °C. 1H NMR (500.17 MHz, CDCl3, 25 °C): δ = 0.91–0.93 (m, 3H, CH3), 1.30–1.46 (m, 4H, CH2), 1.61–1.62 (m, 4H, CH2), 1.98–2.03 (m, 1H, CH) 5.20–5.35 (m, 2H, 2CH). 13C NMR (125.78 MHz, CDCl3, 25 °C): δ = 20.5, 22.6, 22.7, 25.0, 25.2, 31.2, 31.6, 32.0, 32.7, 90.3, 91.2, 113.5. MALDI TOF/TOF, m/z: 219 [M − H]+. Anal. calcd for C9H16O6: C, 49.09; H, 7.32%. Found: C, 49.07; H, 7.30%.Crystal structure determination and refinement
The crystallographic data, coordinates of atoms, and geometric parameters for compounds 4a, 4b, 4d, 4e, 12b were deposited at the Cambridge Crystallographic Data Centre as a CIF deposition with file number CCDC 1905323, 1905327, 1905330, 1905341, 1905334, 1905337, respectively.245 reflections were measured, from which 4644 were independent. The refinement converged to R1 = 0.0608, wR2 = 0.1916, GOF = 0.987.![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 6.4738(3), b = 10.2056(9) and c = 12.4931(10) Å, α = 75.029(7)°, β = 80.670(6)°, γ = 88.090(6)°, V = 786.81(11) Å3, dcalc = 1.365 g cm−3, Z = 2, μ = 0.104 mm−1, 2θmax = 58.558°, 6406 reflections were measured, from which 3602 were independent. The refinement converged to R1 = 0.0472, wR2 = 0.1235, GOF = 1.022.
, a = 6.4738(3), b = 10.2056(9) and c = 12.4931(10) Å, α = 75.029(7)°, β = 80.670(6)°, γ = 88.090(6)°, V = 786.81(11) Å3, dcalc = 1.365 g cm−3, Z = 2, μ = 0.104 mm−1, 2θmax = 58.558°, 6406 reflections were measured, from which 3602 were independent. The refinement converged to R1 = 0.0472, wR2 = 0.1235, GOF = 1.022.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Russian Science Foundation (RSF projects 18-73-00014). The structural studies of the synthesized compounds were performed with the use of Collective Usage Centre “Agidel” at the Institute of Petrochemistry and Catalysis of RAS. The anticancer activity studies of the synthesized compounds were performed in laboratory of molecular design and biological screening of candidate substances for the pharmaceutical industry at the Institute of Petrochemistry and Catalysis of RAS.Notes and references
- (a) D. A. Casteel, Nat. Prod. Rep., 1992, 9, 289 RSC; (b) K. J. McCullough and M. Nojima, Curr. Org. Chem., 2001, 5, 601 CrossRef CAS; (c) C. W. Jefford, in Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Pergamon, Oxford, vol. 6, ch. 20, 1996 Search PubMed; (d) R. K. Haynes and S. C. Vonwiller, Acc. Chem. Res., 1997, 30, 73 CrossRef CAS; (e) A. K. Bhattacharya and R. P. Sharma, Heterocycles, 1999, 51, 1681 CrossRef CAS; (f) Y. Dong, Mini-Rev. Med. Chem., 2002, 2, 113 CrossRef CAS PubMed. For the synthesis of 1,2,4-trioxan-5-ones relevant to the present work, see; (g) C. W. Jefford, J.-C. Rossier and G. D. Richardson, J. Chem. Soc., Chem. Commun., 1983, 1064 RSC; (h) C. W. Jefford, J. Currie, G. D. Richardson and J.-C. Rossier, Helv. Chim. Acta, 1991, 74, 1239 CrossRef CAS.
- K. J. McCullough, H. Tokuhara, A. Masuyama and M. Nojima, Org. Biomol. Chem., 2003, 1, 1522 RSC.
- H.-S. Kim, Y. Nagai, K. Ono, K. Begum, Y. Wataya, Y. Hamada, K. Tsuchiya, A. Masuyama, M. Nojima and K. J. McCullough, J. Med. Chem., 2001, 44, 2357 CrossRef CAS PubMed.
- K. Tsuchiya, Y. Hamada, A. Masuyama, M. Nojima, K. J. McCullough, H.-S. Kim and Y. Wataya, Tetrahedron Lett., 1999, 40, 4077 CrossRef CAS.
- H.-S. Kim, K. Begum, N. Ogura, Y. Wataya, Y. Nonami, Y. Ito, A. Masuyama, M. Nojima and K. J. McCullough, J. Med. Chem., 2003, 46, 1957 CrossRef CAS PubMed.
- Y. Nonami, T. Tokuyasu, A. Masuyama, M. Nojima, K. J. McCullough, H.-S. Kim and Y. Wataya, Tetrahedron Lett., 2000, 41, 4681 CrossRef CAS.
- G.-H. Li, D.-Q. Dong, Q. Deng, S.-Q. Yan and Z.-L. Wang, Synthesis, 2019, 51, 3313 CrossRef CAS.
