
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
TBHP-mediated oxidative synthesis of substituted pyrimido[4,5-d]pyrimidines from N-uracil amidines and methylarenes under metal free conditions†
Pradip Debnath *
*
Department of Chemistry, Maharaja Bir Bikram College, Agartala, Tripura-799004, India. E-mail: pradipchem78@gmail.com; pdn0099@yahoo.co.in; Fax: +3812516728; Tel: +3812526607
First published on 20th September 2019
Abstract
An efficient and operationally simple protocol has been demonstrated for the synthesis of 1,3,5,7-tetrasubstituted pyrimido[4,5-d]pyrimidines via TBHP-mediated direct oxidative coupling of N-uracil amidines and methylarenes under metal-free conditions. Due to the inherent stability of methylarenes compared to aldehydes, the presented synthetic protocol is adaptable to a broad substrate scope, is operationally simple, has no need for stringent protection in the whole preparation process, and has the potential to prepare valuable products that are currently inaccessible or challenging to prepare using conventional methods. It is a significantly important complement to the conventional synthetic methods. The reaction possesses an efficient tandem oxidation–imination–cyclization process.
Introduction
Nitrogen containing heterocycles play leading roles in the field of biological and medicinal chemistry.1 These molecules have also been extensively used in the fields of agrochemicals, pesticides and pharmaceuticals, and in industry.2 Over the past few decades, great efforts have been made to develop novel and efficient methods for the construction of nitrogen containing heterocycles.3 Pyrimidines are one of the most important nitrogen heterocycles, exhibiting remarkable pharmaceutical activities.4 Among them, pyrimido[4,5-d]pyrimidines are very important scaffolds found in many natural products and synthetic drugs or drug candidates exhibiting a wide range of biological activities.5 These compounds have attracted considerable attention in medicinal chemistry due to their significant and diverse biological activities, including anticancer,6 antiviral,7 antifungal,8a antioxidant,8b antitumor,9 and hepatoprotective.10 Some of their derivatives are also useful as bronchodilators,6 vasodilators,11 antiallergic,12 antihypertensive13 agents. Dipyridamole, a pyrimidopyrimidine based heterocycles, is a medicine that inhibits the phosphodiesterase enzyme, lowers pulmonary hypertension,13b and is also used in electro-cardiogram and echocardiography. Recently, pyrimido[4,5-d]pyrimidine analogues of folic acid have also been screened for antitumor activity.14 Despite these enormous applications, the approaches for the preparation of this type of compounds still remain scarce.15,16 According to the literature survey, the previously reported methods for the preparation of pyrimido[4,5-d]pyrimidines are highlighted in Scheme 1. The most frequent routes to pyrimido[4,5-d]pyrimidines typically involve three-component reactions involving 6-aminouracils15 or by the cycloaddition reactions16 between 6-methylideneaminouracils and electron-deficient substrates. The latter method is restricted to electron deficient substrates and does not offer unrestricted scope for the synthesis of products. Other promising methods for the synthesis of pyrimido[4,5-d]pyrimidines involve multistep syntheses starting from 1,3-disubstituted cyanouracils,17 polymer bounded 2-(alkylsulfanyl)-4-aminopyrimidine-5-carbonitrile etc. A new approach to the synthesis of pyrimido[4,5-d]pyrimidines reported by Wamhoff et al.18 is the aza-Wittig type reaction of iminophosphoranes of 5-aminouracils leading to functionalised pyrimido[4,5-d]pyrimidines. Wang and co-workers reported a protocol for the synthesis of tetrasubstituted pyrimido[4,5-d]-pyrimidine derivatives by sequential reactions of 6-aminouracil with hydrazine involving 6 steps.19 Tummaluru and co-workers also developed a one-pot protocol for the synthesis of 5,6-dihydropyrimido[4,5-d]pyrimidines involving 6-amino-N,N-dimethyluracil, phenylisocyanate, and aromatic aldehydes in water solvent.20 In another approach, Ghorbani-Vaghei and Sarmast employed a magnetic nanoparticle Fe3O4@SiO2 as catalyst in water solvent for the synthesis of substituted pyrimido[4,5-d]pyrimidines.21 Nevertheless, these methods are associated with few shortcomings such as – harsh reaction conditions (high temperature, microwave),16a,b use of toxic solvents, use of metal catalysts, or use of complex synthetic pathways,19 and less-environmentally benign, or not readily available substrates.22a,23 So, there is a necessity to develop more effective and sustainable chemical procedures for the synthesis of pyrimido[4,5-d]pyrimidines. More viable and efficient approaches are being tried to alleviate the shortcomings. Recently, Deb and co-workers have developed a new strategy for the synthesis of 5,6-dihydropyrimido[4,5-d]pyrimidines based on a cascade 3-component reaction involving 6-aminouracil, aldehydes and tetrahydroisoquinolines under solvent free reaction conditions.24 Very recently, we have shown that pyrimido[4,5-d]pyrimidines could be achieved by the direct annulation of N-uracil amidines with benzaldehydes under transition metal free conditions.25 Although, this method is highly efficient but the use of aldehydes as the coupling partners could frequently meet some problems such as – (i) the active aldehyde groups may suffer from an oxidation reaction leading to the formation of undesirable by-products, and hence necessitating the requirement for inert conditions;26 (ii) aldehydes could undergo a decarbonylation reaction under harsh reaction conditions resulting in lower yields of the products;27 (iii) cost of some aldehydes are high or not readily available. On the basis of these facts, the search for readily available, in expensive and stable alternatives of aldehydes would provide a new avenue for the synthesis of pyrimido[4,5-d]pyrimidines and is of high importance.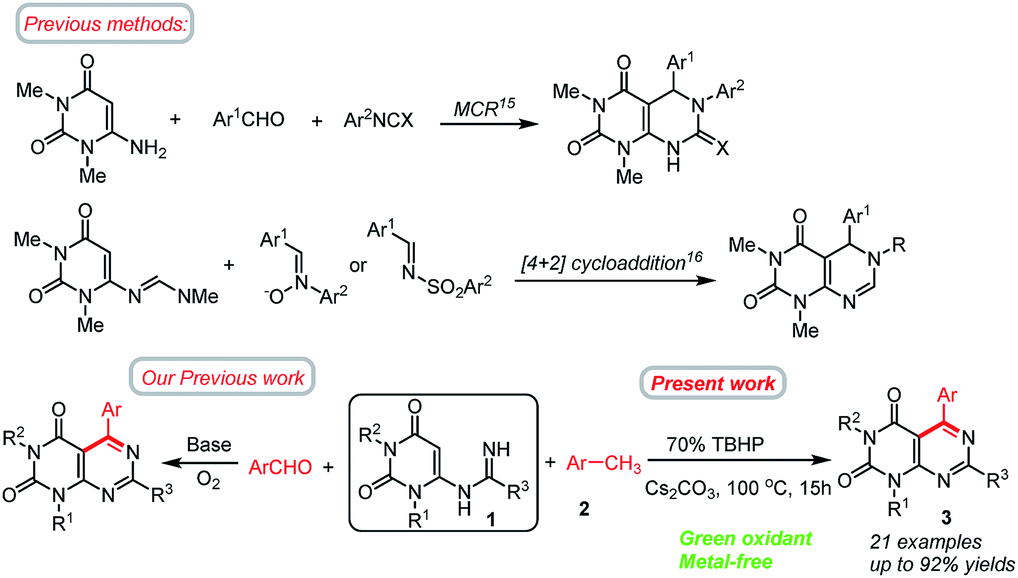 | ||
| Scheme 1 Selected methods for the synthesis of pyrimido[4,5-d]pyrimidines from 6-substituted uracil. | ||
In recent years, the use of abundant and sustainable methylarenes for carbon–carbon (C–C) and carbon–nitrogen (C–N) bond formations have received considerable attentions.28,29 Methylarenes are cheap, stable, less toxic, commercially available, and easy to handle, thus making it advantageous to be used as ideal starting materials. By exploring suitable oxidant systems, the oxidation of methylarenes leading to in situ formation of aldehydes is considered to be the key step for the formation of desired products.30,31 Based on these elegant contributions, we believed that methylarenes could be applied as latent aldehydes32 for the synthesis of aryl substituted pyrimido[4,5-d]pyrimidines. Compared to aldehydes, methylarenes have a variety of advantages such as cost-effectiveness, thermodynamic stability, abundance and sustainability. On the other hand, burgeoning metal-free organo-catalyst C–H functionalization strategies have attracted much attention in recent years in many oxidation processes with TBHP as an oxidant, which have outstanding performances and advantages such as inexpensive, safe, environmentally benign, mild conditions, and ease of purification.32,33
Herein, we wish to develop a new method for the synthesis of N1, N3, C5 and C7 tetrasubstituted pyrimido[4,5-d]pyrimidines directly from N-uracil amidine and methylarenes under meta-free conditions. SNAE of the C6 halogen of 6-chlorouracil by an amidine leads to an N-uracil amidine (1),34 which can be converted to the corresponding pyrimido[4,5-d]pyrimidines (3) by the oxidative coupling with methylarenes (2) using TBHP as a green oxidant (Scheme 1). The reaction possesses an efficient oxidation–imination–cyclization tandem process. The requisite amidine substrates can be easily prepared using a Pinner approach,35 starting from the corresponding nitriles and ammonia, and allowing installation of R at C7 position. To the best of our knowledge, such a synthetic protocol has not been reported.
