
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUltrafine MoO2 nanoparticles encapsulated in a hierarchically porous carbon nanofiber film as a high-performance binder-free anode in lithium ion batteries
Xin Chena,
Guojun Gaoa,
Zhipeng Wua,
Jun Xiang *a,
Xiaoqiang Lia,
Guangguang Guana and
Kaiyin Zhang*b
*a,
Xiaoqiang Lia,
Guangguang Guana and
Kaiyin Zhang*b
aSchool of Science, Jiangsu University of Science and Technology, Zhenjiang 212003, China. E-mail: jxiang@just.edu.cn
bSchool of Physics and Electronic Engineering, Fuyang Normal University, Fuyang 236037, China. E-mail: kyzhang@fync.edu.cn
First published on 18th November 2019
Abstract
Flexible free-standing hierarchically porous carbon nanofibers embedded with ultrafine (∼3.5 nm) MoO2 nanoparticles (denoted as MoO2@HPCNFs) have been synthesized by electrospinning and subsequent heat treatment. When evaluated as a binder-free anode in Li-ion batteries, the as-obtained MoO2@HPCNFs film exhibits excellent capacity retention with high reversible capacity (≥1055 mA h g−1 at 100 mA g−1) and good rate capability (425 mA h g−1 at 2000 mA g−1), which is much superior to most of the previously reported MoO2-based materials. The synergistic effect of uniformly dispersed ultrasmall MoO2 nanoparticles and a three-dimensionally hierarchical porous conductive network constructed by HPCNFs effectively improve the utilization rate of active materials, enhance the transport of both electrons and Li+ ions, facilitate the electrolyte penetration, and promote the Li+ storage kinetics and stability, thus leading to a greatly enhanced electrochemical performance.
1. Introduction
Rechargeable lithium ion batteries (LIBs) have attracted tremendous attention due to their huge successes in electrochemical energy storage systems.1–4 Notwithstanding the remarkable progress achieved so far, it is still a great challenge to develop new high-performance electrode materials with high energy density, excellent cycling stability and rate capability to meet the ever-expanding demands for advanced energy storage and conversion devices.5 Until now, a large number of transition metal oxides have been exploited as promising anode materials for LIBs.6 Among various metal oxide-based electrode materials, molybdenum dioxide (MoO2) has been widely studied because of its rich chemical and structural versatility, and high theoretical specific capacity (838 mA h g−1).7 However, bare MoO2 is not an ideal Li-storage host due to its intrinsically slow electrochemical reaction kinetics, low conductivity, and drastic volume change during cycling, leading to poor rate capability and severe capacity fading.8To overcome these drawbacks and improve the electrochemical properties, nanosized MoO2 combined with carbon materials owning good conductivity and structural stability has been demonstrated to be an effective strategy.9–11 Among diverse carbon materials for LIB anodes, one-dimensional carbon nanofibers (CNFs) and their derivatives have gained special attention because they can be built into a flexible porous three-dimensionally (3D) conductive network for the rapid transport of both electrons and ions, significantly enhancing the electrochemical reaction kinetics and promoting the energy density and cycling stability.12,13 Moreover, some research has indicated that the introduction of various porous structures into CNFs can further improve both the specific capacity and the rate performance of CNFs-based electrodes.14,15 The incorporated pores can probably provide more electrode–electrolyte interface and markedly reduce electronic/ionic diffusion distance, thus further strengthening charge transport capability. In recent years, ultrasmall transition metal oxide nanoparticles loaded porous CNFs (PCNFs) have emerged as novel anode materials for LIBs. For instance, TiO2@PCNFs,16 MOUNPs@HPCNFs (M = Zn, Mn, and Co),17 γ-Fe2O3/PCNFs,18 and MoO3/PCNFs19 have been reported to exhibit excellent electrochemical properties. However, as far as we know, there has been no report as yet on the preparation and electrochemical performance of flexible self-supported PCNFs containing ultrasmall MoO2 nanoparticles. Therefore, it is of great interest to design and synthesize such nanostructure as binder-free anodes in LIBs.
Herein, we have proposed a novel binder-free LIB anode based on ultrafine MoO2 nanoparticles uniformly encapsulated in hierarchically porous carbon nanofibers (denoted as MoO2@HPCNFs) prepared via a facile electrospinning approach using polymethylacrylate (PMMA) as a pore-forming agent, followed by a controlled reduction–carbonization process The MoO2@HPCNFs electrode exhibits high lithium storage capacity and rate capability, showing a great application prospect in high-performance LIBs.
