DOI:
10.1039/C9RA07485F
(Paper)
RSC Adv., 2019,
9, 37162-37170
Synthesis of Bi2O3/g-C3N4 for enhanced photocatalytic CO2 reduction with a Z-scheme mechanism†
Received
17th September 2019
, Accepted 8th November 2019
First published on 13th November 2019
Abstract
Bi2O3/g-C3N4 nanoscale composites with a Z-scheme mechanism were successfully synthesized by high temperature calcination combined with a hydrothermal method. These synthesized composites exhibited excellent photocatalytic performance, especially the 40 wt% Bi2O3/g-C3N4 composite, which produced about 1.8 times the CO yield of pure g-C3N4. The obtained products were characterized by X-ray diffraction (XRD) patterns, X-ray photoelectron spectroscopy (XPS), scanning electron microscope (SEM), transmission electron microscopy (TEM), Brunauer–Emmett–Teller (BET), UV-vis diffuse reflectance spectroscopy (UV-vis DRS) and so on. Characterization results revealed that Bi ions had well covered the surface of g-C3N4, thus restraining the recombination of electron–hole pairs and resulting in a stronger visible-light response and higher CO yield. In addition, the electron transfer process through the Z-scheme mechanism also promoted the photocatalytic activity.
1. Introduction
In recent years, the continuous development of industry and the serious destruction of forest vegetation have led to difficulties in controlling CO2 emission, thus seriously threatening the survival and development of mankind.1–6 Recently, the field of CO2 photocatalytic reduction which translates CO2 into some useful substances has attracted many research teams.7–12 As we all know, many narrow band gap semiconductor based photocatalysts have been used in CO2 photocatalytic reduction.13–15 Among them, g-C3N4 is the most widely used one due to its suitable band gap, simple and convenient preparation and good visible light response.16,17 However, there are still many obvious shortcomings associated with this catalyst, such as its small range of light response, low separation rate of electron–hole pairs and unsatisfactory performance in the photocatalytic reaction. Lots of methods have been developed to improve the photocatalytic performance of g-C3N4, such as element modification,18–20 texture fabrication21,22 and heterojunction formation.23–27 Fabrication of a heterojunction between two semiconductors is a promising way among these methods, in which the formation of an internal electric field and the promoted separation of charge carriers effectively restrains the recombination rate of electron–hole pairs.28
As an important semiconductor material, Bi2O3 has been applied in many fields like electronic ceramics, optoelectronic devices, high-temperature superconductors, catalysts, and sensors due to its special physical properties and crystal structure.29–33 What's more, Bi2O3 also has been used as a normal photocatalyst in water splitting and photodegradation pollutant. Unfortunately, its photoreduction performance is poor owing to the low migration of photo-charges, which leads to a fast combination of photogenerated electron–hole pairs.34–37 For the improvement of photocatalytic ability over Bi2O3, the construction of heterojunction is considered as an effective measure to reduce the probability of electron–hole pairs recombination, like BiOCl/Bi2O3,38 BiVO4/Bi2O3 (ref. 39) and Bi2O3/TiO2.40 It is known that an internal electric field is produced after the formation of a p–n heterojunction. The photocarriers are greatly accelerated by the electric field, which effectively suppresses the reflux of photogenerated carriers and thus improves the photocatalytic performance.41 Due to the presence of heterojunctions, the excitation wavelength of light is also extended at the same time.42
From the above analysis, it can be presumed that there exists a possibility to construct a heterostructure between Bi2O3 and g-C3N4 through their mutual activation. However, the use of Bi2O3/g-C3N4 heterojunctions for CO2 photoreduction has not been reported. Therefore, Bi2O3/g-C3N4 compounds with different Bi2O3 contents were prepared and used in CO2 photocatalytic reduction in this study. It was found that the yields of Bi2O3/g-C3N4 compounds was higher than single Bi2O3 or g-C3N4. Moreover, the visible light response is also enhanced. Based on the results of all the characterization techniques, the possible promotion mechanism is also raised.
2. Experimental
2.1. Chemicals
Urea (H2NCONH3) and bismuth nitrate pentahydrate (Bi(NO3)3·5H2O) were purchased from Sinopharm Chemical Reagent Corp, P. R. China. All of these purchased reagents are of analytical grade and used directly.
2.2. Synthesis
Pure g-C3N4 was obtained by heating urea in air at 550 °C for 2 h. The Bi2O3/g-C3N4 composites with different Bi2O3 mass ratio (0, 20, 40, 60 and 80 wt%) were gained through the following procedure: g-C3N4 (0.5 g) was dissolved in 60 mL of ethylene glycol and sonicated for 30 min to yield the g-C3N4 sheet. Subsequently, a certain amount (0.104, 0.208, 0.312, 0.416 g) of Bi (NO3)3·5H2O and urea (1 g) were added in the solution and stirred for 1 h. The solution was then transfer to Teflon-lined autoclave (100 mL) and hydrothermally treated at 180 °C for 12 h. The obtained product was rinsed several times with deionized water and dried overnight at 80 °C. The hydrothermal product was then calcined in air at 380 °C for 2 h to get the final product. Pure Bi2O3 was obtained by the same procedure without the adjunction of g-C3N4.
2.3. Characterization
The crystal phase of catalyst was determined by X-ray diffraction (XRD, Bruker D8, Cu Kα radiation), and the morphology of the synthesized samples were investigated by scanning electron microscope (SEM, Phillips XL-30 FEG/NEW) and transmission electron microscope (TEM, Phillips Model CM200). The chemical elements of the compositions were analyzed by X-ray photoelectron spectroscopy (XPS, ESCALAB 250xi, USA) and Al Kα radiation sources. The Brunauer–Emmett–Teller (BET, Quantachrome Autosorb-iQ-AG instrument) pore structure and surface area were measured by N2 adsorption–desorption at −196 °C. UV-vis diffuse reflectance spectrum was analyzed in the range of 250–800 nm on a spectrophotometer (SHIMADZU UV-3600, Japan) using BaSO4 as the reflectance standard material. Photoluminescence (PL) was measured by a fluorescence spectrophotometer (Hitachi F-4600, 325 nm excitation wavelength).
2.4. Photoelectrochemical
Photoelectrochemical included electrochemical impedance spectroscopy (EIS) and transient photocurrent responses analysis were carried out on an electrochemical instrument (CHI 660E). A standard three-electrode system was immersed in a Na2SO4 electrolyte solution (0.5 M). The Pt and Ag/AgCl electrodes were used as counter and reference electrodes, respectively. FTO conductive glass covered by the synthesized sample was used as working electrode, and it was gained by the following method: mixture solution was consisted of Nafion (20 μL, 5%) and ethanol (1 mL). After that, 10 mg of the synthesized sample was dropped into the mixed solution and ultrasonically dispersed (2 h), then drop the slurry (0.1 mL) on 1 × 1 cm FTO glass. Therefore, the sample was well attached to the surface of the glass piece after evaporation of the ethanol. In addition, photocurrent and EIS measurements were performed under the illumination of the simulated solar light.
2.5. Photoactivity
The CO2 photoreduction experiment was performed with a reactor (500 mL) in a gas-enclosed circulation system. During the reaction, a Xenon lamp (300 W) was used as the light source. The experimental procedure was designed as follows: 50 mg sample was dispersed in 100 mL deionized water, then magnetic stirring was performed at an appropriate rotation speed. The reactor was vacuum treated and 100 kPa of high purity CO2 was passed into the reactor under the throttling of airflow. Then this pressure was kept for 30 min to obtain the balance of adsorption–desorption. Throughout the experiment, the temperature was maintained at 25 °C. The gas (0.15 mL) in reactor was obtained and analyzed by a gas chromatography (GC-2010 Plus, SHIMADZU, Japan) in the course of the reaction.