Results and discussion
At the outset, we began our study by investigating the TBHP mediated oxidative coupling of N-uracil amidine 1a and toluene 2a under basic medium. We are pleased to obtain the desired product of pyrimido[4,5-d]pyrimidine 3a in 52% isolated yield when the reaction was performed using K2CO3 as base in the presence of 70% TBHP (3.0 equiv.) in toluene (2a) at 100 °C (Table 1, entry 1). For optimizing the reaction conditions, the effect of oxidants was investigated, and we found that oxidant plays an important role in this oxidation process. 70% TBHP was found to be superior to TBHP and PhI(OAc)2. Other oxidant such as H2O2, O2 were proved completely ineffective (Table 1, entries 4 and 5). Various bases such as K3PO4, Cs2CO3, KOAc, Et3N, and DBU were also screened (Table 1, entries 6–10). Cs2CO3 was found to be the optimum choice and provided 82% yield of the desired product (Table 1, entry 7). The reaction did not take place in the presence of organic bases such as Et3N and DBU (entries 8 and 9). No products were obtained in the absence of base or oxidant (Table 1, entries 11 and 12). When the amount of oxidant or base were decreased, dramatic lowering of yields was observed (Table 1, entries 13 and 14). Next, the effect of temperature was studied, and it was observed that 100 °C is the optimum reaction temperature. Increase in the reaction temperature did not have any noticeable effect on this transformation, however, the yield of 3a decreased considerably when the reaction was performed at 90 °C (entries 15 and 16). Moreover, the yield of 3a dropped to 57% when the reaction was performed with a mixture 0.5 mL of toluene and 0.5 mL chlorobenzene (entry 17). Thus, the optimized reaction conditions are summarized as follows: N-uracil benzamidine (1a, 0.5 mmol), toluene (2a, 1 mL), Cs2CO3 (1.0 mmol, 2.0 equiv.), 70% TBHP (3.0 equiv.) at 100 °C for 15 h under air (Table 1, entry 7).
|
|||
|---|---|---|---|
| Entry | Oxidant | Base | Yieldb (%) |
| a Reaction conditions: all reactions were performed with 1a (0.5 mmol, 1.0 equiv.), oxidant (3.0 equiv.) and base (1.0 mmol, 2.0 equiv.) in 1 mL toluene (2a) at 100 °C, 15 h.b Isolated yield of 3a.c 1a was recovered.d 1.5 equiv. of 70% TBHP was used.e 1.5 equiv. of Cs2CO3 was used.f Reaction was carried out at 110 °C (entry 15) and at 90 °C (entry 16).g Reaction was performed with a mixture of 0.5 mL toluene and 0.5 mL chlorobenzene (entry 17). | |||
| 1 | 70% TBHP | K2CO3 | 52 |
| 2 | TBHP | K2CO3 | 37 |
| 3 | PhI(OAc)2 | K2CO3 | 21 |
| 4 | H2O2 | K2CO3 | Tracec |
| 5 | O2 | K2CO3 | 0 |
| 6 | 70% TBHP | K3PO4 | 45 |
| 7 | 70% TBHP | Cs2CO3 | 82 |
| 8 | 70% TBHP | Et3N | 0 |
| 9 | 70% TBHP | DBU | 0 |
| 10 | 70% TBHP | KOAc | 25 |
| 11 | 70% TBHP | — | 0 |
| 12 | — | Cs2CO3 | 0 |
| 13d | 70% TBHP | Cs2CO3 | 28 |
| 14e | 70% TBHP | Cs2CO3 | 40 |
| 15f | 70% TBHP | Cs2CO3 | 79 |
| 16f | 70% TBHP | Cs2CO3 | 64 |
| 17g | 70% TBHP | Cs2CO3 | 57 |
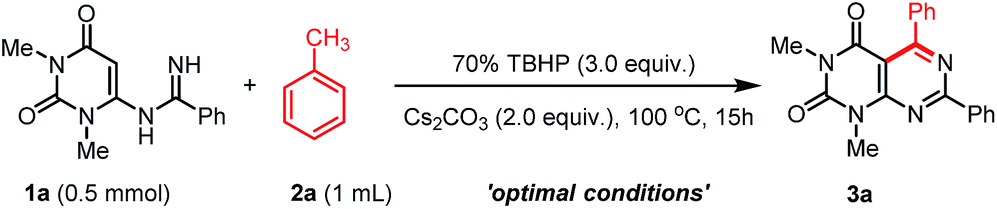
With the optimized conditions in hand, we then tested the generality and limitations of this synthetic protocol. Firstly, the effect of substitution on methylarene was investigated using N-uracil benzamidine 1a as reaction partner and the results are listed in Scheme 2. As shown in Scheme 2, all the reactions using methylarenes proceeded smoothly and furnished desired products in good to excellent isolated yields. It was observed that methylarenes with electron-donating substituents such as OMe, Me, SMe, OH produced respective pyrimido[4,5-d]-pyrimidines 3b–f and 3m in very good yields (69–90%) while methylarenes having strong electron-withdrawing substituents such as F, CN furnished slightly lower yield of the products 3j and 3k. It was also observed that methylarenes having para-substituents afforded the products in higher yields than those with ortho-substituents. We reasoned that steric hindrance by a group at the ortho-position of methylarenes might be playing a major role in lowering the yield (3d, 3i). It is pertinent to mention that when the toluene derivatives were substituted with more than one methyl group, the reaction took place on one methyl group only and the other methyl group remained unreacted (3c,d). This result might be attributed to that, once one of the methyl groups on the aromatic ring was involved in the reaction and its electron-withdrawing property was unfavourable to the subsequent oxidation of other methyl group.31,32 α-Methylnaphthalene could also be employed to react with N-uracil amidine under the optimal reaction conditions giving 3l in 85% yield. Even oxidation-sensitive functional groups such as a thiomethyl group (3e) was compatible with the reaction conditions. Upon a thorough literature investigation, it was found that the synthesis of heteroaryl substituted pyrimido[4,5-d]pyrimidines has not been explored with the conventional methods. Herein, we wish to prepare this type of compounds by employing our synthetic protocol. Representative heterocyclic methylarenes such as 2-methylthiophene and 2-methylpyridine were selected as coupling patterns for the oxidative coupling with N-uracil amidine (1a). Delightfully, both the reactions proceeded smoothly producing the corresponding pyrimido[4,5-d]pyrimidines 3n and 3o in 70% and 63% yields, respectively.
