
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBRET based dual-colour (visible/near-infrared) molecular imaging using a quantum dot/EGFP–luciferase conjugate†
Setsuko Tsuboia and
Takashi Jin *ab
*ab
aRIKEN Center for Biosystems Dynamics Research (BDR), RIKEN Furuedai 6-2-3, Suita, Osaka 565-0874, Japan. E-mail: tjin@riken.jp
bGraduate School of Frontier Biosciences, Osaka University, Yamada-oka 1-3, Suita, Osaka 565-0871, Japan
First published on 29th October 2019
Abstract
Owing to its high sensitivity, bioluminescence imaging is an important tool for biosensing and bioimaging in life sciences. Compared to fluorescence imaging, bioluminescence imaging has a superior advantage that the background signals resulting from autofluorescence are almost zero. In addition, bioluminescence imaging can permit long-term observation of living cells because external excitation is not needed, leading to no photobleaching and photocytotoxicity. Although bioluminescence imaging has such superior properties over fluorescence imaging, observation wavelengths in bioluminescence imaging are mostly limited to the visible region. Here we present bioluminescence resonance energy transfer (BRET) based dual-colour (visible/near-infrared) molecular imaging using a quantum dot (QD) and luciferase protein conjugate. This bioluminescent probe is designed to emit green and near-infrared luminescence from enhanced green fluorescent protein (EGFP) and CdSeTe/CdS (core/shell) QDs, where EGFP–Renilla luciferase (RLuc) fused proteins are conjugated to the QDs. Since the EGFP–RLuc fused protein contains an immunoglobulin binding domain (GB1) of protein G, it is possible to prepare a variety of molecular imaging probes functionalized with antibodies (IgG). We show that the BRET-based QD probe can be used for highly sensitive dual-colour (visible/near-infrared) bioluminescence molecular imaging of membrane receptors in cancer cells.
1. Introduction
Among a variety of optical imaging modalities,1 fluorescence imaging has been widely used for molecular imaging in cells and tissue samples.2,3 This is motivated by the high sensitivity and spatial resolution of fluorescence optical detection. However, fluorescence imaging has several drawbacks such as autofluorescence, photo-bleaching and photocytotoxicity, which result from irradiation of excitation light. In the past decade, to overcome these issues, bioluminescence imaging has been developed as a new optical molecular-imaging modality in life sciences.3,4 In bioluminescence imaging, external excitation is not needed, leading to no photobleaching and photocytotoxicity with very low background signals (e.g., autofluorescence) from numerous fluorescent species.5,6 Thus, bioluminescence imaging is expected to offer high signal to background images with a wider dynamic range of the signal compared to fluorescence imaging.3Despite the highly sensitivity of bioluminescence imaging, its potential use in bioimaging has been restricted at the visible region.4 In the bioluminescence imaging at the visible region, visible-emitting bioluminescence is strongly absorbed and scattered in biological samples.7 To overcome this drawback, the chemical structure of bioluminescent substrates (luciferin) has been modified to emit at longer wavelengths.8–12 Luciferin analogues with extended π-conjugation have been used for bioluminescence imaging with longer wavelengths.8,10,12 Although commonly used luciferase substrates such as D-luciferin and coelenterazine (CTZ) emit at around 560 nm and 480 nm,3,13 a recently developed luciferin analogue, AkaLumine can emit in the near infrared (NIR) region over 700 nm.11 This luciferin analogue has shown the capability of NIR bioluminescence for highly sensitive deep-tissue imaging in living mice.11,13
The use of bioluminescence resonance energy transfer (BRET) is an alternative method to achieve bioluminescence imaging at the longer wavelengths.14,15 BRET consists of a luciferase–luciferin system as a resonance energy donor and a fluorescent protein or dye as a resonance energy acceptor. A NIR fluorescent dye combined with luciferase (RLuc 8.6) has been used for BRET-based NIR imaging in living mice.11,16–18 Recently, Kizaka-Kondoh et al. developed an Alexa Fluor 680 conjugated BRET probe for the detection of ubiquitin-protease system regulated hypoxia-inducible factor.19 They achieved BRET-based NIR imaging of hypoxic liver metastasis in nude mice. Rao et al. also developed a BRET-based NIR imaging system which contains a fluorescent phthalocyanine dye, NIR775 as a resonance energy acceptor.20 They achieved BRET-based in vivo lymph-node mapping and tumour imaging.20 Quantum dots (QDs) can be used as energy acceptors for BRET-based NIR imaging.21–43 The BRET in QD–luciferase conjugates has been applied to NIR tumour imaging in living mice.40 In the BRET-based tumour imaging, a cyclic arginine–glycine–aspartic acid (cRGD) peptide has been used as a targeting ligand to integrin αvβ3, which expresses in many tumour cells20,40
In this paper, we present a BRET-based dual-colour (visible and NIR) molecular-imaging probe, which can be used for the highly sensitive detection of membrane receptors in living cells. To design the BRET-based dual-colour probe, we examined the bioluminescence properties for the conjugates between QDs and several types of EGFP–RLuc fusion proteins. We developed a BRET-based dual-colour probe, where BRET occurs from luciferase/luciferin to QDs and to EGFP. This BRET probe emits green and NIR luminescence from EGFP and CdSeTe/CdS (core/shell) QDs, respectively. Since the present BRET-based dual-colour probe contains an immunoglobulin binding domain (GB1)44 of protein G, this probe is easily functionalized with a variety of antibodies (IgG).43 Here, we demonstrate that the BRET-based dual-colour probe enables highly-sensitive bioluminescence imaging of human epidermal growth factor receptor 2 (HER2)45 and epidermal growth factor receptor (EGFR)45 in breast and skin cancer cells.
