
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAu@Co2P core/shell nanoparticles as a nano-electrocatalyst for enhancing the oxygen evolution reaction
Xiaofang Zhang,
Aixian Shan,
Sibin Duan,
Haofei Zhao,
Rongming Wang * and
Woon-Ming Lau*
* and
Woon-Ming Lau*
Beijing Advanced Innovation Center for Materials Genome Engineering, Center for Green Innovation, Beijing Key Laboratory for Magneto-Photoelectrical Composite and Interface Science, School of Mathematics and Physics, University of Science and Technology Beijing, Beijing 100083, China. E-mail: rmwang@ustb.edu.cn; leolau@ustb.edu.cn
First published on 10th December 2019
Abstract
Core/shell nanoparticles (NPs) of Au@Co2P, each comprising a Au core with a Co2P shell, were prepared, and shown to efficiently catalyze the oxygen evolution reaction (OER). In particular, Au@Co2P has a small overpotential of 321 mV at 10 mA cm−2 in 1 M KOH aqueous solution at room temperature, which is about 95 mV less than pure Co2P. More importantly, the Tafel slope of Au@Co2P, at 57 mV dec−1, is 44 mV dec−1 lower than that of Co2P. Hence, Au@Co2P outperforms Co2P drastically in practical production when a high current density is required.
Introduction
Research on electrochemical water-splitting has been intensive, with an objective of resolving the crisis induced by our consumption and depletion of fossil fuels.1–6 In order to improve the energy efficiency of water-splitting, many electrocatalysts have been designed and applied to catalyze the two half reactions of water electrocatalysis, i.e., the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER). In this context, the kinetically sluggish OER (4OH− → 2H2O + 4e− + O2 in base) is a bottle-neck. Therefore, the search for effective catalysts for OER is particularly critical.Nanoparticles (NPs) with high surface areas are considered to be the preferred form of catalysts. Among them, core/shell NPs composed of hybrid materials at the nanometer scale show remarkable catalytic behavior compared to single-component NPs owing to the cooperative effect of electron transfer between the core and the shell.7–11 In the design of core/shell NPs as effective catalysts, the shell functions as the catalyst and its composition must meet such functionality. For OER, transition metal oxides and hydroxides have been developed as practical catalysts,12–15 to replace the expensive bench-marking catalysts derived from oxides of ruthenium and iridium.16,17 Some of them have indeed demonstrated catalytic activity superior to those ruthenium- and iridium-based catalysts, for a long operation-period at extremely high current densities.18 Recently, transition metal phosphides, particularly those of Ni and Co, have emerged as active electrocatalysts for water splitting.19–23 To extend this trend with the design philosophy of core/shell NPs for enhancing the catalytic efficiency of the bare shell materials, researchers have explored mixing transition-metal-based catalysts with carbon,24–26 and with other transition metal (including noble metal).27,28 Among these trials, the incorporation of gold in catalysts derived from transition-metal-oxides show very promising results.29–32 For example, Au@NiOx, Au@CoOx, and Au@CoFeOx core/shell NPs all exhibit enhanced OER activity in reference to the corresponding oxides without Au.33 Similarly, Au@Ni12P5 NPs all show superior OER activity in reference to Ni12P5 NPs.34 These recent progresses encourage further search for efficient and stable core/shell NPs as OER catalysts.
Herein, we have prepared core/shell NPs each comprising an Au core and a Co2P shell, and examined their OER electrocatalytic performance. The mechanism of the enhancement effect of the Au core over the catalytic performance of Co2P, in the framework of electron transfer from the Co2P shell to the Au core, is elaborated.
Experimental section
Reagents and instrument
Gold(III) chloride hydrate [HAuCl4·4H2O, 99%], cobalt(II) acetylacetonate [Co(acac)2, 97%], and triphenylphosphine (TPP, 97%) were purchased from Sigma-Aldrich. Anhydrous ruthenium(IV) oxide [RuO2, 99.9%] was purchased from Alfa Aesar. Oleylamine [OAm, 70%] and Co2P (98%) were purchased from Aladdin. All chemicals were used without further purification.The crystal structures, morphologies, and chemical compositions of the prepared Au@Co2P NPs were studied using X-ray diffraction (XRD, X'Pert Pro MPD system, Cu-Kα) and transmission electron microscopy (TEM, Titan ETEM G2 with a 300 kV field emission gun). The elemental composition distributions were measured by the TEM with energy dispersive X-ray spectroscopy (EDS, Oxford 80T). X-ray photoelectron spectroscopy (XPS) was performed with a PHI 5000 VersaProbe spectrometer.
Synthesis of Au@Co2P NPS
The Au@Co2P core/shell NPs were synthesized via a method based on our previous work.34,35 Firstly, Co(acac)2 (0.5 g) and oleylamine (OAm, 10 mL) were added into a three-necked flask, and mixed at 100 °C for 60 min. Secondly, the prepared solution of HAuCl4·4H2O (10 mg mL−1 in toluene) was maintained at 100 °C for 120 min under the protection of argon (40 mL min−1). Thirdly, triphenylphosphine (TPP) (1.0 g) was added into the above solution and the mixture was stirred for 30 min. Then the solution was heated to 300 °C for 120 min. Finally, the prepared Au@Co2P core/shell NPs were collected, cleaned, and stored as a colloid in toluene.Electrode preparation
The as-synthesized NPs were firstly dispersed in a Nafion solution (ethanol/H2O/Nafion = 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :199:1) to a concentration of 2 mg mL−1, as a catalyst ink. Then 20 μL of the catalyst ink was drop-casted onto a rotating disc electrode (RDE) with a diameter of 5 mm (the carbon-supported catalyst loading mass was 0.20 mg cm−2).
:199:1) to a concentration of 2 mg mL−1, as a catalyst ink. Then 20 μL of the catalyst ink was drop-casted onto a rotating disc electrode (RDE) with a diameter of 5 mm (the carbon-supported catalyst loading mass was 0.20 mg cm−2).