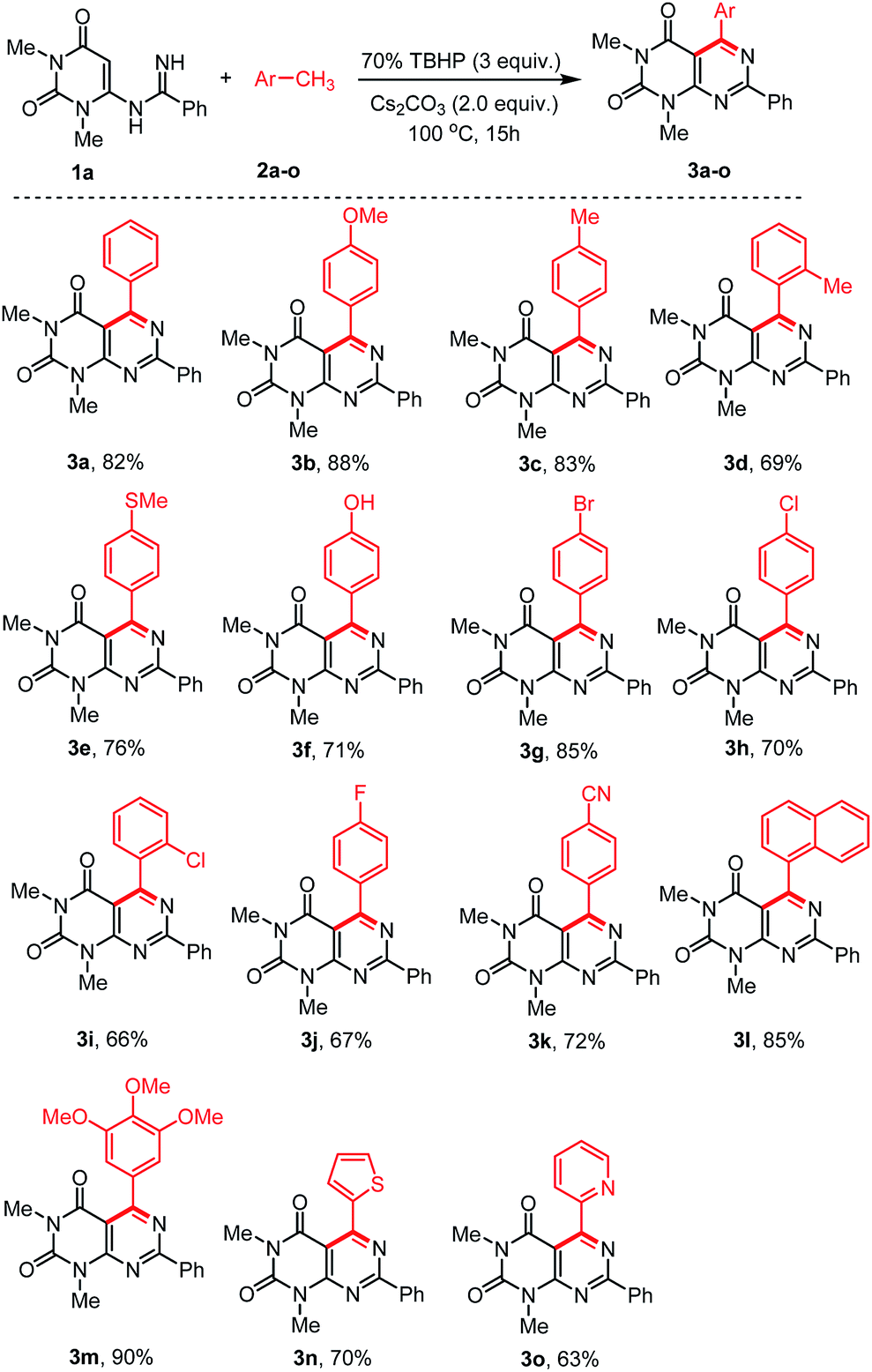 | ||
| Scheme 2 Scope of the arylmethanes (2) in the reaction with N-uracil benzamidine (1a).a,b aReaction conditions: substrate 1a (0.5 mmol), 2 (1 mL), 70% TBHP (3.0 equiv.), and Cs2CO3 (1.0 mmol, 2.0 equiv.), were stirred at 100 °C for 15 h under air. bYield of the isolated product. | ||
To extend the scope of the reaction, we also explored the reactions of different N-uracil benzimidamides (1b–h) with toluene (1a) under the standard reaction conditions (Scheme 3). The protocol tolerated a variety of substituents on the aryl ring of the N-uracil benzimidamide, with both electron-donating and electron-withdrawing groups (3p–u, Scheme 3). Only an exception was observed in case of less stable alkyl amidines such as acetamidine bearing a methyl at the R3 position (1h). No 1h was recovered and its decomposition mechanism is not clear. Notably, the para-bromo substituted pyrimido[4,5-d]pyrimidines 3g and 3s, which can be used for further cross coupling reactions, were obtained in very good yields. As fluorine can have a major influence on the pharmacological properties of a drug,36 we were delighted to see that this protocol is also compatible with this halogen (3j and 3q). Finally, it is also possible to work with N-protected uracil substrates such as N-(1-benzyl-3-methyl-2,6-dioxo-1,2,3,6-tetrahydro-pyrimidin-4-yl)benzimidamide (1f) and N-(1,3-dibenzyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)benz-imidamide (1g) under the optimized reaction conditions. Delightfully, both the reactions proceeded smoothly producing the corresponding pyrimido[4,5-d]pyrimidines 3t and 3u in 88% and 92% yields, respectively. Deprotection of these substrates37 may allowed post-decoration of the uracil moiety, via N-alkylation/arylation or dehydroxychlorination and subsequent consecutive SNAr which is particularly interesting for medicinal chemistry purposes.
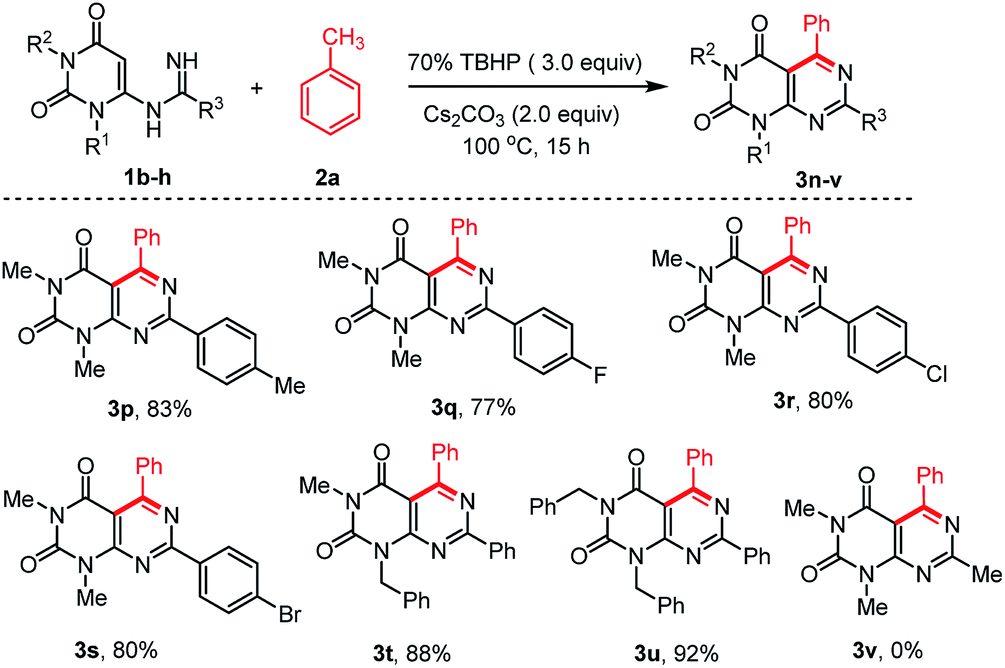 | ||
| Scheme 3 Scope of various N-uracil amidine substrates in the reaction with toluene 2a.a,b aReaction conditions: substrate 1 (0.5 mmol), 2a (1 mL), 70% TBHP (3.0 equiv.), and Cs2CO3 (1.0 mmol, 2.0 equiv.), were stirred at 100 °C for 15 h under air. bYield of the isolated product. | ||
To study the mechanism of the formation of 3, we performed some control experiments (Scheme 4). We isolated compound 3a′ along with 3a when the reaction was stopped after 5 h (Scheme 4a). When compound 3a′ was heated at 100 °C, it produced 3a in excellent yield (Scheme 4b). Therefore, we believed that the reaction proceeded through the formation of 3a′. As our reactions were performed in open air, we thought aerial oxygen might play some crucial role in the cyclization. In order to confirm the possibility of O2 being involved in the oxidation step, a control reaction under N2 atmosphere was performed and trace amount of 3a was observed along with 67% of 3a′ (Scheme 4c). These reactions confirm that oxygen is essential for the oxidation of 3a′ into 3a, however, an external supply of oxygen is not required.
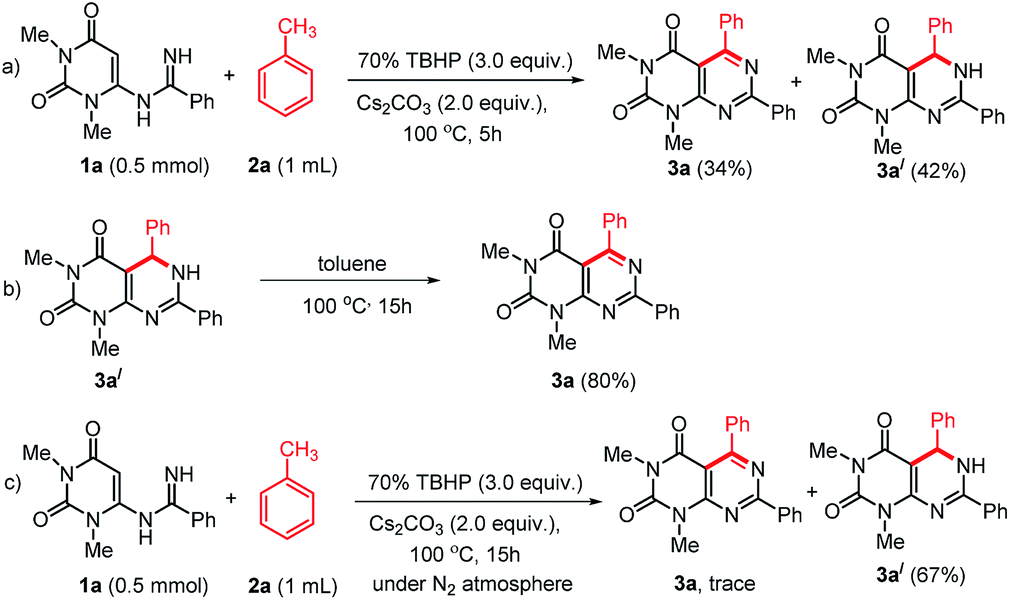 | ||
| Scheme 4 Some control experiments to establish the reaction mechanism. | ||
On the basis of the previous reports on the progresses about TBHP mediated methylarenes oxidation30–32 and based on our control experiments a plausible mechanism for the formation of 3 is proposed in Scheme 5. Initially, the reaction may proceeds with the formation of an aldehyde (A) by TBHP mediated oxidation of toluene.31,32 Then, in situ generated aldehyde A condensed with N-uracil amidine 1a to give azadiene 4.38 The intramolecular [4+2] cycloaddition reaction of 4 followed by a [1,5]-hydrogen shift to form the intermediate 5,6-dihydropyrimido[4,5-d]pyrimidine 3a′, which on oxidation by aerial oxygen affords the desired product 3a.
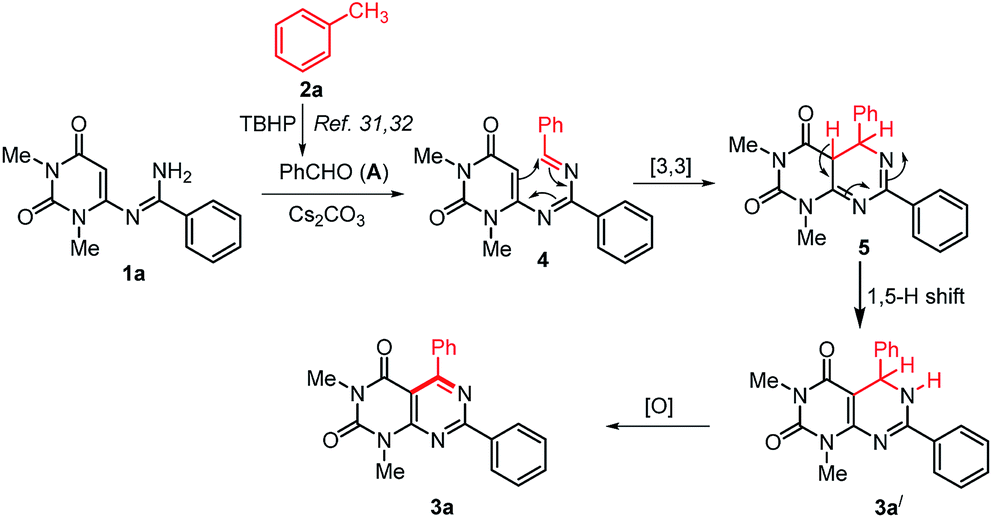 | ||
| Scheme 5 Plausible mechanism for the formation of pyrimido[4,5-d]pyrimidines. | ||
Conclusions
In summary, we have developed an efficient and straightforward method for the synthesis of 1,3,5,7-tetrasubstituted pyrimido[4,5-d]pyrimidines from inexpensive and easily available methylarenes and N-uracil amidines employing a green oxidant under metal-free conditions. Methylarenes are performed as effective aldehyde precursors in the oxidation–imination–cyclization transformation. Compared to other methods, the present protocol has a number of advantages such as – inherent stability of methylarenes compared to aldehydes, operational simplicity, use of green oxidant, avoidance of metal catalysts, and easy accessibility of the starting materials, making it a highly practical approach to access various pyrimido[4,5-d]pyrimidines of biological interest. Hence, it is a significantly superior to the existing methodologies. On the basis of importance of pyrimido[4,5-d]pyrimidines in pharmacological science, this protocol has the potential for applications in both drug discovery and chemical development projects.Experimental
General information
All the chemicals and reagents were purchased from commercial suppliers (Sigma-Aldrich, Alfa-Aesar, Spectrochem, TCI Chemicals) and were used without further purification. Silica gel [(60–120, 230–400 mesh), Spectrochem, India] was used for chromatographic separation. Thin-layer chromatography (TLC) was performed on TLC plates purchase from Merck. Solvents used in extraction and purification were distilled prior to use. Melting points were determined by silicon oil bath in open capillaries and are uncorrected. 1H (13C) NMR spectra were recorded at 400 (100) MHz on a Brucker (Ascend 400 MHz) spectrometer using CDCl3 or DMSO-d6 solvent with tetramethylsilane as internal standard. Chemical shifts were reported in parts per million (ppm, δ) and coupling constants are given in Hertz. Proton coupling patterns are described as singlet (s), doublet (d), triplet (t), multiplet (m). Due to the existence of tautomers, in some cases the amidine NH proton signals in the 1H spectrum and amidine carbon in 13C spectrum was not detected. Only distinct signals are reported. High resolution mass spectra (HRMS) [Make: Waters; Model: Xevo XS QTof mass spectrometer] were obtained by using positive electrospray ionization (ESI) by Time of Flight (TOF) method.General procedure for the TBHP-mediated oxidative synthesis of pyrimido[4,5-d]pyrimidines from N-uracil amidines and methylarenes
An oven-dried microwave vial (10 mL) equipped with a magnetic stirring bar was charged with N-uracil amidine (0.5 mmol, 1.0 equiv.), toluene (1 mL), 70% TBHP (192 mg, 3 equiv.), Cs2CO3 (325 mg, 1.0 mmol, 2.0 equiv.). The reaction mixture was placed in a pre-heated oil bath at 100 °C and stirred for 15 hours with magnetic stirrer. After completion of 15 h reaction time, the mixture was allowed to reach room temperature and extracted with ethyl acetate (2 × 10 mL). The combined organic layers were washed with water and then brine. The organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting residue was purified by column chromatography on silica gel (60–120 mesh) using hexane–ethyl acetate mixture as eluent to give the title compounds.1,3-Dimethyl-5,7-diphenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3a)22a,25
White solid; Rf = 0.4 (SiO2, hexane/ethyl acetate = 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :90); yield: 82% (141 mg, 0.41 mmol), mp 210–211 °C. 1H NMR (400 MHz, CDCl3): δ = 3.41 (s, 3H), 3.85 (s, 3H), 7.48–7.57 (m, 6H), 7.64 (dd, J = 2.0 Hz, 8.4 Hz, 2H), 8.57 (d, J = 8.4 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 28.6, 29.9, 103.7, 127.7, 128.6, 129.1, 129.3, 130.0, 132.3, 136.1, 138.4, 151.3, 157.9, 159.6, 165.2, 170.4. HRMS (ESI): m/z calcd for C20H16N4O2Na [M+ + Na]: 367.1165; found: 367.1158.
:90); yield: 82% (141 mg, 0.41 mmol), mp 210–211 °C. 1H NMR (400 MHz, CDCl3): δ = 3.41 (s, 3H), 3.85 (s, 3H), 7.48–7.57 (m, 6H), 7.64 (dd, J = 2.0 Hz, 8.4 Hz, 2H), 8.57 (d, J = 8.4 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 28.6, 29.9, 103.7, 127.7, 128.6, 129.1, 129.3, 130.0, 132.3, 136.1, 138.4, 151.3, 157.9, 159.6, 165.2, 170.4. HRMS (ESI): m/z calcd for C20H16N4O2Na [M+ + Na]: 367.1165; found: 367.1158.