3. Results and discussion
3.1. SEM, XRD and TEM
The SEM patterns of g-C3N4, Bi2O3 and Bi2O3/g-C3N4 composites are exhibited in Fig. 1. Clearly, the g-C3N4 exhibits a curled layered structure (Fig. 1a), and the Bi2O3 shows a distinct rod-like structure (Fig. 1b). Fig. 1c shows the SEM image of 40 wt% Bi2O3/g-C3N4 sample and its corresponding elemental mapping images. It could be observed from Fig. 1c that 40 wt% Bi2O3/g-C3N4 composite perfectly retains the form of Bi2O3 and g-C3N4, showing a curled sheet structure. In addition, the mapping images of 40 wt% Bi2O3/g-C3N4 composite in Fig. 1c also reveals the coexistence of C, N, O and Bi elements.
 |
| | Fig. 1 SEM of g-C3N4 (a) and Bi2O3 (b); SEM and elemental mapping of C, N, O and Bi in 40 wt% Bi2O3/g-C3N4 composite (c). | |
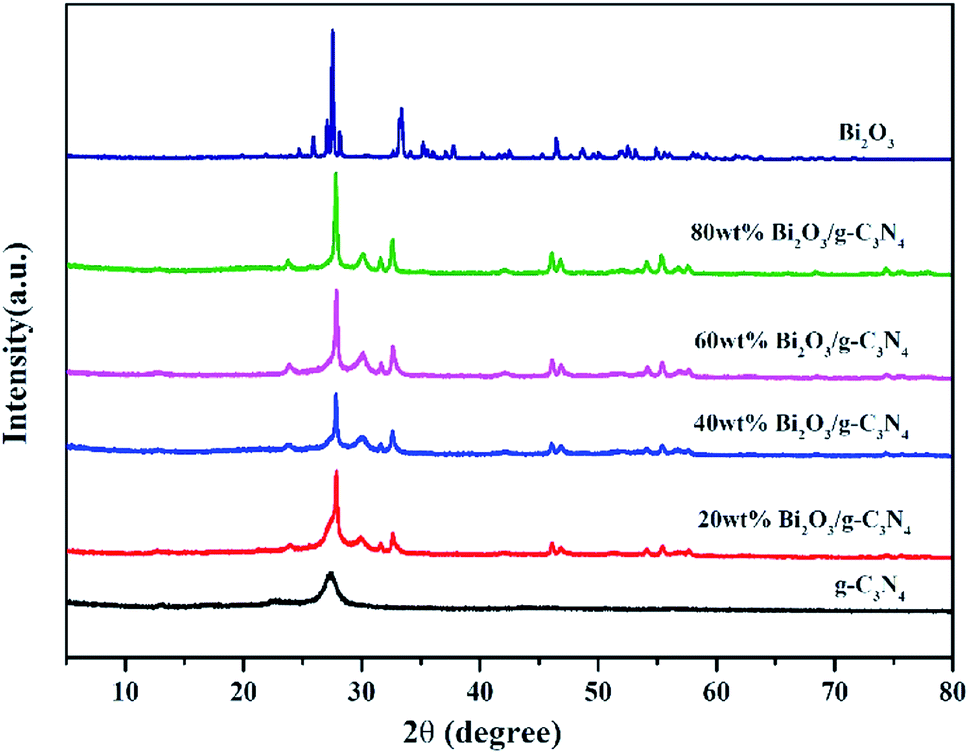
The XRD patterns of all composites are displayed in Fig. 2. In the pattern of g-C3N4, the diffraction peak observed at 27.3° could be attributed to the (002) plane of g-C3N4 (JCPDS 87-1526). The peak at 12.7° is the (100) plane of g-C3N4, corresponding to the in-plane structure packing motif of tri-s-triazine units.43 The synthesized Bi2O3 represents six major peaks at 2θ = 27.8°, 31.7°, 32.6°, 46.2°, 55.4° and 74.4°, corresponding to the (201), (002), (220), (222), (421) and (423) planes respectively, which is in consistent with the β-Bi2O3 (JCPDS 65-1209). As the content of Bi2O3 in Bi2O3/g-C3N4 samples increase, the intensity of g-C3N4 peak becomes weaker. As a contrast, the peak intensity of Bi2O3 becomes stronger, especially for the diffraction peak at 27.8°, revealing the increased crystallinity of Bi2O3.
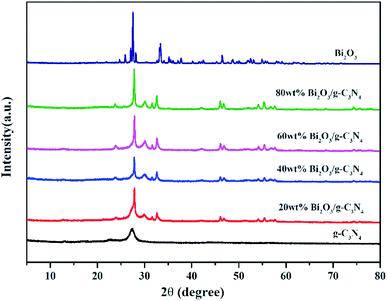 |
| | Fig. 2 XRD of g-C3N4, Bi2O3 and Bi2O3/g-C3N4 composites. | |
The TEM images of samples are demonstrated in Fig. 3. Fig. 3a and b reveal the microscale morphology of g-C3N4 and Bi2O3, which are characterized by a curled edge and a rod-like structure respectively, as also revealed by the SEM patterns. The TEM pattern of 40 wt% Bi2O3/g-C3N4 composite shows that Bi2O3 and g-C3N4 are combined together uniformly (Fig. 3c). In addition, the 0.318 nm interplanar spacing corresponding to the (201) crystal plane of the cubic Bi2O3 is visible in the high resolution TEM image (Fig. 3d).44 The results show that g-C3N4 and the rod-like Bi2O3 have deeply combined together in 40 wt% Bi2O3/g-C3N4 composite.
 |
| | Fig. 3 TEM image of g-C3N4 (a), Bi2O3 (b) and 40 wt% Bi2O3/g-C3N4 (c); HRTEM of 40 wt% Bi2O3/g-C3N4 (d). | |
3.2. XPS, BET and UV
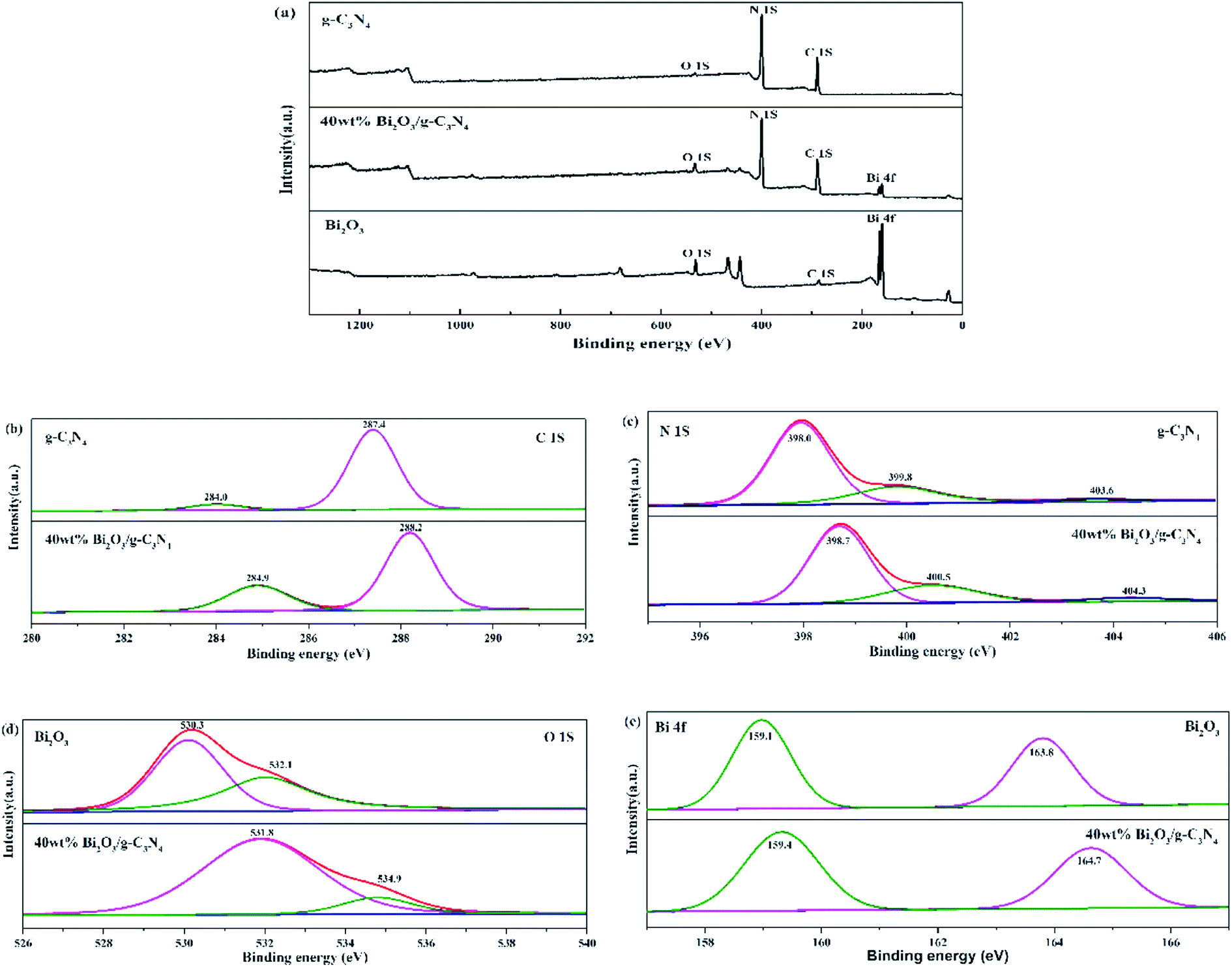
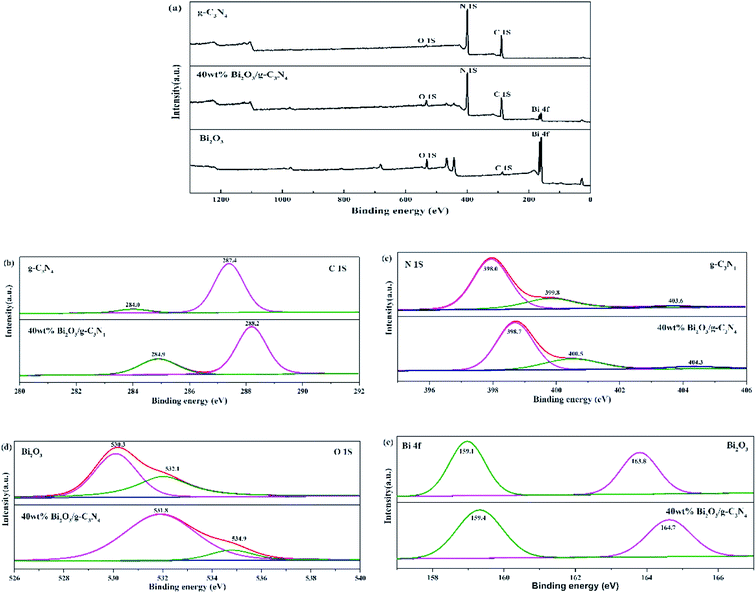
The compositions of chemical elements on g-C3N4, Bi2O3 and 40 wt% Bi2O3/g-C3N4 were analyzed by XPS. As the XPS spectra show (Fig. 4a), C 1s and N 1s signals are available in g-C3N4, Bi2O3 and 40 wt% Bi2O3/g-C3N4, while Bi 4f and O 1s peaks are detected in Bi2O3 and 40 wt% Bi2O3/g-C3N4 respectively, which means that Bi2O3 is successfully doped into g-C3N4. Carbon contained in Bi2O3 sample might be caused by extraneous carbon. The corresponding detailed spectra of C 1s, N 1s, O 1s and Bi 4f are also exhibited in Fig. 4. In the C 