2. Results and discussion
2.1 Design of BRET-based molecular imaging probe
To develop BRET-based dual-colour molecular-imaging probes, we designed three types of probes, where EGFP–RLuc fused proteins are conjugated to NIR-emitting CdSeTe/CdS (core/shell) QDs. The fused proteins consist of RLuc, EGFP and GB1 moieties with six histidine-tags (Fig. 1a–c). In the EGFP–RLuc fused proteins, BRET from CTZ to EGFP can occur in the presence of RLuc. Immuno-globulin binding domain (GB1) of protein G in the fused protein can act as a binding site for the Fc region of the antibody (IgG).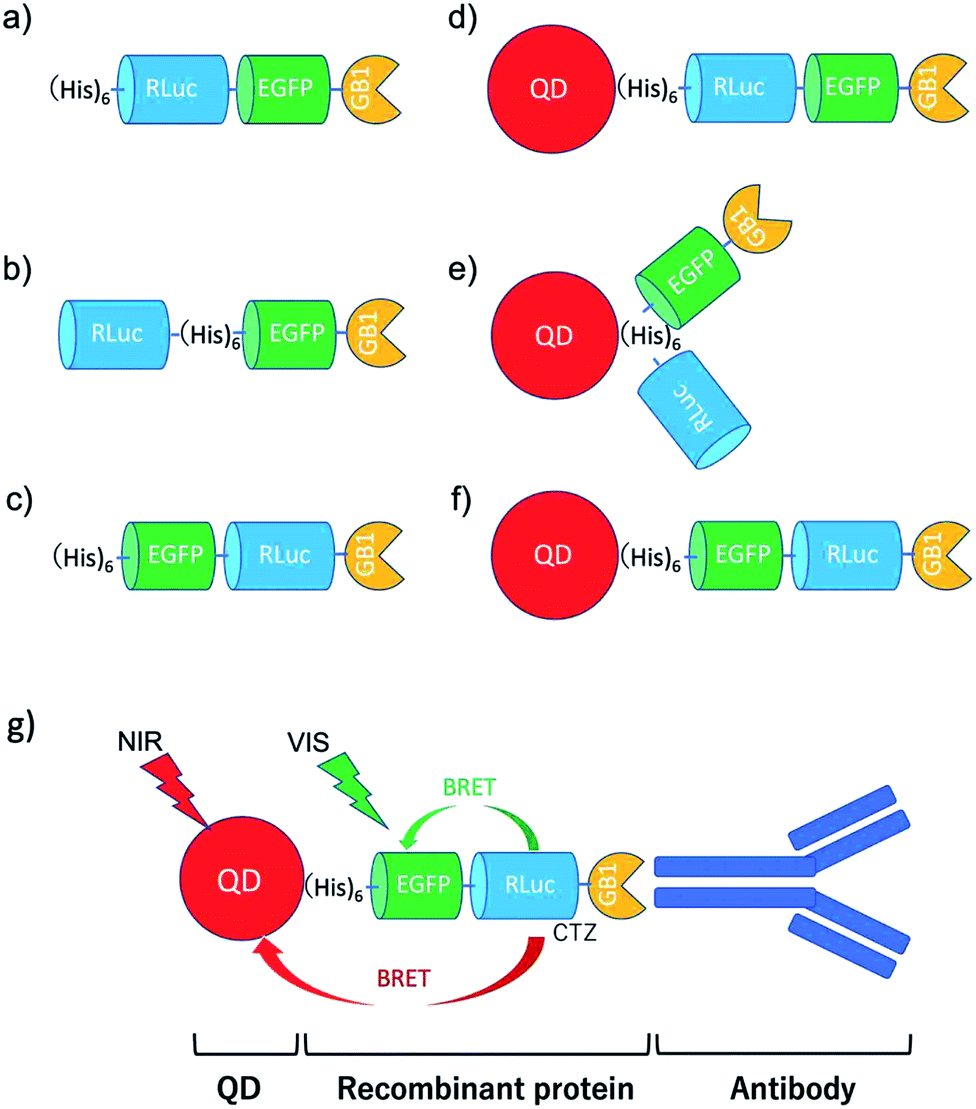 | ||
| Fig. 1 Schematic representation for EGFP–RLuc fused proteins and their conjugates with QDs (CdSeTe/CdS): (a) His–RLuc–EGFP–GB1 (73.6 kDa), (b) RLuc–His–EGFP–GB1 (72.6 kDa), (c) His–EGFP–RLuc–GB1 (73.6 kDa, (d) QD–His–RLuc–EGFP–GB1, (e) QD–RLuc–His–EGFP–GB1, and (f) QD–His–EGFP–RLuc–GB1. (g) Representative structure of a BRET-based dual-colour molecular imaging probe. The probe consists of a NIR-emitting QD, fused protein (His-EGFP–RLuc–GB1), and monoclonal antibodies. BRET is expected to occur from CTZ to EGFP and to QD, leading to visible and NIR dual-colour bioluminescence emission. | ||
The conjugates between QDs and EGFP–RLuc fused proteins (Fig. 1d–f) were designed to occur dual BRET from CTZ to QDs and from CTZ to EGFP (Fig. 1g). In addition, the QD conjugates with EGFP–RLuc fused proteins were designed to act as molecular imaging probes, where antibodies bound to a GB1 domain can recognize targeted biomolecules. We expected that the QD conjugate with EGFP–RLuc fused protein can act as BRET-based dual-colour (visible/NIR) molecular imaging probes in living cells.
2.2 BRET between CTZ and QD
To confirm BRET between CTZ and QD in the conjugate of QD and RLuc protein, we examined bioluminescence property for the mixture of His–RLuc–GB1 protein and QDs, and for the mixture of RLuc–GB1 protein and QDs. Since His–RLuc–GB1 protein has six histidine tags, this protein was expected to bind to the surface of CdSe/ZnS QDs46 and CdSeTe/CdS QDs (Fig. 2a).42,43 This is because histidine molecules have high affinities for Zn2+ and Cd2+ ions.47 In contrast, RLuc–GB1 with no his-tags was expected to show no binding ability to the QD surface (Fig. 2a). Agarose electrophoresis clearly showed that His–RLuc–GB1 protein binds to the surface of CdSeTe/CdS QDs, while RLuc–GB1 protein does not bind to the QDs (Fig. 2b).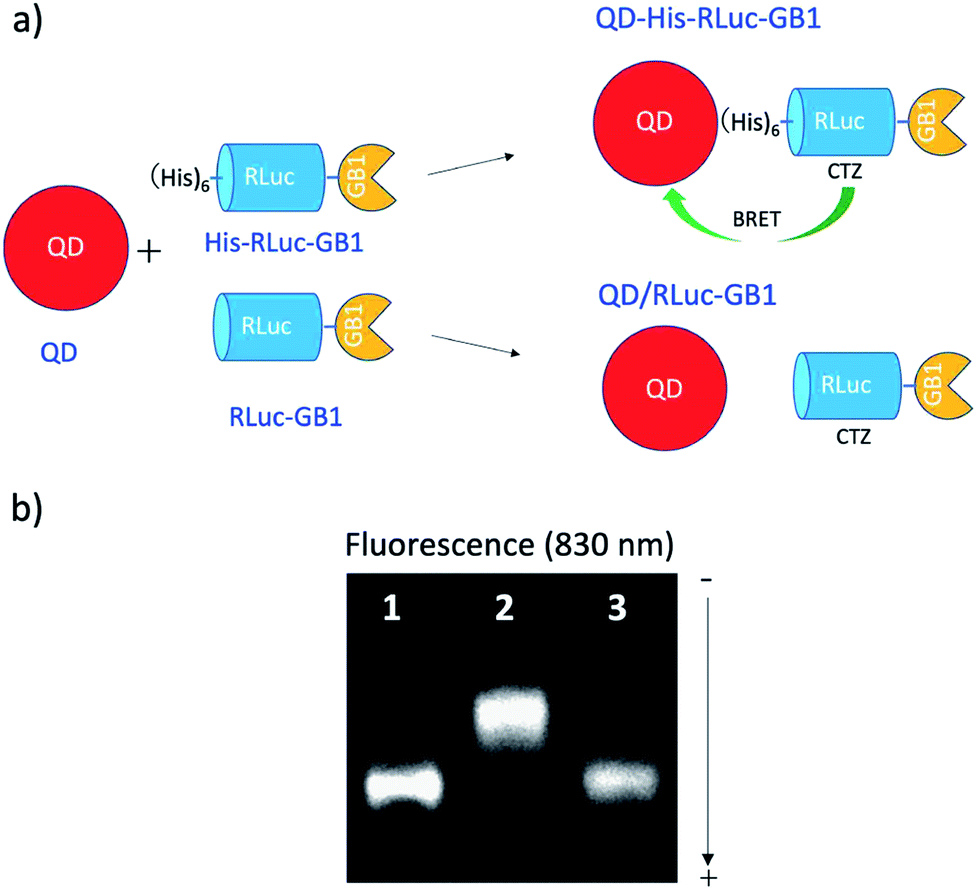 | ||
| Fig. 2 (a) Schematic representation for the conjugation of a CdSeTe/CdS QD to His–RLuc–GB1 and RLuc–GB1 protein. (b) Agarose gel electrophoresis for the QD (1), the mixture of QD and His–RLuc–GB1 (2), and the mixture of QDs and RLuc–GB1 (3). The molar ratio of QD/fused protein was 0.1.‡ The image shows NIR fluorescence detected at the wavelength of 830 ± 20 nm. | ||
Fig. 3a shows the absorption and fluorescence spectrum of QDs together with the bioluminescence spectrum of CTZ catalysed by His–RLuc–GB1. The bioluminescence spectrum of CTZ is overlapped with the absorption spectrum of QDs which emit at the NIR region. This finding suggests that BRET can occur between CTZ and QD in the conjugate of QD and His–RLuc–GB1 protein.