2. Experimental
2.1. Preparation of MoO2@HPCNFs
The MoO2@HPCNFs were prepared via co-electrospinning of polyacrylonitrile (PAN)/polymethylacrylate (PMMA)/Mo precursor combined with a controlled reduction–carbonization process, which was similar to our previous report.20 PMMA was used here as a pore generator that can be removed from the PAN-derived carbon nanofiber matrix under high temperature. 1.76 g of PAN (Mw = 150 k, Aldrich) and 0.44 g of PMMA (Mw = 150 k, Aladdin) were dissolved first in 16.8 g of N,N-dimethylformamide (DMF) at 60 °C for 4 h under magnetic stirring. Then, 1 g of MoCl5 was added into the mixture solution and further stirred approximately 12 h at room temperature to obtain a homogeneous solution for electrospinning. The precursor solution was transferred into a 20 mL plastic syringe and was electrospun by using a TL-01 electrospinning apparatus under the applied voltage of 18 kV, solution feeding rate of 0.4 mL h−1 and collector distance of 18 cm. The as-spun fibers were collected by an aluminum foil enwrapped roller with a rotate speed of about 300 rpm. The formed fiber film was subsequently stabilized at 220 °C for 4 h under air atmosphere and finally carbonized at 600 °C for 5 h in a flowing N2/H2 (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) mixed atmosphere to yield the resulting product.
:1, v/v) mixed atmosphere to yield the resulting product.
2.2. Characterization
The phase structure of as-prepared MoO2@HPCNFs was analyzed by X-ray diffraction (XRD, Shimadzu, XRD-6000). The surface morphology and microstructure of the sample were observed using field-emission scanning electron microscope (FESEM, Zeiss, Merlin Compact) and high-resolution transmission electron microscope (HRTEM, JEOL, JEM-2001F). Raman spectrum was collected at ambient temperature on a Raman spectrometer (Renishaw, inVia Reflex) using 532 nm excitation wavelength. Thermogravimetric analysis (TGA) was conducted on a TGA apparatus (Shimadzu, DTG-60H) in air. X-ray photoelectron spectroscopy (XPS) measurement was performed on a Thermo Fisher Scientific ESCALAB 250Xi XPS System with a monochromatic Al Kα X-ray source. Nitrogen adsorption/desorption isotherms (BET technique) at 77 K were recorded on a Micromeritics ASAP 2020 analyzer to characterize the pore structure of the product.2.3. Electrochemical measurements
The obtained MoO2@HPCNFs film was punched into circular discs and directly utilized as binder-free anodes for electrochemical measurements using CR2025 coin-type cells assembled in a highly pure argon-filled glovebox. Lithium metal foil was employed as the counter electrode, Celgard 2400 microporous polypropylene membrane as the separator, and 1 M LiPF6 in a mixture of ethylene carbonate (EC), ethyl methyl carbonate (EMC) and dimethylcarbonate (DMC) (1:1:1, v/v/v) as the electrolyte. The galvanostatic charge/discharge tests were carried out on a Land CT2001A battery testing system within the voltage range of 0.01–3.0 V vs. Li+/Li at different current densities. Cyclic voltammetry (CV) was measured by using a BioLogic SP-300 electrochemical workstation between 0.01–3.0 V vs. Li+/Li at a scan rate of 0.1 mV S−1. The mass of self-standing MoO2@HPCNFs film electrode is around 3 mg, and all the specific capacities are calculated based on the total mass of MoO2@HPCNFs film in this paper.