1s spectrum of g-C3N4, two sub-bands centered at 287.4 and 284.0 eV could be observed (Fig. 4b), corresponding to the N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group and the C–C bond, respectively.45 Fig. 4c exhibits the N 1s spectra of g-C3N4 and 40 wt% Bi2O3/g-C3N4. The dominant peak at 398.0 eV could be considered as the carbon-bonded sp2-hybrid aromatic N (CN–C),46 while the other two peaks located at 399.8 eV and 403.6 eV could be attributed to the tertiary nitrogen N–(C)3 groups and π excitations.47 The high-resolution O 1s spectrum of Bi2O3 is shown in Fig. 4d, in which the protruding peaks at 530.3 eV and 532.1 eV are derived from the Bi–O bond. Two peaks are observed in high-resolution Bi 4f spectrum of Bi2O3 (Fig. 4e): the peak of Bi 4f at 159.1 eV and 163.8 eV reveal the presence of Bi3+ in Bi2O3.48,49 The band energy shift of most peaks could be found in the spectrum of the 40 wt% Bi2O3/g-C3N4. The shift should be originated from the different electron concentrations,50 which are corresponding to the SEM, XRD and TEM results mentioned above.
N group and the C–C bond, respectively.45 Fig. 4c exhibits the N 1s spectra of g-C3N4 and 40 wt% Bi2O3/g-C3N4. The dominant peak at 398.0 eV could be considered as the carbon-bonded sp2-hybrid aromatic N (CN–C),46 while the other two peaks located at 399.8 eV and 403.6 eV could be attributed to the tertiary nitrogen N–(C)3 groups and π excitations.47 The high-resolution O 1s spectrum of Bi2O3 is shown in Fig. 4d, in which the protruding peaks at 530.3 eV and 532.1 eV are derived from the Bi–O bond. Two peaks are observed in high-resolution Bi 4f spectrum of Bi2O3 (Fig. 4e): the peak of Bi 4f at 159.1 eV and 163.8 eV reveal the presence of Bi3+ in Bi2O3.48,49 The band energy shift of most peaks could be found in the spectrum of the 40 wt% Bi2O3/g-C3N4. The shift should be originated from the different electron concentrations,50 which are corresponding to the SEM, XRD and TEM results mentioned above.
 |
| | Fig. 4 XPS of g-C3N4, Bi2O3 and 40 wt% Bi2O3/g-C3N4 (a); high-resolution C 1s (b) and N 1s (c) of g-C3N4 and 40 wt% Bi2O3/g-C3N4; high-resolution O 1s (d) and Bi 2p (e) of Bi2O3 and 40 wt% Bi2O3/g-C3N4. | |
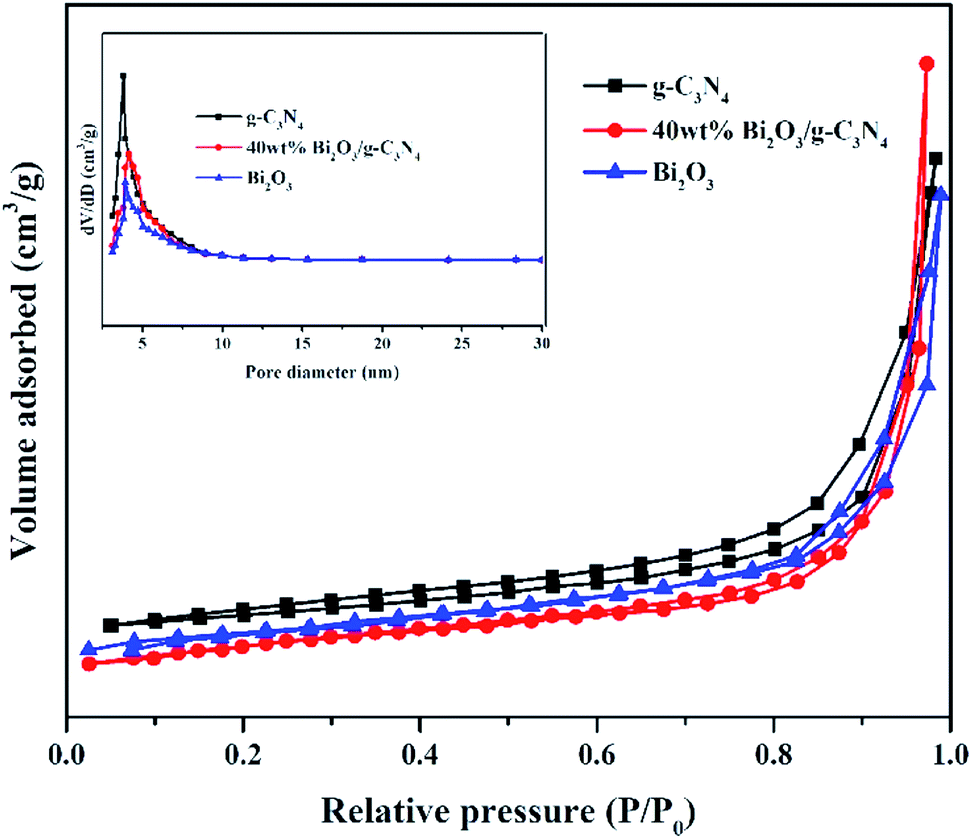
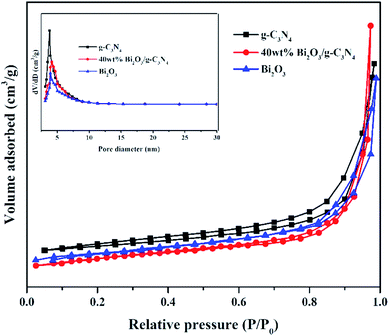
Nitrogen adsorption–desorption measurements were performed at −196 °C to analyze the textural features of the Bi2O3, g-C3N4 and 40 wt% Bi2O3/g-C3N4 composite. All samples show a type IV isotherm with a hysteresis loop (Fig. 5), revealing the presence of mesoporous structure into the composite. The specific surface area and average pore size of photocatalysts are listed in Table 1. Obviously, 40 wt% Bi2O3/g-C3N4 composite possesses the largest specific surface area (136.1 m2 g−1), while that for g-C3N4 and Bi2O3 are 66.7 m2 g−1 and 53.2 m2 g−1 respectively. Correspondingly, more active sites are available on 40 wt% Bi2O3/g-C3N4 composite. The BJH pore size distribution of g-C3N4, Bi2O3 and 40 wt% Bi2O3/g-C3N4 is shown in the inset of Fig. 5. Obviously, all the three samples exhibit a fairly narrow and limited pore size distribution between 3.6–4.1 nm.
