DOI:
10.1039/C9RA07288H
(Paper)
RSC Adv., 2019,
9, 33170-33179
Heterocyclic iodoniums as versatile synthons to approach diversified polycyclic heteroarenes†
Received
11th September 2019
, Accepted 9th October 2019
First published on 16th October 2019
Abstract
Polycyclic heteroarenes are important scaffolds in the construction of pharmaceuticals. We have previously developed a series of novel heterocyclic iodoniums. In our current work, these unique iodoniums were employed to construct various complex polycyclic heteroarenes with structural diversity via tandem dual arylations. As a result, indole, thiophene and triphenylene motifs were fused into these heterocycles with high molecular quality, which might provide promising fragments in drug discovery. Moreover, these heterocycles could be diversified at a late stage.
Introduction
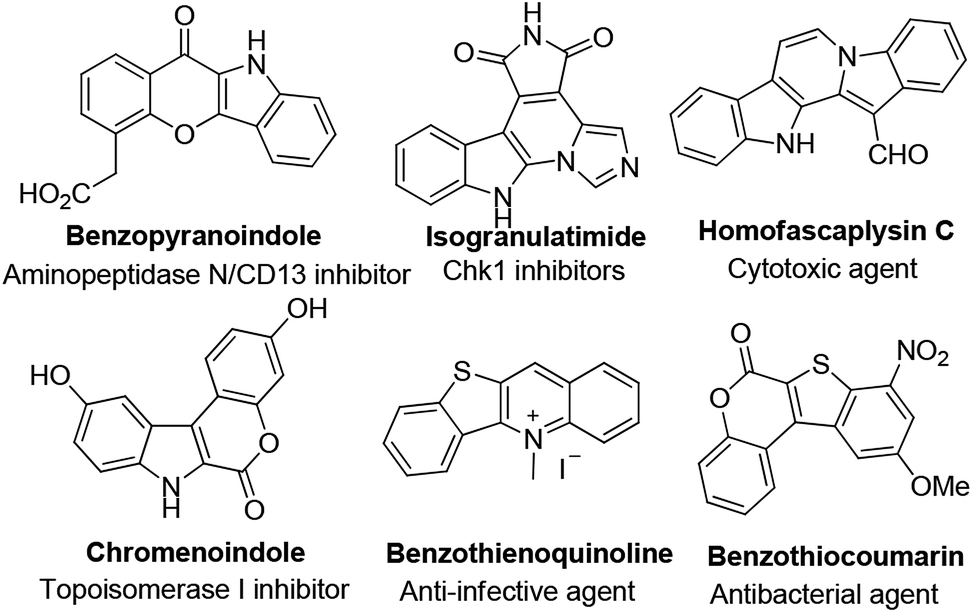
Polycyclic heteroarenes are important scaffolds in the construction of pharmaceuticals.1 Compared with polycyclic aromatic hydrocarbons, heterocycles exhibit improved solubility and bioavailability, which make them promising drug candidates.2 Many heterocycles are reported as kinase inhibitors, anti-infective and antibacterial agents (Fig. 1). Thus, the design and synthesis of polycyclic heteroarenes are highly demanded.
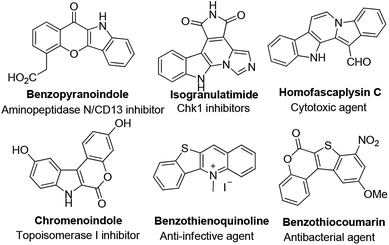 |
| | Fig. 1 Selected examples of heterocyclic natural products and pharmaceuticals. | |
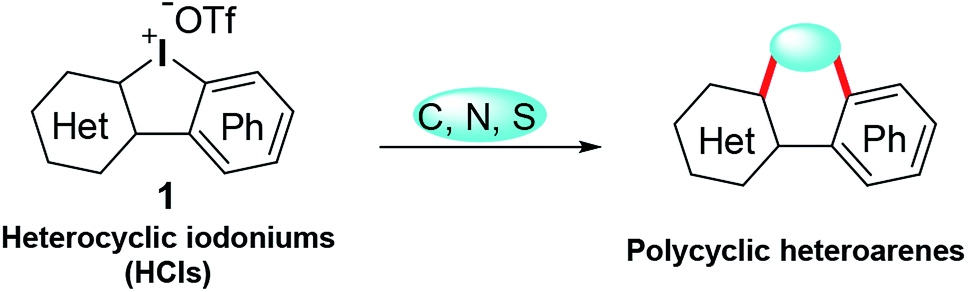
Tandem reactions enable rapid access to complex molecules, avoiding tedious purification steps and minimizing chemical waste generation.3 As a paradigm of tandem reactions, dual arylations with cyclic diphenyl iodoniums (CDPIs) are employed to construct various polycycles, such as acridine,4 carbazole,5 fluorene,6 phenanthrene,7 and dibenzothiophene.8 These obtained polycycles are often heavily hydrocarbon oriented. In the drug discovery field, heterocyclic frameworks are crucial to gain druggability. Heterocyclic iodoniums (HCIs) could be promising alternative reagents to replace CDPIs for the potential construction of heterocycles (Scheme 1). However, HCIs are under-explored and only few of them have been reported.9 Very recently, we have developed a series of new HCIs,10 and now we wish to fully investigate their synthetic application potentials to obtain diverse heterocycles via tandem transformations.
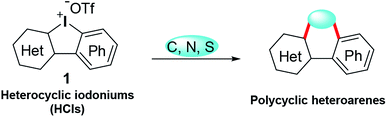 |
| | Scheme 1 Synthesis of polycyclic heteroarenes using HCIs. | |
Indole-fused polyheterocycles are privileged structural motifs.11 Despite various strategies to generate these complex molecules, there still lacks a general approach to rapidly build libraries of indole-fused heterocycles with a skeleton diversity. In our previous work, dual aminations of CDPIs led to the construction of functionalized N-substituted carbazoles.5b Inspired by this work, we hypothesized the amination strategy could be extended to construct indole-fused heterocycles if HCIs were used as starting materials to replace CDPIs. In this current work, we thoroughly investigated tandem dual aminations of HCIs with various amines to produce indole-fused polycyclic scaffolds. In addition, the annulations of HCIs with triethylammonium N-benzyldithiocarbamate and 2-chlorobenzoic acid were also fully investigated. These transformations will provide efficient pathways for rapid generation of complex heterocycles with a structural diversity.
Results and discussion
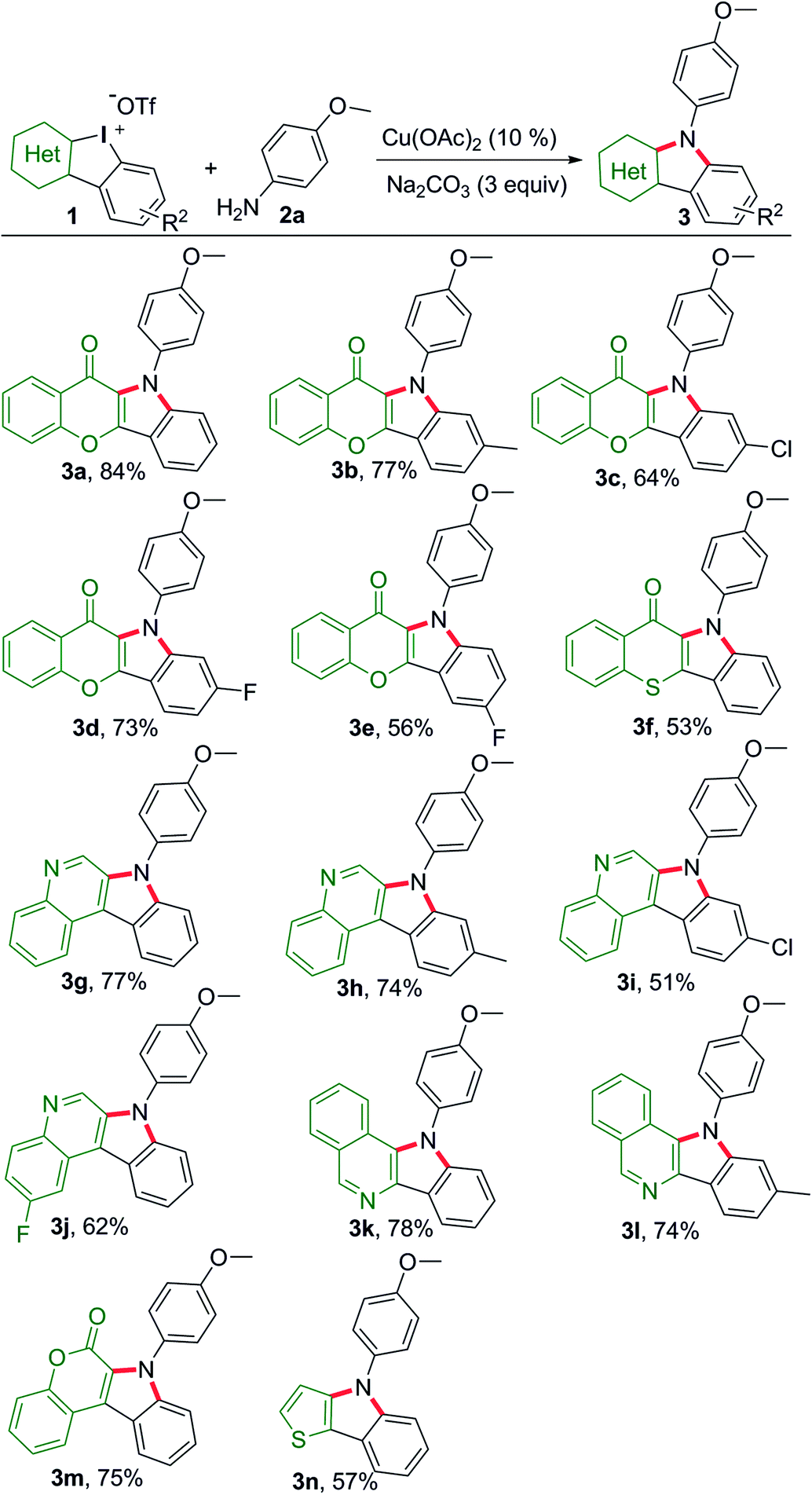
Chromone is a privileged motif for drug design and discovery in the field of medicinal chemistry.12 Thus, we commenced the dual-amination transformation using chromone embedded HCIs 1a–1e as building blocks (Scheme 2). Under catalytic mediation of Cu(OAc)2, p-anisidine underwent dual arylations to complete the amination.
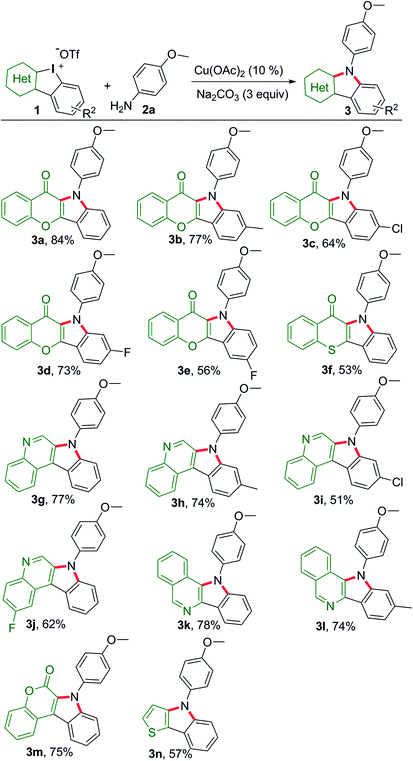 |
| | Scheme 2 Substrate scope of HCIs to synthesize heterocycle-fused indoles. Reaction conditions: 1 (0.1 mmol), p-anisidine (2.5 equiv.), i-PrOH/(CH2OH)2 (0.9/0.1 mL), refluxing, Ar, 16 h. | |
As a result, the desired chromone-fused indoles were obtained at modest to good yields (3a–3e). Then, we explored the substrate scope and generality of other HCIs with different heterocyclic motifs. Thiochromone-fused indole was obtained at a moderate yield (3f). Quinoline, isoquinoline and coumarin are important building blocks for naturally occurring products and pharmaceuticals. These unique heterocyclic fused indoles could be also assembled efficiently (3g–3m). The HCIs bearing chlorine atom usually gave low yields (3a vs. 3c, 3g vs. 3i). Meanwhile, the construction of thieno[3,2-b]indole (3n) was realized, providing a concise method to obtain thiophene-containing materials.
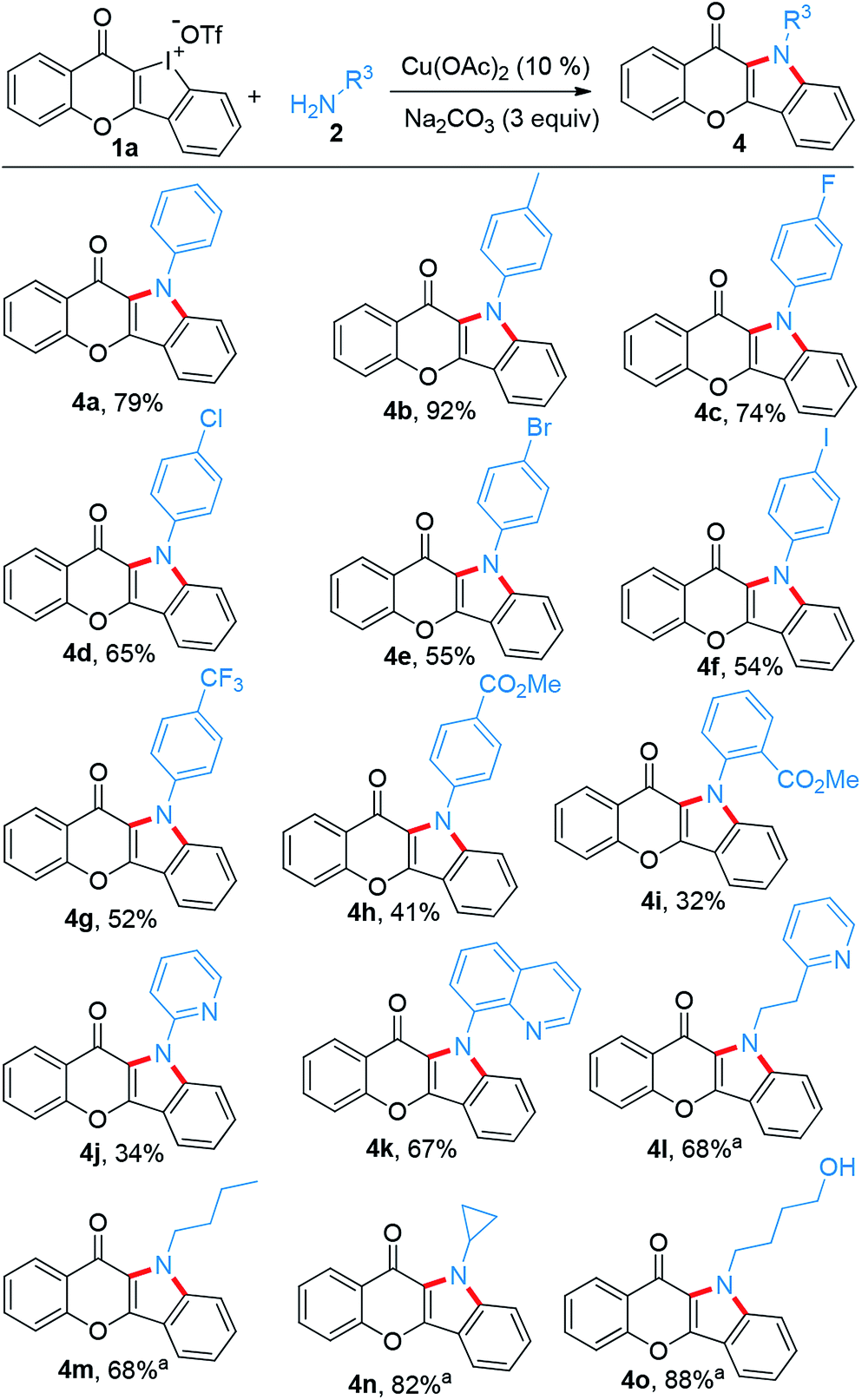
Subsequently, the substrate scope of this reaction was further examined on variation of the amines (Scheme 3). Like p-anisidine, other arylamines bearing different functional groups also enabled their incorporation into chromone-containing HCI 1a to provide diverse chromone-fused indoles (4a–4i). It should be noted that the intact bromo or iodo in products 4e and 4f could serve as a potential transformation platform for late-stage diversification. Meanwhile, arylamines with electron-deficient groups disfavored these reactions and provided the products in low yields (4h–4j). Amines tethering on pyridine and quinoline also performed smoothly (4j–4k). However, additional CuI (0.1 equiv.) was required while alkyl amines were used. Under the modified condition, the desirable products were successfully obtained (4l–4o). Again, pyridine motif in the alkylamine did not disrupt the reaction (4l).
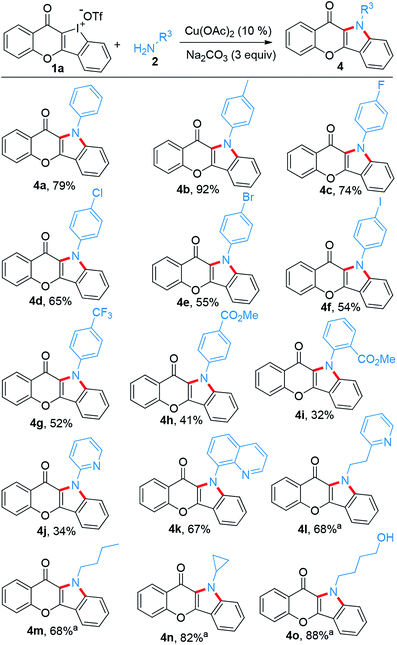 |
| | Scheme 3 Scope of amines reacting with 1a to construct chromone-fused indoles. Reaction conditions: 1a (0.1 mmol), amine (2.5 equiv.), i-PrOH/(CH2OH)2 (0.9/0.1 mL), refluxing, Ar, 16 h. aAdditional CuI (0.1 equiv.) added. | |
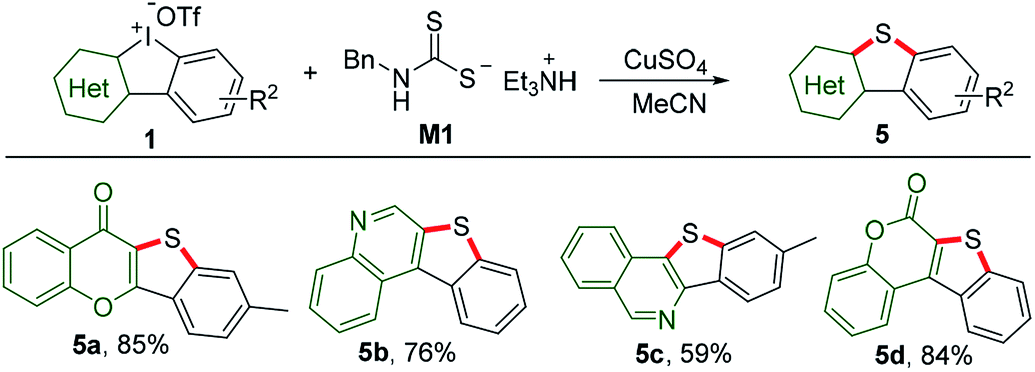
Sulfur containing heterocycles have found considerable utility particularly in material science because of high resonance energy of sulfur atom.13 Traditional methods for the introduction of sulfur suffer from several disadvantages such as catalyst poisoning, over-oxidization, and stinky smell. Using our recently discovered odor-free triethylammonium N-benzyldithiocarbamate (M1) as the sulfur source donor,8a reactions of HCIs and M1 under mediation of copper sulfate could smoothly furnish benzothiophene-fused heterocyclic frameworks, including chromone (5a), quinoline (5b), isoquinoline (5c) and coumarin (5d), as shown in Scheme 4.
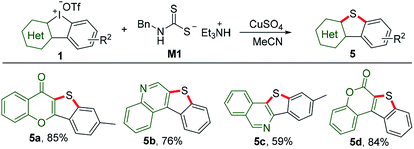 |
| | Scheme 4 Sulfur insertion of HCIs with triethylammonium N-benzyldithiocarbamate M1. Reaction conditions: 1 (1 equiv.), M1 (2 equiv.), CuSO4 (0.1 equiv.), MeCN, 60 °C, Ar, 6 h. | |
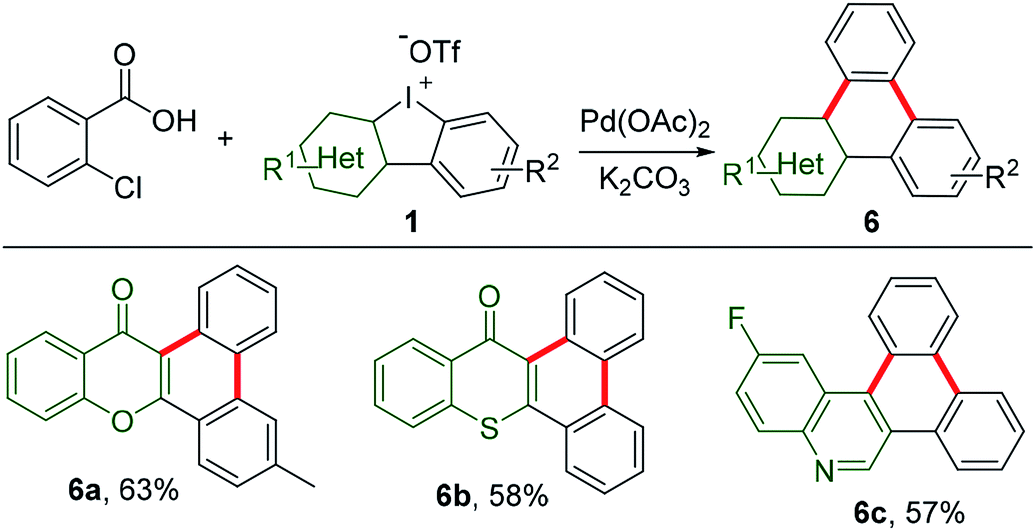
Decarboxylation of commercially available carboxylic acids is emerging as a novel strategy for aromatic functionalization.14 A pioneering work has recently been extended to decarboxylation of 2-chlorobenzoic acid for in situ generation of benzyne to construct triphenylenes.15 As counterparts of these hydrocarbons, the heteroatom-containing triphenylene analogues exhibit distinct chemical and physical properties.16 However, they have been so far less touched due to limited synthetic protocols. Thus, HCIs could be very promising synthons to construct such unique triphenylene. In our study, HCIs reacted with 2-chlorobenzoic acid to effectively afford the desirable annulated heterocycles containing chromone (6a), thiochromone (6b), or quinoline (6c), as shown in Scheme 5.