3. Results and discussion
Fig. 1a shows the XRD pattern of MoO2@HPCNFs. All the distinct diffraction peaks can be indexed to the monoclinic structure of MoO2 (JCPDS no. 73-1249), and no other distinguishable crystalline peaks belonging to molybdenum carbide/nitride are detected in the pattern, suggesting the high purity of the sample. In addition, a broad peak at low angle rang in the baseline can be clearly observed, demonstrating that the HPCNFs matrix is mainly amorphous. The structural information of carbon is further investigated using Raman spectra as presented in Fig. 1b. The as-prepared sample displays two wide peaks located at about 1324 and 1586 cm−1, known as the D-band and G-band, corresponding to the disordered carbon and graphite carbon, respectively.21 This result indicates that the sample is partially graphitized during the carbonization process. A large number of defects and vacancies in amorphous carbon can offer more diffusion channels and intercalation sites for Li+ storage, and meanwhile the appearance of graphite carbon can endow the whole film electrode with better conductivity, both of which would contribute to the overall capacity.17,22 The mass fraction of MoO2 in the MoO2@HPCNFs is determined by TGA (Fig. 1c). A small weight loss of 6.5 wt% before 170 °C in the TG curve may be attributed to the evaporation of the absorbed water. The large weight loss of 72.7 wt% from 290 to 510 °C is caused by the combustion of carbon. Consequently, the content of MoO2 in the MoO2@HPCNFs is estimated to be about 22.4 wt%.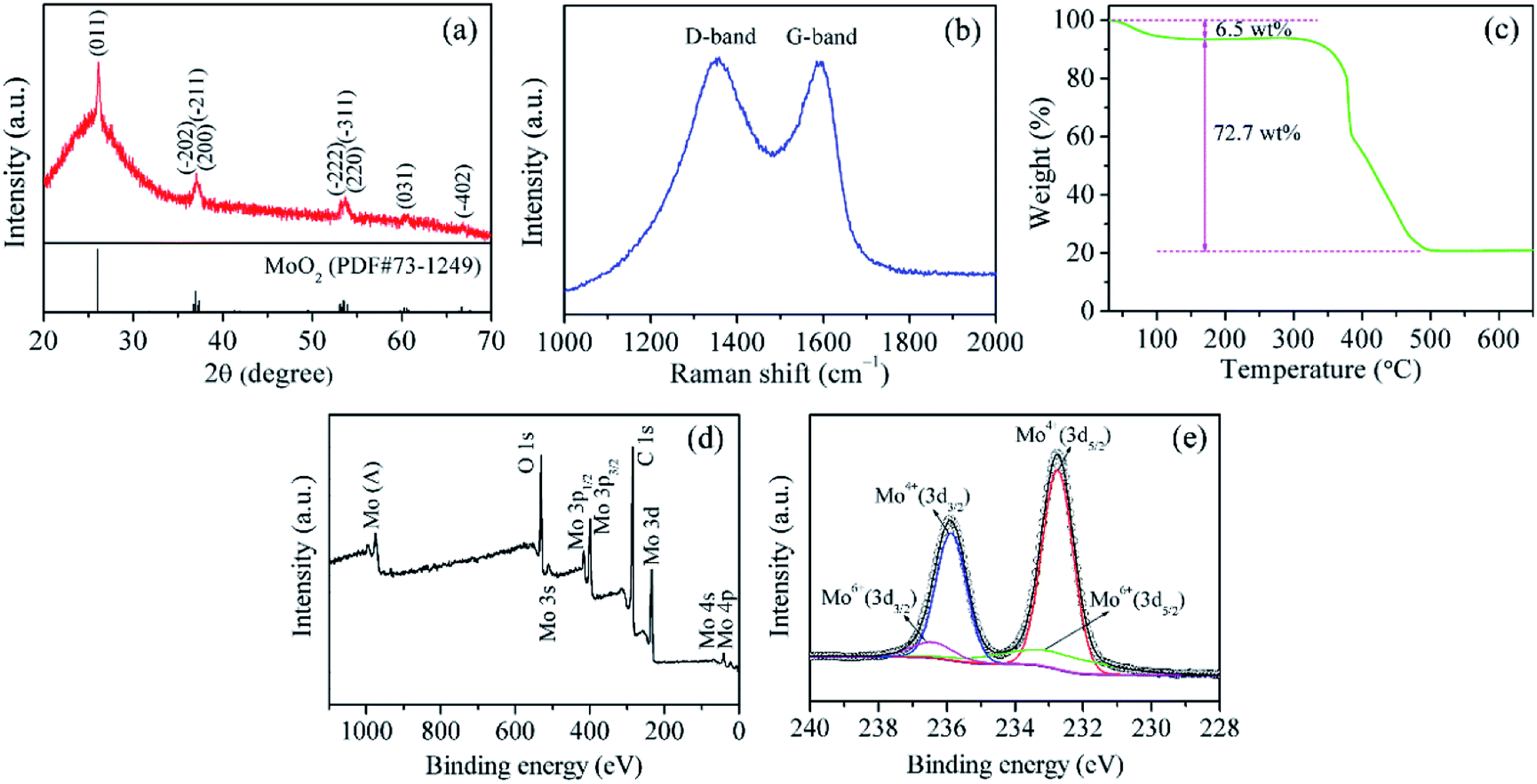 | ||
| Fig. 1 (a) XRD pattern, (b) Raman spectrum, (c) TG curve, (d) XPS survey spectrum, and (e) high-resolution Mo 3d spectrum of MoO2@HPCNFs. | ||
XPS technique was utilized to analyze the composition and valence states of MoO2@HPCNFs. As shown in Fig. 1d, the survey spectrum reveals that the composite is composed of O, C and Mo elements. The high-resolution Mo 3d spectrum (Fig. 1e) can be fitted to two doublets.23 One doublet with low energy and high intensity centered at 235.9 and 232.8 eV are ascribed to Mo(IV) 3d3/2 and Mo(IV) 3d5/2, respectively, further confirming the formation of MoO2. The other doublet with high energy and low intensity at 236.5 and 233.4 eV arise from Mo(VI) 3d3/2 and Mo(VI) 3d5/2 of MoO3, probably due to the slight surface oxidation of metastable MoO2 in air. Nevertheless, the XRD spectrum in Fig. 1a do not display any peaks coming from MoO3, which may be because of the low content of MoO3 or its amorphous nature in the product.