 |
| | Fig. 5 N2 adsorption–desorption isotherms and pore size distribution curves (inset) of g-C3N4, Bi2O3 and 40 wt% Bi2O3/g-C3N4 composite. | |
Table 1 Textural properties of g-C3N4, Bi2O3 and Bi2O3/g-C3N4 composites
| Samples |
SBET (m2 g−1) |
Pore size (nm) |
| g-C3N4 |
66.7 |
3.839 |
| 20 wt% Bi2O3/g-C3N4 |
109.2 |
3.834 |
| 40 wt% Bi2O3/g-C3N4 |
136.1 |
3.821 |
| 60 wt% Bi2O3/g-C3N4 |
98.3 |
3.849 |
| 80 wt% Bi2O3/g-C3N4 |
78.2 |
3.815 |
| Bi2O3 |
53.2 |
3.856 |
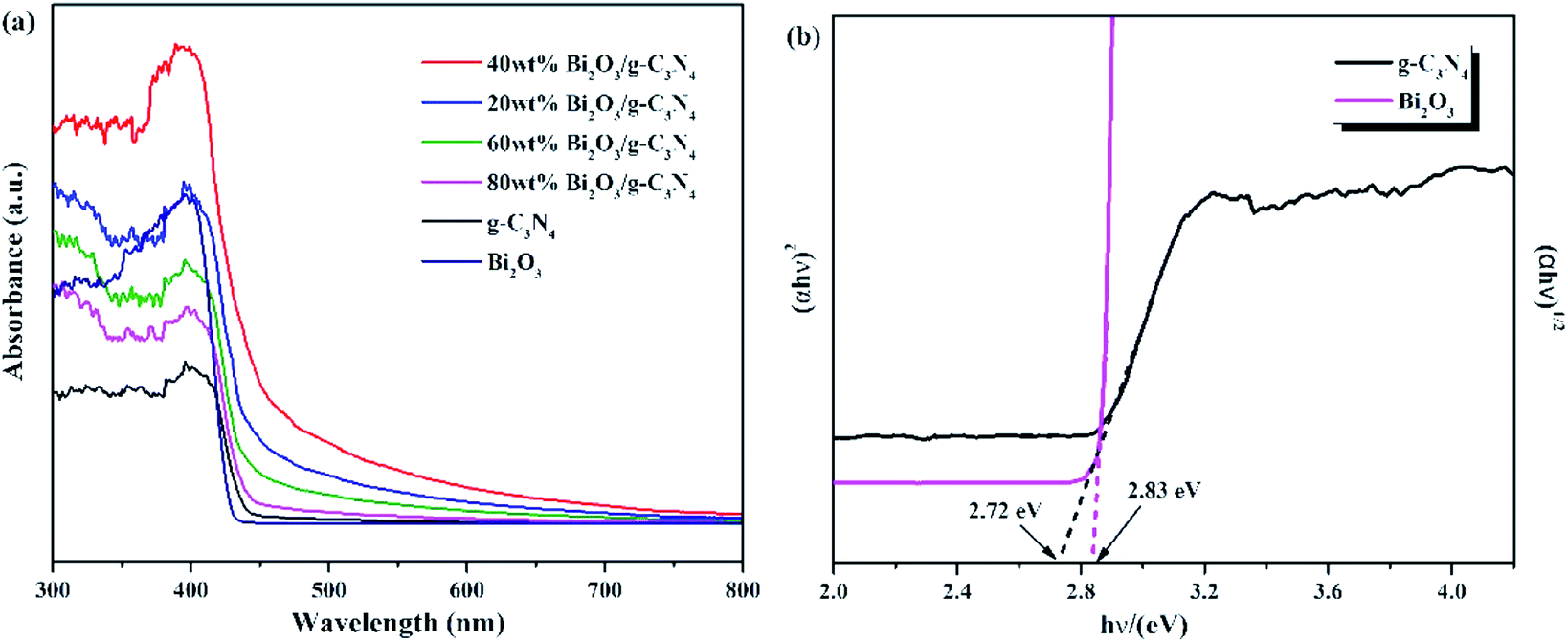
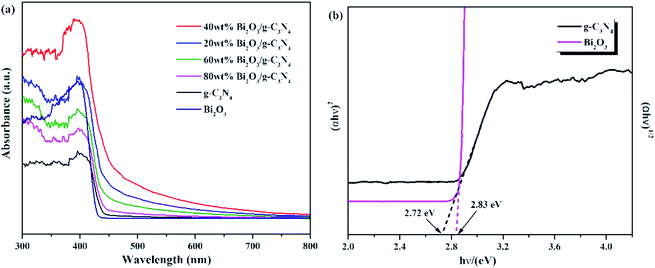
The optical properties of Bi2O3, g-C3N4, and Bi2O3/g-C3N4 composites were tested by UV-vis measurement, and the relevant data were converted from the Kubelka–Munk equation. The 450 nm absorption edge of g-C3N4 could be observed in Fig. 6a, while the Bi2O3 shows a similar absorption properties. It can be found that Bi2O3/g-C3N4 samples could obtain more photons during the reaction according to the vertical coordinates, which is good to the photoreduction of CO2. The band gap energy of Bi2O3 and g-C3N4 could be figured out by the formula: αhv = A (hν − Eg)n/2, where α, h, ν, A and Eg are the absorption coefficient, Planck constant, light frequency, band gap energy and constant related to the catalyst. Among them, n relies on optical transition type of the semiconductor (n = 4 for indirect transition and n = 1 for direct transition). For Bi2O3 and g-C3N4, the values of n are 1 and 4, respectively.51–53 According to Fig. 6b, the Eg values of Bi2O3 and g-C3N4 are counted to be 2.83 and 2.72 eV respectively.