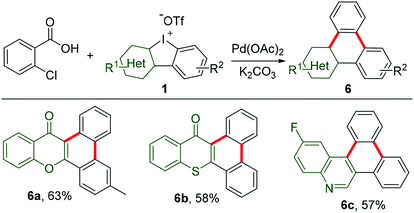 |
| | Scheme 5 Annulation of 2-chlorobenzoic acid with HCIs. Reaction conditions: 2-chorobenzoic acid (1 equiv.), 1 (1.2 equiv.), K2CO3 (2.2 equiv.), Pd(OAc)2 (0.1 equiv.), 1-methyl-2-pyrrolidinone, 140 °C, 16 h. | |
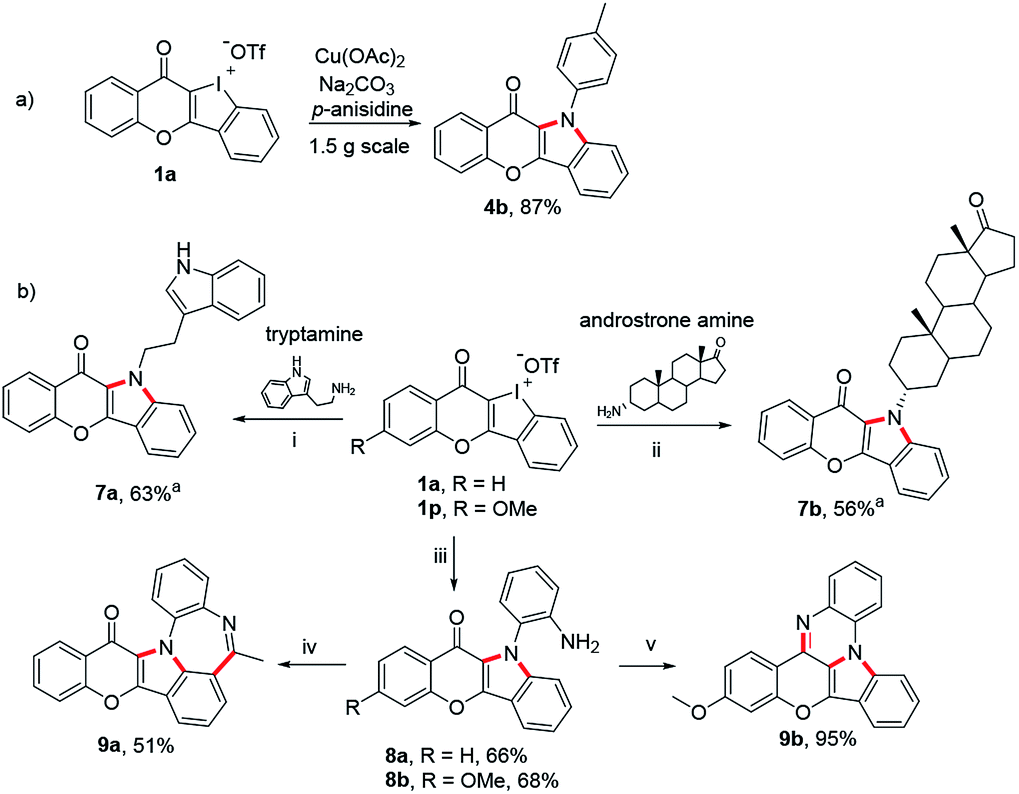
Finally, we have taken several applications to further demonstrate the robustness of these unique heterocyclic iodoniums as synthons (Scheme 6). Firstly, the copper-catalyzed dual aminations of HCIs performed well in a gram-scale reaction without a compromised yield (Scheme 6a). The increasing emergence of drug resistance in treating diseases demands an urgent need to develop new drugs. One effective strategy has been pursued combining two different drugs to form a new hybrid molecule.17 Herein, tryptamine, a serotonin receptor agonist, and an amine derived from androstrone (an endogenous steroid hormone) were employed to react with chromone-fused HCI 1a under the standard conditions. Both transformations successfully provided the expected fused hybrids 7a and 7b. In a final venture to establish the generality of this strategy, benzene-1,2-diamine was also used to prepare 8a and 8b which were readily for further transformation to obtain more complex heteropolycycles 9a and 9b with potential drug-likeness (Scheme 6b).
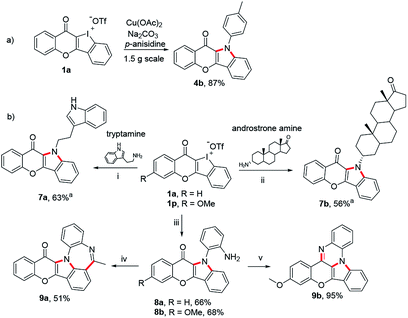 |
| | Scheme 6 (a) Scale-up synthesis of 4b under the standard condition. (b) Synthesis of drug-like hybrids and late-stage diversification with 1a and 1p. Reaction conditions: (i)–(iii) 1a or 1p (0.2 mmol), amine (2.5 equiv.), Na2CO3 (3 equiv.), Cu(OAc)2 (0.1 equiv.), i-PrOH/(CH2OH)2 (1.8/0.2 mL), Ar, refluxing, 16 h. (iv) AcCl (1.2 equiv.), Et3N (2.0 equiv.), CH2Cl2, rt; PPA (0.2 mL), POCl3 (10 equiv.), 120 °C. (v) TsOH-H2O (0.1 equiv.), EtOH, reflux. aAdditional CuI (0.1 equiv.) added. | |
Conclusions
In conclusion, we have fully explored the synthetic application of our recently reported heterocyclic iodoniums (HCIs). These unique iodoniums may gain more attention to build complex polycyclic heteroarenes which are widely present in naturally occurring products and pharmaceuticals. All the transformations with HCIs underwent a cyclization to build structurally diverse indole-fused, thiophene-fused, and triphenylene-fused heterocycles. Our current thorough investigation of HCIs highlights their value as versatile building blocks in synthetic chemistry, which may provide novel structures for drug development. These particular heterocycles are currently under our anticancer drug screening.
Experimental
The 1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance spectrometer 400 at 400 MHz and 100 MHz, respectively. Chemical shifts are given in ppm (δ) referenced to CDCl3 with 7.26 for 1H and 77.10 for 13C, and to d6-DMSO with 2.50 for 1H and 39.5 for 13C. In the case of multiplet, signals are reported as intervals and abbreviated as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Coupling constants are expressed in hertz. High-resolution mass spectra (HRMS) were recorded on a BRUKER VPEXII spectrometer with ESI mode unless otherwise stated. Melting point was measured by BUCHI Melting Point B-540. The progress of the reactions was monitored by thin-layer chromatography on a glass plate coated with silica gel with fluorescent indicator (GF254). Column chromatography was performed on silica gel (200–300 mesh).
General procedure A for the synthesis of 3a–4k
The synthesis of 10-(4-methoxyphenyl)chromeno[3,2-b]indol-11(10H)-one (3a) is exemplified. To a stirred solution of iodonium 1a (0.1 mmol) in i-PrOH (0.9 mL), was added ethylene glycol (0.1 mL), p-anisidine (2.5 equiv.), Na2CO3 (3 equiv.), and Cu(OAc)2 (0.1 equiv.). The reaction proceeded at reflux for 16 h under argon atmosphere before i-PrOH was removed by a rotary evaporation. The remained mixture was extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and evaporated in a vacuo. The residue was purified by column chromatography (PE/EtOAc = 15/1–5/1) to provide 3a as a white solid (29 mg, 84% yield), mp 199.1–200.6 °C. 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.74–7.68 (m, 2H), 7.51–7.45 (m, 1H), 7.44–7.37 (m, 3H), 7.32 (dd, J = 13.2, 7.9 Hz, 2H), 7.11–7.04 (m, 2H), 3.91 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.2, 159.3, 155.5, 146.0, 139.4, 132.8, 130.0, 129.1, 128.6, 126.4, 124.6, 124.0, 121.1, 120.9, 120.0, 118.1, 115.6, 114.3, 111.6, 55.6 ppm. HRMS (ESI) m/z calcd for C22H16NO3 [M + H]+: 342.1125, found: 342.1112.
10-(4-Methoxyphenyl)-8-methylchromeno[3,2-b]indol-11(10H)-one (3b). 3b (27 mg, 77% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 195.6–196.3 °C. 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J = 8.2 Hz, 1H), 7.95 (d, J = 8.2 Hz, 1H), 7.69 (d, J = 2.2 Hz, 2H), 7.40 (dd, J = 10.9, 5.9 Hz, 3H), 7.14 (d, J = 8.3 Hz, 1H), 7.11–7.04 (m, 3H), 3.91 (s, 3H), 2.48 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 168.9, 159.3, 155.5, 146.3, 140.0, 139.5, 132.6, 130.2, 129.2, 126.4, 124.7, 123.9, 123.2, 120.7, 119.7, 118.0, 114.3, 113.5, 111.1, 55.7, 22.5 ppm. HRMS (ESI) m/z calcd for C23H18NO3 [M + H]+: 356.1281, found: 356.1284.
8-Chloro-10-(4-methoxyphenyl)chromeno[3,2-b]indol-11(10H)-one (3c). 3c (24 mg, 74% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 215.1–216.4 °C. 1H NMR (400 MHz, CDCl3) δ 8.34 (d, J = 7.8 Hz, 1H), 7.99 (d, J = 8.5 Hz, 1H), 7.75–7.64 (m, 2H), 7.41 (dd, J = 11.2, 8.1 Hz, 3H), 7.30 (s, 1H), 7.27 (d, J = 7.0 Hz, 1H), 7.08 (d, J = 8.7 Hz, 2H), 3.91 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.1, 159.6, 155.6, 145.6, 139.6, 134.8, 133.1, 129.5, 129.0, 126.4, 124.5, 124.3, 122.2, 121.4, 121.1, 118.1, 114.5, 114.2, 111.6, 55.7 ppm. HRMS (ESI) m/z calcd for C22H15ClNO3 [M + H]+: 376.0735, found: 376.0742.
8-Fluoro-10-(4-methoxyphenyl)chromeno[3,2-b]indol-11(10H)-one (3d). 3d (26 mg, 73% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 169.3–170.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.35 (d, J = 7.5 Hz, 1H), 8.02 (dd, J = 8.8, 5.3 Hz, 1H), 7.69 (dd, J = 12.3, 4.9 Hz, 2H), 7.41 (t, J = 7.8 Hz, 3H), 7.05 (dd, J = 14.9, 5.4 Hz, 3H), 6.97 (dd, J = 9.7, 1.8 Hz, 1H), 3.91 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 168.7, 165.0, 162.6, 159.6, 155.5, 145.9, 140.1, 140.0, 132.9, 129.7, 128.9, 126.5, 124.5, 124.2, 121.7, 121.6, 121.5, 118.1, 114.5, 112.4, 111.0, 110.8, 98.0, 97.7, 55.7 ppm. 19F NMR (376 MHz, CDCl3) δ −109.8, −109.8, −109.8, −109.8, −109.8, −109.9 ppm. HRMS (ESI) m/z calcd for C22H15FNO3 [M + H]+: 360.1030, found: 360.1014.
7-Fluoro-10-(4-methoxyphenyl)chromeno[3,2-b]indol-11(10H)-one (3e). 3e (20 mg, 56% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 208.9–209.6 °C. 1H NMR (400 MHz, CDCl3) δ 8.40–8.30 (m, 2H), 7.53–7.47 (m, 2H), 7.46–7.41 (m, 2H), 7.40–7.35 (m, 2H), 7.32 (d, J = 8.4 Hz, 1H), 7.08 (d, J = 8.9 Hz, 2H), 3.92 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 159.7, 155.0, 151.2, 142.5, 129.5, 129.2, 127.7, 124.6, 123.6, 122.7, 122.4, 122.3, 121.8, 121.6, 118.8, 117.3, 114.5, 112.5, 55.6 ppm. HRMS (ESI) m/z calcd for C22H15FNO3 [M + H]+: 360.1030, found: 360.1036.
10-(4-Methoxyphenyl)thiochromeno[3,2-b]indol-11(10H)-one (3f). 3f (19 mg, 53% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 224.9–225.5 °C. 1H NMR (400 MHz, CDCl3) δ 8.64 (d, J = 8.1 Hz, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.65–7.56 (m, 1H), 7.48 (dd, J = 18.4, 7.5 Hz, 2H), 7.35 (d, J = 8.8 Hz, 2H), 7.33–7.29 (m, 1H), 7.25 (d, J = 9.7 Hz, 1H), 7.08 (d, J = 8.8 Hz, 2H), 3.92 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 172.3, 159.4, 141.2, 135.9, 132.8, 131.4, 130.8, 129.2, 129.2, 128.6, 128.2, 126.9, 126.2, 123.2, 121.2, 120.6, 120.3, 114.3, 112.2, 55.6 ppm. HRMS (ESI) m/z calcd for C22H16NO2S [M + H]+: 358.0896, found: 358.0889.
7-(4-Methoxyphenyl)-7H-indolo[2,3-c]quinolone (3g). 3g (25 mg, 77% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 130.4–131.2 °C. 1H NMR (400 MHz, CDCl3) δ 9.08 (s, 1H), 8.77 (d, J = 8.2 Hz, 1H), 8.64 (d, J = 8.0 Hz, 1H), 8.31 (d, J = 8.3 Hz, 1H), 7.76 (t, J = 7.5 Hz, 1H), 7.69 (t, J = 7.6 Hz, 1H), 7.59–7.54 (m, 1H), 7.48 (dd, J = 18.5, 7.9 Hz, 4H), 7.17 (d, J = 8.7 Hz, 2H), 3.95 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 159.7, 143.5, 141.4, 137.4, 134.1, 130.5, 129.1, 128.9, 127.4, 127.2, 125.9, 124.7, 123.5, 123.4, 122.2, 121.4, 121.3, 115.5, 111.3, 55.8 ppm. HRMS (ESI) m/z calcd for C22H17N2O [M + H]+: 325.1335, found: 325.1330.
7-(4-Methoxyphenyl)-9-methyl-7H-indolo[2,3-c]quinolone (3h). 3h (25 mg, 74% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 177.9–179.1 °C. 1H NMR (400 MHz, CDCl3) δ 9.02 (s, 1H), 8.71 (d, J = 8.1 Hz, 1H), 8.47 (d, J = 8.7 Hz, 1H), 8.28 (d, J = 8.2 Hz, 1H), 7.72 (t, J = 7.4 Hz, 1H), 7.65 (t, J = 7.5 Hz, 1H), 7.47 (d, J = 8.8 Hz, 2H), 7.26 (t, J = 7.6 Hz, 2H), 7.15 (d, J = 8.8 Hz, 2H), 3.93 (s, 3H), 2.53 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 159.6, 143.5, 141.9, 137.8, 137.3, 134.1, 130.5, 129.2, 128.9, 127.1, 125.8, 124.6, 123.5, 123.1, 123.1, 121.5, 120.0, 115.4, 111.1, 55.8, 22.3 ppm. HRMS (ESI) m/z calcd for C23H19N2O [M + H]+: 339.1492, found: 339.1487.
9-Chloro-7-(4-methoxyphenyl)-7H-indolo[2,3-c]quinolone (3i). 3i (18 mg, 51% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 204.7–205.6 °C. 1H NMR (400 MHz, CDCl3) δ 9.02 (s, 1H), 8.65 (d, J = 8.0 Hz, 1H), 8.48 (d, J = 8.5 Hz, 1H), 8.30 (d, J = 8.1 Hz, 1H), 7.74 (t, J = 7.4 Hz, 1H), 7.68 (t, J = 7.4 Hz, 1H), 7.46 (d, J = 8.5 Hz, 3H), 7.40 (d, J = 8.6 Hz, 1H), 7.17 (d, J = 8.7 Hz, 2H), 3.95 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 160.0, 143.3, 141.9, 137.1, 134.4, 133.4, 130.4, 128.8, 128.4, 127.6, 126.4, 124.3, 123.3, 122.1, 121.2, 120.7, 115.6, 111.3, 55.8 ppm. HRMS (ESI) m/z calcd for C22H16ClN2O [M + H]+: 359.0946, found: 359.0940.
2-Fluoro-7-(4-methoxyphenyl)-7H-indolo[2,3-c]quinolone (3j). 3j (21 mg, 62% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 161.1–161.8 °C. 1H NMR (400 MHz, CDCl3) δ 9.01 (s, 1H), 8.52 (d, J = 7.9 Hz, 1H), 8.28 (ddd, J = 15.1, 9.5, 4.2 Hz, 2H), 7.60–7.53 (m, 1H), 7.52–7.45 (m, 4H), 7.41 (td, J = 8.8, 2.7 Hz, 1H), 7.17 (d, J = 8.8 Hz, 2H), 3.95 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 162.7, 160.3, 159.8, 141.3, 140.4, 136.7, 134.2, 132.8, 132.7, 128.9, 128.8, 127.3, 125.4, 125.3, 122.9, 122.0, 121.6, 121.0, 115.6, 115.5, 115.3, 111.4, 107.6, 107.4, 55.8 ppm. 19F NMR (376 MHz, CDCl3) δ −112.2, −112.2, −112.2, −112.3 ppm. HRMS (ESI) m/z calcd for C22H16FN2O [M + H]+: 343.1241, found: 343.1229.
11-(4-Methoxyphenyl)-11H-indolo[3,2-c]isoquinoline (3k). 3k (25 mg, 78% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 185.8–186.9 °C. 1H NMR (400 MHz, CDCl3) δ 9.17 (s, 1H), 8.49 (d, J = 7.4 Hz, 1H), 8.12 (d, J = 8.0 Hz, 1H), 7.53 (t, J = 7.3 Hz, 1H), 7.50–7.39 (m, 6H), 7.21 (d, J = 8.0 Hz, 1H), 7.16 (d, J = 8.7 Hz, 2H), 3.97 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 160.2, 146.2, 142.0, 135.1, 131.6, 130.2, 129.4, 129.1, 128.4, 127.7, 126.1, 125.7, 124.5, 122.9, 121.2, 121.0, 119.9, 115.4, 110.4, 55.8 ppm. HRMS (ESI) m/z calcd for C22H17N2O [M + H]+: 325.1335, found: 325.1327.
11-(4-Methoxyphenyl)-9-methyl-11H-indolo[3,2-c]isoquinoline (3l). 3l (25 mg, 74% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 173.8–175.2 °C. 1H NMR (400 MHz, CDCl3) δ 9.15 (s, 1H), 8.37 (d, J = 8.0 Hz, 1H), 8.11 (d, J = 8.1 Hz, 1H), 7.54–7.49 (m, 1H), 7.48–7.42 (m, 3H), 7.39 (d, J = 8.4 Hz, 1H), 7.26–7.22 (m, 1H), 7.17 (d, J = 8.8 Hz, 2H), 6.99 (s, 1H), 3.98 (s, 3H), 2.50 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 160.1, 145.8, 142.5, 136.6, 135.1, 131.7, 130.3, 129.4, 129.2, 128.2, 127.5, 125.4, 124.6, 122.8, 121.1, 120.6, 119.7, 115.4, 110.4, 55.8, 22.3 ppm. HRMS (ESI) m/z calcd for C23H19N2O [M + H]+: 339.1492, found: 339.1489.
7-(4-Methoxyphenyl)chromeno[3,4-b]indol-6(7H)-one (3m). 3m (26 mg, 75% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 231.1–232.5 °C. 1H NMR (400 MHz, CDCl3) δ 8.35 (dd, J = 8.0, 1.3 Hz, 1H), 7.76–7.64 (m, 3H), 7.44–7.35 (m, 3H), 7.29–7.18 (m, 3H), 7.07 (d, J = 8.9 Hz, 2H), 3.91 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.4, 159.5, 155.6, 145.5, 136.0, 133.1, 129.8, 129.1, 126.4, 124.5, 124.1, 122.1, 118.1, 117.8, 117.5, 115.5, 115.4, 114.4, 113.1, 113.0, 104.7, 104.4, 55.7 ppm. HRMS (ESI) m/z calcd for C22H16NO3[M + H]+: 342.1125, found: 342.1124.
4-(4-Methoxyphenyl)-4H-thieno[3,2-b]indole (3n). 3n (16 mg, 57% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 125.1–126.9 °C. 1H NMR (400 MHz, CDCl3) δ 7.80 (d, J = 7.4 Hz, 1H), 7.48 (t, J = 8.6 Hz, 3H), 7.36 (d, J = 5.2 Hz, 1H), 7.29–7.19 (m, 2H), 7.08 (d, J = 8.8 Hz, 2H), 7.04 (d, J = 5.2 Hz, 1H), 3.91 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 158.5, 145.6, 141.8, 131.8, 126.9, 126.8, 124.4, 123.0, 122.2, 120.1, 119.0, 117.4, 115.0, 114.7, 111.4, 111.0, 55.7 ppm. HRMS (ESI) m/z calcd for C17H14NOS [M + H]+: 280.0791, found: 280.0787.
10-Phenylchromeno[3,2-b]indol-11(10H)-one (4a). 4a (24 mg, 79% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 190.7–191.6 °C. 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 7.8 Hz, 1H), 8.10 (d, J = 8.1 Hz, 1H), 7.72 (d, J = 3.0 Hz, 2H), 7.60–7.54 (m, 2H), 7.53–7.46 (m, 4H), 7.45–7.40 (m, 1H), 7.38 (d, J = 8.5 Hz, 1H), 7.33 (t, J = 7.5 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.1, 155.5, 146.3, 139.0, 137.1, 132.8, 129.0, 128.7, 128.1, 128.0, 126.4, 124.5, 124.0, 121.2, 120.6, 120.0, 118.1, 115.7, 111.5 ppm. HRMS (ESI) m/z calcd for C21H14NO2 [M + H]+: 312.1019, found: 312.1017.
10-(p-Tolyl)chromeno[3,2-b]indol-11(10H)-one (4b). 4b (30 mg, 92% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 205.2–206.6 °C. 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 7.9 Hz, 1H), 8.09 (d, J = 8.1 Hz, 1H), 7.71 (d, J = 3.0 Hz, 2H), 7.51–7.45 (m, 1H), 7.44–7.40 (m, 1H), 7.41–7.34 (m, 5H), 7.31 (t, J = 7.5 Hz, 1H), 2.48 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.1, 155.5, 146.1, 139.1, 138.0, 134.5, 132.8, 129.7, 128.6, 127.7, 126.4, 124.5, 124.0, 121.1, 120.7, 120.0, 118.0, 115.6, 111.6, 21.4 ppm. HRMS (ESI) m/z calcd for C22H16NO2 [M + H]+: 326.1176, found: 326.1171.
10-(4-Fluorophenyl)chromeno[3,2-b]indol-11(10H)-one (4c). 4c (24 mg, 74% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 227.5–228.6 °C. 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 7.9 Hz, 1H), 8.09 (d, J = 8.1 Hz, 1H), 7.72 (t, J = 5.8 Hz, 2H), 7.57–7.40 (m, 4H), 7.38–7.30 (m, 2H), 7.26 (dd, J = 11.0, 6.0 Hz, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.2, 163.4, 161.0, 155.6, 146.3, 139.2, 133.2, 133.0, 129.8, 129.7, 128.9, 126.3, 124.5, 124.2, 121.4, 120.7, 120.2, 118.2, 116.1, 115.9, 115.8, 111.4 ppm. 19F NMR (376 MHz, CDCl3) δ −113.4, −113.5, −113.5, −113.5, −113.5, −113.5, −113.5 ppm. HRMS (ESI) m/z calcd for C21H13FNO2 [M + H]+: 330.0925, found: 330.0934.
10-(4-Chlorophenyl)chromeno[3,2-b]indol-11(10H)-one (4d). 4d (23 mg, 65% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 265.6–266.2 °C. 1H NMR (400 MHz, CDCl3) δ 8.40–8.30 (m, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.72 (dd, J = 5.7, 1.4 Hz, 2H), 7.53 (d, J = 8.6 Hz, 2H), 7.51–7.48 (m, 1H), 7.47–7.40 (m, 3H), 7.39–7.31 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.2, 155.6, 146.5, 139.0, 135.7, 133.9, 133.1, 129.3, 129.0, 126.4, 124.5, 124.3, 121.6, 120.6, 120.2, 118.2, 116.0, 111.4 ppm. HRMS (ESI) m/z calcd for C21H13ClNO2 [M + H]+: 346.0629, found: 346.0621.
10-(4-Bromophenyl)chromeno[3,2-b]indol-11(10H)-one (4e). 4e (22 mg, 65% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 283.5–284.7 °C. 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J = 8.0 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.78–7.64 (m, 4H), 7.50 (t, J = 7.7 Hz, 1H), 7.46–7.29 (m, 5H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.2, 155.6, 146.5, 138.9, 136.2, 133.1, 132.3, 129.6, 129.1, 126.4, 124.5, 124.3, 121.9, 121.6, 120.5, 120.3, 118.2, 116.1, 111.4 ppm. HRMS (ESI) m/z calcd for C21H13BrNO2 [M + H]+: 390.0124, found: 390.0127.
10-(4-Iodophenyl)chromeno[3,2-b]indol-11(10H)-one (4f). 4f (24 mg, 54% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 241.1–242.4 °C. 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J = 7.7 Hz, 1H), 8.10 (d, J = 8.0 Hz, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.72 (t, J = 6.1 Hz, 2H), 7.54–7.48 (m, 1H), 7.46–7.41 (m, 1H), 7.35 (dd, J = 17.3, 8.1 Hz, 2H), 7.27 (d, J = 6.7 Hz, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.1, 155.5, 146.6, 138.8, 138.2, 136.9, 133.0, 129.8, 129.0, 126.4, 124.5, 124.3, 121.6, 120.5, 120.3, 118.2, 116.1, 111.4, 93.3 ppm. HRMS (ESI) m/z calcd for C21H13INO2 [M + H]+: 437.9986, found: 437.9986.
10-(4-(Trifluoromethyl)phenyl)chromeno[3,2-b]indol-11(10H)-one (4g). 4g (20 mg, 52% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 247.8–248.5 °C. 1H NMR (400 MHz, CDCl3) δ 8.40–8.33 (m, 1H), 8.12 (d, J = 8.1 Hz, 1H), 7.83 (d, J = 8.4 Hz, 2H), 7.74 (dt, J = 8.0, 4.0 Hz, 2H), 7.65 (d, J = 8.3 Hz, 2H), 7.52 (dd, J = 11.4, 4.1 Hz, 1H), 7.47–7.43 (m, 1H), 7.43–7.34 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.2, 155.5, 147.0, 140.2, 138.8, 133.2, 129.3, 128.3, 126.4, 126.2, 124.4, 121.9, 120.4, 118.2, 116.3, 111.3 ppm. 19F NMR (376 MHz, CDCl3) δ −62.4 ppm. HRMS (ESI) m/z calcd for C22H13F3NO2 [M + H]+: 380.0893, found: 380.0908.
Methyl 4-(11-oxochromeno[3,2-b]indol-10(11H)-yl)benzoate (4h). 4h (15 mg, 41% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 232.4–233.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.37 (d, J = 7.5 Hz, 1H), 8.24 (d, J = 8.5 Hz, 2H), 8.11 (d, J = 8.0 Hz, 1H), 7.78–7.70 (m, 2H), 7.60 (d, J = 8.5 Hz, 2H), 7.55–7.49 (m, 1H), 7.48–7.40 (m, 2H), 7.36 (t, J = 7.5 Hz, 1H), 3.98 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.1, 166.6, 155.6, 147.0, 141.2, 138.8, 133.1, 130.5, 129.5, 129.2, 127.8, 126.5, 124.5, 124.3, 121.8, 120.5, 120.3, 118.2, 116.3, 111.5, 52.4 ppm. HRMS (ESI) m/z calcd for C23H16NO4 [M + H]+: 370.1074, found: 370.1074.
Methyl 2-(11-oxochromeno[3,2-b]indol-10(11H)-yl)benzoate (4i). 4i (12 mg, 32% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 219.9–221.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.33 (d, J = 7.9 Hz, 1H), 8.20 (dd, J = 7.8, 1.4 Hz, 1H), 8.10 (d, J = 8.0 Hz, 1H), 7.78–7.66 (m, 3H), 7.61 (td, J = 7.7, 1.1 Hz, 1H), 7.57 (d, J = 7.8 Hz, 1H), 7.50–7.43 (m, 1H), 7.43–7.37 (m, 1H), 7.31 (t, J = 7.5 Hz, 1H), 7.15 (d, J = 8.5 Hz, 1H), 3.46 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.3, 165.6, 155.7, 146.0, 139.2, 137.3, 132.9, 132.8, 131.6, 130.5, 129.5, 128.8, 128.7, 126.3, 124.4, 124.0, 121.5, 121.2, 120.1, 118.2, 115.8, 111.1, 52.2 ppm. HRMS (ESI) m/z calcd for C23H16NO4 [M + H]+: 370.1074, found: 370.1078.
10-(Pyridin-2-yl)chromeno[3,2-b]indol-11(10H)-one (4j). 4j (11 mg, 34% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 191.3–192.5 °C. 1H NMR (400 MHz, CDCl3) δ 8.67 (s, 1H), 8.39 (d, J = 7.9 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.93 (t, J = 7.2 Hz, 1H), 7.84 (d, J = 8.4 Hz, 1H), 7.72 (s, 2H), 7.54 (t, J = 7.6 Hz, 2H), 7.45 (dd, J = 14.0, 6.4 Hz, 1H), 7.42–7.33 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.0, 155.5, 150.6, 148.4, 147.7, 138.8, 137.9, 133.0, 129.5, 126.5, 124.6, 124.3, 122.6, 122.5, 122.1, 120.1, 120.0, 118.2, 116.7, 113.0 ppm. HRMS (ESI) m/z calcd for C20H13N2O2 [M + H]+: 313.0972, found: 313.0963.
10-(Quinolin-8-yl)chromeno[3,2-b]indol-11(10H)-one (4k). 4k (24 mg, 67% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 213.5–214.5 °C. 1H NMR (400 MHz, CDCl3) δ 8.76 (d, J = 3.0 Hz, 1H), 8.28 (dd, J = 18.7, 7.9 Hz, 2H), 8.15 (d, J = 8.0 Hz, 1H), 8.03 (d, J = 8.2 Hz, 1H), 7.91 (d, J = 7.1 Hz, 1H), 7.73 (dd, J = 15.9, 8.4 Hz, 3H), 7.48–7.30 (m, 4H), 7.08 (d, J = 8.4 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.3, 155.8, 150.9, 146.2, 145.2, 139.9, 136.7, 135.1, 132.7, 129.8, 129.3, 129.0, 128.5, 126.3, 126.3, 124.7, 123.9, 122.6, 121.9, 121.2, 120.2, 118.2, 116.2, 111.8 ppm. HRMS (ESI) m/z calcd for C24H15N2O2 [M + H]+: 363.1128, found: 363.1113.
General procedure B for the synthesis of 4l–4o
The synthesis of 10-(2-(pyridin-2-yl)ethyl)chromeno[3,2-b]indol-11(10H)-one (4l) is exemplified. To a stirred solution of iodonium 1a (0.1 mmol) in i-PrOH (0.9 mL), was added ethylene glycol (0.1 mL), 2-(pyridin-2-yl)ethan-1-amine (2.5 equiv.), Na2CO3 (3 equiv.), Cu(OAc)2 (0.1 equiv.), and CuI (0.1 equiv.). The reaction proceeded at reflux for 16 h under argon atmosphere before i-PrOH was removed by a rotary evaporation. The remained mixture was extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and evaporated in a vacuo. The residue was purified by column chromatography (PE/EtOAc = 15/1–5/1) to provide 4l as a white solid (23 mg, 68% yield), mp 139.7–140.8 °C. 1H NMR (400 MHz, CDCl3) δ 8.55 (d, J = 4.3 Hz, 1H), 8.45 (d, J = 7.9 Hz, 1H), 7.95 (d, J = 8.0 Hz, 1H), 7.75–7.59 (m, 2H), 7.43 (t, J = 7.5 Hz, 2H), 7.34 (dd, J = 20.0, 7.8 Hz, 2H), 7.17 (t, J = 7.4 Hz, 1H), 7.06 (t, J = 8.4 Hz, 2H), 5.08 (t, J = 7.3 Hz, 2H), 3.37 (t, J = 7.2 Hz, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ 170.1, 158.8, 155.6, 149.5, 145.2, 138.0, 136.5, 132.7, 128.1, 126.1, 124.4, 123.9, 123.9, 121.7, 120.2, 120.0, 119.9, 118.1, 114.7, 110.5, 45.0, 39.9 ppm. HRMS (ESI) m/z calcd for C22H17N2O2 [M + H]+: 341.1285, found: 341.1278.
10-Butylchromeno[3,2-b]indol-11(10H)-one (4m). 4m (20 mg, 68% yield) was generated following a procedure for the synthesis of 4l as a white solid, mp 87.4–88.6 °C. 1H NMR (400 MHz, CDCl3) δ 8.45 (dd, J = 8.0, 1.3 Hz, 1H), 8.02 (d, J = 8.1 Hz, 1H), 7.74–7.61 (m, 2H), 7.57–7.46 (m, 2H), 7.45–7.40 (m, 1H), 7.25 (ddd, J = 7.9, 5.3, 1.1 Hz, 1H), 4.76 (t, J = 7.3 Hz, 2H), 1.94–1.76 (m, 2H), 1.41 (dd, J = 15.3, 7.5 Hz, 2H), 0.95 (t, J = 7.4 Hz, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 170.3, 155.6, 145.0, 137.9, 132.7, 128.1, 126.3, 124.5, 123.9, 120.4, 120.2, 118.2, 115.0, 110.6, 44.8, 33.1, 20.2, 14.0 ppm. HRMS (ESI) m/z calcd for C19H18NO2 [M + H]+: 292.1332, found: 292.1337.
10-Cyclopropylchromeno[3,2-b]indol-11(10H)-one (4n). 4n (23 mg, 82% yield) was generated following a procedure for the synthesis of 4l as a white solid, mp 167.9–168.4 °C. 1H NMR (400 MHz, CDCl3) δ 8.46 (dd, J = 8.0, 1.4 Hz, 1H), 8.03–7.98 (m, 1H), 7.73 (d, J = 8.5 Hz, 1H), 7.71–7.63 (m, 2H), 7.52 (ddd, J = 8.4, 5.4, 1.2 Hz, 1H), 7.43 (ddd, J = 8.1, 6.8, 1.4 Hz, 1H), 7.30–7.23 (m, 1H), 3.63–3.54 (m, 1H), 1.40–1.33 (m, 2H), 1.17 (qd, J = 5.6, 4.5 Hz, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.5, 155.4, 145.5, 139.1, 132.7, 128.1, 126.4, 124.8, 124.0, 121.6, 120.6, 120.1, 118.0, 115.2, 112.3, 26.6, 9.5 ppm. HRMS (ESI) m/z calcd for C18H14NO2 [M + H]+: 276.1019, found: 276.1011.
10-(4-Hydroxybutyl)chromeno[3,2-b]indol-11(10H)-one (4o). 4o (27 mg, 88% yield) was generated following a procedure for the synthesis of 4l as a white solid, mp 110.7–111.3 °C. 1H NMR (400 MHz, CDCl3) δ 8.44 (d, J = 7.8 Hz, 1H), 8.03 (d, J = 8.1 Hz, 1H), 7.76–7.62 (m, 2H), 7.57–7.46 (m, 2H), 7.44 (t, J = 7.2 Hz, 1H), 7.30–7.18 (m, 1H), 4.81–4.73 (m, 2H), 3.75 (t, J = 6.2 Hz, 2H), 2.01 (dt, J = 14.8, 7.4 Hz, 2H), 1.74–1.55 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ 170.3, 155.5, 145.2, 137.9, 132.8, 128.4, 126.2, 124.2, 123.9, 120.3, 120.2, 118.1, 114.9, 110.4, 62.1, 44.2, 29.4, 27.2 ppm. HRMS (ESI) m/z calcd for C19H18NO3 [M + H]+: 308.1281, found: 308.1278.
Procedure for the synthesis of triethylammonium benzylcarbamodithioate (M1). To a solution of benzylamine (1.11 g, 10.38 mmol, 1.05 equiv.) and Et3N (1.0 g, 9.88 mmol, 1 equiv.) in CH2Cl2 (25 mL), CS2 (0.83 g, 10.87 mmol, 1.1 equiv.) was dropped slowly. The solution was stirred at room temperature for 3 h, concentrated by a rotary evaporator, and finally dried by a high vacuum to give M1 (2.75 g, 98% yield) as a white solid.
The reaction procedure of heterocyclic iodoniums and M1 to provide benzothiophene-fused heterocycles 5. Syntheses of 8-methyl-11H-benzo[4,5]thieno[3,2-b]chromen-11-one (5a) is exemplified. To a stirred solution of iodonium 1b (0.1 mmol) in MeCN (2.0 mL), was added M1 (2 equiv.) and CuSO4 (0.1 equiv.). The reaction proceeded at 70 °C for 6 h under argon atmosphere before MeCN was removed by a rotary evaporation. The remained mixture was extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and evaporated in a vacuo. The residue was purified by column chromatography (PE/EtOAc = 20/1–5/1) to provide 5a as a white solid (22 mg, 85% yield), mp 191.2–192.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.39 (dd, J = 8.0, 1.5 Hz, 1H), 8.07 (d, J = 8.2 Hz, 1H), 7.79–7.72 (m, 1H), 7.72–7.64 (m, 2H), 7.48 (dd, J = 11.0, 3.9 Hz, 1H), 7.35 (d, J = 8.2 Hz, 1H), 2.54 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 173.3, 156.0, 153.9, 140.4, 140.2, 133.7, 127.0, 126.0, 124.9, 123.6, 122.7, 122.1, 119.9, 118.1, 22.1 ppm. HRMS (ESI) m/z calcd for C16H11O2S[M + H]+: 267.0474, found: 267.0481.
Benzo[4,5]thieno[2,3-c]quinolone (5b). 5b (18 mg, 76% yield) was generated following a procedure for the synthesis of 5a as a white solid, mp 129.7–131.2 °C. 1H NMR (400 MHz, CDCl3) δ 9.40 (s, 1H), 8.98–8.93 (m, 1H), 8.91 (dd, J = 5.8, 2.9 Hz, 1H), 8.49–8.38 (m, 1H), 8.10 (dd, J = 5.5, 3.6 Hz, 1H), 7.82 (dd, J = 5.7, 3.9 Hz, 2H), 7.75–7.63 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ 145.5, 145.4, 141.5, 135.6, 135.2, 133.4, 130.7, 127.9, 127.7, 126.1, 125.5, 123.9, 123.0 ppm. HRMS (ESI) m/z calcd for C15H10NS [M + H]+: 236.0528, found: 236.0520.
9-Methylbenzo[4,5]thieno[3,2-c]isoquinoline (5c). 5c (15 mg, 59% yield) was generated following a procedure for the synthesis of 5a as a white solid, mp 108.8–110.3 °C. 1H NMR (400 MHz, CDCl3) δ 9.31 (s, 1H), 8.53 (d, J = 8.1 Hz, 1H), 8.16 (d, J = 8.1 Hz, 1H), 8.09 (d, J = 8.3 Hz, 1H), 7.85 (t, J = 7.6 Hz, 1H), 7.76 (s, 1H), 7.69 (t, J = 7.6 Hz, 1H), 7.42 (d, J = 8.2 Hz, 1H), 2.57 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 150.2, 138.7, 137.9, 133.5, 132.0, 131.2, 129.5, 129.1, 127.9, 127.3, 127.2, 126.9, 126.8, 123.7, 123.6, 123.0, 122.8, 122.7, 122.4, 22.0 ppm. HRMS (ESI) m/z calcd for C16H12NS [M + H]+: 250.0685, found: 250.0677.
6H-Benzo[4,5]thieno[2,3-c]chromen-6-one (5d). 5d (21 mg, 84% yield) was generated following a procedure for the synthesis of 5a as a white solid, mp 205.0–206.8 °C. 1H NMR (400 MHz, CDCl3) δ 8.69–8.59 (m, 1H), 8.50 (d, J = 8.0 Hz, 1H), 8.06–7.97 (m, 1H), 7.67–7.60 (m, 2H), 7.56 (q, J = 8.3 Hz, 2H), 7.46 (t, J = 7.2 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ 158.0, 152.6, 143.6, 138.6, 134.9, 130.0, 128.4, 126.0, 125.6, 124.8, 124.0, 123.5, 118.3, 118.0 ppm. HRMS (ESI) m/z calcd for C15H9O2S [M + H]+: 253.0318, found: 253.0313.
General procedure for synthesis of 6
The synthesis of 6-methyl-14H-dibenzo[a,c]xanthen-14-one (6a) is exemplified. To a stirred solution of 2-chlorobenzoic acid (0.1 mmol) in 1-methyl-2-pyrrolidinone (1.5 mL), was added heterocyclic iodonium (1.2 equiv.), Pd(OAc)2 (0.1 equiv.), and K2CO3 (2.2 equiv.). The reaction mixture was sealed in a tube. The reaction proceeded at 140 °C for 16 h before it was cooled to rt. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and evaporated in a vacuo. The residue was purified by column chromatography (PE/EtOAc = 20/1–5/1) to provide 6a as a white solid (20 mg, 63% yield), mp 207.5–209.2 °C. 1H NMR (400 MHz, CDCl3) δ 10.08 (dd, J = 8.4, 0.9 Hz, 1H), 8.49 (d, J = 8.1 Hz, 1H), 8.39 (dd, J = 8.0, 1.8 Hz, 2H), 8.26 (s, 1H), 7.69 (dddd, J = 12.7, 8.3, 7.1, 1.4 Hz, 2H), 7.62–7.56 (m, 1H), 7.54 (d, J = 8.2 Hz, 1H), 7.45–7.40 (m, 1H), 7.38 (d, J = 8.3 Hz, 1H), 2.55 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 178.2, 155.3, 154.3, 141.0, 133.9, 133.6, 129.2, 128.9, 128.4, 127.7, 127.2, 126.7, 126.5, 124.6, 124.0, 123.9, 122.7, 122.3, 121.6, 117.5, 112.0, 22.4 ppm. HRMS (ESI) m/z calcd for C22H15O2 [M + H]+: 311.1067, found: 311.1062.
14H-Dibenzo[a,c]thioxanthen-14-one (6b). 6b (18 mg, 58% yield) was generated following a procedure for the synthesis of 6a as a white solid, mp 198.6–199.4 °C. 1H NMR (400 MHz, CDCl3) δ 9.59–9.43 (m, 1H), 8.67 (d, J = 8.3 Hz, 1H), 8.64–8.59 (m, 1H), 8.53 (d, J = 7.9 Hz, 1H), 8.46 (d, J = 8.2 Hz, 1H), 7.77 (t, J = 7.6 Hz, 1H), 7.73–7.59 (m, 5H), 7.54 (t, J = 7.5 Hz, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ 182.9, 139.2, 134.1, 132.3, 131.7, 131.6, 130.1, 129.9, 129.5, 129.4, 128.0, 127.7, 127.5, 127.4, 127.3, 127.2, 125.6, 124.9, 124.7, 123.5, 122.6 ppm. HRMS (ESI) m/z calcd for C21H13OS [M + H]+: 313.0682, found: 313.0671.
9-Fluorodibenzo[i,k]phenanthridine (6c). 6c (17 mg, 57% yield) was generated following a procedure for the synthesis of 6a as a white solid, mp 151.9–152.5 °C. 1H NMR (400 MHz, CDCl3) δ 10.04 (s, 1H), 8.88 (d, J = 8.2 Hz, 1H), 8.82–8.64 (m, 3H), 8.54 (dd, J = 11.2, 2.3 Hz, 1H), 8.31 (dd, J = 9.0, 6.0 Hz, 1H), 7.86–7.69 (m, 4H), 7.61–7.46 (m, 1H) ppm. 13C NMR (100 MHz, CDCl3) δ 162.6, 160.2, 146.2, 143.7, 132.6, 132.0, 131.9, 130.5, 129.1, 129.0, 128.5, 128.3, 128.1, 127.6, 127.2, 124.0, 123.5, 123.2, 122.6, 118.4, 118.1, 112.1, 111.8 ppm. 19F NMR (376 MHz, CDCl3) δ −112.6 ppm. HRMS (ESI) m/z calcd for C21H13FN [M + H]+: 298.1027, found: 298.1024.
General procedure for synthesis of 7
10-(2-(1H-Indol-3-yl)ethyl)chromeno[3,2-b]indol-11(10H)-one (7a). 7a (48 mg, 63% yield) was generated following a procedure for the synthesis of 4l as a white solid, mp 157.5–158.6 °C. 1H NMR (400 MHz, CDCl3) δ 8.55–8.45 (m, 1H), 8.01 (d, J = 8.1 Hz, 1H), 7.97 (s, 1H), 7.81 (d, J = 7.0 Hz, 1H), 7.76–7.66 (m, 2H), 7.50–7.44 (m, 1H), 7.43–7.38 (m, 1H), 7.35 (d, J = 7.0 Hz, 1H), 7.29 (d, J = 8.6 Hz, 1H), 7.25–7.16 (m, 3H), 6.97 (d, J = 2.0 Hz, 1H), 5.09–4.96 (m, 2H), 3.43–3.25 (m, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ 170.3, 155.6, 145.2, 138.1, 136.4, 132.8, 128.1, 127.6, 126.3, 124.4, 124.0, 122.5, 122.2, 120.2, 120.1, 119.7, 118.9, 118.2, 114.9, 112.8, 111.3, 110.4, 100.1, 45.8, 27.1 ppm. HRMS (ESI) m/z calcd for C25H19N2O2 [M + H]+: 379.1441, found: 379.1442.
10-((3R)-10,13-Dimethyl-17-oxohexadecahydro-1H-cyclopenta[a]phenanthren-3-yl)chromeno[3,2-b]indol-11(10H)-one (7b). 7b (57 mg, 56% yield) was generated following a procedure for the synthesis of 4l as a white solid, mp 167.5–168.6 °C. 1H NMR (400 MHz, CDCl3) δ 8.47 (d, J = 7.6 Hz, 1H), 8.07 (d, J = 7.5 Hz, 1H), 7.70 (dd, J = 14.6, 7.1 Hz, 2H), 7.61 (d, J = 8.2 Hz, 1H), 7.51 (s, 1H), 7.45 (s, 1H), 7.27 (d, J = 9.2 Hz, 1H), 5.99 (d, J = 6.3 Hz, 1H), 2.76–2.64 (m, 1H), 2.48 (dd, J = 19.2, 8.4 Hz, 1H), 2.27 (s, 1H), 2.10 (dd, J = 19.3, 9.6 Hz, 1H), 2.05–1.91 (m, 3H), 1.84 (t, J = 14.4 Hz, 2H), 1.71 (s, 5H), 1.51 (dt, J = 26.0, 13.5 Hz, 5H), 1.30 (d, J = 8.6 Hz, 4H), 1.12 (s, 3H), 1.07 (s, 1H), 0.93 (s, 3H), 0.89 (d, J = 11.4 Hz, 2H) ppm. 13C NMR (100 MHz, CDCl3) δ 170.2, 155.2, 145.5, 136.9, 132.8, 127.8, 126.4, 124.5, 123.9, 120.5, 120.3, 120.0, 118.0, 115.7, 112.5, 56.3, 51.6, 50.4, 48.1, 39.6, 36.9, 36.0, 35.6, 34.4, 33.0, 31.9, 30.6, 28.4, 26.3, 21.9, 20.8, 17.4, 14.0 ppm. 13C NMR (100 MHz, dept 90, CDCl3) δ 132.7, 127.7, 126.3, 123.8, 120.4, 120.0, 117.9, 112.4, 56.2, 51.4, 50.3, 39.5, 35.5 ppm. 13C NMR (100 MHz, dept 135, CDCl3) δ 132.7, 127.7, 126.3, 123.8, 120.4, 119.9, 117.9, 112.4, 56.2, 51.4, 50.3, 39.5, 36.8, 35.9, 35.5, 32.9, 31.8, 30.5, 28.3, 26.2, 21.8, 20.7, 17.3, 13.9 ppm. HRMS (ESI) m/z calcd for C34H38NO3 [M + H]+: 508.2846, found: 508.2855.
10-(2-Aminophenyl)chromeno[3,2-b]indol-11(10H)-one (8a). 8a (43 mg, 66% yield) was generated following a procedure for the synthesis of 3a as a pink solid, mp 215.5–216.8 °C. 1H NMR (500 MHz, CDCl3) δ 8.33 (d, J = 7.2 Hz, 1H), 8.09 (d, J = 8.0 Hz, 1H), 7.77–7.66 (m, 2H), 7.49 (t, J = 7.6 Hz, 1H), 7.40 (dd, J = 14.6, 6.9 Hz, 1H), 7.34 (dd, J = 9.3, 5.4 Hz, 2H), 7.23 (d, J = 8.5 Hz, 1H), 7.19 (d, J = 7.6 Hz, 1H), 7.01 (d, J = 6.8 Hz, 1H), 6.91 (s, 1H), 2.71 (s, 2H). ppm. 13C NMR (100 MHz, CDCl3) δ 169.3, 155.8, 146.3, 139.0, 133.0, 130.0, 129.6, 128.9, 126.5, 124.6, 124.2, 121.5, 120.1, 119.1, 118.2, 116.9, 115.9, 112.1 ppm. HRMS (ESI) m/z calcd for C21H15N2O2 [M + H]+: 327.1128, found: 327.1127.
10-(2-Aminophenyl)-3-methoxychromeno[3,2-b]indol-11(10H)-one (8b). 8b (48 mg, 68% yield) was generated following a procedure for the synthesis of 3a as a white solid, mp 224.2–226.1 °C. 1H NMR (400 MHz, CDCl3) δ 8.38 (d, J = 8.8 Hz, 1H), 8.20 (d, J = 8.0 Hz, 1H), 7.62 (t, J = 7.6 Hz, 1H), 7.47 (t, J = 7.6 Hz, 2H), 7.41 (s, 1H), 7.35 (dd, J = 12.6, 8.3 Hz, 2H), 7.13 (d, J = 8.9 Hz, 1H), 7.09 (d, J = 8.0 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H), 4.11 (s, 3H). ppm. 13C NMR (100 MHz, CDCl3) δ 169.1, 163.7, 157.5, 145.9, 144.2, 138.6, 130.0, 129.6, 128.5, 127.7, 123.5, 121.3, 121.0, 119.8, 118.9, 118.4, 116.7, 115.9, 113.4, 112.0, 100.6, 56.0 ppm. HRMS (ESI) m/z calcd for C22H17N2O3 [M + H]+: 357.1234, found: 357.1237.
Procedure for the synthesis of 9a
To a stirred solution of 8a (40 mg) in dichloromethane (3 mL), was added acetyl chloride (1.2 equiv.) and triethylamine (2.0 equiv.). The reaction proceeded at rt for 4 h before dichloromethane was removed by a rotary evaporation. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and evaporated in a vacuo. The residue was purified by column chromatography (PE/EtOAc = 10/1–5/1) to provide N-(2-(11-oxochromeno[3,2-b] indol-10(11H)-yl)phenyl) acetamide (42 mg) as a yellow solid. Then, to this obtained solid was added polyphosphoric acid (0.2 mL) and POCl3 (10 equiv.). The reaction proceeded in a sealed tube at 120 °C for 3 h. The reaction mixture was neutralized with NaHCO3 (Sat.) and extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and evaporated in a vacuo. The residue was purified by column chromatography (PE/EtOAc = 10/1–3/1) to provide 6-methyl-15H-benzo[2,3][1,4] diazepino[6,7,1-h,i]chromeno [3,2-b]indol-15-one 9a as a yellow solid (33 mg, 51% yield over two steps), mp 198.2–199.4 °C. 1H NMR (400 MHz, CDCl3) δ 8.47 (dd, J = 8.0, 1.5 Hz, 1H), 7.93 (d, J = 7.8 Hz, 1H), 7.81–7.74 (m, 1H), 7.69 (d, J = 8.4 Hz, 1H), 7.54 (d, J = 7.3 Hz, 1H), 7.53–7.48 (m, 1H), 7.33 (t, J = 7.8 Hz, 2H), 7.17–7.10 (m, 2H), 6.75–6.65 (m, 1H), 2.60 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 169.4, 167.1, 155.5, 151.9, 151.0, 139.7, 135.2, 133.5, 129.5, 129.4, 127.5, 126.9, 125.9, 125.8, 124.9, 124.6, 124.4, 123.8, 122.6, 122.2, 119.8, 118.1, 28.5 ppm. HRMS (ESI) m/z calcd for C23H15N2O2 [M + H]+: 351.1128, found: 351.1127.
Procedure for the synthesis of 9b
To a stirred solution of 8b (45 mg) in EtOH (4 mL), was added TsOH·H2O (0.1 equiv.). The reaction proceeded at a reflux overnight before EtOH was removed by a rotary evaporation. The reaction mixture was extracted with EtOAc. The combined organic layers were washed with H2O and brine, dried over anhydrous Na2SO4, and evaporated in a vacuo. The residue was purified by column chromatography (PE/EtOAc = 10/1–5/1) to provide 8-methoxy-10-oxa-5,14b-diazaindeno [1,2,3-g,h]tetraphene 9b as a yellow solid (41 mg, 95% yield), mp 173.5–174.4 °C. 1H NMR (400 MHz, CDCl3) δ 8.33 (d, J = 8.0 Hz, 1H), 8.24 (d, J = 8.6 Hz, 1H), 8.02 (dd, J = 17.2, 8.0 Hz, 2H), 7.75 (d, J = 7.3 Hz, 1H), 7.58 (t, J = 7.8 Hz, 1H), 7.44 (t, J = 7.5 Hz, 1H), 7.32 (t, J = 7.6 Hz, 1H), 7.27–7.21 (m, 1H), 6.92 (d, J = 10.1 Hz, 2H), 3.93 (s, 3H) ppm. 13C NMR (100 MHz, CDCl3) δ 163.3, 158.1, 148.0, 139.5, 132.8, 131.2, 130.3, 129.1, 126.3, 125.8, 125.1, 124.6, 121.6, 119.2, 118.1, 116.7, 114.8, 114.0, 113.6, 112.4, 102.2, 55.8 ppm. HRMS (ESI) m/z calcd for C22H15N2O2 [M + H]+: 339.1128, found: 339.1122.
The general synthesis of heterocyclic iodoniums 1
All the synthetic heterocyclic idoniums are reported in our previous work, and they are prepared conveniently using reported procedure.10,18
11-Oxo-11H-benzo[b]chromeno[2,3-d]iodol-10-ium triflate (1a). 1H NMR (400 MHz, DMSO) δ 8.50–8.35 (m, 2H), 8.22 (dd, J = 7.9, 1.4 Hz, 1H), 8.11–8.02 (m, 2H), 8.01–7.91 (m, 2H), 7.72 (t, J = 7.5 Hz, 1H) ppm. 13C NMR (100 MHz, DMSO) δ 172.3, 164.1, 155.2, 135.9, 135.2, 134.7, 131.4, 131.3, 128.9, 127.0, 125.2, 122.5, 119.6, 118.8, 108.4 ppm.
8-Methyl-11-oxo-10λ3-benzo[b]chromeno[2,3-d]iodol-10(11H)-yl trifluoromethanesulfonate (1b). 1H NMR (400 MHz, DMSO) δ 8.31 (d, J = 8.0 Hz, 1H), 8.23 (s, 1H), 8.21 (dd, J = 8.0, 1.5 Hz, 1H), 8.03 (ddd, J = 8.6, 7.1, 1.7 Hz, 1H), 7.95 (d, J = 7.9 Hz, 1H), 7.88 (d, J = 7.7 Hz, 1H), 7.74–7.68 (m, 1H), 2.58 (s, 3H) ppm. 13C NMR (100 MHz, DMSO) δ 172.2, 164.0, 155.1, 146.2, 135.8, 132.5, 132.2, 131.2, 128.5, 126.9, 125.2, 122.4, 119.7, 118.7, 107.4, 21.8 ppm.
8-Chloro-11-oxo-10λ3-benzo[b]chromeno[2,3-d]iodol-10(11H)-yl trifluoromethanesulfonate (1c). 1H NMR (400 MHz, DMSO) δ 8.42 (d, J = 1.8 Hz, 1H), 8.39 (d, J = 8.4 Hz, 1H), 8.21 (dd, J = 7.9, 1.2 Hz, 1H), 8.14 (dd, J = 8.4, 1.8 Hz, 1H), 8.07–8.00 (m, 1H), 7.95 (d, J = 8.3 Hz, 1H), 7.71 (t, J = 7.5 Hz, 1H) ppm. 13C NMR (100 MHz, DMSO) δ 172.2, 163.2, 155.1, 138.5, 136.0, 134.3, 131.9, 131.3, 130.9, 129.8, 128.5, 127.1, 125.3, 122.5, 120.1, 118.8, 109.1 ppm.
8-Fluoro-11-oxo-10λ3-benzo[b]chromeno[2,3-d]iodol-10(11H)-yl trifluoromethanesulfonate (1d). 1H NMR (400 MHz, DMSO) δ 8.50–8.44 (m, 1H), 8.22 (ddd, J = 8.0, 5.9, 2.3 Hz, 2H), 8.09–8.00 (m, 1H), 8.01–7.91 (m, 2H), 7.71 (dd, J = 10.6, 4.3 Hz, 1H) ppm. 13C NMR (100 MHz, DMSO) δ 172.1, 165.2, 163.2, 162.7, 155.2, 136.0, 132.1, 130.8, 130.7, 127.1, 125.3, 122.5, 120.3, 120.2, 112.0, 119.7, 119.3, 119.0, 118.8, 108.5 ppm.
7-Fluoro-11-oxo-10λ3-benzo[b]chromeno[2,3-d]iodol-10(11H)-yl trifluoromethanesulfonate (1e). 1H NMR (400 MHz, DMSO) δ 8.50–8.43 (m, 1H), 8.39 (dd, J = 5.6, 2.7 Hz, 1H), 8.23 (d, J = 7.9 Hz, 1H), 8.06 (t, J = 7.5 Hz, 1H), 7.95 (dd, J = 8.2, 2.3 Hz, 1H), 7.87 (ddd, J = 8.9, 6.2, 2.7 Hz, 1H), 7.73 (t, J = 6.2 Hz, 1H) ppm. 13C NMR (100 MHz, DMSO) δ 172.3, 164.9, 163.0, 162.4, 155.1, 137.5, 137.4, 136.1, 133.7, 133.6, 127.1, 125.3, 122.4, 122.3, 122.0, 118.8, 115.9, 115.7, 113.6, 109.7 ppm.
11-Oxo-10λ3-benzo[b]thiochromeno[2,3-d]iodol-10(11H)-yl trifluoromethanesulfonate (1f). 1H NMR (400 MHz, DMSO) δ 8.46 (t, J = 9.1 Hz, 2H), 8.34 (d, J = 7.8 Hz, 1H), 8.22 (d, J = 8.1 Hz, 1H), 8.04 (d, J = 7.6 Hz, 1H), 8.01–7.96 (m, 1H), 7.92 (t, J = 7.9 Hz, 1H), 7.87 (t, J = 7.6 Hz, 1H) ppm. 13C NMR (100 MHz, DMSO) δ 174.9, 150.6, 143.4, 135.8, 134.0, 132.1, 131.5, 129.7, 128.9, 128.8, 128.1, 128.0, 123.3, 122.8 ppm.
7H-7λ3-Benzo[4,5]iodolo[2,3-c]quinolin-7-yl trifluoromethanesulfonate (1g). To a stirred solution of 3-iodo-4-phenylquinoline 1g–l (2.0 g, 6.04 mol) in anhydrous DCM (20 mL) was added TfOH (1.60 mL, 3.0 equiv.) and followed by the slow addition of m-CPBA (85%, 1.84 g, 1.5 equiv.). The solution was stirred for 2 h at rt before DCM was removed by rotary evaporation. Et2O (20 mL) was added to the remained solid. The mixture was stirred for 30 min, and then filtered. The obtained solid was washed with Et2O three times and dried in high vacuo to provide 1g (2.43 g, 84% yield) as a yellow solid. 1H NMR (400 MHz, DMSO) δ 9.53 (s, 1H), 9.14 (d, J = 7.9 Hz, 1H), 9.09 (d, J = 8.8 Hz, 1H), 8.45 (d, J = 7.6 Hz, 1H), 8.30 (d, J = 7.9 Hz, 1H), 8.03 (t, J = 7.6 Hz, 2H), 7.97–7.86 (m, 2H) ppm. 13C NMR (100 MHz, DMSO) δ 148.6, 147.9, 144.8, 141.2, 132.4, 132.3, 131.2, 131.1, 131.1, 131.0, 130.6, 129.1, 126.4, 124.2, 123.9, 117.7 ppm.
9-Methyl-7H-7λ3-benzo[4,5]iodolo[2,3-c]quinolin-7-yl trifluoromethanesulfonate (1h). 1H NMR (400 MHz, DMSO) δ 9.48 (s, 1H), 9.04 (d, J = 8.4 Hz, 1H), 8.99 (d, J = 8.4 Hz, 1H), 8.28 (dd, J = 8.4, 1.0 Hz, 1H), 8.20 (s, 1H), 8.02 (dd, J = 11.3, 4.0 Hz, 1H), 7.95–7.88 (m, 1H), 7.81 (d, J = 7.5 Hz, 1H), 2.56 (s, 3H) ppm. 13C NMR (100 MHz, DMSO) δ 148.4, 147.7, 144.8, 143.2, 138.4, 132.0, 131.9, 131.1, 131.0, 130.5, 129.0, 126.2, 124.2, 124.0, 122.4, 119.1, 117.0, 21.2 ppm.
9-Chloro-7H-7λ3-benzo[4,5]iodolo[2,3-c]quinolin-7-yl trifluoromethanesulfonate (1i). 1H NMR (400 MHz, DMSO) δ 9.46 (s, 1H), 9.03 (d, J = 8.9 Hz, 1H), 8.94 (d, J = 8.5 Hz, 1H), 8.38 (d, J = 2.2 Hz, 1H), 8.27 (dd, J = 8.4, 1.1 Hz, 1H), 8.05–7.97 (m, 2H), 7.90 (ddd, J = 8.4, 7.0, 1.3 Hz, 1H) ppm. 13C NMR (100 MHz, DMSO) δ 148.4, 147.9, 143.8, 140.1, 136.2, 133.2, 131.4, 130.7, 130.5, 129.3, 126.2, 124.9, 124.1, 122.4, 119.1, 118.2 ppm.
2-Fluoro-7H-7λ3-benzo[4,5]iodolo[2,3-c]quinolin-7-yl trifluoromethanesulfonate (1j). 1H NMR (400 MHz, DMSO) δ 9.54 (d, J = 3.4 Hz, 1H), 9.15 (dd, J = 7.7, 2.7 Hz, 1H), 8.89–8.72 (m, 1H), 8.46 (dd, J = 8.1, 2.3 Hz, 1H), 8.40 (ddd, J = 9.3, 6.0, 3.4 Hz, 1H), 8.08–7.85 (m, 3H) ppm. 13C NMR (100 MHz, DMSO) δ 162.4, 159.9, 148.0, 145.3, 144.5, 144.4, 140.7, 133.7, 133.6, 132.4, 132.2, 131.2, 131.2, 127.0, 126.9, 123.7, 122.3, 121.2, 121.0, 119.1, 118.9, 108.8, 108.6 ppm.
Benzo[4,5]iodolo[3,2-c]isoquinolin-11-ium triflate (1k). 1H NMR (400 MHz, DMSO) δ 9.73 (s, 1H), 8.59–8.51 (m, 2H), 8.43 (dd, J = 10.6, 8.3 Hz, 2H), 8.18–8.07 (m, 1H), 7.97 (t, J = 7.5 Hz, 2H), 7.90–7.81 (m, 1H) ppm. 13C NMR (100 MHz, DMSO) δ 155.6, 153.4, 140.6, 134.5, 133.6, 132.6, 131.1, 130.7, 130.1, 129.5, 129.3, 126.7, 122.3, 121.4, 120.5 ppm.
9-Methyl-11H-11λ3-benzo[4,5]iodolo[3,2-c]isoquinolin-11-yl trifluoromethanesulfonate (1l). 1H NMR (400 MHz, DMSO) δ 9.58 (s, 1H), 8.40 (d, J = 8.2 Hz, 1H), 8.34 (d, J = 8.1 Hz, 1H), 8.24 (d, J = 7.9 Hz, 1H), 8.07 (s, 1H), 8.06–8.01 (m, 1H), 7.89 (t, J = 7.3 Hz, 1H), 7.66 (d, J = 7.8 Hz, 1H), 2.51 (s, 3H) ppm. 13C NMR (100 MHz, DMSO) δ 155.4, 153.2, 143.2, 137.8, 134.3, 133.5, 132.0, 130.2, 129.8, 129.4, 129.2, 128.6, 126.4, 121.2, 119.5, 21.5 ppm.
6-Oxo-7λ3-benzo[b]chromeno[4,3-d]iodol-7(6H)-yl trifluoromethanesulfonate (1m). 1H NMR (400 MHz, DMSO) δ 9.12–9.02 (m, 1H), 8.81 (d, J = 7.5 Hz, 1H), 8.53 (dd, J = 8.3, 1.0 Hz, 1H), 8.10–8.01 (m, 1H), 8.00–7.93 (m, 1H), 7.92–7.85 (m, 1H), 7.71 (dd, J = 8.3, 0.9 Hz, 1H), 7.66–7.56 (m, 1H) ppm. 13C NMR (100 MHz, DMSO) δ 157.2, 153.6, 152.7, 141.0, 133.5, 133.4, 133.0, 132.2, 131.4, 126.2, 125.6, 125.2, 118.2, 117.9, 117.3 ppm.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the grant support from National Natural Science Foundation of China (81672952, 81872440), Guangdong Science and Technology Program (2017A020215198, 2018A030310240), and Guangzhou Science and Technology Program (201807010041).
Notes and references
-
(a) H. Ito, K. Ozaki and K. Itami, Angew. Chem. Int. Ed., 2017, 56, 11144–11164 (Angew. Chem., 2017, 129, 11296–11317) CrossRef CAS;
(b) J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS.
- D. A. Erlanson, S. W. Fesik, R. E. Hubbard, W. Jahnke and H. Jhoti, Nat. Rev. Drug Discovery, 2016, 15, 605–619 CrossRef CAS.
-
(a) G. B. Deng, Z. Q. Wang, J. D. Xia, P. C. Qian, R. J. Song, M. Hu, L. B. Gong and J. H. Li, Angew. Chem. Int. Ed., 2013, 52, 1535–1538 (Angew. Chem., 2013, 125, 1575–1578) CrossRef CAS;
(b) J. H. Kim, J. Bouffard and S. G. Lee, Angew. Chem. Int. Ed., 2014, 53, 6435–6438 (Angew. Chem., 2014, 126, 6553–6556) CrossRef CAS PubMed;
(c) J. H. Kim, Y. O. Ko, J. Bouffard and S. G. Lee, Chem. Soc. Rev., 2015, 44, 2489–2507 RSC;
(d) Y. Liu and J.-P. Wan, Org. Biomol. Chem., 2011, 9, 6873–6894 RSC.
- M. Wang, Q. Fan and X. Jiang, Org. Lett., 2018, 20, 216–219 CrossRef CAS.
-
(a) Y. Wu, X. Peng, B. Luo, F. Wu, B. Liu, F. Song, P. Huang and S. Wen, Org. Biomol. Chem., 2014, 12, 9777–9780 RSC;
(b) D. Zhu, Q. Liu, B. Luo, M. Chen, R. Pi, P. Huang and S. Wen, Adv. Synth. Catal., 2013, 355, 2172–2178 CrossRef CAS.
-
(a) X. Peng, H. Luo, F. Wu, D. Zhu, A. Ganesan, P. Huang and S. Wen, Adv. Synth. Catal., 2017, 359, 1152–1156 CrossRef CAS;
(b) D. Zhu, Y. Wu, B. Wu, B. Luo, A. Ganesan, F. H. Wu, R. Pi, P. Huang and S. Wen, Org. Lett., 2014, 16, 2350–2353 CrossRef CAS.
- Z. Liu, D. Zhu, B. Luo, N. Zhang, Q. Liu, Y. Hu, R. Pi, P. Huang and S. Wen, Org. Lett., 2014, 16, 5600–5603 CrossRef CAS PubMed.
-
(a) D. Zhu, M. Li, Z. Wu, Y. Du, B. Luo, P. Huang and S. Wen, Eur. J. Org. Chem., 2019, 4566–4571 CrossRef CAS;
(b) B. Luo, Q. Cui, H. Luo, Y. Hu, P. Huang and S. Wen, Adv. Synth. Catal., 2016, 358, 2733–2738 CrossRef CAS;
(c) Q. Tan, D. Zhou, T. Zhang, B. Liu and B. Xu, Chem. Commun., 2017, 53, 10279–10282 RSC;
(d) M. Wang, J. Wei, Q. Fan and X. Jiang, Chem. Commun., 2017, 53, 2918–2921 RSC;
(e) P. S. Postnikov, O. A. Guselnikova, M. S. Yusubov, A. Yoshimura, V. N. Nemykin and V. V. Zhdankin, J. Org. Chem., 2015, 80, 5783–5788 CrossRef CAS;
(f) Y. A. Vlasenko, P. S. Postnikov, M. E. Trusova, A. Shafir, V. V. Zhdankin, A. Yoshimura and M. S. Yusubov, J. Org. Chem., 2018, 83, 12056–12070 CrossRef CAS.
- J. Letessier and H. Detert, Synthesis, 2012, 44, 290–296 CAS.
- D. Zhu, Z. Wu, B. Luo, Y. Du, P. Liu, Y. Chen, Y. Hu, P. Huang and S. Wen, Org. Lett., 2018, 20, 4815–4818 CrossRef CAS PubMed.
-
(a) S. U. Dighe, S. Khan, I. Soni, P. Jain, S. Shukla, R. Yadav, P. Sen, S. M. Meeran and S. Batra, J. Med. Chem., 2015, 58, 3485–3499 CrossRef CAS;
(b) T. K. Mazu, J. R. Etukala, M. R. Jacob, S. I. Khan, L. A. Walker and S. Y. Ablordeppey, Eur. J. Med. Chem., 2011, 46, 2378–2385 CrossRef CAS;
(c) G. Van Baelen, S. Hostyn, L. Dhooghe, P. Tapolcsányi, P. Mátyus, G. Lemière, R. Dommisse, M. Kaiser, R. Brun, P. Cos, L. Maes, G. Hajós, Z. Riedl, I. Nagy, B. U. W. Maes and L. Pieters, Bioorg. Med. Chem., 2009, 17, 7209–7217 CrossRef CAS.
-
(a) J. Reis, A. Gaspar, N. Milhazes and F. Borges, J. Med. Chem., 2017, 60, 7941–7957 CrossRef CAS;
(b) R. S. Keri, S. Budagumpi, R. K. Pai and R. G. Balakrishna, Eur. J. Med. Chem., 2014, 78, 340–374 CrossRef CAS.
-
(a) Shaveta, S. Mishra and P. Singh, Eur. J. Med. Chem., 2016, 124, 500–536 CrossRef CAS;
(b) C. de Bodinat, B. Guardiola-Lemaitre, E. Mocaer, P. Renard, C. Munoz and M. J. Millan, Nat. Rev. Drug Discovery, 2010, 9, 628–642 CrossRef CAS;
(c) F. H. Havaldar, S. Bhise and S. Burudkar, J. Serb. Chem. Soc., 2004, 69, 527–532 CrossRef CAS;
(d) L. Duan, J. Qiao, Y. Sun and Y. Qiu, Adv. Mater., 2011, 23, 1137–1144 CrossRef CAS PubMed.
-
(a) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS;
(b) N. Rodriguez and L. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC.
-
(a) S. Yang, F. Wang, Y. Wu, W. Hua and F. Zhang, Org. Lett., 2018, 20, 1491–1495 CrossRef CAS;
(b) S. Yang, W. Hua, Y. Wu, T. Hu, F. Wang, X. Zhang and F. Zhang, Chem. Commun., 2018, 54, 3239–3242 RSC.
-
(a) S. Sergeyev, W. Pisulab and Y. Geerts, Chem. Soc. Rev., 2007, 36, 1902–1929 RSC;
(b) S. Laschat, A. Baro, N. Steinke, F. Giesselmann, C. Hgele, G. Scalia, R. Judele, E. Kapatsina, S. Sauer, A. Schreivogel and M. Tosoni, Angew. Chem. Int. Ed., 2007, 46, 4832–4887 (Angew. Chem., 2007, 119, 4916–4973) CrossRef CAS.
- J. Wei, B. Han, Q. Guo, X. Shi, W. Wang and N. Wei, Angew. Chem. Int. Ed., 2010, 49, 8209–8213 (Angew. Chem., 2010, 122, 8385–8389) CrossRef CAS.
-
(a) C. Zhou, A. Dubrovsky and R. Larock, J. Org. Chem., 2006, 71, 1626–1632 CrossRef CAS;
(b) Q. Huang, J. Hunter and R. Larock, J. Org. Chem., 2002, 67, 3437–3444 CrossRef CAS;
(c) M. Reddy, N. Thirupathi, M. Babu and S. Puri, J. Org. Chem., 2013, 78, 5878–5888 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07288h |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a