Fig. 2a presents the SEM image of MoO2@HPCNFs. The HPCNFs have smooth surface and continuous 1D structure with a mean diameter of 420 ± 50 nm and length of up to several hundred microns, which are randomly oriented and interlinked into a porous 3D conductive network. Such architecture can endow the electrode with improved cell reaction kinetics and structural integrity for lithium insertion/extraction.24 Furthermore, the cross-section SEM image (Fig. 2b) reveals that there are numerous mesopores and macropores inside the HPCNFs. The TEM image (Fig. 2d) further verifies this result, and we can also see that the partial MoO2@HPCNFs have a hollow structure possibly due to the connection of some channel-like macropores. The nitrogen adsorption–desorption isotherm of MoO2@HPCNFs (Fig. 2c) shows a typical type-IV behavior with an obvious hysteresis loop corresponding to the mesoporous structure, and meanwhile the isotherm displays a steep uptake at high relative pressures (P/P0 > 0.8), also indicating the existence of macroporous structure in the material,25 which are well accord with the FESEM and TEM observations. By using the BET method, the total pore volume and specific surface area of MoO2@HPCNFs are approximately 0.13 cm3 g−1 and 56.6 m2 g−1, respectively. The abundant mesopores and macropores throughout the HPCNFs not only enable the electrode more fully accessible to the electrolyte and take full advantage of the dispersed high-capacity MoO2 nanoparticles, but also effectively tolerate the mechanical stress caused by volume change and prevent the nanoparticle pulverization/aggregation during cycling, which will be conducive to achieving an enhanced and stable electrochemical performance.26 The HRTEM image (Fig. 2e) reveals that the obtained ultrasmall MoO2 nanoparticles have a uniform particle size of about 3.5 nm and are homogeneously embedded in the HPCNFs. In addition, the distinct lattice fringes are observed in the HRTEM image, suggesting a comparatively high crystallinity of MoO2 nanoparticles. The measured interplanar spacings of 0.215, 0.240 and 0.246 nm can be assigned to the (210), (200) and (−202) planes of monoclinic MoO2, respectively. Moreover, the clear lattice fringes belonging to the graphitic carbon can be observed, which further validates the existence of graphitized carbon in HPCNFs and is well consistent with the Raman results. As shown in Fig. 2f–i, the scanning TEM (STEM) and corresponding elemental mapping images demonstrate the homogeneous distribution O and Mo elements along the HPCNFs, confirming that the MoO2 nanoparticles with ultrasmall size are evenly dispersed throughout the HPCNFs from another angle. It should be emphasized that such a homogeneous distribution of ultrafine MoO2 nanoparticles inside the HPCNFs can facilitate the charge transfer and Li+ ion diffusion kinetics due to the shortened conducting and diffusion paths.17
 | ||
| Fig. 2 (a) Surface and (b) cross-sectional SEM images, (c) nitrogen adsorption–desorption isotherm, (d) TEM, (e) HRTEM, (f) STEM and (g–i) corresponding elemental mapping images of MoO2@HPCNFs. | ||
The typical CV curves of MoO2@HPCNFs film for the initial five cycles are presented in Fig. 3a. In the first cathodic sweep, a broad and large peak at approximately 0.4 V is observed, which is ascribed to the irreversible formation of solid electrolyte interphase (SEI) film on the HPCNFs surfaces.27 During the subsequent cycles, this peak disappears and one redox couple at about 1.31/1.05 V corresponding to the reversible phase transition of partially lithiated LixMoO2 become relatively distinct.28,29 At the potential above 1 V, Li+ is intercalated into MoO2 to form the solid solution LixMoO2.23 As the electrode is discharged below 1 V, the as-formed LixMoO2 would reaction with Li+ and be gradually converted into metallic Mo and Li2O. From the second cycles onwards, a clear reduction peak appears at around 0.12 V, possibly indicating a more complete conversion reaction from LixMoO2 to Mo and Li2O.9 It is worth pointing out that after the first cycle, the CV curves nearly overlap, suggesting the good Li insertion/extraction reversibility of the MoO2@HPCNFs electrode. In addition, no visible lithiation/delithiation peaks related to the carbon matrix are observed below 0.3 V, implying its small contribution to the capacity.