- F.-L. Zeng, X.-L. Chen, S.-Q. He, K. Sun, Y. Liu, R. Fu, L.-B. Qu, Y.-F. Zhao and B. Yu, Org. Chem. Front., 2019, 6, 1476 RSC.
- K. Sun, Z. Liu, B. Luan, J. Zhu and Y. Xue, Org. Lett., 2018, 20, 6687 CrossRef CAS PubMed.
- X. Huang, N. Rong, P. Li, G. Shen, Q. Li, N. Xin, C. Cui, J. Cui, B. yang, D. Li, C. Zhao, J. Dou and B. Wang, Org. Lett., 2018, 20, 3332 CrossRef CAS PubMed.
- C. Wu, X. Xin, Z.-M. Fu, L.-Y. Xie, K.-J. Liu, Z. Wang, W. Li, Z.-H. Yuan and W.-M. He, Green Chem., 2017, 19, 1983 RSC.
- L.-H. Lu, Z. Wang, P. Cheng, B. Zhang, Z. Cao and W.-M. He, Chin. Chem. Lett., 2019, 20, 1237 CrossRef.
- J. Fayos, D. Lokensgard, J. Clardy, R. J. Cole and J. W. Kirksey, J. Am. Chem. Soc., 1974, 96, 6785 CrossRef CAS PubMed.
- R. J. Cole, J. W. Kirsey, J. W. Dorner, D. M. Wilson, J. Johnson Jr, N. Johnson, D. M. Dedell, J. P. Springer, K. K. Chexel, J. Clardy and R. H. Cox, J. Agric. Food Chem., 1977, 25, 826 CrossRef CAS PubMed.
- C. Madelaine, O. Buriez, B. Crousse, I. Florent, Ph. Grellier, P. Retailleau and Y. Six, Org. Biomol. Chem., 2010, 8, 5591 RSC.
- D. A. Casteel, Nat. Prod. Rep., 1999, 16, 55 RSC.
- N. N. Makhmudiyarova, G. M. Khatmullina, R. Sh. Rakhimov, A. G. Ibragimov and U. M. Dzhemilev, ARKIVOC, 2016, 427 Search PubMed.
- N. N. Makhmudiyarova, G. M. Khatmullina, R. Sh. Rakhimov, E. S. Meshcheryakova, A. G. Ibragimov and U. M. Dzhemilev, Tetrahedron, 2016, 72, 3277 CrossRef CAS.
- T. V. Tyumkina, N. N. Makhmudiyarova, G. M. Kiyamutdinova, E. S. Meshcheryakova, K. Sh. Bikmukhametov, M. F. Abdullin, L. M. Khalilov, A. G. Ibragimov and U. M. Dzhemilev, Tetrahedron, 2018, 74, 1749 CrossRef CAS.
- (a) N. N. Makhmudiyarova, I. R. Ishmukhametova, T. V. Tyumkina, A. G. Ibragimov and U. M. Dzhemilev, Tetrahedron Lett., 2018, 59, 3161 CrossRef CAS; (b) N. N. Makhmudiyarova, I. R. Ishmukhametova, L. U. Dzhemileva, T. V. Tyumkina, V. A. D'yakonov, A. G. Ibragimov and U. M. Dzhemilev, RSC Adv., 2019, 9, 18923 RSC.
- (a) G. Verardo, A. G. Giumanini, G. Favret and P. Strazzolini, Synthesis, 1991, 447 CrossRef CAS; (b) B. Li, S. Liu, M. Wu, Q. Lin, W. Deng, Sh. Jiang and L. Chen, Tetrahedron Lett., 2018, 59, 3467 CrossRef CAS.
- (a) C. Denekamp, L. Gottlieb, T. Tamiri, A. Tsoglin, R. Shilav and M. Karon, Org. Lett., 2005, 7, 2461 CrossRef CAS PubMed; (b) N. Haroune, A. Crowson and B. Campbell, Sci. Justice, 2011, 51, 50 CrossRef CAS PubMed.
- N. N. Makhmudiyarova, R. Sh. Rakhimov, T. V. Tyumkina, E. S. Meshcheryakova, A. G. Ibragimov and U. M. Dzhemilev, Russ. J. Org. Chem., 2019, 5, 620 CrossRef.
- (a) M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta Jr, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision D.01), Gaussian, Inc., Wallingford, CT, 2013 Search PubMed; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS PubMed; (d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
- Agilent Technologies Ltd, CrysAlis PRO, Yarnton, Oxfordshire, England, 2012 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. Van De Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS.
- A. O. Terent'ev, M. M. Platonov, E. J. Sonneveld, R. Peschar, V. V. Chernyshev, Z. A. Starikova and G. I. Nikishin, J. Org. Chem., 2007, 72, 7237c CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1905323, 1905327, 1905330, 1905341, 1905334, 1905337. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra06372b |
| This journal is © The Royal Society of Chemistry 2019 |