 |
| | Fig. 6 (a) UV-vis DRS for g-C3N4, Bi2O3 and Bi2O3/g-C3N4 composites. (b) (αhν)1/2 and (αhν)2 versus energy (hν) for the band gap energies of g-C3N4 and Bi2O3, respectively. | |
3.3. Photocatalytic activity, PL and photoelectrochemical
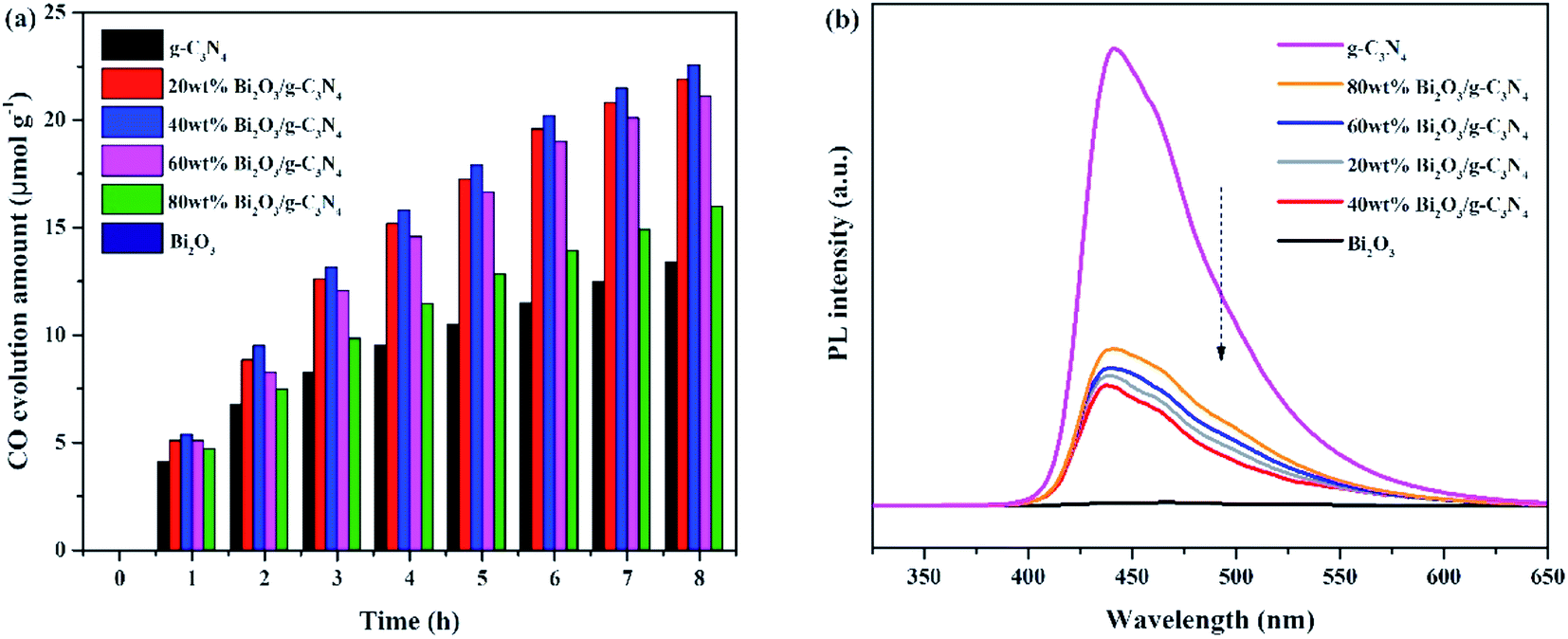
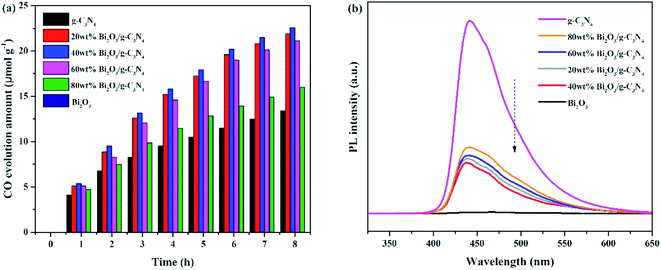
The photocatalytic activities of all samples are tested and CO was found as the main product. In addition, the control experiments were also conducted. No hydrocarbon products are tested without simulated solar light or sample, revealing that the conditions mentioned above are essential for the CO2 photocatalytic reduction. Fig. 7a demonstrated the amount of CO production over the photocatalysts during the light irradiation. It could be clearly seen that the pure Bi2O3 sample has not produced any CO, which means that pure Bi2O3 cannot reduce CO2 to CO under photocatalytic conditions, due to the fact that the CB edge of Bi2O3 is lower than the reduction potential of CO2/CO (−0.52 V vs. NHE).54 The yield of g-C3N4 is also not high, indicating that the photocatalytic performance of pure catalyst is not very well, which can be ascribed to the fast recombination rate of electron–hole pairs in reaction. A great enhancement of CO yield is detected in terms of the Bi2O3/g-C3N4 composites. Moreover, the photocatalytic activity enhances as the increasing Bi2O3 content in the composites, which is owing to the increased quantity of the heterojunctions. During the reaction, the highest CO yield (22.5 μmol g−1) is achieved on 40 wt% Bi2O3/g-C3N4, which is about 1.8 times the CO yield of g-C3N4. However, the photocatalytic performance would decline along with the excessive addition of Bi2O3, which might be aroused by the reduce of the specific surface area, and the excessive Bi2O3 might act as the recombination core of electron–hole pairs during the reaction.55,56 The photocatalytic stability of all composites and g-C3N4 are shown in the Fig. S1,† the yield of CO keeps stable during three experimental runs, revealing that the performance of all composites and g-C3N4 are stable during the reaction. To further probe the truth, the separation efficiency was characterized by PL. As shown in Fig. 7b, the emission peak of Bi2O3 is almost invisible in the 450–500 nm range, which may be due to a trace of surface defects or surface oxygen vacancies of Bi2O3. Compared with Bi2O3, a strong peak concentrated at 450 nm could be noticed in the g-C3N4 spectrum, which reflects the fast recombination rate of electrons–hole pairs.57 Different from g-C3N4, the Bi2O3/g-C3N4 composites exhibit a moderate peak intensity in the pattern, suggesting that heterojunctions are built among Bi2O3 and g-C3N4, so the holes and electrons are effectively separated. Therefore, the recombination rate is greatly reduced, which is good to enhance the activity of CO2 photoreduction. In addition, the 40 wt% Bi2O3/g-C3N4 has the lowest peak intensity among all the synthesized Bi2O3/g-C3N4 samples.
 |
| | Fig. 7 (a) Yield of CO and (b) PL spectra of all samples. | |
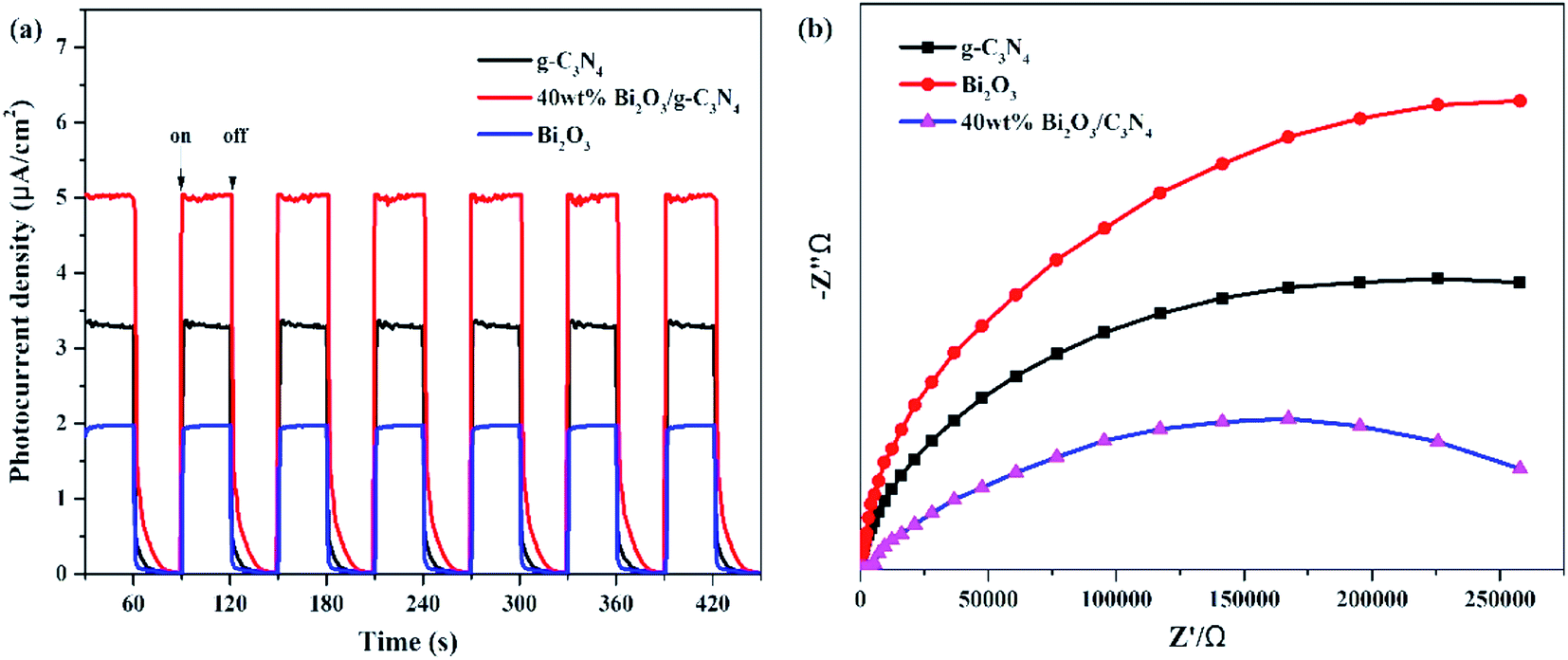
In addition to PL measurements, photo-electrochemical measurements are used to further analyze the properties of photoelectrons in Bi2O3, g-C3N4 and 40 wt% Bi2O3/g-C3N4. Fig. 8a shows the photocurrent response of the samples under visible light absorption. Transient photocurrent measurements could show more evidence for the rapid electron transfer efficiency in the photocatalyst. In the light-on and light-off tests, all the photocatalysts show rapid and intense photocurrent response, indicating that the photocatalytic activity is relatively stable. Both Bi2O3 and g-C3N4 exhibit a rapidly increasing current when light is on and an abruptly decreasing signal when the light is off. This transient response behavior might be resulted from the capture and release of electrons caused by surface defects.58 In addition, the photocurrent intensity of 40 wt% Bi2O3/g-C3N4 heterostructured composite is 2.5 and 1.5 times the intensity of Bi2O3 and g-C3N4, respectively, indicating that 40 wt% Bi2O3/g-C3N4 has higher charge separation efficiency and better photocatalytic properties than pure catalysts. To confirm this inference, the EIS experiment was also performed. The semicircular Nynquist plots of the samples are presented in Fig. 8b. Obviously, a smaller semicircle diameter could be detected on the plot of 40 wt% Bi2O3/g-C3N4 than that of pure Bi2O3 and pure g-C3N4, indicating that the electron transfer rate is faster in 40 wt% Bi2O3/g-C3N4,59 which is corresponding to the previous photocatalytic activity results.
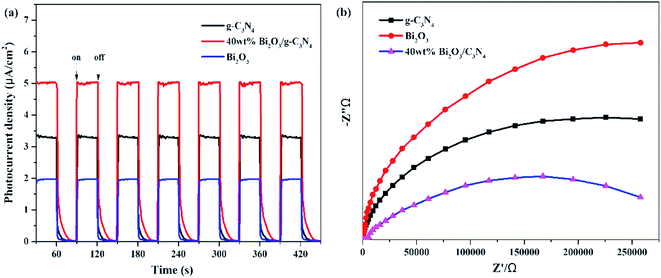 |
| | Fig. 8 (a) Transient photocurrent density and (b) electrochemical impedance spectra of Bi2O3, g-C3N4 and 40 wt% Bi2O3/g-C3N4 composite. | |
3.4. Mechanism
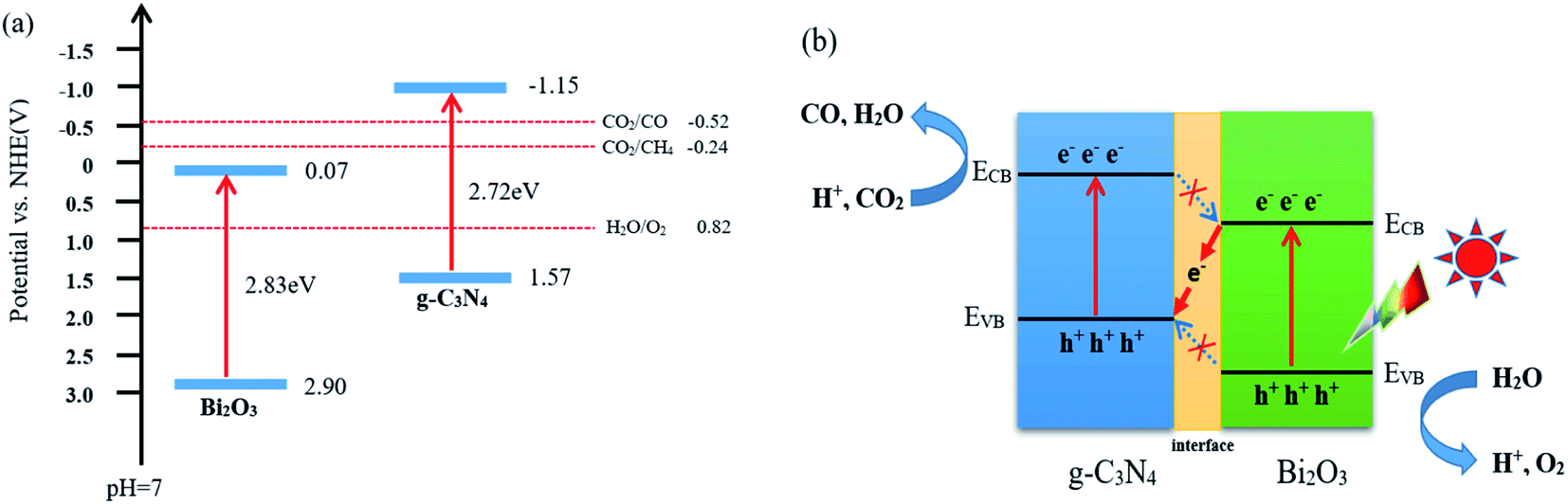
According to the characterization and CO2 photocatalytic reduction test results of the prepared samples, the reaction mechanism of CO2 reduction over Bi2O3/g-C3N4 composites were proposed. As shown in Fig. 9a, the valence band (VB) and conduction band (CB) of a semiconductor could be determined using the following equation:60 E0CB = χ − EC − 1/2Eg. Where χ is the absolute electronegativity of the semiconductor (χ for Bi2O3 is 5.986 eV), EC is the energy of free electrons in the hydrogen size (4.5 eV) and Eg is the band gap of the semiconductor.61,62 Combined with the results in Fig. 6b, the conduction band (CB) and valence band (VB) values for the pure Bi2O3 are 0.07 eV and 2.90 eV, respectively. For g-C3N4, the VB is −1.57 eV, and the CB is 1.15 eV.63,64 The charge transfer in the interface can follow a double-transfer mode or a Z-scheme transfer mechanism. Double-transfer means that the CO2 reduction reaction would happen on the CB of Bi2O3, and the H2O oxidation reaction would occur on the VB of g-C3N4. In fact, the CB of Bi2O3 is lower than the reduction potential level of CO2/CO (−0.52 V vs. NHE), thus the reduction of CO2 would not proceed and CO would not be generated, which means that double-transfer mechanism is not suitable here to explain. Z-scheme transfer mechanism is different from the double-transfer mechanism, as shown in Fig. 9b, the photo-induced electrons on the surface of Bi2O3 can easily be transferred to g-C3N4 through the interface, the holes on VB of Bi2O3 are captured by H2O molecules to produce O2 and protons. At the same time, the CO2 molecules react with the electrons on the CB of g-C3N4 to generate CO and H2O with the participation of protons. Moreover, the formation of other products including CH4, H2, and O2 were also observed in the experimental process, and the charge balance table is shown in Table 2. The enhanced activity of 40 wt% Bi2O3/g-C3N4 in CO2 reduction is mainly due to the Z-scheme heterostructure, which effectively delays the rapid recombination of the electron–hole pairs in Bi2O3 and g-C3N4, therefore improving the photocatalytic activity.
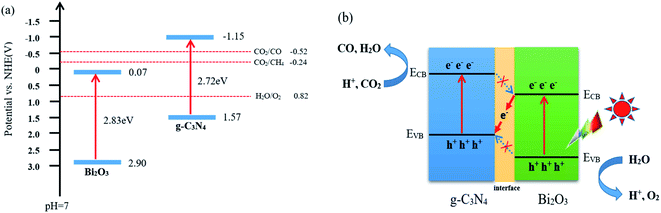 |
| | Fig. 9 (a) Band gap energy and band positions of Bi2O3 and g-C3N4 together with CO2/CO, CO2/CH4 and H2O/O2 redox potentials at pH = 7 and (b) Z-type mechanism of CO2 photoreduction route on Bi2O3/g-C3N4 composite. | |
Table 2 The production rate of redox products for g-C3N4 and 40 wt% Bi2O3/g-C3N4
| Sample |
Reduction products (μmol g−1 h−1) |
Oxidation products (μmol g−1 h−1) |
Ratio |
| CO |
CH4 |
H2 |
O2 |
e−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :h+ :h+ |
| g-C3N4 |
1.68 |
0.38 |
1.11 |
2.42 |
0.89:1 |
| 40 wt% Bi2O3/g-C3N4 |
2.81 |
1.34 |
1.38 |
6.95 |
0.88:1 |
4. Conclusions
In general, Bi2O3/g-C3N4 nanoscale composites with heterojunction was successfully synthesized by the combination of high temperature calcination and hydrothermal method. The 40 wt% Bi2O3/g-C3N4 composite has the highest CO yield in the CO2 photocatalytic reduction, which is 1.8 times the yield of that g-C3N4. This obvious improvement in photoactivity is mainly due to the effective separation of electron–hole pairs and successive charge transfer through the interface. Moreover, the enhancement of specific surface area and visible light response also upgrade the photocatalytic performance of Bi2O3/g-C3N4 composite. This study gives some valuable opinion for the research of g-C3N4-related photocatalysts.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by the National Key R&D Program of China (2018YFB0605002).
References
- S. Zhou, Y. Liu, J. M. Li, Y. J. Wang, G. Y. Jiang, Z. Zhao, D. X. Wang, A. J. Duan, J. Liu and Y. C. Wei, Appl. Catal., B, 2014, 158–159, 20–29 CrossRef CAS.
- G. G. Zhang, G. S. Li, T. Heil, S. Zafeiratos, F. L. Lai, A. Savateev, M. Antonietti and X. C. Wang, Angew. Chem., Int. Ed., 2019, 58, 3433–3437 CrossRef CAS PubMed.
- H. B. Yu, B. B. Huang, H. Wang, X. Z. Yuan, L. B. Jiang, Z. B. Wu, J. Zhang and G. M. Zeng, J. Colloid Interface Sci., 2018, 522, 82–94 CrossRef CAS PubMed.
- H. B. Yu, L. B. Jiang, H. Wang, B. B. Huang, X. Z. Yuan, J. H. Huang, J. Zhang and G. M. Zeng, Small, 2019, 15, 1901008 CrossRef PubMed.
- Y. Yang, Z. T. Zeng, G. M. Zeng, D. L. Huang, R. Xiao, C. Zhang, C. Y. Zhou, W. P. Xiong, W. J. Wang, M. Cheng, W. J. Xue, H. Guo, X. Tang and D. H. He, Appl. Catal., B, 2019, 258, 117956 CrossRef CAS.
- H. Yi, M. Jiang, D. L. Huang, G. M. Zeng, C. Lai, L. Qin, C. Y. Zhou, B. S. Li, X. G. Liu, M. Cheng, W. J. Xue, P. Xu and C. Zhang, J. Taiwan Inst. Chem. Eng., 2018, 93, 184–192 CrossRef CAS.
- J. Xu, K. K. Wang, T. Liu, Y. Peng and B. G. Xu, CrystEngComm, 2017, 19, 5001–5007 RSC.
- S. M. Lopez, V. Vaiano, M. C. Hidalgo, J. A. Navio and D. Sannino, Photochem. Photobiol. Sci., 2015, 14, 678–685 RSC.
- J. J. Wang, L. Tang, G. M. Zeng, Y. C. Deng, Y. N. Liu, L. L. Wang, Y. Y. Zhou, Z. Guo, J. J. Wang and C. Zhang, Appl. Catal., B, 2017, 209, 285–294 CrossRef CAS.
- H. F. Cheng, B. B. Huang, Y. Y. Liu, Z. Y. Wang, X. Y. Qin, X. Y. Zhang and Y. Dai, Chem. Commun., 2012, 48, 9729–9731 RSC.
- M. Sierra, E. Borges, P. Esparza, J. M. Ramos, J. M. Gil and P. M. Ramos, Sci. Technol. Adv. Mater., 2016, 17, 659–668 CrossRef CAS PubMed.
- C. Y. Liu, H. W. Huang, X. Du, T. R. Zhang, N. Tian, Y. X. Guo and Y. H. Zhang, J. Phys. Chem. C, 2015, 119, 17156–17165 CrossRef CAS.
- H. H. Yang, Y. Bai, T. Chen, X. Shi and Y. C. Zhu, Phys. E, 2016, 78, 100–104 CrossRef CAS.
- X. Y. Liu, R. T. Guo, H. Qin, Z. Y. Wang, X. Shi, W. G. Pan, J. Y. Tang, P. Y. Jia, Y. F. Miao and J. W. Gu, Colloids Surf., A, 2019, 580, 123782 CrossRef CAS.
- R. T. Guo, X. Y. Liu, H. Qin, Z. Y. Wang, X. Shi, W. G. Pan, Z. G. Fu, J. Y. Tang, P. Y. Jia, Y. F. Miao and J. W. Gu, Appl. Surf. Sci., 2020, 500, 144069 Search PubMed.
- J. C. Wang, H. C. Yao, Z. Y. Fan, L. Zhang, J. S. Wang, S. Y. Zang and Z. J. Li, ACS Appl. Mater. Interfaces, 2016, 8, 3765–3775 CrossRef CAS PubMed.
- N. Tian, H. W. Huang, Y. X. Guo, Y. He and Y. H. Zhang, Appl. Surf. Sci., 2014, 322, 249–254 CrossRef CAS.
- G. P. Gao, Y. Jiao, E. R. Waclawik and A. J. Du, J. Am. Chem. Soc., 2016, 138, 6292–6297 CrossRef CAS PubMed.
- X. F. Chen, J. S. Zhang, X. Z. Fu, M. Antonietti and X. C. Wang, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS PubMed.
- K. Wang, Q. Li, B. S. Liu, B. Cheng, W. K. Ho and J. G. Yu, Appl. Catal., B, 2015, 176–177, 44–52 CAS.
- D. D. Zheng, C. J. Huang and X. C. Wang, Nanoscale, 2015, 7, 465–470 RSC.
- X. J. Bai, J. Li, C. B. Cao and S. Hussain, Mater. Lett., 2011, 65, 1101–1104 CrossRef CAS.
- X. S. Zhou, B. Jin, L. D. Li, F. Peng, H. J. Wang, H. Yu and Y. P. Fang, J. Mater. Chem., 2012, 22, 17900–17905 RSC.