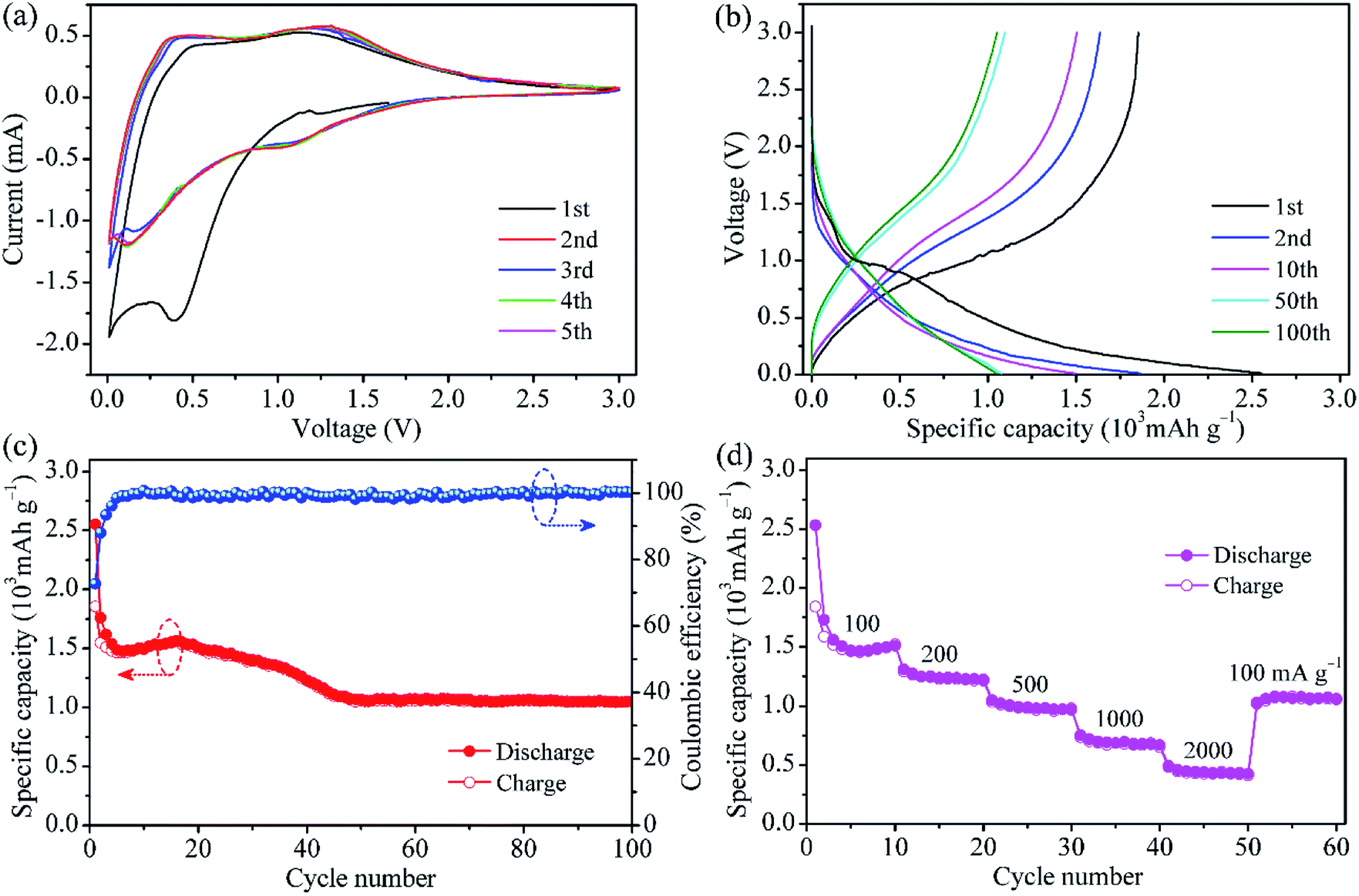 | ||
| Fig. 3 (a) Cyclic voltammograms, (b) galvanostatic discharge/charge curves for selected cycles at 100 mA g−1, (c) cycling performance and coulombic efficiency at 100 mA g−1, and (d) rate capacities at various current densities for the MoO2@HPCNFs film electrode. | ||
Fig. 3b presents the galvanostatic charge/discharge profiles of MoO2@HPCNFs at 100 mA g−1. During the first cycle, the MoO2@HPCNFs electrode delivers large discharge/charge capacities of 2552/1854 mA h g−1 with a relatively high initial coulombic efficiency of 72.6%, which indicated that the detrimental reactions between MoO2 and electrolyte can be significantly alleviated in the MoO2@HPCNFs. The initial capacity loss is generally attributed to the formation of SEI film and the trapping of Li+ in the lattices of MoO2.9,11 Furthermore, a discharge plateau corresponding to SEI formation is clearly observed and then disappears after the first cycle, which is in good agreement with the CV results. From the 50th cycle, the discharge/charge curves mostly overlap and the capacity remains nearly constant.