Electrochemical measurements
All the electrochemical measurements were conducted in a typical three-electrode setup with an Hg/HgO (in 1 M KOH solution) electrode as the reference electrode, and a Pt foil electrode (1 cm2) as the counter electrode. The potentials measured were then calibrated against the reversible hydrogen electrode (RHE). Linear sweep voltammetry (0.05 to −0.8 V) with a scan rate of 2 mV s−1 in 1 M KOH was performed and the data were used to deduce the catalyst-performance for OER.Results and discussion
Fabrication and growth process of Au@Co2P NPs
The mechanism in synthesizing the Au@Co2P NPs in this work is basically a seed-mediated growth, and the reaction processes are illustrated in Fig. 1a. Briefly, Au NPs are firstly formed by reducing HAuCl4 in an OAm solution to yield Au-nuclei and by using TPP as a surfactant to curb any excessive growth of these Au-nuclei (Fig. 1b). Subsequently, Co ions are reduced and metallic Co is deposited onto the Au-nuclei for the formation of Au@Co NPs (Fig. 1c). The gradual phosphating of Au@Co NPs to yield Au@Co2P NPs is traced by the changes in the selected area electron diffraction (SAED) patterns and the lattice images, and the results are shown in Fig. 1c and d. In addition, the high resolution transmission electron microscopy (HR-TEM) images at different growth-stages also show that spherical Au NPs are firstly formed with an average diameter of 8.2 ± 2.5 nm (Fig. 1b), with the subsequently formation of Au@Co and Au@Co2P NPs (Fig. 1c and d). The size range of Au@Co2P NPs spans from 15–25 nm.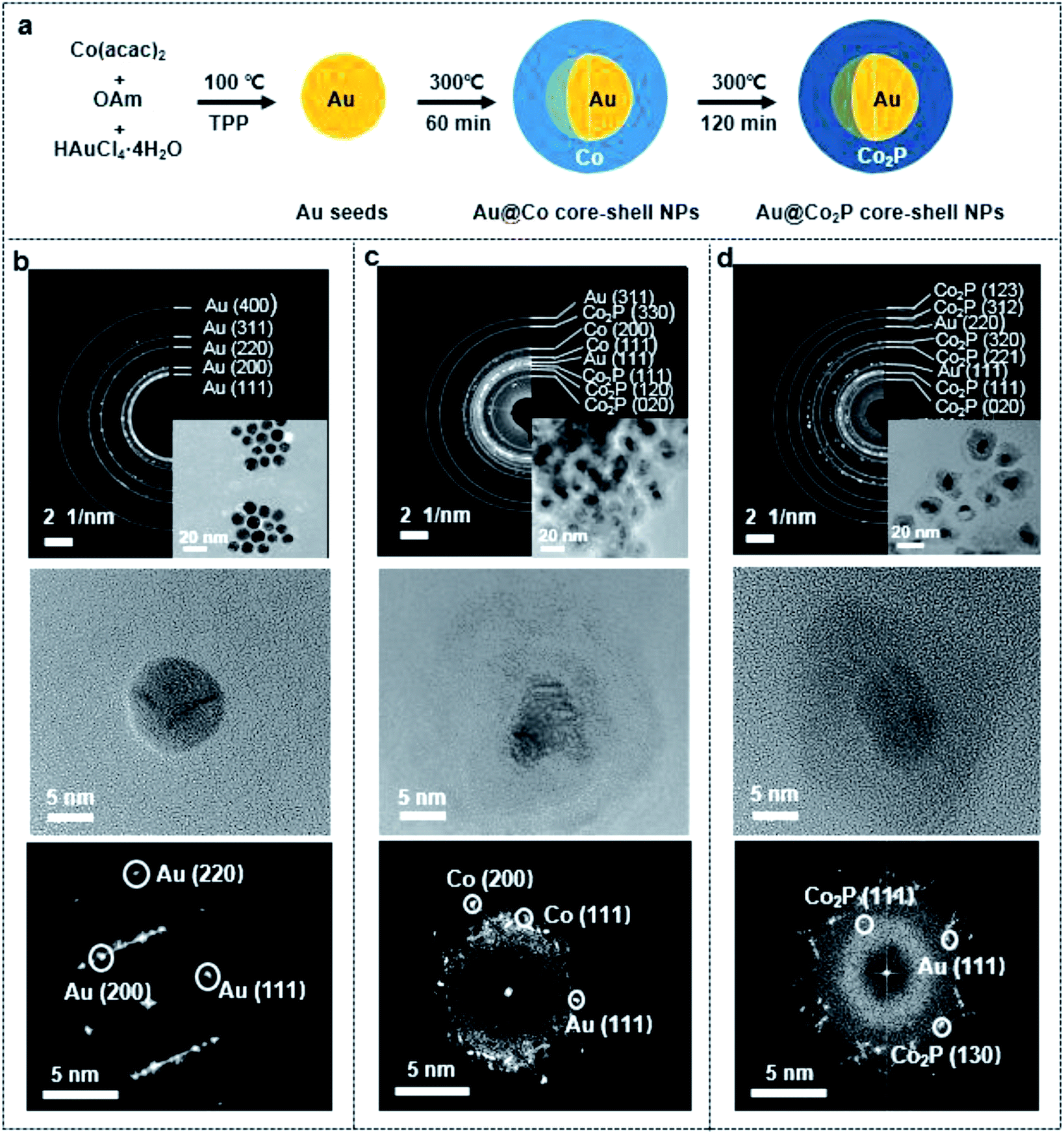 | ||
| Fig. 1 (a) Schematic fabrication process of the Au@Co2P core–shell NPs via seed-mediated growth. (b–d) The selected area electron diffraction (SAED) patterns, HR-TEM images and FFT images of the reaction intermediate/product in the growth of Au@Co2P NPs: (b) core of Au NPs; (c) Co-shell enclosing the Au core; (d) Au@Co2P. The insets in SAED of (b)–(d) are the corresponding TEM images. | ||
Morphology and composition characterization