5-(4-Methoxyphenyl)-1,3-dimethyl-7-phenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3b)25
White solid; Rf = 0.4 (SiO2, hexane/ethyl acetate = 10:90); yield: 88% (164 mg, 0.44 mmol), mp 235–236 °C. 1H NMR (400 MHz, CDCl3): δ = 3.43 (s, 3H), 3.85 (s, 3H), 3.89 (s, 3H), 7.01 (dd, J = 2.0 Hz, 6.8 Hz, 2H), 7.49–7.58 (m, 3H), 7.69–7.72 (m, 2H), 8.58 (dd, J = 1.6 Hz, 8.4 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 28.7, 29.9, 55.4, 103.2, 113.1, 128.6, 129.2, 130.4, 131.6, 132.2, 136.2, 151.4, 158.1, 159.9, 161.5, 164.9, 169.7. HRMS (ESI): m/z calcd for C21H18N4O3Na [M+ + Na]: 397.1271; found: 397.1263.
1,3-Dimethyl-5-(4-methylphenyl)-7-phenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3c)25
White solid; Rf = 0.4 (SiO2, hexane/ethyl acetate = 5:95); yield: 83% (149 mg, 0.415 mmol), mp 226–228 °C. 1H NMR (400 MHz, CDCl3): δ = 2.45 (s, 3H), 3.41 (s, 3H), 3.85 (s, 3H), 7.30 (d, J = 8.0 Hz, 2H) 7.48–7.58 (m, 5H), 8.57 (dd, J = 1.6 Hz, 8.4 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 21.6, 28.6, 29.9, 103.5, 128.5, 128.6, 129.3, 132.2, 135.5, 136.1, 140.3, 151.3, 157.9, 159.7, 165.1, 170.4. HRMS (ESI): m/z calcd for C21H18N4O2Na [M+ + Na]: 381.1322; found: 381.1316.
1,3-Dimethyl-5-(2-methylphenyl)-7-phenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3d)
White solid; Rf = 0.45 (SiO2, hexane/ethyl acetate = 5:95); yield: 69% (123 mg, 0.345 mmol), mp 246 °C. 1H NMR (400 MHz, CDCl3): δ = 2.19 (s, 3H), 3.38 (s, 3H), 3.87 (s, 3H), 7.20 (d, J = 7.6 Hz, 1H), 7.32 (t, J = 7.2 Hz, 2H), 7.39–7.42 (m, 1H) 7.48–7.58 (m, 3H), 8.56 (t, J = 1.6 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 19.7, 28.5, 29.8, 104.7, 125.5, 127.4, 128.7, 129.0, 129.4, 130.0, 132.3, 134.8, 136.2, 138.9, 151.4, 157.5, 159.3, 165.8, 171.1. HRMS (ESI): m/z calcd for C21H19N4O2 [M+ + H]: 359.1508; found: 359.1492.
1,3-Dimethyl-5-(4-thiomethylphenyl)-7-phenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3e)
Off-white solid; Rf = 0.4 (SiO2, hexane/ethyl acetate = 20:80); yield: 76% (148 mg, 0.38 mmol), mp 270 °C. 1H NMR (400 MHz, CDCl3): δ = 2.55 (s, 3H), 3.42 (s, 3H), 3.85 (s, 3H), 7.34 (d, J = 8.4 Hz, 2H), 7.49–7.56 (m, 3H) 7.63 (d, J = 8.4 Hz, 2H), 8.56 (t, J = 1.2 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 15.2, 28.6, 29.9, 103.4, 124.9, 128.6, 129.3, 130.0, 132.3, 134.5, 136.1, 141.9, 151.3, 158.0, 159.8, 165.1, 169.6. HRMS (ESI): m/z calcd for C21H18N4O2SNa [M+ + Na]: 413.1043; found: 413.1036.
5-(4-Hydroxyphenyl)-1,3-dimethyl-7-phenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3f)25
White solid; Rf = 0.4 (SiO2, hexane/ethyl acetate = 20:80); yield: 71% (128 mg, 0.355 mmol), mp 246–248 °C. 1H NMR (400 MHz, DMSO): δ = 3.22 (s, 3H), 3.66 (s, 3H), 6.84 (d, J = 8.8 Hz, 2H), 7.54–7.62 (m, 5H), 8.47 (d, J = 6.8 Hz, 2H), 10.0 (br s, 1H, OH). 13C NMR (100 MHz, DMSO): δ = 28.7, 30.0, 103.8, 114.7, 129.0, 129.2, 129.3, 132.3, 132.6, 136.4, 151.4, 158.3, 160.0, 163.6, 168.8. HRMS (ESI): m/z calcd for C20H17N4O3 [M+ + H]: 361.1301; found: 361.1294.
5-(4-Bromophenyl)-1,3-dimethyl-7-phenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3g)25
White solid; Rf = 0.45 (SiO2, hexane/ethyl acetate = 5:95); yield: 85% (179 mg, 0.425 mmol), mp 258 °C. 1H NMR (400 MHz, CDCl3): δ = 3.41 (s, 3H), 3.86 (s, 3H), 7.49–7.59 (m, 6H), 7.63 (d, J = 8.4 Hz, 1H), 8.58 (d, J = 7.2 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 28.6, 30.0, 103.6, 124.7, 128.7, 129.3, 130.9, 131.0, 132.5, 135.9, 137.2, 151.2, 157.9, 159.6, 165.4, 169.3. HRMS (ESI): m/z calcd for C20H16BrN4O2 [M+ + H]: 423.0457; found: 423.0477.
5-(4-Chlorophenyl)-1,3-dimethyl-7-phenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3h)25
White solid; Rf = 0.4 (SiO2, hexane/ethyl acetate = 10:90); yield: 70% (132 mg, 0.35 mmol), mp 242 °C. 1H NMR (400 MHz, CDCl3): δ = 3.41 (s, 3H), 3.86 (s, 3H), 7.46–7.61 (m, 7H), 8.55 (t, J = 1.6 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 28.6, 30.0, 103.6, 128.0, 128.7, 129.3, 130.7, 132.5, 136.0, 136.3, 136.7, 151.2, 157.9, 159.6, 165.4, 169.2. HRMS (ESI): m/z calcd for C20H15ClN4O2Na [M+ + Na]: 401.0776; found: 401.0766.
1,3-Dimethyl-5-(2-chlorophenyl)-7-phenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3i)
White solid; Rf = 0.4 (SiO2, hexane/ethyl acetate = 10:90); yield: 66% (125 mg, 0.33 mmol), mp 258–259 °C. 1H NMR (400 MHz, CDCl3): δ = 3.39 (s, 3H), 3.87 (s, 3H), 7.37–7.58 (m, 7H), 8.56 (t, J = 1.2 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 28.5, 29.8, 105.0, 126.8, 128.7, 128.9, 129.2, 129.4, 130.2, 132.0, 132.4, 136.1, 138.2, 151.3, 157.3, 159.3, 166.2, 167.9. HRMS (ESI): m/z calcd for C20H16ClN4O2 [M+ + H]: 379.0962; found: 379.0971.
5-(4-Fluorophenyl)-1,3-dimethyl-7-phenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3j)25
White solid; Rf = 0.45 (SiO2, hexane/ethyl acetate = 10:90); yield: 67% (121 mg, 0.335 mmol), mp 233 °C. 1H NMR (400 MHz, CDCl3): δ = 3.41 (s, 3H), 3.85 (s, 3H), 7.18 (t, J = 8.8 Hz, 2H), 7.50–7.56 (m, 3H), 7.66–7.69 (m, 2H), 8.55 (t, J = 1.6 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 28.6, 29.5, 103.6, 114.8 (d, J = 22.0 Hz), 128.7, 129.3, 131.6 (d, J = 8.0 Hz), 132.4, 134.22, 134.25, 136.0, 151.2, 158.0, 159.7, 164.0 (d, J = 250 Hz), 169.2. HRMS (ESI): m/z calcd for C20H16FN4O2 [M+ + H]: 363.1257; found: 363.1268.
1,3-Dimethyl-5-(4-cyanophenyl)-7-phenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3k)
Off-white solid; Rf = 0.4 (SiO2, hexane/ethyl acetate = 10:90); yield: 72% (133 mg, 0.36 mmol), mp 252 °C. 1H NMR (400 MHz, CDCl3): δ = 3.41 (s, 3H), 3.87 (s, 3H), 7.52 (t, J = 6.8 Hz, 2H), 7.65 (d, J = 8.4 Hz, 1H), 7.71 (d, J = 8.4 Hz, 2H), 7.79 (d, J = 8.0 Hz, 2H), 8.55 (d, J = 6.8 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 28.6, 30.0, 103.7, 118.7, 127.0, 128.8, 129.4, 129.8, 131.5, 132.3, 132.7, 135.7, 142.9, 151.1, 157.9, 159.5, 165.8. HRMS (ESI): m/z calcd for C21H15N5O2Na [M+ + Na]: 392.1118; found: 392.1111.
1,3-Dimethyl-5-(naphthalen-1-yl)-7-phenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3l)
Pale yellow solid; Rf = 0.35 (SiO2, hexane/ethyl acetate = 10:90); yield: 85% (167 mg, 0.425 mmol), mp 264 °C. 1H NMR (400 MHz, CDCl3): δ = 3.29 (s, 3H), 3.91 (s, 3H), 7.36–7.40 (m, 2H) 7.44–7.62 (m, 7H), 7.94 (d, J = 8.4 Hz, 1H), 8.0 (d, J = 8.4 Hz, 1H), 8.56 (t, J = 1.2 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ = 28.5, 29.8, 105.5, 124.6, 125.1, 125.4, 126.0, 126.5, 128.7, 129.1, 129.3, 129.5, 130.8, 132.4, 133.3, 136.1, 136.8, 151.3, 157.6, 159.0, 165.9, 170.2. HRMS (ESI): m/z calcd for C24H18N4O2Na [M+ + Na]: 417.1322; found: 417.1315.
1,3-Dimethyl-7-phenyl-5-(3,4,5-trimethoxyphenyl)pyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3m)
White solid; Rf = 0.35 (SiO2, hexane/ethyl acetate = 20:80); yield: 90% (195 mg, 0.45 mmol), mp 208–209 °C. 1H NMR (400 MHz, CDCl3): δ = 3.45 (s, 3H), 3.89 (s, 3H), 3.93 (s, 6H), 3.97 (s, 3H), 6.92 (s, 2H), 7.52–7.61 (m, 3H), 8.60 (t, J = 1.6 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 28.7, 30.0, 56.3, 61.0, 103.6, 106.9, 128.7, 129.3, 132.4, 133.6, 136.0, 139.7, 151.3, 152.6, 158.0, 159.5, 165.1, 170.0. HRMS (ESI): m/z calcd for C23H23N4O5 [M+ + H]: 435.1663; found: 435.1658.
1,3-Dimethyl-7-phenyl-5-(thiophen-2-yl)pyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3n)
Pale yellow solid; Rf = 0.4 (SiO2, hexane/ethyl acetate = 10:90); yield: 70% (122 mg, 0.35 mmol), mp 260–261 °C. 1H NMR (400 MHz, CDCl3): δ = 3.48 (s, 3H), 3.79 (s, 3H), 7.21 (t, J = 4.0 Hz, 1H), 7.53–7.58 (m, 3H), 7.66 (d, J = 4.4 Hz, 1H), 8.54 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3): δ = 28.8, 30.2, 101.8, 128.2, 128.6, 129.0, 132.3, 132.9, 135.0, 135.9, 141.8, 151.0, 158.4, 160.0, 161.3, 164.3. HRMS (ESI): m/z calcd for C18H14N4O2SNa [M+ + Na]: 373.0730; found: 373.0723.
1,3-Dimethyl-7-phenyl-5-(pyridin-2-yl)pyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3o)
Pale yellow solid; Rf = 0.45 (SiO2, hexane/ethyl acetate = 50:50); yield: 63% (109 mg, 0.315 mmol), mp 254 °C. 1H NMR (400 MHz, CDCl3): δ = 3.38 (s, 3H), 3.86 (s, 3H), 7.48–7.56 (m, 4H), 7.63 (d, J = 8.0 Hz, 1H), 7.89 (d, J = 7.6 Hz, 1H), 8.56 (d, J = 7.2 Hz, 2H), 8.73 (d, J = 4.4 Hz, 1H). 13C NMR (100 MHz, CDCl3): δ = 28.5, 29.8, 104.3, 122.8, 124.0, 128.7, 129.4, 132.4, 136.0, 136.5, 149.0, 151.3, 156.7, 157.6, 159.3, 165.9, 168.5. HRMS (ESI): m/z calcd for C19H16N5O2 [M+ + H]: 346.1298; found: 346.1293.
1,3-Dimethyl-5-phenyl-7-(p-tolyl)pyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3p)25
White solid; Rf = 0.4 (SiO2, hexane/ethyl acetate = 10:90); yield: 83% (148 mg, 0.415 mmol), mp 247 °C. 1H NMR (400 MHz, CDCl3): δ = 2.44 (s, 3H), 3.40 (s, 3H), 3.84 (s, 3H), 7.30 (d, J = 8.4 Hz, 2H), 7.47–7.53 (m, 3H), 7.64 (dd, J = 2.0 Hz, 8.0 Hz, 2H), 8.46 (t, J = 8.0 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 21.7, 28.6, 29.9, 103.4, 127.7, 128.6, 129.1, 129.3, 129.4, 129.9, 133.5, 138.5, 143.0, 157.8, 159.7, 165.3, 170.3. HRMS (ESI): m/z calcd for C21H19N4O2 [M+ + H]: 359.1508; found: 359.1492.
7-(4-Fluorophenyl)-1,3-dimethyl-5-phenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3q)25
White solid; Rf = 0.4 (SiO2, hexane/ethyl acetate = 10:90); yield: 77% (139 mg, 0.385 mmol), mp 243–245 °C. 1H NMR (400 MHz, CDCl3): δ = 3.41 (s, 3H), 3.85 (s, 3H), 7.18 (t, J = 8.8 Hz, 2H), 7.48–7.54 (m, 3H), 7.63 (dd, J = 1.6 Hz, 8.0 Hz, 2H), 8.60 (dd, J = 5.6 Hz, 8.4 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 28.6, 29.9, 103.6, 115.7 (d, J = 21.0 Hz), 127.7, 129.1, 130.0, 131.7 (d, J = 10.0 Hz), 132.4, 138.3, 151.3, 157.9, 159.6, 164.4, 165.6 (d, J = 161.0 Hz), 170.5. HRMS (ESI): m/z calcd for C20H15FN4O2Na [M+ + Na]: 385.1071; found: 385.1063.
7-(4-Chlorophenyl)-1,3-dimethyl-5-phenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3r)25
White solid; Rf = 0.4 (SiO2, hexane/ethyl acetate = 10:90); yield: 80% (151 mg, 0.40 mmol), mp 259–260 °C. 1H NMR (400 MHz, CDCl3): δ = 3.41 (s, 3H), 3.85 (s, 3H), 7.46–7.54 (m, 5H), 7.62 (t, J = 1.6 Hz, 2H), 8.52 (d, J = 8.8 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 28.6, 29.9, 103.8, 127.8, 128.9, 129.1, 130.1, 130.6, 134.6, 138.3, 138.7, 151.3, 157.9, 159.5, 164.3, 170.5. HRMS (ESI): m/z calcd for C20H15ClN4O2Na [M+ + Na]: 401.0776; found: 401.0766.
7-(4-Bromophenyl)-1,3-dimethyl-5-phenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3s)25
Off-white solid; Rf = 0.45 (SiO2, hexane/ethyl acetate = 10:90); yield: 80% (169 mg, 0.40 mmol), mp 255–256 °C. 1H NMR (400 MHz, CDCl3): δ = 3.41 (s, 3H), 3.85 (s, 3H), 7.48–7.54 (m, 3H), 7.61–7.64 (m, 4H), 8.44 (dd, J = 1.6 Hz, J = 6.8 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 28.7, 30.0, 103.9, 127.4, 127.8, 129.1, 130.1, 130.8, 131.9, 135.1, 138.2, 151.3, 157.9, 159.5, 164.4, 170.6. HRMS (ESI): m/z calcd for C20H16BrN4O2 [M+ + H]: 423.0451; found: 423.0441.
1-Benzyl-3-methyl-5,7-diphenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3t)25
White solid; Rf = 0.4 (SiO2, hexane/ethyl acetate = 5:95); yield: 88% (185 mg, 0.44 mmol), mp 211–212 °C. 1H NMR (400 MHz, CDCl3): δ = 3.41 (s, 3H), 5.71 (s, 2H), 7.28–7.36 (m, 3H), 7.48–7.57 (m, 6H), 7.60–7.65 (m, 4H), 8.58 (t, J = 6.8 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 28.7, 45.9, 103.7, 127.7, 128.0, 128.70, 128.74, 128.8, 129.1, 129.4, 130.0, 132.4, 136.2, 136.6, 138.4, 151.2, 157.6, 159.5, 165.3, 170.7. HRMS (ESI): m/z calcd for C26H20N4O2Na [M+ + Na]: 443.1478; found: 443.1471.
1,3-Dibenzyl-5,7-diphenylpyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3u)25
White solid; Rf = 0.5 (SiO2, hexane/ethyl acetate = 5:95); yield: 92% (228 mg, 0.46 mmol), mp 215 °C. 1H NMR (400 MHz, CDCl3): δ = 4.70 (s, 2H), 5.21 (s, 2H), 7.26–7.38 (m, 6H), 7.46–7.60 (m, 10H), 7.65 (t, J = 1.6 Hz, 2H), 8.56 (t, J = 2.0 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 45.0, 46.0, 103.9, 127.8, 127.9, 128.5, 128.68, 128.7, 129.1, 129.4, 130.2, 132.3, 136.2, 136.5, 136.6, 138.3, 151.1, 157.7, 159.4, 165.2, 170.7. HRMS (ESI): m/z calcd for C32H24N4O2Na [M+ + Na]: 519.1791; found: 519.1782.
1,3-Dimethyl-5,7-diphenyl-5,6-dihydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (3a′)
Yellow solid; Rf = 0.4 (SiO2, hexane/ethyl acetate = 50:50); yield: 42% (73 mg, 0.21 mmol), 1H NMR (400 MHz, CDCl3): δ = 3.28 (s, 3H), 3.68 (s, 3H), 5.80 (d, J = 2.8 Hz, 1H), 6.81 (br s, 1H, NH), 7.30–7.37 (m, 3H), 7.46–7.53 (m, 4H), 7.61 (t, J = 7.6 Hz, 1H), 7.89 (t, J = 1.2 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ = 27.8, 29.4, 53.4, 89.8, 126.7, 127.2, 128.5, 128.96, 129.0, 132.6, 133.0, 144.2, 150.6, 152.3, 159.4, 161.4.
Conflicts of interest
The author declares no conflicts of financial interest.Acknowledgements
The author P. Debnath is thankful to DST (New Delhi) for financial assistance (Grant no. YSS/2015/001554).Notes and references
- (a) T. C. Kühler, J. Fryklund, N.-Å. Bergman, J. Weilitz, A. Lee and H. Larsson, J. Med. Chem., 1995, 38, 4906 CrossRef PubMed; (b) P. Madsen, L. B. Knudsen, F. C. Wiberg and R. D. Carr, J. Med. Chem., 1998, 41, 5150 CrossRef CAS PubMed; (c) L. Llauger, H. He, J. Kim, J. Aguirre, N. Rosen, U. Peters, P. Davies and G. Chiosis, J. Med. Chem., 2005, 48, 2892 CrossRef CAS PubMed; (d) N. Pietrancosta, A. Moumen, R. Dono, P. Lingor, V. Planchamp, F. Lamballe, M. Bähr, J.-L. Kraus and F. Maina, J. Med. Chem., 2006, 49, 3645 CrossRef CAS PubMed; (e) A. T. Mavrova, D. Vuchev, K. Anichina and N. Vassilev, Eur. J. Med. Chem., 2010, 45, 5856 CrossRef CAS.
- For review: (a) M. Asif, Int. J. Bioorg. Chem., 2017, 2, 146 Search PubMed; (b) H. A. Al-Khamees, S. M. Bayomi, H. A. Kandil and K. E. H. El-Tahir, Eur. J. Med. Chem., 1990, 25, 103 CrossRef CAS; (c) A. T. Chaviara, P. J. Cox, K. H. Repana, R. M. Papi, K. T. Papazisis, D. Zambouli, A. H. Kortsaris, D. A. Kyriakidis and C. A. J. Bolos, Inorg. Biochem., 2004, 98, 1271 CrossRef CAS; (d) G. Deepika, P. Gopinath, G. Kranthi, C. Nagamani, Y. V. Jayasree, N. V. Naidu and S. Enaganti, J. Pharm. Res., 2012, 5, 1105 Search PubMed.
- For review on synthesis of nitrogen heterocycles: (a) L. Yu, R. Qian, X. Deng, F. Wang and Q. Xu, Sci. Bull., 2018, 63, 1010 CrossRef CAS; (b) L. Yu, M. Liu, F. Chen and Q. Xu, Org. Biomol. Chem., 2015, 13, 8379 RSC; (c) K. C. Majumdar, P. Debnath, N. De and B. Roy, Curr. Org. Chem., 2011, 15, 1760 CrossRef CAS; (d) K. C. Majumdar, B. Roy, P. Debnath and A. Taher, Curr. Org. Chem., 2010, 14, 846 CrossRef CAS; (e) C. Allais, J.-M. Grassot, J. Rodriguez and T. Constantieux, Chem. Rev., 2014, 114, 10829 CrossRef CAS PubMed; (f) Y. Yamamoto, Chem. Soc. Rev., 2014, 43, 1575 RSC; (g) B. Zhang and A. Studer, Chem. Soc. Rev., 2015, 44, 3505 RSC.
- (a) G. M. Ziarani, N. H. Nasaba and N. Lashgarib, RSC Adv., 2016, 6, 38827 RSC; (b) T. P. Selvam, C. R. James, P. V. Dniandev and S. K. Valzita, Res. Pharm., 2012, 4, 01 Search PubMed.
- (a) J. D. Brown, in Comprehensive Heterocyclic Chemistry, ed. A. R. Katrizky and C. W. Rees, Pergamon, Oxford, 1984, vol. 3, p. 57 Search PubMed; (b) E. D. Clercq and R. Beraaerts, J. Biol. Chem., 1987, 262, 14905–14911 Search PubMed; (c) V. S. Dinakaran, B. Bomma and K. K. Srinivasan, Pharma Chem., 2012, 4, 255 CAS; (d) A. Y. Aksinenko, T. V. Goreva, T. A. Epishina, S. V. Trepalin and V. B. Sokolov, J. Fluorine Chem., 2016, 188, 191 CrossRef CAS.
- (a) W. J. Coates, Eur. Pat., 351058, 1990Chem. Abstr., 1990, 113, 40711; (b) A. A. Ramsey, US Pat., 3830812, FMC Corp., 1974Chem. Abstr., 1974, 81, 136174.
- Y. S. Sanghvi, S. B. Larson, S. S. Matsumoto, L. D. Nord, D. F. Smee, R. C. Willis, T. L. Avery, R. K. Robins and G. R. Revankar, J. Med. Chem., 1989, 32, 629 CrossRef CAS PubMed.
- (a) P. Sharma, N. Rane and V. Gurram, Bioorg. Med. Chem. Lett., 2004, 14, 4185 CrossRef CAS PubMed; (b) J. De La Cruz, T. Carrasco, G. Ortega and G. F. S. De La Cuesta, Lipids, 1992, 27, 192 CrossRef CAS PubMed.
- (a) D. W. Fry, M. A. Becker and R. L. Switzer, Mol. Pharmacol., 1995, 47, 810 CAS; (b) P. Sharma, N. Rane and P. Pandey, Arch. Pharm., 2006, 339, 572 CrossRef CAS PubMed; (c) H. Wang, C. Yang, J. R. Doherty, W. R. Roush, J. L. Cleveland and T. D. Bannister, J. Med. Chem., 2014, 57, 7317 CrossRef CAS PubMed.
- V. J. Ram, A. Goel, S. Sarkhel and P. R. Maulik, Bioorg. Med. Chem., 2002, 10, 1275 CrossRef CAS PubMed.
- J. D. Figueroa-Villar, C. L. Carneiro and E. R. Cruz, Heterocycles, 1992, 34, 891 CrossRef CAS.
- N. Kitamura and A. Onishi, Eur. Pat., 163599, 1984Chem. Abstr., 1984, 104, 186439.
- (a) P. Raddatz and R. Bergmann, Ger. Pat., 360731, 1988Chem. Abstr., 1988, 109, 54786; (b) R. C. Dukarm, F. C. Morin, J. A. Russell and R. H. Steinhorn, Pediatr. Res., 1998, 44, 831 CrossRef CAS PubMed.
- T. J. Delia, M. Baumann and A. Bunker, Heterocycles, 1993, 35, 1397 CrossRef CAS.
- Synthesis of 5,6-dihydropyrimido[4,5-d]pyrimidine by three component reactions: (a) M. Dabiri, H. Arvin-Nezhad, H. R. Khavasi and A. Bazgir, Tetrahedron, 2007, 63, 1770 CrossRef CAS; (b) S. Majumder, P. Borah and P. J. Bhuyan, Tetrahedron Lett., 2014, 55, 1168 CrossRef CAS; (c) M. Kidwai, K. Singhal and S. Kukreja, Z. Naturforsch., 2007, 62b, 732 Search PubMed.
- Synthesis of 5,6-dihydropyrimido[4,5-d]pyrimidine by [4+2] cycloaddition reactions: (a) D. Prajapati, M. Gohain and A. Thakur, Bioorg. Med. Chem. Lett., 2006, 16, 3537 CrossRef CAS PubMed; (b) I. Devi, H. N. Borah and P. J. Bhuyan, Tetrahedron Lett., 2004, 45, 2405 CrossRef CAS; (c) M. Gohain, D. Prajapati, B. J. Gogoi and J. S. Sandhu, Synlett, 2004, 1179 CAS; (d) M. El-Moghazy, D. A. Ibrahim, N. M. Abdelgawad, N. A. H. Farag and A. S. El-Khouly, Sci. Pharm., 2011, 79, 429 CrossRef PubMed; (e) D. Prajapati and A. J. Thakur, Tetrahedron Lett., 2005, 46, 1433 CrossRef CAS.
- K. Hirota, Y. Kitade, H. Sajiki and Y. Maki, J. Chem. Soc., Perkin Trans. 1, 1990, 123 RSC.
- H. Wamhoff and J. Muhr, Synthesis, 1998, 919 Search PubMed.
- H. Wang, C. Wang and T. D. Bannister, Tetrahedron Lett., 2015, 56, 1949–1952 CrossRef CAS PubMed.
- S. Badvel, R. R. Gopireddy, T. B. Shaik, S. Hasti, V. R. Tummaluru and N. R. Chamarthi, Chem. Heterocycl. Compd., 2015, 51, 749 CrossRef CAS.
- R. Ghorbani-Vaghei and N. Sarmast, Appl. Organomet. Chem., 2018, 32, e4003 CrossRef.
- (a) M. F. Moghaddam, M. R. Khodabakhshi and M. Aminaee, Tetrahedron Lett., 2014, 55, 4720 CrossRef; (b) A. Hamed, Synthesis, 1992, 591 CrossRef CAS.
- S. K. Srivastava, W. Haq and P. M. S. Chauhan, Bioorg. Med. Chem. Lett., 1999, 9, 965 CrossRef CAS PubMed.
- M. L. Deb, P. J. Borpatra and P. K. Baruah, Green Chem., 2019, 21, 69 RSC.
- P. Debnath, G. Sahu and U. C. De, ChemistrySelect, 2019, 4, 2327 CrossRef CAS.
- G. V. R. Sharama and A. Robert, Res. Chem. Intermed., 2013, 39, 3251 CrossRef.
- (a) A. Modak, A. Deb, S. Patra, S. Rana, S. Maity and D. Maiti, Chem. Commun., 2012, 4253 RSC; (b) T. Iwai, T. Fujihara and Y. Tsuji, Chem. Commun., 2008, 6215 RSC.
- (a) S. Michlik and R. Kempe, Nat. Chem., 2013, 5, 140 CrossRef CAS PubMed; (b) S. Michlik and R. Kempe, Angew. Chem., Int. Ed., 2013, 52, 6326 CrossRef CAS PubMed; (c) D. Srimani, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2013, 52, 4012 CrossRef CAS PubMed; (d) M. Zhang, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 597 CrossRef CAS PubMed; (e) M. Zhang, X. Fang, H. Neumann and M. Beller, J. Am. Chem. Soc., 2013, 135, 11384 CrossRef CAS PubMed; (f) R. H. Crabtree, Organometallics, 2011, 30, 17 CrossRef CAS; (g) T. D. Nixon, M. K. Whittlesey and J. M. J. Williams, Dalton Trans., 2009, 753 RSC; (h) G. Guillena, D. J. Ramón and M. Yus, Chem. Rev., 2009, 110, 1611 CrossRef PubMed; (i) M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555 CrossRef CAS.
- (a) M. Zhang, S. Imm, S. Bähn, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 11197 CrossRef CAS PubMed; (b) S. Imm, S. Bähn, M. Zhang, L. Neubert, H. Neumann, F. Klasovsky, J. Pfeffer, T. Haas and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 7599 CrossRef CAS PubMed; (c) S. Bähn, S. Imm, L. Neubert, M. Zhang, H. Neumann and M. Beller, ChemCatChem, 2011, 3, 1853 CrossRef; (d) G. Guillena, D. J. Ramón and M. Yus, Angew. Chem., Int. Ed., 2007, 46, 2358–2364 CrossRef CAS PubMed; (e) F.-X. Yan, M. Zhang, X.-T. Wang, F. Xie, M.-M. Chen and H.-F. Jiang, Tetrahedron, 2014, 70, 1193 CrossRef CAS; (f) X.-T. Wang, M.-M. Chen, F. Xie and M. Zhang, RSC Adv., 2014, 4, 14744 RSC; (g) M. Zhang, F. Xie, X.-T. Wang, F.-X. Yan, T. Wang, M.-M. Chen and Y.-Q. Ding, RSC Adv., 2013, 3, 6022 RSC.
- Y. Wu, P. Y. Choy, F. Mao and F. Y. Kwong, Chem. Commun., 2013, 49, 689 RSC.
- W. Guo, K. Huang, F. Ji, W. Wu and H. Jiang, Chem. Commun., 2015, 51, 8857 RSC.
- (a) W. Guo, Org. Biomol. Chem., 2015, 13, 10285 RSC; (b) Z. Yin and P. Sun, J. Org. Chem., 2012, 77, 11339 CrossRef CAS PubMed; (c) F. Xie, M. Chen, X. Wang, H. Jiang and M. Zhang, Org. Biomol. Chem., 2014, 12, 2761 RSC.
- (a) X.-F. Wu, J.-L. Gong and X. Qi, Org. Biomol. Chem., 2014, 12, 5807 RSC; (b) Y. F. Li, Synlett, 2007, 2922 CrossRef CAS; (c) T. K. Achar and P. Mal, J. Org. Chem., 2015, 80, 666 CrossRef CAS PubMed; (d) X. Zhang, M. Wang, P. Li and L. Wang, Chem. Commun., 2014, 50, 8006 RSC; (e) L. Gao, H. Tang and Z. Wang, Chem. Commun., 2014, 50, 4085 RSC; (f) W.-P. Mai, H.-H. Wang, Z.-C. Li, J.-W. Yuan, Y.-M. Xiao, L.-R. Yang, P. Mao and L.-B. Qu, Chem. Commun., 2012, 48, 10117 RSC.
- B. Morel, P. Franck, J. Bidange, S. Sergeyev, D. Smith, J. Moseley and B. U. W. Maes, ChemSusChem, 2017, 10, 624 CrossRef CAS PubMed.
- M. Noe, A. Perosa and M. Selva, Green Chem., 2013, 15, 2252 RSC.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- (a) M. Cernova, I. Cerna, R. Pohl and M. Hocek, J. Org. Chem., 2011, 76, 5309 CrossRef CAS PubMed; (b) C. E. Muller, M. Thorand, R. Qurishi, M. Diekmann, K. A. Jacobson, W. L. Padgett and J. W. Daly, J. Med. Chem., 2002, 45, 3440 CrossRef PubMed.
- (a) M. Wang, Y. Meng, W. Wei, J. Wu, W. Yu and J. Changa, Adv. Synth. Catal., 2018, 360, 86 CrossRef CAS; (b) A. R. Tiwari and B. M. Bhanage, ChemistrySelect, 2016, 3, 343 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed of experimental procedure and analytical data for starting materials, and copies of 1H NMR and 13C NMR for all synthesized compounds. See DOI: 10.1039/c9ra06625j |
| This journal is © The Royal Society of Chemistry 2019 |