Fig. 3c shows the cycling performance of MoO2@HPCNFs at 100 mA g−1. The slight rise in discharge capacity from 6th to 16th cycle is ascribed to the infiltration of electrolyte and activation of materials. Then, the discharge capacity decreases gradually upon continuous cycling, which may be due to the SEI layer unstability and electrolyte degradation.30 The capacity finally remains stable with negligible fading after 50 cycles. The anode can deliver a high reversible capacity of 1055 mA h g−1 at the 100th cycle, which is obviously higher than the theoretical capacity (838 mA h g−1) of neat MoO2, demonstrating the efficiently synergistic effect between ultrafine MoO2 nano-particles and HPCNFs. Furthermore, it needs to be mentioned that the coulombic efficiency reaches approximately 99% after only a few cycles and maintains relatively stable in the subsequent cycles, implying a high electrochemical reversibility and structural stability of MoO2@HPCNFs.
The rate capability of the MoO2@HPCNFs electrode is illustrated in Fig. 3d. When the current density is increased, the MoO2@HPCNFs electrode displays a gradual reduction in the capacity. The specific reversible capacities are about 1510, 1224, 968, 669 and 425 mA h g−1 at 100, 200, 500, 1000 and 2000 mA g−1, respectively. More importantly, as the current density is returned to 100 mA g−1 even after a high-rate test, the electrode still maintains stable cycling with a reversible capacity of 1060 mA h g−1, indicating the excellent rate performance and strong tolerance for the rapid Li insertion/extraction reaction.
To evaluate the morphological change in the MoO2@HPCNFs film electrode after 100 charging/discharging cycles at 100 mA g−1, SEM analysis was conducted as shown in Fig. 4. The SEM image clearly reveals that the fibrous morphology of MoO2@HPCNFs and the formed 3D porous network structure are well-preserved after cycling, which further demonstrates that the as-prepared MoO2@HPCNFs film electrode has good structural integrity during continuous lithiation and delithiation processes.
 | ||
| Fig. 4 SEM image of the MoO2@HPCNFs film electrode after 100 cycles at 100 mA g−1. | ||
Considering the specific capacity and rate capability, the MoO2@HPCNFs film electrode is clearly superior to the previously reported MoO2/CNFs prepared without using pore-forming agent and many other MoO2-based anode materials (Table 1). We believe the enhanced electrochemical performance of MoO2@HPCNFs should be mainly attribute to the 3D hierarchical structure and synergistic effect. The ultrasmall MoO2 nanoparticles can be more effectively utilized for Li-storage and can enhance the Li uptake/release kinetics due to the short diffusion pathways. Meanwhile, the abundant meso/macropores in the HPCNFs are able to offer more active sites for Li-storage, and also facilitate the electrolyte penetrate into the inner part of HPCNFs, improving the electrolyte/electrode contacting area. Moreover, the porous 3D conductive network interconnected by HPCNFs is able to provide an efficient and rapid 3D channel for the transportation of both Li ions and electrons, which would increase the electrochemical conductivity and decrease the internal resistance of the MoO2@HPCNFs electrode. In addition, the combination of hierarchical pores with flexible CNFs matrix may mitigate the pulverization/aggregation of MoO2 nanoparticles caused by volume change and preserve the structural integrity of the whole film electrode. The combined action of all the above factors leads to the high Li-storage performance of MoO2@HPCNFs film.