- J. L. Zhao, Z. Y. Ji, X. P. Shen, H. Zhou and L. B. Ma, Ceram. Int., 2015, 41, 5600–5606 CrossRef CAS.
- M. L. Li, L. X. Zhang, X. Q. Fan, M. Y. Wu, M. Wang, R. L. Cheng, L. L. Zhang, H. L. Yao and J. L. Shi, Appl. Catal., B, 2017, 201, 629–635 CrossRef CAS.
- M. L. Li, L. X. Zhang, M. Y. Wu, Y. Y. Du, X. Q. Fan, M. Wang, L. L. Zhang, Q. L. Kong and J. L. Shi, Nano Energy, 2016, 19, 145–155 CrossRef CAS.
- M. Wang, M. Shen, L. X. Zhang, J. J. Tian, X. X. Jin, Y. J. Zhou and J. L. Shi, Carbon, 2017, 120, 23–31 CrossRef CAS.
- S. S. Yi, X. Z. Yue, D. D. Xu, Z. P. Liu, F. Zhao, D. J. Wang and Y. H. Lin, New J. Chem., 2015, 39, 2917–2924 RSC.
- J. Lee, H. Lee and M. Kang, Mater. Lett., 2016, 178, 316–319 CrossRef CAS.
- M. Ahila, J. Dhanalakshmi, J. Selvakumari and D. Padiyan, Mater. Res. Express, 2016, 3, 105025 CrossRef.
- M. X. Ji, J. Di, Y. P. Ge, J. X. Xia and H. M. Li, Appl. Surf. Sci., 2017, 413, 372–380 CrossRef CAS.
- C. Chang, H. C. Yang, N. Gao and S. Y. Lu, J. Alloys Compd., 2018, 738, 138–144 CrossRef CAS.
- E. Diez, O. Monnereau, L. Tortet, G. Vacquier, P. Llewellin and F. Rouquerol, J. Optoelectron. Adv. Mater., 2000, 2, 552–556 CAS.
- Y. P. Li, S. L. Wu, L. Y. Huang, H. Xu, R. X. Zhang, M. L. Qu, Q. Gao and H. M. Hua, J. Phys. Chem. Solids, 2015, 76, 112–119 CrossRef CAS.
- W. J. Shan, Y. Hu, Z. G. Bai, M. M. Zheng and C. H. Wei, Appl. Catal., B, 2016, 188, 1–12 CrossRef CAS.
- J. F. Zhang, Y. F. Hu, X. L. Jiang, S. F. Chen, S. G. Meng and X. L. Fu, J. Hazard. Mater., 2014, 280, 713–722 CrossRef CAS PubMed.
- J. J. Wang, L. Tang, G. M. Zeng, Y. N. Liu, Y. Y. Zhou, Y. C. Deng, J. J. Wang and B. Peng, ACS Sustainable Chem. Eng., 2017, 5, 1062–1072 CrossRef CAS.
- S. Y. Chai, Y. J. Kim, M. H. Jung, A. K. Chakraborty, D. W. Jung and W. I. Lee, J. Catal., 2009, 262, 144–149 CrossRef CAS.
- M. L. Guan, D. K. Ma, S. W. Hu, Y. J. Chen and S. M. Huang, Inorg. Chem., 2010, 50, 800–805 CrossRef PubMed.
- J. Zhu, S. H. Wang, J. G. Wang, D. Q. Zhang and H. X. Li, Appl. Catal., B, 2011, 102, 120–125 CrossRef CAS.
- L. X. Yang, S. L. Luo, Y. Li, Y. Xiao, Q. Kang and Q. Y. Cai, Environ. Sci. Technol., 2010, 44, 7641–7646 CrossRef CAS PubMed.
- C. Y. Tsai, C. W. Liu, C. H. Fan, H. C. Hsi and T. Y. Chang, J. Phys. Chem. C, 2017, 121, 6050–6059 CrossRef CAS.
- X. H. Li, J. S. Chen, X. C. Wang, J. H. Sun and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 8074–8077 CrossRef CAS PubMed.
- L. B. Jiang, X. Z. Yuan, G. M. Zeng, J. Liang, X. H. Chen, H. B. Yu, H. Wang, Z. B. Wu, J. Zhang and T. Xiong, Appl. Catal., B, 2018, 227, 376–385 CrossRef CAS.
- Y. B. Li, H. M. Zhang, P. R. Liu, D. Wang, Y. Li and H. J. Zhao, Small, 2013, 9, 3336–3344 CAS.
- C. Z. Zhang, R. Hao, H. B. Liao and Y. L. Hou, Nano Energy, 2013, 2, 88–97 CrossRef CAS.
- S. Thaweesak, M. Lyu, P. Peerakiatkhajohn, T. Butburee, B. Luo, H. J. Chen and L. Z. Wang, Appl. Catal., B, 2017, 202, 184–190 CrossRef CAS.
- Z. H. Wu, J. Liu, Q. Y. Tian and W. Wu, ACS Sustainable Chem. Eng., 2017, 5, 5008–5017 CrossRef CAS.
- F. Dong, T. Xiong, R. Wang, Y. J. Sun and Y. K. Jiang, Dalton Trans., 2014, 43, 6631–6642 RSC.
- Y. Liu, S. Yu, Z. Y. Zhao, F. Dong, X. A. Dong and Y. Zhou, J. Phys. Chem. C, 2017, 121, 12168–12177 CrossRef CAS.
- J. R. Jin, Y. J. Wang and T. He, RSC Adv., 2015, 5, 100244–100250 RSC.
- J. J. Hu, G. Q. Xu, J. W. Wang, J. Lv, X. Y. Zhang, T. Xie, Z. X. Zheng and Y. C. Wu, Dalton Trans., 2015, 44, 5386–5395 RSC.
- J. S. Chen, S. Y. Qin, G. X. Song, T. Y. Xiang, F. Xin and X. H. Yin, Dalton Trans., 2013, 42, 15133–15138 RSC.
- D. Wu, L. Q. Ye, H. Y. Yip and P. K. Wong, Catal. Sci. Technol., 2017, 7, 265–271 RSC.
- F. Li, L. Zhang, X. Chen, Y. L. Liu, S. G. Xua and S. K. Cao, Phys. Chem. Chem. Phys., 2017, 19, 21862–21868 RSC.
- C. Xue, T. X. Zhang, S. J. Ding, J. J. Wei and G. D. Yang, ACS Appl. Mater. Interfaces, 2017, 9, 16091–16102 CrossRef CAS PubMed.
- T. Xiong, W. L. Cen, Y. X. Zhang and F. Dong, ACS Catal., 2016, 6, 2462–2472 CrossRef CAS.
- D. Jiang, W. Z. Wang, S. M. Sun, L. Zhang and Y. L. Zheng, ACS Catal., 2015, 5, 613–621 CrossRef CAS.
- D. Wu, L. Q. Ye, H. Y. Yip and P. K. Wong, Catal. Sci. Technol., 2017, 7, 265–271 RSC.
- H. Xu, Y. G. Xu, H. M. Li, J. X. Xia, J. Xiong, S. Yin, C. J. Huang and H. L. Wan, Dalton Trans., 2012, 41, 3387–3394 RSC.
- H. M. Fan, H. Y. Li, B. K. Liu, Y. C. Lu, T. F. Xie and D. J. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 4853–4857 CrossRef CAS PubMed.
- H. L. Li, Y. Gao, X. Y. Wu, P. H. Lee and K. Shih, Appl. Surf. Sci., 2017, 402, 198–207 CrossRef CAS.
- S. C. Yan, S. B. Lv, Z. S. Li and Z. G. Zou, Dalton Trans., 2010, 39, 1488–1491 RSC.
- H. Qin, R. T. Guo, X. Y. Liu, W. G. Pan, Z. Y. Wang, X. Shi, J. Y. Tang and C. Y. Huang, Dalton Trans., 2018, 47, 15155–15163 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07485f |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *bcd,
He Lin*a and
Xing-Yu Liubc
*bcd,
He Lin*a and
Xing-Yu Liubc