Further, High-Angle Annular Dark Field (HAADF) TEM images reveal that the prepared Au@Co2P NPs are near spherical in morphology (Fig. 2a). Furthermore, the distribution of atomic components in the core and the shell can be well revealed by the TEM image. The Au core has a larger atomic number than Co2P shell, hence, appears in a dark contrast in this image. As shown in Fig. 2b, the HR-TEM image exhibits that the Au@Co2P NPs are well-crystallized. The interplanar d-spacing of about 0.22 nm of Au reveals the presence of the (111) lattice fringes of metallic Au in the core. On the other hand, the interplanar d-spacing of about 0.18 nm confirms the presence of the (130) lattice fringes of Co2P. The lattice spacing of Au in the core is slightly less than its common values of 0.23–0.24 nm, indicating that the Au core is strained and suppressed by the Co2P shell. The HRTEM also suggests the presence of an amorphous overlayer at the outer surface of the Co2P shell, with a thickness of around 1–2 nm. Naturally, Co2P is expected to be oxidized and passivated when it is exposed to oxygen or air. Hence, this amorphous overlayer is likely formed due to the oxidation of Co2P. Such oxidation is later elaborated with the following composition analyses of Au@Co2P NPs.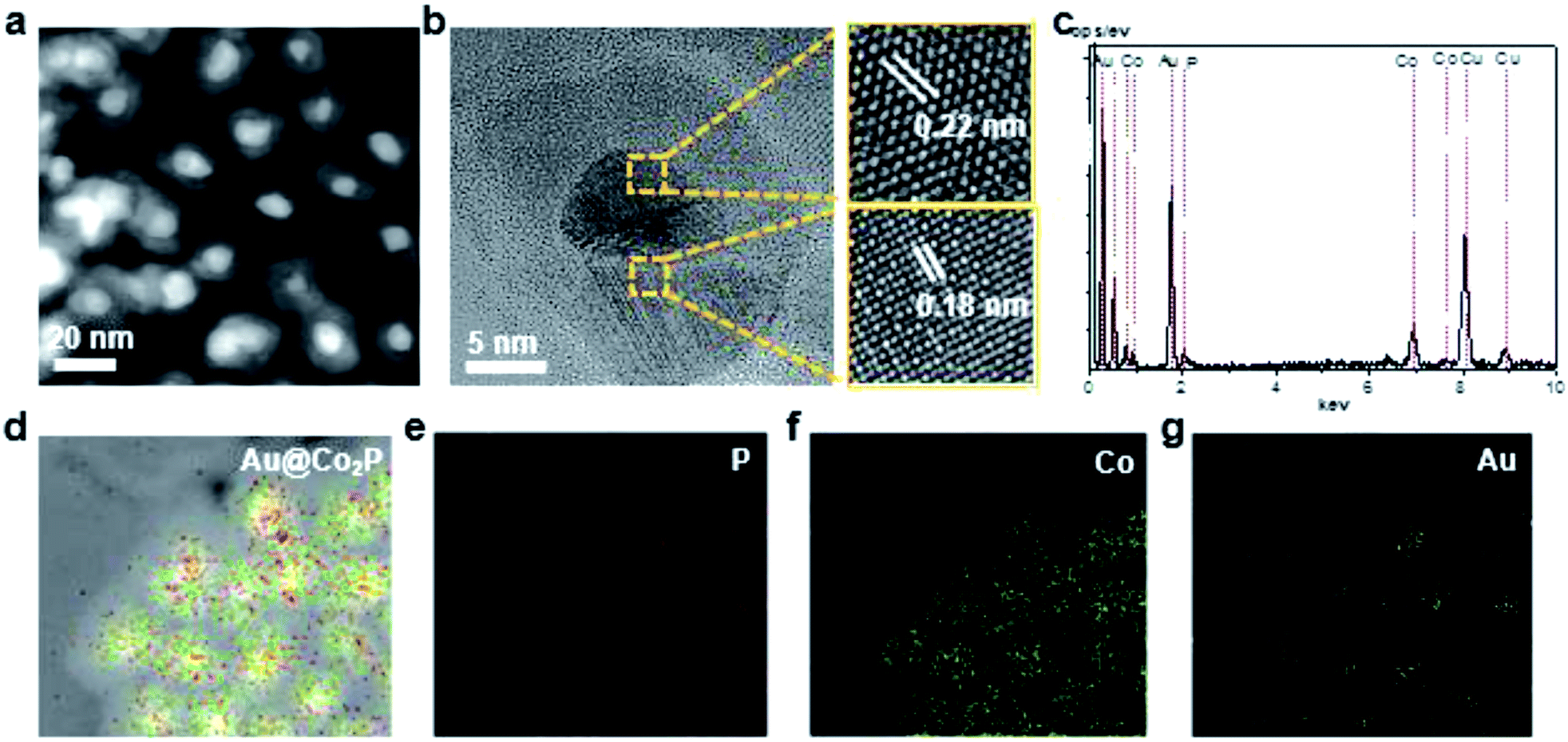 | ||
| Fig. 2 (a) HAADF TEM images of the prepared Au@Co2P core/shell NPs. (b) HR-TEM image of well-crystallized Au@Co2P NPs, revealing the lattice fringes of core/shell NPs. (c) EDS spectrum of Au@Co2P NPs, confirming the Co2P stoichiometry. (d–g) EDS elemental mapping images of Au@Co2P NPs. | ||
The composition of Au@Co2P is first identified with EDS (Fig. 2c–g). The stoichiometry of Co2P in the shell and the presence of pure Au in the core are confirmed. X-ray diffraction (XRD) patterns of the prepared Au@Co2P NPs are shown in Fig. 3a. The four observable peaks at 38.2°, 44.4°, 64.6°, and 77.5° are respectively assigned to the Au (111), (200), (220) and (311) lattice planes (PDF#04-0784). The peaks at 40.7°, 44.5°, 48.81°, 52.15°, and 56.27° are respectively assigned to the Co2P (121), (130), (031), (131), (320) lattice planes (PDF#32-306).36,37
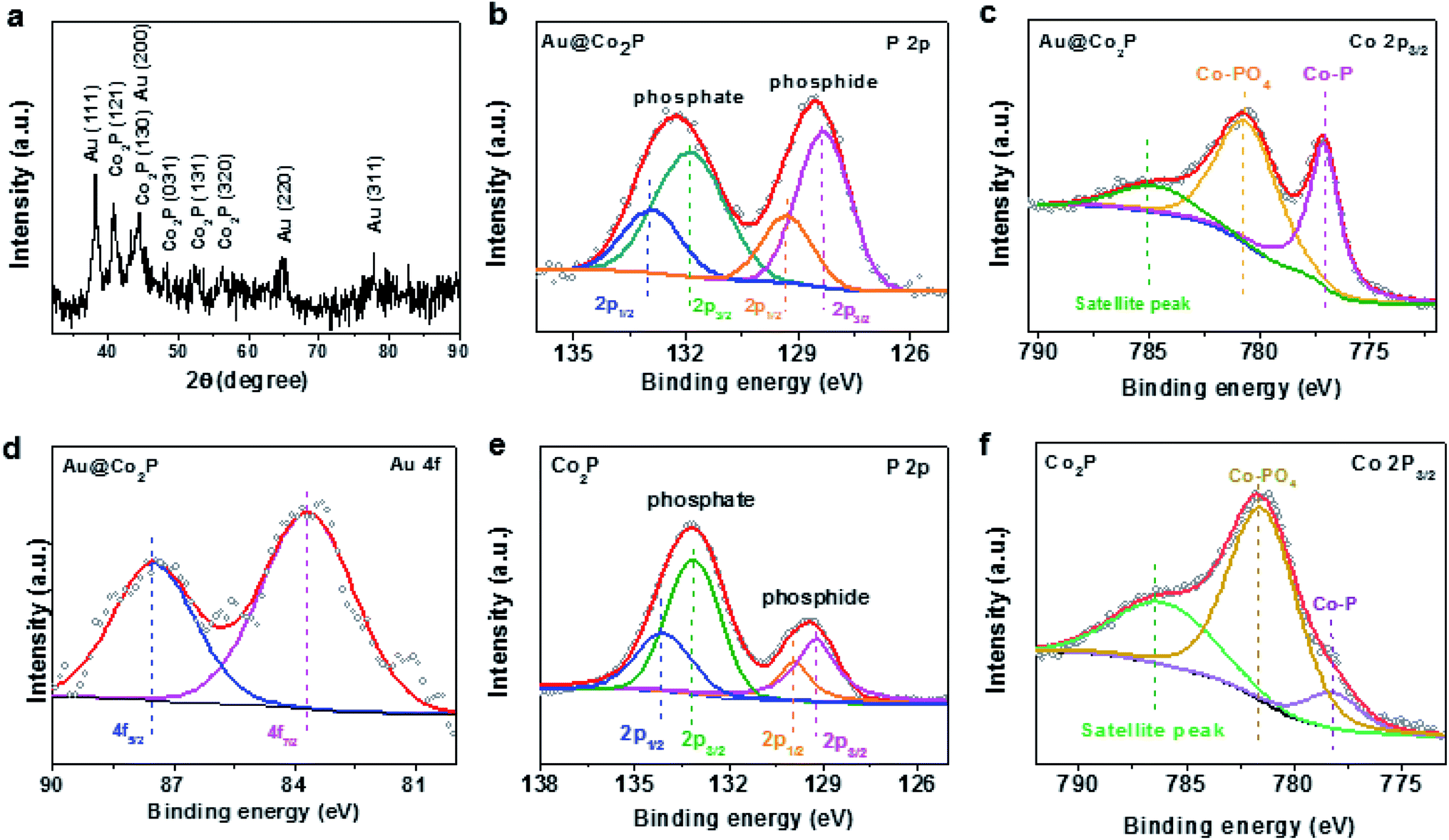 | ||
| Fig. 3 (a) XRD patterns of the prepared Au@Co2P NPs. (b) P 2p, (c) Co 2p3/2, and (d) Au 4f XPS spectra of Au@Co2P core/shell NPs, respectively. (e) P 2p, (f) Co 2p3/2 XPS spectra of Co2P, respectively. | ||
Additional compositional and electronic information are revealed by XPS. All high-resolution spectra are calibrated against the C 1s line at 284.8 eV arising from the presence of adventitious C. Briefly, the presence of Co, P and Au are confirmed by XPS. Details in chemical states and electron-transfer are revealed by those high resolution spectra included in Fig. 3b–d. First, the P 2p spectrum clearly displays two spectral groups each of which can be fitted to a pair of P 2p3/2 and P 2p1/2 (2:1 ratio in relative intensity and spin–orbit splitting of 0.86 eV) (Fig. 3b). The spectral group of P 2p3/2 at 128.4 eV is attributed to the presence of Co2P, and that at 131.9 eV to phosphate derived from the surface oxidation and passivation of Co2P.38–42 By the intensity ratio of phosphate/phosphide and with the standard XPS overlayer-measurement method applicable to the present situation: thickness = 3 nm × ln (phosphate/phosphide + 1), we estimate that the phosphate overlayer thickness is ∼2 nm. Although this method merely gives a rough estimate due to the particulate-nature of the XPS sample, the presence of a phosphate overlayer is evident and its thickness of ∼2 nm is consistent to the TEM result in Fig. 2b. Second, the high resolution Co 2p3/2 spectrum shows the presence of Co2P at 777.5 eV (Fig. 3c)38 and Co-phosphate at 781.6 eV.29 The surface oxidation of Co2P due to air exposure and due to OER may also lead to the formation of various Co-oxides which have XPS binding energies (BE) around 781.6 eV. Hence, the scientific identity of Co-phosphate in the present work is not exact and is likely a composite of Co-phosphate and various forms of Co-oxides/hydroxides. The broad peak at about 785 eV is commonly known as XPS satellite in association with Co 2p3/2. In short, both the P 2p and Co 2p3/2 XPS results clarify that the Co2P of Au@Co2P is naturally oxidized to yield an intriguing structure comprising an Au core, a Co2P inner shell, and a Co-phosphate outer shell. Obviously, OER takes place on the outer shell of Co-phosphate, hence, Co-phosphate is the actual OER catalyst. While Co2P is metallic, Co-phosphate and Co-oxides/hydroxides are semiconducting. On a semiconductor, OER can only proceed with an external voltage-bias such that the OER electrochemical potential is close to the valence band maximum (VMB) of the semiconductor for electrons releasing from OER to drain to the empty electron states at VBM.
The following XPS spectra give some hints on how this mechanism works. First, the Au 4f7/2 and Au 4f5/2, spectra in Fig. 3d reveal their respective BE at 83.7 eV and 87.5 eV. These are slightly smaller than those for pure gold (4f7/2: 84 eV, 4f5/2: 87.8 eV). Hence, the core of Au in Au 4f7/2 and Au 4f5/2 is slightly more electron-rich than pure-gold. Second, the P 2p and Co 2p3/2 of pure Co2P (in NPs) are also quite different from those of Au@Co2P. The differences are: (a) the phosphate/phosphide ratio is much higher in Co2P than that in Au@Co2P; hence, the presence of electron-withdrawing gold as the core constituent in Au@Co2P retards the surface oxidation of Co2P. We speculate that this passivation effect may raise the stability of Au@Co2P in OER. (b) All peaks in Co2P are up-shifted in XPS BE by about 0.7 eV in reference to those in Au@Co2P. Since in the present analysis case, XPS spectra data are collected with a semiconductor (phosphate or oxide/hydroxide) connecting to the XPS spectrometer and with other sample-constituents (phosphide and gold) inside the semiconductor, all spectral peak positions are affected by the Fermi level position of the semiconductor. Take a simple example of a semiconductor with a bandgap energy of 1 eV, the Fermi level of an n-type semiconductor is located near the conduction band minimum and that of a p-type semiconductor is near the VBM, with a difference of 1 eV. Hence, the XPS peaks of the p-type semiconductor are expected to down-shift in BE by about 1 eV in reference to those of the n-type semiconductor. With this simple example, we hypothesize that the Fermi level of the Co-phosphate semiconductor of Au@Co2P is closer to the VBM of Co-phosphate by 0.7 eV than that of Co2P. Since OER is facilitated by electron-drainage to the VBM of Co-phosphate, the OER overpotential of Au@Co2P must be significantly lower than that of Co2P. The Fermi level movement from Co2P to Au@Co2P, with a movement direction towards the VBM of the phosphate-semiconductor is driven by the incorporation of gold as a core constituent in Au@Co2P. In our hypothesis, gold has a work-function higher than those of Co2P and Co-phosphate, and this induces the observed Fermi level movement in favour of OER catalysis.
Electrochemical properties of Au@Co2P NPs
The OER activities of the Au@Co2P NPs are best expressed by the polarization curves in Fig. 4a. The results show that Au@Co2P has an overpotential of 321 mV at a current density of 10 mA cm−2. This overpotential is about 95 mV less than that of pure Co2P (416 mV) and nearly the same as that of RuO2 (320 mV). At a potential of 1.60 V vs. RHE, Au@Co2P and Co2P reach current densities of 41.08 mA cm−2 and 3.47 mA cm−2, respectively. The superiority of Au@Co2P to Co2P is obvious.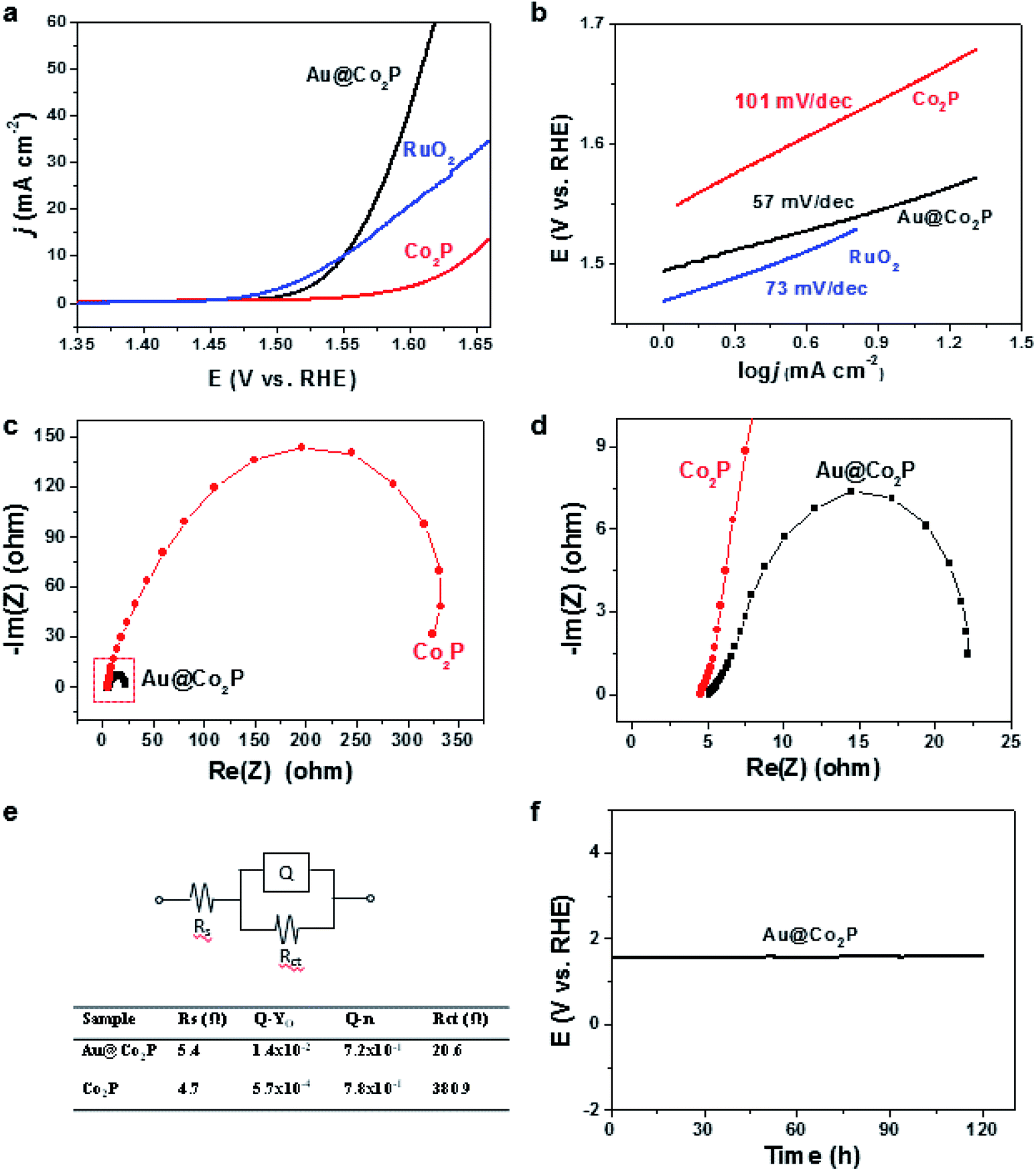 | ||
| Fig. 4 (a) Polarization curves of Au@Co2P core/shell NPs, Co2P and RuO2. (b) Tafel plots of Au@Co2P core/shell NPs, Co2P and RuO2. (c) Nyquist plots of Au@Co2P and Co2P. (d) The enlarged view of the dash line area of the plot (c). (e) The equivalent circuit and the corresponding data. (f) Stability of Au@Co2P (chronopotentiometry curve). | ||
As shown in Fig. 4b, Au@Co2P and Co2P display small Tafel slopes, which are about 57 and 101 mV dec−1, respectively. Hence, Au@Co2P outperforms Co2P, particularly in conditions when high current densities are demanded. The Tafel slope of Au@Co2P is also smaller than that of RuO2 (73 mV dec−1).
Nyquist plots of Au@Co2P and Co2P (Fig. 4c–e) indicate that the charge-transfer resistance of Au@Co2P (20.6 Ω) is much smaller than Co2P (380.9 Ω). The long term stability test for the OER of Au@Co2P in 1 M KOH at 10 mA cm−2 confirms, as shown in Fig. 4f, that the Au@Co2P catalytic effect is stable in the testing duration of 122 hours.
Finally, the critical presence of phosphate after the long stability test is confirmed by XPS, as shown in Fig. 5. For both Au@Co2P and Co2P, XPS analyses give evidence of the presence of phosphate and the growth of phosphate in the expense of phosphide oxidation (at least in the nanometer-thickness scale of XPS) after a prolonged OER of 122 hours. A comparison of the XPS atomic compositional measurements, expressed in Co, P, and O, before and after such a prolonged trial of OER shows that although the presence of phosphate is evident after the prolonged trial, the P/Co and P/O concentration-ratios decrease greatly after the trial. Hence, Co-phosphide is effectively converted to Co-phosphate and various forms of Co-oxides/hydroxides, and the oxides/hydroxides become the dominant oxidation products after a prolonged trial of OER. Dynamic evolution like this has also been found and reported.43 In addition, the conversion of Co-phosphate to Co-oxides/hydroxides in alkaline OER conditions has also been previously reported.13,20,23,27,28,38,42 However, a comparison of the published works on OER performance of Co-phosphate and Co-oxides/hydroxides reveals no large differences in OER overpotential among them.12,27,33,38 As such, this comparative analysis also explains the seemingly-contradicting observations in this work that although the presence of Co phosphate in Au@Co2P in the prolonged OER trial is dynamically overtaken by the presence of Co oxides/hydroxides, the catalytic performance of such nominal Au@Co2P remains virtually unchanged and stable as shown in Fig. 4f. In short, this examination confirms that phosphide is merely a precursor for the formation of the real OER catalyst of Co-phosphate and/or Co-oxides/hydroxides. Further studies of the exact roles of Co-phosphate, Co-oxides, Co-hydroxides, and the composites and derivatives of these species in OER are critical.
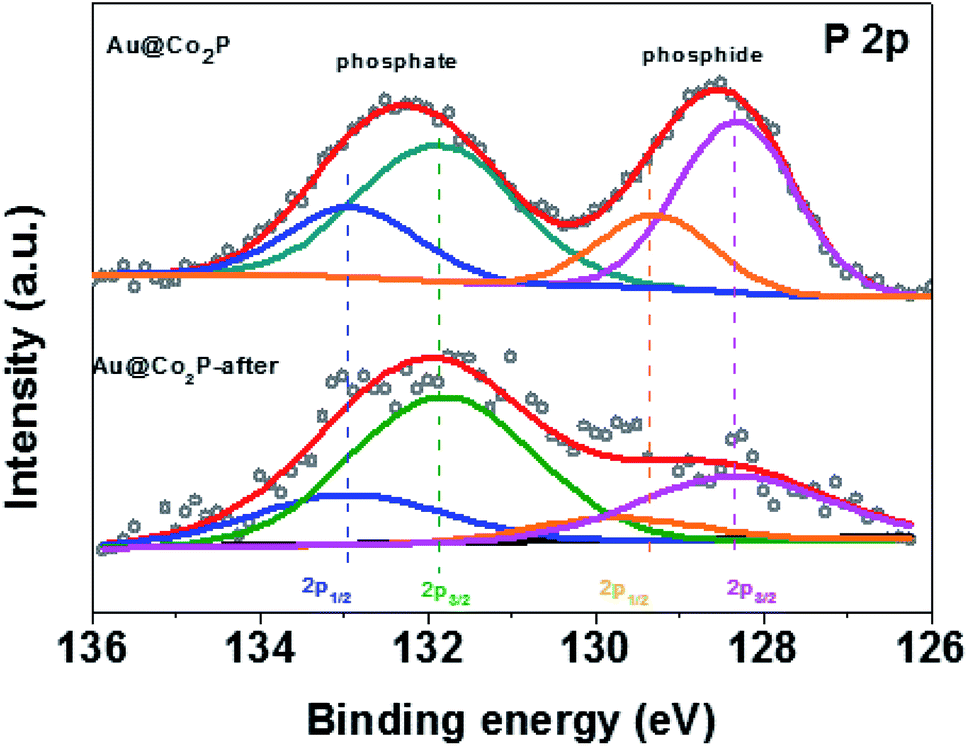 | ||
| Fig. 5 P 2p of Au@Co2P after OER tests in comparison with those before OER tests. | ||
Conclusions
Core–shell Au@Co2P NPs are synthesized via a wet chemical method and their catalytic OER performance is examined. A relatively small OER overpotential of 321 mV at 10 mA cm−2, with a long stability of over 122 hours, is evident. The Tafel slope of 57 mV dec−1 is smaller than that of Ru-oxide. In comparison, Co2P NPs with no core-Au show an OER overpotential of 416 mV and Tafel slope of 101 mV dec−1. A set of comprehensive analyses reveals that the natural surface-oxidation processes of Co2P to form Co-phosphate and Co-oxides/hydroxides are critical. As such, the phosphide (Co2P) is merely a precursor. The real OER catalyst is derived from the oxidation of Co2P and is a semiconductor. In accord to the basic science of a semiconducting catalyst in electrochemistry, the Fermi level position of the semiconducting catalyst is affected by the core-Au of the semiconducting catalyst, and this affects the OER activity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China no. 21601013, no. 11674023, 111 Project no. B170003 and the Fundamental Research Funds for the Central Universities no. FRF-TP-17-072A1.References
- W. Leitner, J. Klankermayer, S. Pischinger, H. Pitsch and K. Kohse-Hoinghaus, Advanced biofuels and beyond: chemistry solutions for propulsion and production, Angew. Chem., Int. Ed., 2017, 56, 5412 CrossRef CAS PubMed
.
- S. E. Habas, H. A. Platt, M. F. van Hest and D. S. Ginley, Low-cost inorganic solar cells: from ink to printed device, Chem. Rev., 2010, 110, 6571 CrossRef CAS PubMed
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Recent advances in bulk heterojunction polymer solar cells, Chem. Rev., 2015, 115, 12666 CrossRef CAS PubMed
- G. Tan, L. D. Zhao and M. G. Kanatzidis, Rationally designing high-performance bulk thermoelectric materials, Chem. Rev., 2016, 116, 12123 CrossRef CAS PubMed
- J. R. McKone, S. C. Marinescu, B. S. Brunschwig, J. R. Winkler and H. B. Gray, Earth-abundant hydrogen evolution electrocatalysts, Chem. Sci., 2014, 5, 865 RSC
- M. S. Dresselhaus and I. L. Thomas, Alternative energy technologies, Nature, 2001, 414, 332 CrossRef CAS PubMed
- C. J. Zhong and M. M. Maye, Core–shell assembled nanoparticles as catalysts, Adv. Mater., 2001, 13, 1507 CrossRef CAS
- T. Hirakawa and P. V. Kamat, Charge separation and catalytic activity of Ag@TiO2 core–shell composite clusters under UV-irradiation, J. Am. Chem. Soc., 2005, 127, 3928 CrossRef CAS PubMed
- K. Maeda, K. Teramura, D. L. Lu, N. Saito, Y. Inoue and K. Domen, Noble-metal/Cr2O3 core/shell nanoparticles as a cocatalyst for photocatalytic overall water splitting, Angew. Chem., Int. Ed., 2006, 45, 7806 CrossRef CAS PubMed
- V. Mazumder, M. F. Chi, K. L. More and S. H. Sun, Core/shell Pd/FePt nanoparticles as an active and durable catalyst for the oxygen reduction reaction, J. Am. Chem. Soc., 2010, 132, 7848 CrossRef CAS PubMed
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. F. Yu, Z. C. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Lattice-strain control of the activity in dealloyed core–shell fuel cell catalysts, Nat. Chem., 2010, 2, 454 CrossRef CAS PubMed
- Z. Zhuang, W. Sheng and Y. Yan, Synthesis of monodisperse Au@Co3O4 core–shell nanocrystals and their enhanced catalytic activity for oxygen evolution reaction, Adv. Mater., 2014, 26, 3950 CrossRef CAS PubMed
- Z. Wu, Q. Gan, X. Li, Y. Zhong and H. Wang, Elucidating surface restructuring-induced catalytic reactivity of cobalt phosphide nanoparticles under electrochemical conditions, J. Phys. Chem. C, 2018, 122, 2848 CrossRef CAS
- J. Xin, H. Tan, Z. Liu, L. Zhao, J. Xie, Y. Sang, W. Zhou, A. Wang, H. Liu and J. Wang, Facile synthesis of hierarchical porous NixCo1−xSeO3 networks with controllable composition as a new and efficient water oxidation catalyst, Nanoscale, 2019, 11, 3268 RSC
- F. Yu, L. Yu, I. Mishra, Y. Yu, Z. Ren and H. Zhou, Recent developments in earth-abundant and non-noble electrocatalysts for water electrolysis, Materials Today Physics, 2018, 7, 121 CrossRef
- F. I. Mattos-Costa, P. de Lima-Neto, S. A. S. Machado and L. A. Avaca, Characterisation of surfaces modified by sol-gel derived RuxIr1–xO2 coatings for oxygen evolution in acid medium, Electrochim. Acta, 1998, 44, 1515 CrossRef CAS
- H. Over, Surface chemistry of ruthenium dioxide in heterogeneous catalysis and electrocatalysis: from fundamental to applied research, Chem. Rev., 2012, 112, 3356 CrossRef CAS PubMed
- H. Zhou, F. Yu, Q. Zhu, J. Sun, F. Qin, L. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Water splitting by electrolysis at high current densities under 1.6 volts, Energy Environ. Sci., 2018, 11, 2858 RSC
- Y. Shi and B. Zhang, Recent advances in transition metal phosphide nanomaterials: synthesis and applications in hydrogen evolution reaction, Chem. Soc. Rev., 2016, 45, 1529 RSC
- J. Chang, Y. Xiao, M. Xiao, J. Ge, C. Liu and W. Xing, Surface oxidized cobalt-phosphide nanorods as an advanced oxygen evolution catalyst in alkaline solution, ACS Catal., 2015, 5, 6874 CrossRef CAS
- L. A. Stern, L. Feng, F. Song and X. Hu, Ni2P as a Janus catalyst for water splitting: the oxygen evolution activity of Ni2P nanoparticles, Energy Environ. Sci., 2015, 8, 2347 RSC
- J. Sun, M. Ren, L. Yu, Z. Yang, L. Xie, F. Tian, Y. Yu, Z. Ren, S. Chen and H. Zhou, Highly efficient hydrogen evolution from a mesoporous hybrid of nickel phosphide nanoparticles anchored on cobalt phosphosulfide/phosphide nanosheet arrays, Small, 2019, 15, 1804272 CrossRef PubMed
- F. Yu, H. Zhou, Y. Huang, J. Sun, F. Qin, J. Bao, W. A. GoddardIII, S. Chen and Z. Ren, High-performance bifunctional porous non-noble metal phosphide catalyst for overall water splitting, Nat. Commun., 2018, 9, 2551 CrossRef PubMed
- A. Mendoza-Garcia, H. Zhu, Y. Yu, Q. Li, L. Zhou, D. Su, M. J. Kramer and S. Sun, Controlled anisotropic growth of Co-Fe-P from Co-Fe-O nanoparticles, Angew. Chem., Int. Ed., 2015, 54, 9642 CrossRef CAS PubMed
- J. Xu, H. Zhang, P. Xu, R. Wang, Y. Tong, Q. Lu and F. Gao, In situ construction of hierarchical Co/MnO@graphite carbon composites for highly supercapacitive and OER electrocatalytic performances, Nanoscale, 2018, 10, 13702 RSC
- D. Yang, W. Hou, Y. Lu, W. Zhang and Y. Chen, Scalable synthesis of self-assembled bimetallic phosphide/N-doped graphene nanoflakes as an efficient electrocatalyst for overall water splitting, Nanoscale, 2019, 11, 12837 RSC
- Y. Hou, Y. Liu, R. Gao, Q. Li, H. Guo, A. Goswami, R. Zboril, M. B. Gawande and X. Zou, Ag@CoxP core–shell heterogeneous nanoparticles as efficient oxygen evolution reaction catalysts, ACS Catal., 2017, 7, 7038 CrossRef CAS
- D. Li, H. Baydoun, C. N. Verani and S. L. Brock, Efficient water oxidation using CoMnP nanoparticles, J. Am. Chem. Soc., 2016, 138, 4006 CrossRef CAS PubMed
- H. Liao, Y. Sun, C. Dai, Y. Du, S. Xi, F. Liu, L. Yu, Z. Yang, Y. Hou, A. C. Fisher, S. Li and Z. J. Xu, An electron deficiency strategy for enhancing hydrogen evolution on CoP nano-electrocatalysts, Nano Energy, 2018, 50, 273 CrossRef CAS
- M. Görlin, J. F. de Araújo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, Tracking catalyst redox states and reaction dynamics in Ni-Fe oxyhydroxide oxygen evolution reaction electrocatalysts: the role of catalyst support and electrolyte pH, J. Am. Chem. Soc., 2017, 139, 2070 CrossRef PubMed
- S. Klaus, L. Trotochaud, M.-J. Cheng, M. Head-Gordon and A. T. Bell, Experimental and computational evidence of highly active Fe impurity sites on the surface of oxidized Au for the electrocatalytic oxidation of water in basic media, ChemElectroChem, 2016, 3, 66 CrossRef CAS
- B. S. Yeo and A. T. Bell, Enhanced activity of gold-supported cobalt oxide for the electrochemical evolution of oxygen, J. Am. Chem. Soc., 2011, 133, 5587 CrossRef CAS PubMed
- A. L. Strickler, M. Escudero-Escribano and T. F. Jaramillo, Core–shell Au@metal-oxide nanoparticle electrocatalysts for enhanced oxygen evolution, Nano Lett., 2017, 17, 6040 CrossRef CAS PubMed
- Y. Xu, S. Duan, H. Li, M. Yang, S. Wang, X. Wang and R. Wang, Au/Ni12P5 core/shell single-crystal nanoparticles as oxygen evolution reaction catalyst, Nano Res., 2017, 10, 3103 CrossRef CAS
- S. Duan and R. Wang, Au/Ni12P5 core/shell nanocrystals from bimetallic heterostructures: in situ synthesis, evolution and supercapacitor properties, NPG Asia Mater., 2014, 6, e00 Search PubMed
- P. E. R. Blanchard, A. P. Grosvenor, R. G. Cavell and A. Mar, X-ray photoelectron and absorption spectroscopy of metal-rich phosphides M2P and M3P (M=Cr-Ni), Chem. Mater., 2008, 20, 7081 CrossRef CAS
- A. P. Grosvenor, S. D. Wik, R. G. Cavell and A. Mar, Examination of the bonding in binary transition-metal monophosphides MP (M=Cr, Mn, Fe, Co) by X-ray photoelectron spectroscopy, Inorg. Chem., 2005, 44, 8988 CrossRef CAS PubMed
- A. Dutta, A. K. Samantara, S. K. Dutta, B. K. Jena and N. Pradhan, Surface-oxidized dicobalt phosphide nanoneedles as a nonprecious, durable, and efficient OER catalyst, ACS Energy Lett., 2016, 1, 169 CrossRef CAS
- R. Wu, Y. Dong, P. Jiang, G. Wang, Y. Chen and X. Wu, Electrodeposited synthesis of self-supported Ni-P cathode for efficient electrocatalytic hydrogen generation, Prog. Nat. Sci.: Mater. Int., 2016, 26, 303 CrossRef CAS
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni, Appl. Surf. Sci., 2011, 257, 2717 CrossRef CAS
- Y.-J. Yuan, Z.-K. Shen, S. Song, J. Guan, L. Bao, L. Pei, Y. Su, S. Wu, W. Bai, Z.-T. Yu, Z. Ji and Z. Zou, Co–P bonds as atomic-level charge transfer channel to boost photocatalytic H2 production of Co2P/black phosphorus nanosheets photocatalyst, ACS Catal., 2019, 9, 7801 CrossRef CAS
- Z. Jin, P. Lia and D. Xiao, Metallic Co2P ultrathin nanowires distinguished from CoP as robust electrocatalysts for overall water-splitting, Green Chem., 2016, 18, 1459 RSC
- L. Yu, Q. Zhu, S. Song, B. McElhenny, D. Wang, C. Wu, Z. Qin, J. Bao, Y. Yu, S. Chen and Z. Ren, Non-noble metal-nitride based electrocatalysts for high-performance alkaline seawater electrolysis, Nat. Commun., 2019, 10, 5106 CrossRef PubMed
| This journal is © The Royal Society of Chemistry 2019 |