| Sampleref. | Current density (mA g−1) | Capacity (mA h g−1) | Cycle number |
|---|---|---|---|
| MoO2/CNFs18 | 100 | 811 | 100 |
| Carbon coated MoO2 nanoflowers31 | 90 | 784.7 | 60 |
| Hierarchical MoO2 microspheres32 | 100 | 709 | 60 |
| Carbon-coated MoO2 nanocrystals33 | 200 | 629 | 50 |
| Carbon coated MoO2 nanofibers34 | 50 | 762.7 | 100 |
| Hierarchical MoO2 monolith35 | 200 | 719.1 | 20 |
| MoO2/Mo2C/C spheres36 | 100 | 800 | 100 |
| MoO2/graphene37 | 60 | 765.3 | 40 |
| Mo2N nanolayer coated MoO2 hollow nanostructures38 | 100 | 815 | 100 |
| Meso-MoO2/rGO39 | 100 | 801 | 50 |
| Present work | 100 | 1055 | 100 |
4. Conclusions
In summary, a novel binder-free anode made of flexible self-supporting MoO2@HPCNFs film has been prepared by the combination of electrospinning with controlled reduction–carbonization process, in which uniform ultrasmall MoO2 nanoparticles (∼3.5 nm) and plentiful meso/macropores are simultaneously formed and homogenously distributed inside the HPCNFs. Such unique structure endows the composite with a relatively higher reversible capacity and improved rate capability compared to many other MoO2-based anode materials. The MoO2@HPCNFs electrode can deliver a high reversible capacity of 1055 mA h g−1 after 100 cycles at 100 mA g−1, and it still remains 425 mA h g−1 even at a high current rate of 2000 mA g−1. The enhanced electrochemical performance for the MoO2@HPCNFs film is the end product of the synergistic effect of well-distributed ultrafine MoO2 nanoparticles and flexible porous 3D conductive network interlinked by HPCNFs, making it a promising anode candidate for LIBs worthy of in-depth study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of Jiangsu Province, China (Grant No. BK20171307) and the Postgraduate Research & Practice Innovation Program of Jiangsu Province (Grant No. SJKY19-2634).Notes and references
- T. Kim, W. T. Song, D. Y. Son, L. K. Ono and Y. B. Qi, J. Mater. Chem. A, 2019, 7, 2942 RSC.
- B. Huang, Z. F. Pan, X. Y. Su and L. An, J. Power Sources, 2018, 399, 274 CrossRef CAS.
- X. Y. Deng, X. F. Chen, Y. Huang, B. B. Xiao and H. Y. Du, J. Phys. Chem. C, 2019, 123, 4721 CrossRef CAS.
- Y. Yan, F. H. Du, X. P. Shen, Z. Y. Ji, H. Zhou and G. X. Zhu, Dalton Trans., 2014, 43, 17544 RSC.
- L. M. Zhou, K. Zhang, Z. Hu, Z. L. Tao, L. Q. Mai, Y. M. Kang, S. L. Chou and J. Chen, Adv. Energy Mater., 2018, 8, 1701415 CrossRef.
- M. B. Zheng, H. Tang, L. L. Li, Q. Hu, L. Zhang, H. G. Xue and H. Pang, Adv. Sci., 2018, 5, 1700592 CrossRef.
- X. C. Pan, S. T. Li, Z. M. Wang, L. Y. Yang, K. J. Zhu, L. Ren, M. Lei and J. Liu, Mater. Lett., 2017, 199, 139 CrossRef CAS.
- H. J. Zhang, K. X. Wang, X. Y. Wu, Y. M. Jiang, Y. B. Zhai, C. Wang, X. Wei and J. S. Chen, Adv. Funct. Mater., 2014, 24, 3399 CrossRef CAS.
- L. C. Yang, W. Sun, Z. W. Zhong, J. W. Liu, Q. S. Gao, R. Z. Hu and M. Zhu, J. Power Sources, 2016, 306, 78 CrossRef CAS.
- X. Wang, Y. Xiao, J. Q. Wang, L. N. Sun and M. H. Cao, J. Power Sources, 2015, 274, 142 CrossRef CAS.
- X. X. Liu, H. H. Xu, Y. H. Huang and X. L. Hu, Phys. Chem. Chem. Phys., 2016, 18, 19832 RSC.
- Y. L. Chen, Y. Hu, Z. Shen, R. Z. Chen, X. He, X. W. Zhang, Y. Q. Li and K. S. Wu, J. Power Sources, 2017, 342, 467 CrossRef CAS.
- T. T. Yu, H. L. Liu, M. Huang, J. H. Zhang, D. Q. Su, Z. H. Tang, J. F. Xie, Y. J. Liu, A. H. Yuan and Q. H. Kong, RSC Adv., 2017, 7, 51807 RSC.
- W. H. Li, M. S. Li, K. R. Adair, X. L. Sun and Y. Yu, J. Mater. Chem. A, 2017, 5, 13882 RSC.
- X. Y. Qian, L. Jin, S. W. Wang, S. S. Yao, D. W. Rao, X. Q. Shen, X. M. Xi and J. Xiang, RSC Adv., 2016, 6, 94629 RSC.
- L. Zungia, V. Agubra, D. Flores, H. Campos, J. Villareal and M. Alcoutlabi, J. Alloys Compd., 2016, 686, 733 CrossRef.