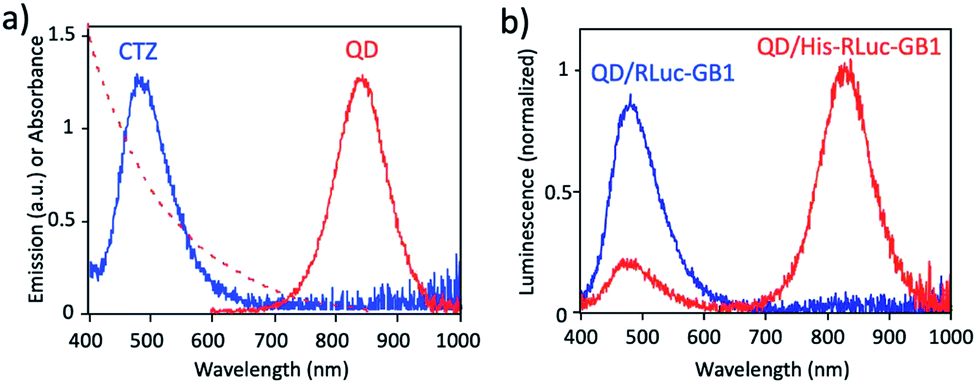 | ||
| Fig. 3 (a) Bioluminescence spectrum (blue line) of CTZ catalyzed by His–RLuc–GB1, and absorption (red dotted line) and emission spectrum (red solid line) of CdSeTe/CdS QDs in PBS. (b) Bioluminescence spectra for the mixture of QD and RLuc–GB1 protein (QD/RLuc–GB1), and the mixture of QD and His–RLuc–GB1 protein (QD/His–RLuc–GB1) in the presence of CTZ. The molar ratio of QD/fused protein was 0.1. | ||
To demonstrate BRET from CTZ to QD in the conjugate of QD and RLuc protein, we examined bioluminescence spectra for the mixture of QD/His–RLuc–GB1 and the mixture of QD/RLuc–GB1 in phosphate buffered saline (PBS). In the presence of CTZ, the mixture of QD/His–RLuc–GB1 showed bioluminescence spectrum with two peaks resulting from CTZ and QD at the visible and NIR region. (Fig. 3b). In contrast, the mixture of QD/RLuc–GB1 showed only a visible-emitting bioluminescence which corresponds to the emission of CTZ. This difference in the bioluminescence spectra clearly shows that the intramolecular BRET from CTZ to QD occurs in the conjugate of QD and His–RLuc–GB1 protein.
For the conjugate of QD and His–RLuc–GB1 protein, BRET efficiency (E) was estimated to about 63%, which was calculated from the following equation: E = 1 − (IDA/ID) × 100%,48,49 where IDA and ID represent the emission intensity of CTZ in the presence and absence of acceptor (QD), respectively. It should be noted that NIR luminescence at 700–1000 nm weakly emitted for the mixture of QD/RLuc–GB1. This NIR emission may have resulted from the reabsorption of the bioluminescence of CTZ by QDs.
2.3 BRET in EGFP–RLuc fused proteins
To develop a BRET-based dual-colour imaging probe, we prepared three types of EGFP–RLuc fused protein (Fig. 1a–c). The purity and molecular weight of these proteins were checked by SDS-PAGE (Fig. S1†). The absorption and emission spectra of EGFP in His–EGFP–GB1 (ref. 50) protein are shown in Fig. 4a with the bioluminescence spectrum of CTZ catalysed by His–RLuc–GB1. Since the emission spectrum of CTZ overlapped with the absorption spectrum of EGFP, it was expected that BRET form CTZ to EGFP can occur in the three types of EGFP–RLuc fused proteins, His–RLuc–EGFP–GB1, RLuc–His–EGFP–GB1, and His–EGFP–RLuc–GB1. In all three proteins, BRET emissions from CTZ to EGFP were observed (Fig. 4b–d). Among the EGFP–RLuc fused proteins, His–EGFP–RLuc–GB1 showed most efficient BRET from CTZ to EGFP (Fig. 4d). The value of BRET efficiency was estimated to 55% for His–RLuc–EGFP–GB1, 49% for RLuc–His–EGFP–GB1, and 65% for His–EGFP–RLuc–GB1. The BRET efficiency was not proportional to the linker length between EGFP and RLuc. The linker between His–RLuc–EGFP–GB1, RLuc–EGFP–GB1, and His–EGFP–RLuc–GB1 was two-, nine-, and two-amino acids, respectively (Fig. S2†).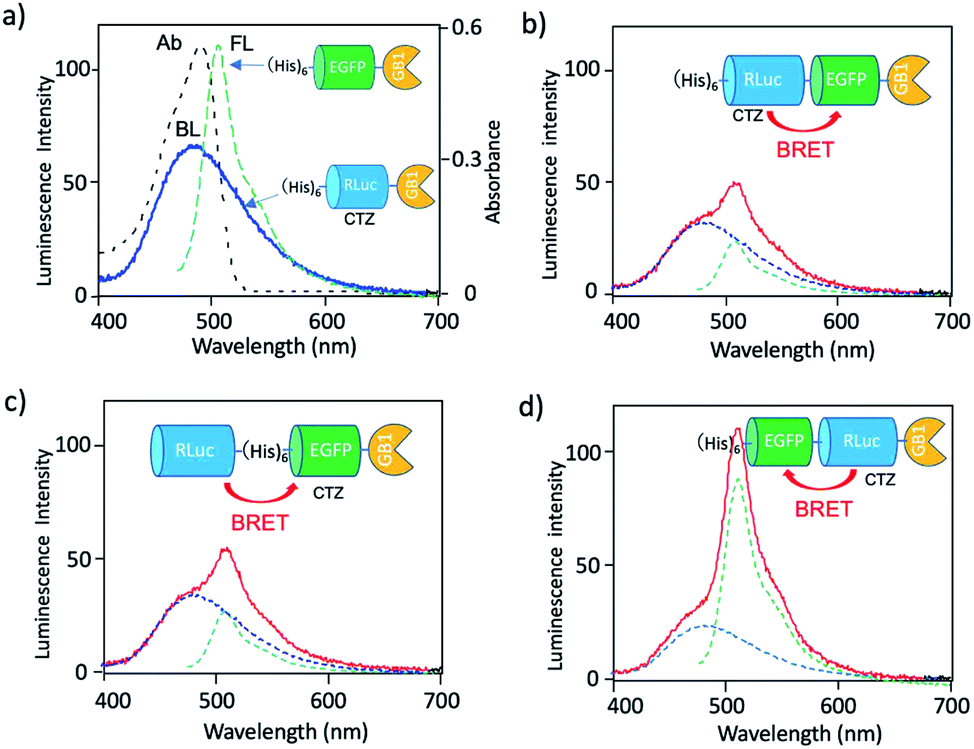 | ||
| Fig. 4 (a) Absorption (Ab: black dotted line) and fluorescence (FL: green dotted line) spectrum of His–EGFP–GB1 protein. A blue solid line shows bioluminescence (BL) of CTZ catalyzed by His–RLuc–GB1. (b–d) Bioluminescence spectra (red line) for His–RLuc–EGFP–GB1, RLuc–His–EGFP–GB1, and His–EGFP–RLuc–GB1. Blue and green dotted lines show the spectral contribution from CTZ and EGFP, respectively. | ||
The BRET efficiency can be influenced by the emission quantum yield (QY) of acceptor as well as the distance between donor and acceptor. The QYs of EGFP in three types of EGFP–RLuc fused proteins are shown in Table 1. The QY values of EGFP emission in the fused proteins are almost the same as that of His–EGFP–GB1. In addition, fluorescence lifetime of EGFP emission for the three fused proteins also showed similar values (τ = 2.63–2.65 ns) (Fig. S3†). These findings indicate that the chemical environment of EGFP is not significantly different in the three EGFP–RLuc fused proteins. Thus, the difference in the BRET efficiency in the fused proteins may be attributed to the difference in the conformation between CTZ and EGFP moieties in the protein. It is well-known that the conformation of the transition dipoles between donor and acceptor significantly affects the efficiency of resonance energy transfer.51
| Protein | QY | τ(ns) |
|---|---|---|
| His–EGFP–GB1 | 0.77 | 2.62 |
| His–RLuc–EGFP–GB1 | 0.78 | 2.63 |
| RLuc–His–EGFP–GB1 | 0.76 | 2.64 |
| His–EGFP–RLuc–GB1 | 0.77 | 2.65 |
2.4 BRET in QD conjugates with EGFP–RLuc fused proteins
Since the EGFP–RLuc proteins (Fig. 1a–c) have six histidine tags, these proteins can bind to the surface of CdSeTe/CdS QDs. The binding of these proteins to the QDs was confirmed by agarose gel electrophoresis (Fig. 5a–c). The binding affinities of EGFP–RLuc fused proteins to the QDs were almost the same between His–RLuc–EGFP–GB1 (Fig. 5a) and His–EGFP–RLuc–GB1 (Fig. 5c). In contrast, the binding affinity of RLuc–His–EGFP–GB1 to the QD was two times lower than that of His–RLuc–EGFP–GB1 and His–EGFP–RLuc–GB1 (Fig. 5b). This difference may be attributed to the steric hindrance of histidine tags of RLuc–His–EGFP–GB1 in their accessibility to the surface of QDs.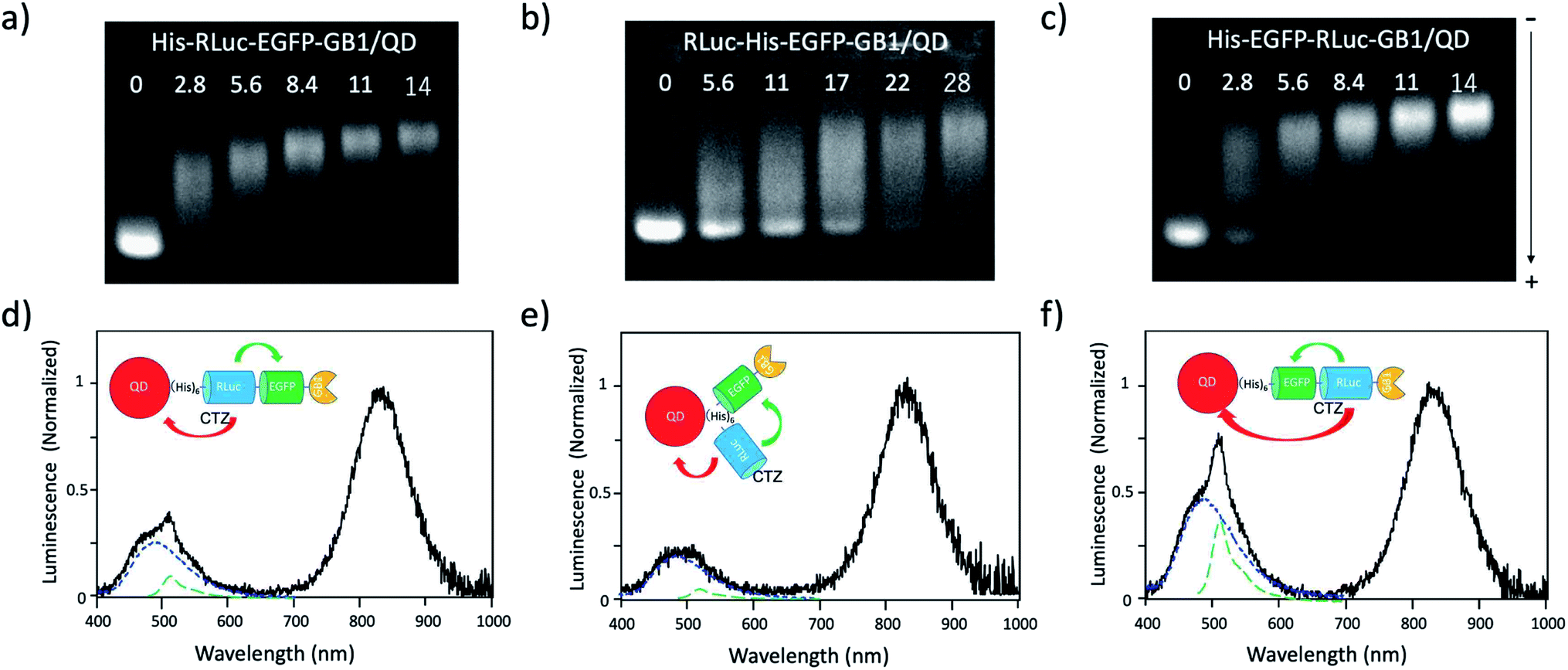 | ||
| Fig. 5 (a)–(c) Agarose gel electrophoresis for the mixtures of His–RLuc–EGFP–GB1 and QD, RLuc–His–EGFP–GB1 and QD, and His–EGFP–RLuc–GB1 and QD. The molar ratios of fused protein/QD are described in the images. The gel images were obtained by the NIR fluorescence of QDs at 830 nm. (d)–(f) Bioluminescence spectra of QD conjugates with His–RLuc–EGFP–GB1, RLuc–His–EGFP–GB1, and His–EGFP–RLuc–GB1 in the presence of CTZ. The molecular ratios of fused protein/QD were 14.‡Blue and green dotted lines show the spectral contribution from CTZ and EGFP, respectively. | ||
For all three types of the conjugates between QDs and EGFP–RLuc fused proteins, strong NIR emissions owing to BRET from CTZ to QDs were observed (Fig. 5d–f). At the same time, EGFP emissions around at 515 nm were observed for the three types of QD conjugates. The overall BRET efficiency from CTZ to QDs and EGFP was estimated to 83% for a QD/His–RLuc–EGFP–GB1 conjugate, 91% for a QD/RLuc–His–EGFP–GB1 conjugate, 69% for a QD/His–RLuc–EGFP–GB1 conjugate (Fig. S4†). It should be noted that the intensity of QD emission (700–1000 nm) is much higher than that of EGFP emission (480–600 nm) in the QD/protein conjugates. The QYs (0.76–0.78) of EGFP emission in the three fused proteins were not significantly different from the QY (0.58) of NIR-emitting QDs. Thus, the brighter QD emission due to BRET should be resulted from the large extinction coefficient (ca. 3 × 106)§ of QDs compared to EGFP (5.5 × 104).52 Among the QD conjugates with EGFP–RLuc fused proteins, a QD/His–RLuc–EGFP–GB1 conjugate showed comparable intensities for the visible (400–600 nm) and NIR emission (700–1000 nm) (Fig. 5f). As a BRET probe for dual-colour molecular imaging, we used a QD/His–RLuc–EGFP–GB1 conjugate. In this QD/His–RLuc–EGFP–GB1 conjugate, the half-life time of BRET emissions was observed to ca. 100 s (Fig. S5†).
2.5 Binding ability of His–EGFP–RLuc–GB1 conjugated QDs for antibodies
The binding ability of His–EGFP–RLuc–GB1 conjugated QDs for antibodies was examined by fluorescence correlation spectroscopy (FCS).51,53 FCS gives the value of diffusion time of fluorescent particles from the fluctuation of their fluorescence intensity at the single molecule level. FCS is very sensitive to the change in the particle size, which affects the diffusion time of particles in solution. By FCS, the diffusion time of QDs in PBS was estimated to be 0.40 ± 0.01 ms (Fig. 6a). The addition of His–EGFP–RLuc–GB1 to QDs increased the diffusion time of QDs by a factor of 1.8 (Fig. 6a and b), showing that His–EGFP–RLuc–GB1 protein binds to the surface of the QDs. The number of the His–EGFP–RLuc–GB1 molecule bound to one QD particle was estimated to be 6.4 by using the size-exclusion chromatography (Fig. S6†). His–EGFP–RLuc–GB1 conjugated QDs were mono-dispersed particles with the core size of 4.0 ± 0.6 nm (Fig. S7†). Further addition of Herceptin (monoclonal anti-HER2 antibody) to the mixture of QDs and His–EGFP–RLuc–GB1 increased the diffusion time of QDs to be 1.31 ± 0.09 ms (Fig. 6a and b), indicating the binding of the antibody to the surface of His–EGFP–RLuc–GB1 conjugated QDs. The binding ability of His–EGFP–RLuc–GB1 conjugated QDs for the antibody was also confirmed by agarose gel electrophoresis (Fig. S8†).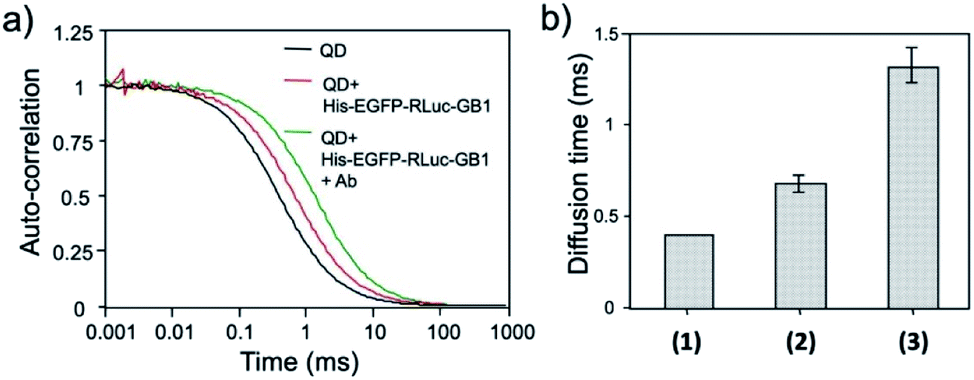 | ||
| Fig. 6 (a) Fluorescence autocorrelation curves for QDs, the mixture of QDs and His–EGFP–RLuc–GB1, and the mixture of QDs, His–EGFP–RLuc–GB1 and antibody (Ab) in PBS. (b) Diffusion time of QDs (1), the mixture of QDs and His–EGFP–RLuc–GB1 (2), and the mixture of QDs, His–EGFP–RLuc–GB1 and Ab (3). The values of diffusion time were obtained by using a single-component diffusion model.53 | ||
Further, the binding ability of a QD/His–EGFP–RLuc–GB1 conjugate to the antibody was examined in human breast tumour cells (KPL-4),54,55¶using fluorescence activated cell sorter (FACS). KPL-4 cells are known to overexpress HER2 on their plasma membranes, and the expression level of HER2 in KPL-4 cell is higher than that to EGFR.54 FACS shows that Herceptin strongly binds to the KPL-4 cell in the presence of QD/His–EGFP–RLuc–GB1, while Erbitux (anti-EGFR monoclonal antibody) does not bind to the cell (Fig. 7). Normal human IgG was used as a negative control. Control data (none) was also obtained by using QD/His–EGFP–RLuc–GB1 conjugate. The FACS result indicated that the QD/His–EGFP–RLuc–GB1 conjugate functionalized with the antibody can act as a molecular imaging probe in living cells.
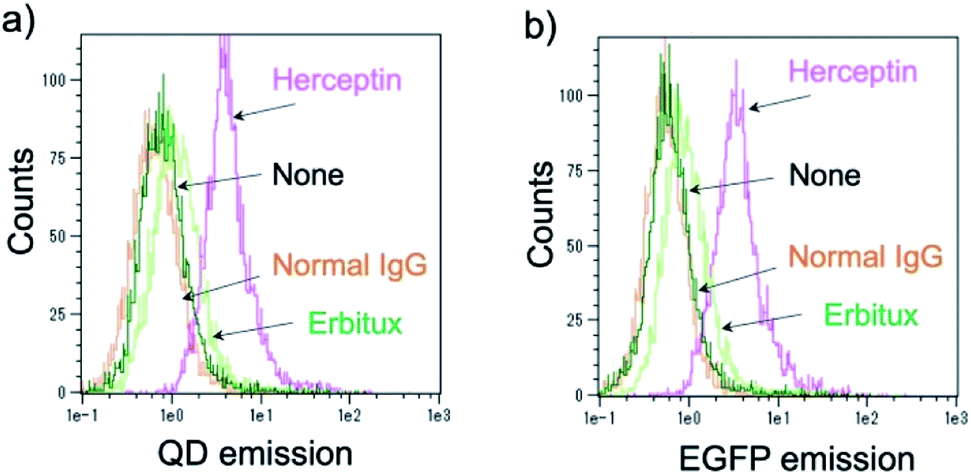 | ||
| Fig. 7 Flow cytometric analysis of KPL-4 cells treated with QD–His–EGFP–RLuc–GB1 (none), normal human IgG and QD–His–EGFP–RLuc–GB1 (normal IgG), Herceptin and QD–His–EGFP–RLuc–GB1 (Herceptin), Erbitux and QD–His–EGFP–RLuc–GB1 (Erbitux). Fluorescence signals were detected at >750 nm for QD emission (a) and at 525 nm for EGFP emission (b). | ||
2.6 BRET-based dual-colour molecular imaging
Next, we examined the capability of a QD/His–EGFP–RLuc–GB1 conjugate for the dual-colour bioluminescence molecular imaging of membrane receptors in cancer cells. We performed BRET-based molecular imaging for KPL-4 and A431 (human skin cancer cells)|| cell pellets (∼106 cells) for the detection of HER2 and EGFR on their cell surface. In this molecular imaging, we first incubated cancer cells with antibodies, and then, His–EGFP–RLuc–GB1 conjugated QDs were added to the cancer cells to prevent the aggregation of QDs.** BRET and fluorescence (FL) images for KPL-4 and A431 cells were taken immediately after the addition of CTZ to the cell pellets.As shown in Fig. 8a, BRET emissions were observed both at 830 and 530 nm for KPL-4 cells treated with Herceptin and Erbitux. In the case of A431 cells, BRET emissions were observed only for the cells treated with Erbitux (Fig. 8b). The BRET emissions in the control (none in Fig. 8a) may have resulted from the nonspecific binding of QD-protein conjugates to the KPL-4 cell surface. The results of BRET imaging for KPL-4 and A431 cells were consistent with the results of western blotting analysis (Fig. 8c). For the KPL-4 cells, both the expression of HER2 and EGFR was observed, while for the A431 cells, only the expression of EGFR was observed.
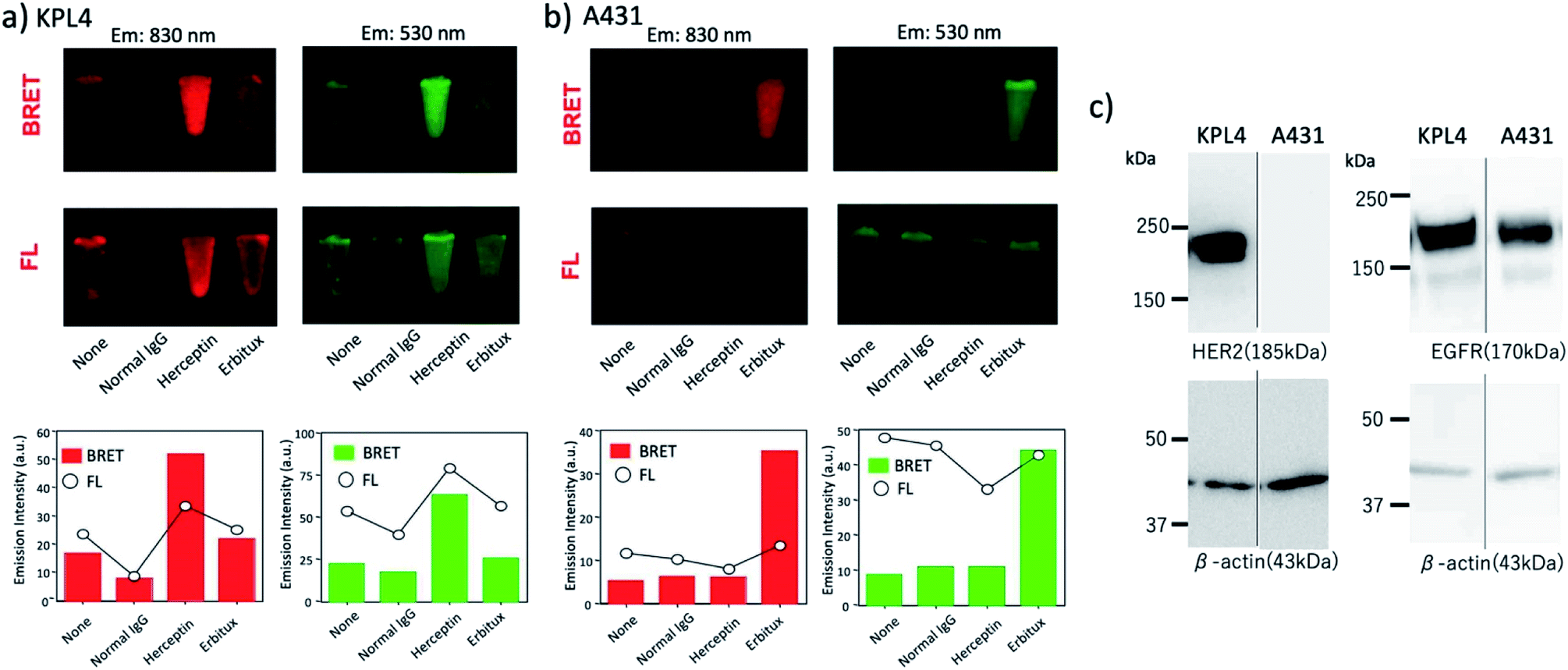 | ||
| Fig. 8 (a) BRET and fluorescence (FL) images for the pellets of (a) KPL-4 cell (∼106 cells) and (b) A431 cell (∼106 cells) treated with QD–His–EGFP–RLuc–GB1 (none), normal human IgG/QD–His–EGFP–RLuc–GB1 (normal IgG), Herceptin/QD–His–EGFP–RLuc–GB1 (Herceptin), and Erbitux/QD–His–EGFP–RLuc–GB1 (Erbitux). The lower graphs show the emission intensity of BRET and FL for the above images. BRET and FL images were taken at 830 ± 20 nm and 530 ± 20 nm, respectively. Exposure time was 10 min both for BRET and FL imaging. (c) Western blotting analysis for the expression level of HER2 and EGFR in KPL-4 and A431 cells. | ||
The comparison of BRET and fluorescence (FL) images (Fig. 8a and b) of KPL-4 and A431 cells showed higher detection sensitivity for the BRET imaging of HER2 and EGFR (lower graphs in Fig. 8a and b). The higher signal to background ratios in the BRET images compared to the FL images should be attributed to the low signals of background emissions, which resulted from sample autofluorescence.
Finally, we performed bioluminescence cellular imaging based on BRET. For the bioluminescence cellular imaging, we used a highly sensitive imaging microscopy equipped with a cooled EM-CCD camera,56 because of the low photon-counts in BRET-based cellular imaging. In the BRET imaging, we needed an exposure time of more than 3 min to get clear images with high signal to background ratios. Fig. 9 shows the bioluminescence images for KPL-4 cells treated with a Herceptin and QD/His–EGFP–RLuc–GB1 conjugate. The localization of HER2 at the plasma membrane was clearly visualized by the BRET imaging both at the visible and NIR region (Fig. 9b). This result shows that the present BRET-based probe can be used for the dual-colour bioluminescence molecular imaging of membrane receptors in living cells. Although the BRET-based QD probe has a dose-dependent cytotoxicity (Fig. S9†),57 significant cytotoxicities were not found under the concentration of QDs (<50 nM) used for the BRET-based cellular imaging.
 | ||
| Fig. 9 (a) Bright field (BF) and bioluminescence images of KLP-4 cells treated with Herceptin and His–EGFP–RLuc–GB1 conjugated QDs. (b) Magnification images for the square regions in the above images. Bioluminescence images are taken at the wavelength of >715 nm for QD emission and 495–540 nm for EGFP emission. Exposure time for the bioluminescence imaging was set to 3 min. | ||
3. Conclusions
In this paper, we have presented a BRET-based molecular imaging probe for the highly sensitive detection of membrane receptors in cancer cells. The present BERT probe consists of a NIR-emitting QD and EGFP–RLuc fusion protein. In addition, this BRET probe has an immunoglobulin binding domain (GB1). Thus, the BRET probe is applicable to a variety of bioluminescence molecular imaging by the functionalization with antibodies. We demonstrated that the BRET-based molecular imaging has higher detection sensitivity for HER2 and EGFR compared to fluorescence-based molecular imaging. We further demonstrated that the present BRET probe can be used for dual-colour molecular imaging at the visible and NIR region.Although bioluminescence imaging has a great potential as an optical molecular imaging, its application has been restricted to protein expression and protein–protein interaction using their reporter genes. To date, there have been no reports of BRET based dual-colour probes which can be used for a variety of bioluminescence molecular imaging. We believe that the present BRET probe will add a significant progress in the BRET-based molecular imaging at the cellular and tissue level.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Sayumi Yamada and Satoko Masa for their help in protein synthesis, cell culture, and viability test. The authors also thank Dr Takao Sakata and Prof. Hidehiro Yasuda for their help in TEM measurements. The authors also thank Dr Mahadeva Swamy for his critical reading of the manuscript. This work is partly supported by the Ministry of Education, Science, Sport, and Culture of Japan (Grant-in-Aid for Scientific Research (B) 19H04459 to TJ).Notes and references
- A. P. Dhawan, B. D'Alessandro and X. Fu, IEEE Rev. Biomed. Eng., 2010, 3, 69–92 Search PubMed.
- T. Ozawa, H. Yoshimura and S. Kim, Anal. Chem., 2013, 85, 590–609 CrossRef CAS.
- V. Ntziachristos, Annu. Rev. Biomed. Eng., 2006, 8, 1–33 CrossRef CAS.
- C. E. Badr and B. A. Tannous, Trends Biotechnol., 2011, 29, 624–633 CrossRef CAS.
- M. Monici, Biotechnol. Annu. Rev., 2005, 11, 227–256 CAS.
- J. M. Menter, Photochem. Photobiol. Sci., 2006, 5, 403–410 RSC.
- R. Weissleder, Nat. Biotechnol., 2001, 19, 316–317 CrossRef CAS.
- R. Kojima, H. Takakura, T. Ozawa, Y. Tada, T. Nagano and Y. Urano, Angew. Chem., Int. Ed., 2013, 52, 1175–1179 CrossRef CAS.
- S. Iwano, R. Obata, C. Miura, M. Kiyama, K. Hama, M. Nakamura, Y. Amano, S. Kojima, T. Hirano, S. Maki and H. Niwa, Tetrahedron, 2013, 69, 3847–3856 CrossRef CAS.
- A. P. Jathoul, H. Grounds, J. C. Anderson and M. A. Pule, Angew. Chem., Int. Ed., 2014, 53, 13059–13063 CrossRef CAS.
- T. Kuchimaru, S. Iwano, M. Kiyama, S. Mitsumata, T. Kadonosono, H. Niwa, S. Maki and S. Kizaka-Kondoh, Nat. Commun., 2016, 7, 11856 CrossRef CAS.
- J. C. Anderson, H. Grounds, A. P. Jathoul, J. A. H. Murray, S. J. Pacman and L. Tisi, RSC Adv., 2017, 7, 3975–3982 RSC.
- S. Iwano, M. Sugiyama, H. Hama, A. Watakabe, N. Hasegawa, T. Kuchimaru, K. Z. Tanaka, M. Takahashi, Y. Ishida, J. Hata, S. Shimozono, K. Namiki, T. Fukano, M. Kiyama, H. Okano, S. Kizaka-Kondoh, T. J. McHugh, T. Yamamori, H. Hioki, S. Maki and A. Miyawaki, Science, 2018, 359, 935–939 CrossRef CAS.
- C. Wu, K. Mino, H. Akimoto, M. Kawabata, K. Nakamura, M. Ozaki and Y. Ohmiya, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15599–15603 CrossRef CAS.
- K. Saito, Y. F. Chang, K. Horikawa, N. Hatsugai, Y. Higuchi, M. Hashida, Y. Yoshida, T. Matsuda, Y. Arai and T. Nagai, Nat. Commun., 2012, 3, 1262 CrossRef.
- A. Takai, M. Nakano, K. Saito, R. Haruno, T. M. Watanabe, T. Ohyanagi, T. Jin, Y. Okada and T. Nagai, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4352–4356 CrossRef CAS.
- B. R. Branchini, D. M. Ablamsky and J. C. Rosenberg, Bioconjugate Chem., 2010, 21, 2023–2030 CrossRef CAS.
- K. A. Rumyantsev, K. K. Turoverov and V. V. Verkhusha, Sci. Rep., 2016, 6, 36588 CrossRef CAS.
- T. Kuchimaru, T. Suka, K. Hirota, T. Kadonosono and S. Kizaka-Kondoh, Sci. Rep., 2016, 6, 34311 CrossRef CAS.
- L. Xiong, A. J. Shuhendler and J. Rao, Nat. Commun., 2012, 3, 1193 CrossRef.
- M. K. So, C. Xu, A. M. Loening, S. S. Gambhir and J. Rao, Nat. Biotechnol., 2006, 24, 339–343 CrossRef CAS PubMed.
- M. K. So, A. M. Loening, S. S. Gambhir and J. Rao, Nat. Protoc., 2006, 1, 1160–1164 CrossRef CAS.
- Y. Zhang, M. K. So, A. M. Loening, H. Yao, S. S. Gambhir and J. Rao, Angew. Chem., Int. Ed., 2006, 45, 4936–4940 CrossRef CAS.
- H. Yao, Y. Zhang, F. Xiao, Z. Xia and J. Rao, Angew. Chem., Int. Ed., 2007, 46, 4346–4349 CrossRef CAS.
- Y. Xing, M. K. So, A. L. Koh, R. Sinclair and J. Rao, Biochem. Biophys. Res. Commun., 2008, 372, 388–394 CrossRef CAS.
- K. A. Cissell, S. Campbell and S. K. Deo, Anal. Bioanal. Chem., 2008, 391, 2577–2581 CrossRef CAS.
- Z. Xia, Y. Xing, M. K. So, A. L. Koh, R. Sinclair and J. Rao, Anal. Chem., 2008, 80, 8649–8655 CrossRef CAS.
- J. Du, C. Yu, D. Pan, J. Li, W. Chen, M. Yan, T. Segura and Y. Lu, J. Am. Chem. Soc., 2010, 132, 12780–12781 CrossRef CAS.
- N. Ma, A. F. Marshall and J. Rao, J. Am. Chem. Soc., 2010, 132, 6884–6885 CrossRef CAS.
- C. Wu, K. Kawasaki, S. Ohgiya and Y. Ohmiya, Photochem. Photobiol. Sci., 2011, 10, 1531–1534 RSC.
- M. Kumar, D. Zhang, D. Broyles and S. K. Deo, Biosens. Bioelectron., 2011, 30, 133–139 CrossRef CAS.
- G. A. Quinones, S. C. Miller, S. Bhattacharyya, D. Sobek and J. P. Stephan, J. Cell. Biochem., 2012, 113, 2397–2405 CrossRef CAS.
- Q. Wu and M. Chu, Int. J. Nanomed., 2012, 7, 3433–3443 CAS.
- R. Alam, D. M. Fontaine, B. R. Branchini and M. M. Maye, Nano Lett., 2012, 12, 3251–3256 CrossRef CAS.
- L. Xiong, A. J. Shuhendler and J. Rao, Nat. Commun., 2012, 3, 1193 CrossRef.
- M. Hasegawa, Y. Tsukasaki, T. Ohyanagi and T. Jin, Chem. Commun., 2013, 49, 228–230 RSC.
- R. Alam, J. Zylstra, D. M. Fontaine, B. R. Branchini and M. M. Maye, Nanoscale, 2013, 5, 5303–5306 RSC.
- R. Alam, L. M. Karam, T. L. Doane, J. Zylstra, D. M. Fontaine, B. R. Branchini and M. M. Maye, Nanotechnology, 2014, 25, 495606 CrossRef PubMed.
- A. Samanta, S. A. Walper, K. Susumu, C. L. Dwyer and I. L. Medintz, Nanoscale, 2015, 7, 7603–7614 RSC.
- A. Kamkaew, H. Sun, C. G. England, L. Cheng, Z. Liu and W. Cai, Chem. Commun., 2016, 52, 6997–7000 RSC.
- R. Alam, L. M. Karam, T. L. Doane, K. Coopersmith, D. M. Fontaine, B. R. Branchini and M. M. Maye, ACS Nano, 2016, 10, 1969–1977 CrossRef CAS.
- S. Tsuboi and T. Jin, ChemBioChem, 2017, 18, 2231–2235 CrossRef CAS.
- S. Tsuboi and T. Jin, Bioconjugate Chem., 2018, 29, 1466–1474 CrossRef CAS.
- A. M. Gronenborn, D. R. Filpula, N. Z. Essig, A. Achari, M. Whitlow, P. T. Wingfield and G. M. Clore, Science, 1991, 253, 657–661 CrossRef CAS.
- L. T. Jia, L. H. Zhang, C. J. Yu, J. Zhao, Y. M. Xu, J. H. Gui, M. Jin, Z. L. Ji, W. H. Wen, C. J. Wang, S. Y. Chen and A. G. Yang, Cancer Res., 2003, 63, 3257–3262 CAS.
- J. Zylstra, J. Amey, N. J. Miska, L. Pang, C. R. Hine, J. Langer, R. P. Doyle and M. M. Maye, Langmuir, 2011, 27, 4371–4379 CrossRef CAS PubMed.
- (a) P. J. Morris and R. B. Martin, J. Inorg. Nucl. Chem., 1970, 32, 2891–2897 CrossRef CAS; (b) U. L. Lao, A. Mulchandani and W. Chen, J. Am. Chem. Soc., 2006, 128, 14756–14757 CrossRef CAS; (c) R. Alam, L. M. Karam, T. L. Doane, J. Zylstra, D. M. Fontaine, B. R. Branchini and M. M. Maye, Nanotechnology, 2014, 25, 495606 CrossRef.
- S. Tsuboi, A. Sasaki, T. Sakata, H. Yasuda and T. Jin, Chem. Commun., 2017, 53, 9450–9453 RSC.
- A. Samanta, S. A. Walper, K. Susumu, C. L. Dwyer and I. L. Medintz, Nanoscale, 2015, 7, 7603–7614 RSC.
- S. Tsuboi and T. Jin, ChemBioChem, 2019, 20, 568–575 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- S. R. McRae, C. L. Brown and G. R. Bushell, Protein Expression Purif., 2005, 41, 121–127 CrossRef CAS.
- A. A. de Thomaz, D. B. Almeida and C. L. Cesar, Methods Mol. Biol., 2014, 1199, 85–91 CrossRef CAS.
- J. Kurebayashi, T. Otsuki, C. K. Tang, M. Kurosumi, S. Yamamoto, K. Tanaka, M. Mochizuki, H. Nakamura and H. Sonoo, Br. J. Cancer, 1999, 79, 707–717 CrossRef CAS.
- K. Fujimoto-Ouchi, F. Sekiguchi and Y. Tanaka, Cancer Chemother. Pharmacol., 2002, 49, 211–216 CrossRef CAS.
- D. J. Denvir and C. G. Coates, Proc. SPIE, 2002, 4626, 502–512 CrossRef CAS.
- T. Jin, F. Fujii, Y. Komai, J. Seki, A. Seiyama and Y. Yoshioka, Int. J. Mol. Sci., 2008, 9, 2044–2061 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and supporting figures. See DOI: 10.1039/c9ra07011g |
| ‡ The molar ratios of QD/fused protein were estimated from the concentrations and volumes of their aqueous solutions used for the gel electrophoresis. The concentration of QDs was 1 μM, which was estimated by using fluorescence correlation spectroscopy for the aqueous solution of QDs (see, ESI†). The concentration of aqueous solutions of His–RLuc (Mw: 40.0 kDa) and His–Rluc–GB1 (Mw: 45.5 kDa) was 25 and 22 μM, respectively. |
| § Molar extinction coefficient (ε) of a QD solution was calculated by the equation: ε = C × L/A, where C, L, A is the concentration of QDs, path length, and the absorbance of a QD solution. The concentration of QD was estimated by FCS (see, ESI†). |
| ¶ KPL-4 human breast cancer cells were kindly provided by Dr Kurebayashi (Kawasaki Medical school, Kurashiki, Japan). |
| || A413 human skin cancer cells were purchased from RIKEN BRC CELL BANK. |
| ** When the mixture of antibodies and His–EGFP–RLuc–GB1 conjugated QDs was directly added to a cell culture dish, QD aggregation was observed probably due to electric interaction between antibodies and QDs. |
| This journal is © The Royal Society of Chemistry 2019 |