- G. L. Xia, L. J. Zhang, F. Fang, D. L. Sun, Z. P. Guo, H. K. Liu and X. B. Yu, Adv. Funct. Mater., 2016, 26, 6188 CrossRef CAS.
- Y. J. Chen, X. T. Yuan, C. Yang, Y. B. Lian, A. A. Razzaq, R. Shah, J. Guo, X. H. Zhao, Y. Peng and Z. Deng, J. Alloys Compd., 2019, 777, 127 CrossRef CAS.
- X. Liu, Y. Liu, X. D. Yan, J. L. Lan, Y. H. Yu and X. P. Yang, Mater. Chem. Front., 2019, 9, 120 RSC.
- J. Xiang, Z. P. Wu, X. K. Zhang and S. S. Yao, Mater. Res. Bull., 2018, 100, 254 CrossRef CAS.
- P. Fan, T. S. Mu, S. F. Lou, X. Q. Cheng, Y. Z. Gao, C. Y. Du, P. J. Zuo, Y. L. Ma and G. P. Yin, Electrochim. Acta, 2019, 306, 590 CrossRef CAS.
- Z. W. Xu, L. Y. Wang, W. Wang, N. Li, C. Chen, C. Y. Li, C. Y. Yang, H. J. Fu and L. Y. Kuang, Electrochim. Acta, 2016, 222, 385 CrossRef CAS.
- A. Bhaskar, M. Deepa and T. N. Rao, ACS Appl. Mater. Interfaces, 2013, 5, 2555 CrossRef CAS.
- F. L. Yan, X. Tang, Y. H. Wei, L. B. Chen, G. Z. Cao, M. Zhang and T. H. Wang, J. Mater. Chem. A, 2015, 3, 12672 RSC.
- P. T. Xie, H. Y. Li, B. He, F. Dang, J. Lin, R. H. Fan, C. X. Hou, H. Liu, J. X. Zhang, Y. Ma and Z. H. Guo, J. Mater. Chem. C, 2018, 6, 8812 RSC.
- Q. L. Wei, F. Y. Xiong, S. S. Tan, L. Huang, E. H. Lan, B. Dunn and L. Q. Mai, Adv. Mater., 2017, 29, 1602300 CrossRef.
- Y. C. Liu, N. Zhang, C. M. Yu, L. F. Jiao and J. Chen, Nano Lett., 2016, 16, 3321 CrossRef CAS.
- Z. Chen, T. Yang, H. M. Shi, T. H. Wang, M. Zhang and G. Z. Cao, Adv. Mater. Interfaces, 2017, 4, 1600816 CrossRef.
- Y. M. Sun, X. L. Hu, W. Luo and Y. H. Huang, ACS Nano, 2011, 5, 7100 CrossRef CAS.
- Y. H. Dou, J. T. Xu, B. Y. Ruan, Q. N. Liu, Y. D. Pan, Z. Q. Sun and S. X. Dou, Adv. Energy Mater., 2016, 6, 1501835 CrossRef.
- S. S. Tang, L. Y. Yang, J. Liu and D. Fichou, Mater. Res. Bull., 2018, 102, 277 CrossRef CAS.
- W. K. Zhai, Y. M. Xu, X. L. Cheng, S. Gao, X. F. Zhang, H. Zhao and L. H. Huo, Mater. Lett., 2015, 145, 287 CrossRef CAS.
- L. Zhou, H. B. Wu, Z. Y. Wang and X. W. Lou, ACS Appl. Mater. Interfaces, 2011, 3, 4853 CrossRef CAS.
- W. Luo, X. L. Hu, Y. M. Sun and Y. H. Huang, Phys. Chem. Chem. Phys., 2011, 13, 16735 RSC.
- Y. M. Sun, X. L. Hu, J. C. Yu, Q. Li, W. Luo, L. X. Yuan, W. X Zhang and Y. H. Huang, Energy Environ. Sci., 2011, 4, 2870 RSC.
- M. Ihsan, H. Q. Wang, S. R. Majid, J. P. Yang, S. J. Kennedy, Z. P. Guo and H. K. Liu, Carbon, 2016, 96, 1200 CrossRef CAS.
- Q. Yang, Q. Liang, J. Liu, S. Q. Liang, S. S. Tang, P. J. Lu and Y. K. Lu, Mater. Lett., 2014, 127, 32 CrossRef CAS.
- J. Liu, S. S. Tang, Y. K. Lu, G. M. Cai, S. Q. Liang, W. J. Wang and X. L. Chen, Energy Environ. Sci., 2013, 6, 2691 RSC.
- S. S. Wang, B. C. Liu, G. L. Zhi, G. R. Xu, Q. Wang and J. Zhang, Microporous Mesoporous Mater., 2017, 246, 14 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |