
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The effects of trace metal impurities on Ga-68-radiolabelling with a tris(3-hydroxy-1,6-dimethylpyridin-4-one) (THP) chelator†
Ruslan Cusnirab,
Andrew Cakebreadc,
Margaret S. Coopera,
Jennifer D. Younga,
Philip J. Blower a and
Michelle T. Ma*a
a and
Michelle T. Ma*a
aSchool of Biomedical Engineering and Imaging Sciences, King's College London, St Thomas' Hospital, London SE1 7EH, UK. E-mail: michelle.ma@kcl.ac.uk
bLaboratory of Radiochemistry, Paul Scherrer Institute, 5232 Villigen-PSI, Switzerland
cMass Spectrometry Facility, King's College London, Franklin Wilkins Building, 150 Stamford St, London SE1 9NH, UK
First published on 14th November 2019
Abstract
GMP-grade 68Ge/68Ga generators provide access to positron-emitting 68Ga, enabling preparation of Positron Emission Tomography (PET) tracers and PET imaging at sites that do not have access to cyclotron-produced radionuclides. Radiotracers based on tris(3-hydroxy-1,6-dimethylpyridin-4-one) (THP) chelators enable simple one-step preparations of 68Ga PET radiopharmaceuticals from pre-fabricated kits without pre-processing of generator eluate or post-purification. However, trace metal impurities eluted along with 68Ga could compete for THP and reduce radiochemical yields (RCY). We have quantified trace metal impurities in 68Ga eluate from an Eckert & Ziegler (E&Z) generator using ICP-MS. The metals Al, Fe, natGa, Pb, Ti and natZn were present in generator eluate in significantly higher concentrations than in the starting eluent solution. Concentrations of Fe and natGa in eluate were in the range of 0.01–0.1 μM, Al, Zn and Pb in the range of 0.1–1 μM, and Ti in the range of 0.9–1.5 μM. To assess the ability of THP to chelate 68Ga in the presence of such metal ions, radiolabelling reactions were undertaken in which selected metal ions were added to make them equimolar with THP, or higher. Al3+, Fe3+, natGa3+ and Ti4+ reduced RCY at concentrations equimolar with THP and higher, but at lower concentrations they did not affect RCY. Pb2+, Zn2+, Ni2+ and Cr3+ had no effect on RCY (even under conditions in which each metal ion was present in 100-fold molar excess over THP). The multi-sample ICP-MS analysis reported here is (to date) the most comprehensive and robust quantification of metal impurities in the widely used E&Z 68Ga generator. 68Ga from an E&Z generator enables near-quantitative radiolabelling of THP at chelator concentrations as low as 5 μM (lower than other common gallium chelators) without pre-processing. The combination of Al3+, Fe3+, natGa3+ and Ti4+ in unprocessed 68Ga eluate is likely to decrease RCY of 68Ga radiolabelling if a lower amount of THP chelator is used, and future kit design should take this into account. To increase specific activities by using even lower THP concentrations, purification of 68Ga from trace metal ions will likely be required.
Background
Molecular positron emission tomography (PET) imaging with radiolabelled receptor-targeted peptides and proteins has been transformative in management of cancer patients in clinical centres where it is available.1,2 Gallium-68 (68Ga) is a metallic radionuclide that emits positrons (t1/2 = 68 min, β+ 90%, Emax 1880 keV) suitable for diagnostic imaging with 68Ga-labelled chelator-peptide conjugates.3 The short half-life of 68Ga is compatible with the fast clearance from blood and rapid target localisation of 68Ga-labelled chelator-peptide conjugates. However, it also necessitates fast radiolabelling approaches.One-step, kit-based radiolabelling protocols that provide a radiopharmaceutical in quantitative radiochemical yield (RCY), and in pyrogen-free, physiologically-compatible solutions are the ideal for 68Ga radiopharmaceuticals.4,5 Such kit-based radiosyntheses reduce the need for expensive infrastructure and highly trained personnel. Kit-based radiosynthesis protocols have been used for formulation of 99mTc radiopharmaceuticals for almost five decades. These “one-step” kits consist of a single pre-fabricated vial that contains all non-radioactive components and reagents. For simple kit-based radiosynthesis of 68Ga radiopharmaceuticals to be realised, the nuclear medicine community needs chelators that bind 68Ga rapidly (<5 min) and quantitatively (RCY > 95%) at low chelator concentrations (μM range) and near physiological pH (6–7), preferably avoiding heating or other additional manipulations.
DOTA and HBED-CC chelators are used in the radiopharmaceuticals 68Ga-DOTA-TATE for imaging neuroendocrine tumours6 and 68Ga-HBED-PSMA for imaging prostate tumours,7 respectively. To date, these chelators have typically required pre-processing of 68Ga generator eluate to remove competing trace metal impurities and concentrate 68Ga3+, heating to reproducibly incorporate 68Ga3+ into the chelator, and post-synthetic purification to remove unreacted 68Ga3+ and physiologically-incompatible buffer components.8–10 Hence, they do not meet the ideal requirements of kit-based radiolabelling.
We have recently developed a THP (tris(3-hydroxy-1,6-dimethylpyridin-4-one)) chelator (Fig. 1) that rapidly and quantitatively incorporates 68Ga3+ at room temperature, neutral pH, and low chelator concentrations, giving a single species.11–13 It is consequently well-suited to kit-based 68Ga-radiopharmceutical synthesis, unlike other common gallium chelators such as DOTA and HBED-CC. THP has demonstrated superior radiolabelling properties compared to many other chelators, with higher RCYs achievable for THP at very low concentration compared to e.g. DOTA and HBED.14 THP has been attached to a number of peptides and proteins that target cancer cell surface receptors and the conjugates demonstrate favourable biodistribution and targeting properties in in vivo PET imaging studies.5,11,15–19 A THP bioconjugate, THP-PSMA, that targets the prostate specific membrane antigen (PSMA) has been evaluated recently for one-step kit-based radiolabelling with 68Ga.5 These resulting GalliProst™ kits can be radiolabelled in 2–5 min, simply by addition of unprocessed 68Ga generator eluate (typically 5 mL of 0.1 M aqueous HCl containing 170–270 MBq of 68Ga) to a vial containing THP-PSMA and sodium bicarbonate buffer.5,16 A new homologue of THP has been developed with further improved radiolabelling properties and affinity and selectivity for Ga3+.13
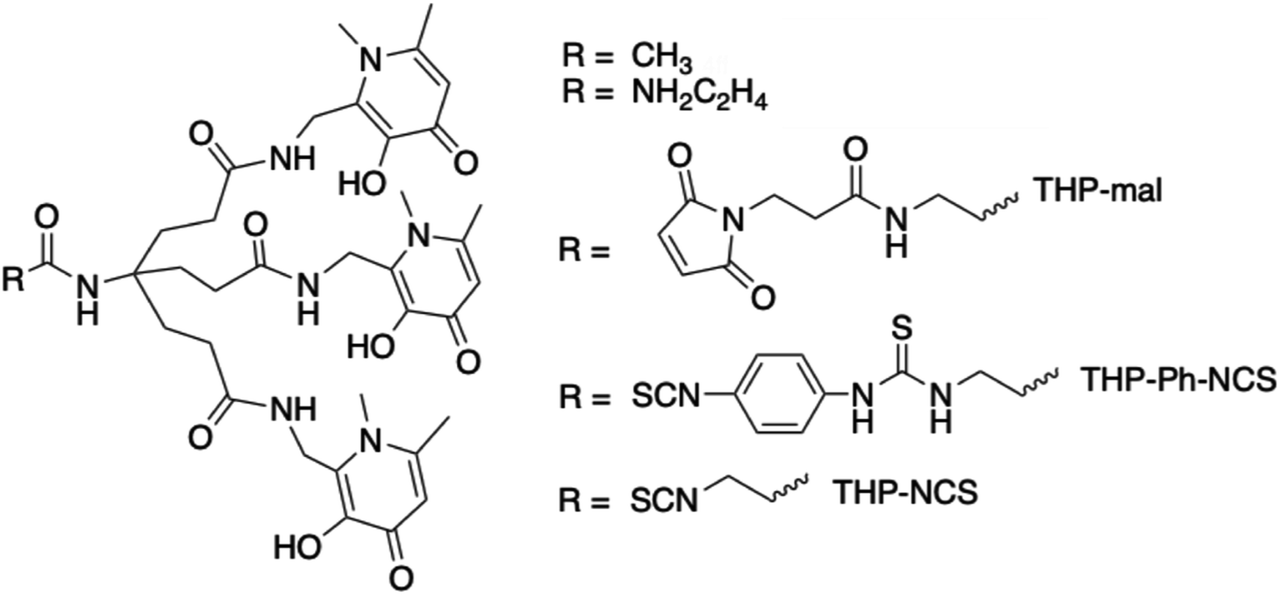 | ||
| Fig. 1 THP chelator (R = CH3) and its bifunctional chelator derivatives. | ||
Currently, 68Ga is usually produced from pharmaceutical grade 68Ge/68Ga generators.20,21 In these generators, the parent radionuclide 68Ge (t1/2 = 271 days) is adsorbed on a stationary phase, typically TiO2. 68Ge decay yields 68Ga, which is normally eluted with an acidic solution (typically aqueous hydrochloric acid) as 68Ga3+. Frequent elutions and continuous use of acid can result in leaching of other metal ions from the generator stationary phase. These can significantly reduce radiolabelling efficiency due to competition with the radiometal ion for chelator binding.22 Additionally, the concentration of 68Ga3+ is very low: 1 GBq of carrier-free 68Ga contains only 9.8 pmol of 68Ga3+, equivalent to approx. 2 nM concentration in 5 mL of generator eluate. Most chelators do not exclusively bind a single metal ion, and so both the selectivity of a chelator and the purity of 68Ga eluate can influence the RCYs of these reactions. Some 68Ga chelators, such as HBED,23 NOTA24 and TRAP24 have demonstrated selectivity for Ga3+ over divalent metal ions.
THP ligands were originally developed as iron-sequestering chelators,25,26 and so as well as having high affinity for Ga3+, THP also has high affinity for Fe3+, Al3+ and other oxiphilic transition metal ions.12,13,27–29 The highest molar activity that we have achieved for a THP conjugate to date is 80 MBq nmol−1,11,19 equating to <0.1% 68Ga3+ occupancy of THP in these solutions. This is comparatively low compared to other radiometallated conjugates – for example, some DTPA- or DOTA-conjugates with 111In can achieve specific activities up to 1 GBq nmol−1,30 and TRAP-conjugates with 68Ga can achieve over 4.8 GBq nmol−1, although the latter is obtained under reaction conditions that result in <70% RCY.31 It is likely that competition from interfering metal ions in 68Ga generator eluate limits achievement of higher molar activities for THP.
The objectives of this study were to (i) quantify trace metal impurities in 68Ga eluate from an Eckert & Ziegler generator and (ii) evaluate the competition effects of the most common contaminant metal ions on 68Ga radiolabelling with THP.
Methods
Reagents
Sterile ultrapure 0.1 M hydrochloric acid aqueous solution for elution of 68Ga from 68Ge/68Ga Eckert & Ziegler generator was supplied by ABX GmbH (Radeberg, Germany). One-year old generators (yielding 350–450 MBq) were used in this study. Lead, titanium and zinc standard solutions (1000 mg L−1) for AAS were purchased from VWR. Gallium and nickel standards for AAS (TraceCERT TM, 1000 mg L−1) were purchased from Sigma-Aldrich. Chromium (as Cr3+) solution for ICP (1000 mg L−1) was purchased from Fisher Chemical. Aluminium standard for ICP (1000 mg L−1) was purchased from Fluka. Iron AAS standard solution (Specpure®, 1000 mg L−1) was from Alfa Aesar. Sodium chloride ultrapure bioreagent was obtained from Fisher Scientific. (+)-Sodium L-ascorbate BioXtra was obtained from Sigma-Aldrich. Hydrochloric acid 20% Primar for trace metal analysis from Fisher Chemical was used for conditioning SCX columns. THP was synthesised according to a previously described procedure.32 Stock solutions of THP in water (UltraPure™ distilled water from Invitrogen) were stored at −20 °C. Instant thin layer chromatography strips impregnated with silica gel (iTLC-SG) were obtained from Agilent Technologies and used to determine RCYs. Ethanol and acetone were of HPLC-grade.Determination of trace metals in 68Ga generator eluate
68Ga3+ was eluted from an E&Z 68Ge/68Ga generator with ultrapure HCl (5 mL, 0.1 M) in a single fraction. Eluates were allowed to decay for at least 24 h prior to analysis by ICP-MS. Eluent (ultrapure 0.1 M HCl) was also analysed as a blank. All samples were stored in polypropylene tubes (Elkay, cat. no. 2086) with low trace metal content. The concentration of trace metals was determined on a PerkinElmer NexION 350D inductively coupled plasma mass spectrometer running Syngistix v1.0 software (London Metallomics Facility, King's College London, UK). The acquisition mode included 5 replicates averaged to give reported values for 27Al, 137Ba, 52Cr, 65Cu, 56Fe, 69Ga, 55Mn, 60Ni, 23Na, 208Pb, 45Sc, 118Sn, 47Ti, 51V, 66Zn and 68Zn. The dwell time was 50 ms per isotope, with 18 L min−1 main argon flow, 1.2 L min−1 auxiliary argon flow, 0.97 L min−1 nebuliser argon flow (optimised daily), 1600 W RF power, 0.2 mL min−1 sample flow, and KED cell mode with 4.2 mL min−1 helium flow. Concentrations of the individual isotopes, 66Zn and 68Zn, were determined by ICP-MS. natZn was calculated based on natural abundance of 66Zn (27.9%). 68Zn arising from decay of 68Ga (decay68Zn) was calculated by subtracting naturally occurring 68Zn (18.75%) from 68Zn determined by ICP-MS.Competition experiments
To evaluate competition effects of trace metals, THP (5 μM) in sodium bicarbonate buffer was radiolabelled with 68Ga in the presence of additional Al3+, Fe3+, natGa3+, Ti4+, Pb2+, Zn2+, Ni2+ and Cr3+ (0.05–500 μM), using the following solutions:Radiolabelling with solutions 68Ga3+ pre-processed using cation exchange methodologies
Radiolabelling of THP was tested with 68Ga “pre-processed” on a SCX Bond Elut 100 mg cartridge using methods that use acidified NaCl,33 ethanol9 or acetone10 solutions to elute the trapped 68Ga3+ from the cation exchange resin. The amount of NaHCO3 required to neutralise processed 68Ga was determined experimentally by titration (in order to account for extra H+ released from the SCX column). Processed eluate (50 μL, 7–19 MBq 68Ga) was added to a solution of THP (5 μM in a final volume of 300 μL at pH 7). After incubation at RT for 10 min, RCY was determined by iTLC.Results
Trace metal impurities
Using ICP-MS, we determined the concentrations of common trace metal impurities (Al, Cr, Cu, Fe, natGa, Mn, Ni, Pb, Sc, Sn, Ti, V, 66Zn and 68Zn) in 68Ga eluate. The concentrations of trace metals are known to correlate with the amount of time between generator elutions.14,22 To probe this, we obtained generator eluate (5 mL in 0.1 M HCl) either (i) 2 hours after a previous generator elution (sample set A), or (ii) 1 day after a previous elution (sample set B). These samples were collected within a period of 20 days, from a generator eluting 330–480 MBq. The mean concentrations of Cr, Cu, Mn, Ni, Sc, Sn and V were all very low (below 10 nM) in both sample sets A and B, and in blank eluent solutions of 0.1 M HCl. We have therefore focused on transition and main group metals Al, Fe, natGa, Pb, Ti and Zn (Fig. 2). The mean concentrations of metals Al, Fe, natGa, Pb, Ti and natZn were all higher in sample sets A and B than in eluent (blank) solution. These concentration differences between eluate and blank solution were statistically significant in all cases (p < 0.1, Table SI-1†), with the exception of the difference between mean natZn concentrations in sample set A and blank solutions.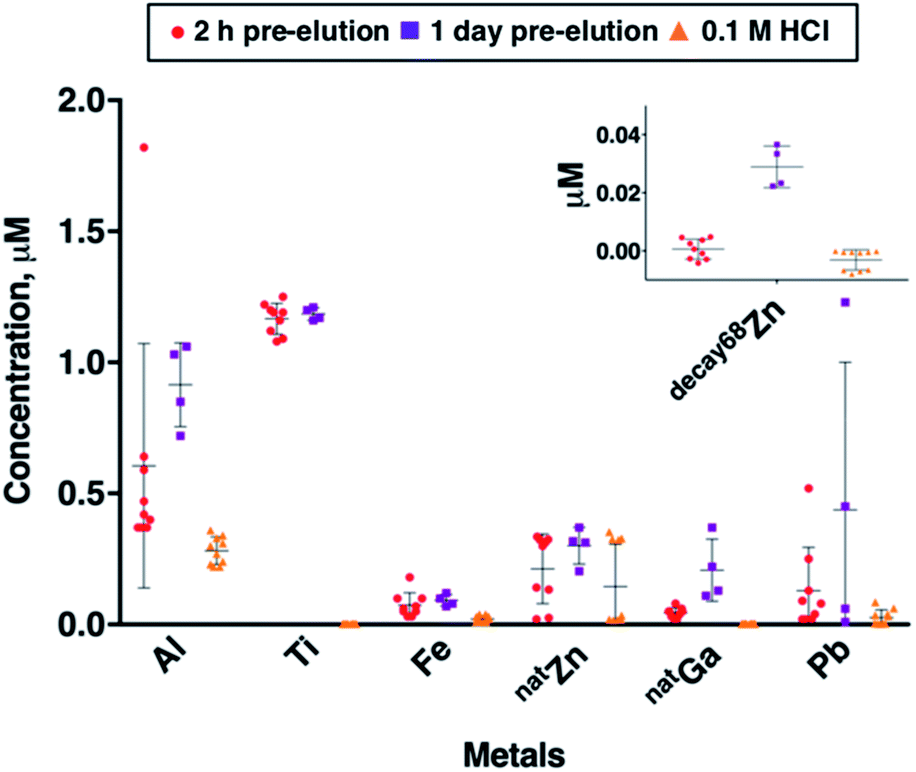 | ||
| Fig. 2 Concentrations of selected trace metals in 68Ga generator eluate measured by ICP-MS. Generator eluate (5 mL in 0.1 M HCl) was obtained from a single generator either 2 hours after a previous generator elution (sample set A, red circles, n = 9), or 1 day after a previous elution (sample set B, purple squares, n = 4). “Blank” eluent (0.1 M HCl solution from ABX Gmb) was also measured (yellow triangles, n = 10). These samples were collected within a period of 20 days, from a generator eluting 330–480 MBq. Error bars represent standard deviation of concentrations in the samples. | ||
The metal present at highest concentrations in generator eluate was Ti. Mean Ti concentration was 1.17 ± 0.37 μM in sample set A, and 0.95 ± 0.53 μM in sample set B. There was no significant difference in mean Ti concentration between sample sets A and B. The high concentration of Ti in eluate has been previously observed by us and others: it arises from the solid phase titanium dioxide material (contained in a borosilicate glass column) on which 68Ge4+ is immobilised.14,34
The trivalent metal ions Al3+ and Fe3+ bind hydroxypyridinones with high affinities,12,13,27–29 and high concentrations of Al, Fe and non-radioactive natGa (all likely to be present as trivalent cations) might therefore be expected to decrease specific activities of [68Ga(THP)] and its bioconjugates. Al was present in highest concentration, measuring 0.61 ± 0.47 μM in sample set A, and 0.91 ± 0.16 μM in sample set B. Al mean concentrations in blank eluent solutions (0.28 ± 0.05 μM) were relatively high compared to other metal impurities in blank eluent solutions. Fe mean concentrations were 0.07 ± 0.05 μM in sample set A and 0.09 ± 0.02 μM in sample set B. Whilst Al and Fe mean concentrations were both higher in sample set B compared with sample set A, neither of these observed differences were statistically significant. We observed that increased time between 68Ga generator elutions resulted in significantly increased amounts of non-radioactive natGa in generator eluate. Mean natGa concentration measured 0.04 ± 0.02 μM in sample set A, and 0.21 ± 0.12 μM in sample set B (mean difference = 0.16 μM, p = 1.4 × 10−3).
Many chelators used for 68Ga radiolabelling, including DOTA,35 have high affinity for divalent cations such as Zn2+ and Pb2+. Zn impurities arise from the eluent solution, leaching from generator components and decay of 68Ga. The presence of Zn (occurring as Zn2+) in generator eluate can compromise 68Ga radiolabelling of DOTA-based radiopharmaceuticals.21,36,37 In some clinical 68Ga radiosynthesis protocols, the generator eluate undergoes pre-processing to separate Zn2+ from 68Ga prior to biomolecule radiolabelling.10,38 68Ga decays exclusively to 68Zn, and this contributes to the amount of Zn2+ present in eluate solution. Factors that directly correlate with the amount of 68Zn in generator eluate include the activity of the generator, and the amount of time between elutions of the generator. Here, we quantified (i) natZn concentration (using 66Zn ICP-MS signal), and (ii) the amount of 68Zn arising from decay of 68Ga, “decay68Zn” (using 68Zn ICP-MS signal, and subtracting known proportions of nat68Zn from this measurement).
Mean natZn concentration measured 0.21 ± 0.13 μM in sample set A, and 0.30 ± 0.07 μM in sample set B and was higher in both sample sets compared to blank eluent solution (0.15 ± 0.16 μM), although this was only statistically significant for sample set B (mean difference = 0.15, p = 0.09). Mean decay68Zn concentration measured 6.1 × 10−4 ± 3.4 × 10−3 μM in sample set A (in which samples were collected 2 hours after a previous elution), and 2.9 ± 0.7 × 10−2 μM in sample set B (in which samples were collected 1 day after a previous elution) (mean difference = 2.8 × 10−2, p < 10−4).
Mean eluate Pb concentrations in both sample set A (0.13 ± 0.16 μM) and sample set B (0.44 ± 0.56 μM) were higher than that observed in blank eluent solution (0.03 ± 0.03 μM), and there was high variability in Pb concentration between individual samples in both sample sets A and B. There was no statistically significant difference between Pb concentrations in sample sets A and B. It is likely that Pb content arises from lead structures used to the shield the generator. Others have also observed significant Pb content in generator eluate.34
Changes in Al, Fe, Ga and Ti concentrations in eluate over six months
To further probe Al, Fe, Ga and Ti concentration in generator eluate, eluate samples were collected six months apart from each other, from a second E&Z generator (Fig. 3). These samples were all obtained 2 hours after a previous elution. During this six month period, between our two sample collection campaigns, the generator was eluted 130 times.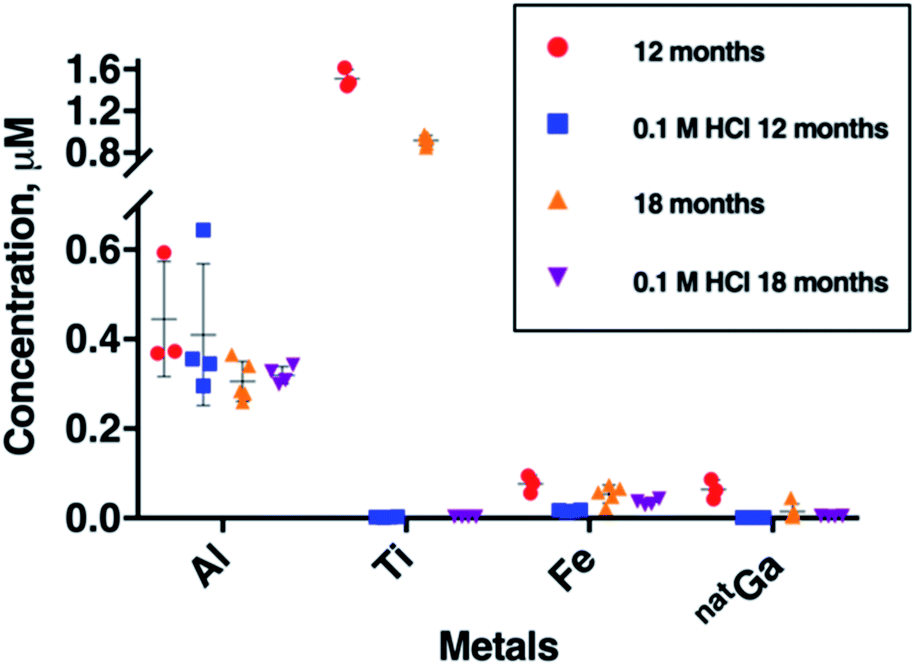 | ||
| Fig. 3 Concentrations of Al, Ti, Fe and natGa in 68Ga eluate as a function of generator age. Two sets of eluate samples were collected from the same 68Ga E&Z generator, six months apart from each other. “Blank” eluent (0.1 M HCl solution from ABX Gmb) used in these two sets of samples was also measured. During this period, the generator was eluted 130 times. All eluates were sampled with a pre-elution window of 2 h. Error bars represent standard deviation of concentrations in the samples. | ||
The most remarkable change was observed for mean Ti breakthrough, which decreased from 1.51 ± 0.09 μM to 0.92 ± 0.05 μM (mean difference = 0.59 μM, p = 1.8 × 10−5) within six months (Fig. 3 and Table SI-3†). Concentrations of Al and natGa over this time also decreased. Mean Al concentration decreased from 0.45 ± 0.13 μM to 0.31 ± 0.05 μM (mean difference = 0.14 μM, p = 6.11 × 10−2) and mean natGa concentration decreased from 0.064 ± 0.022 μM to 0.015 ± 0.018 μM (mean difference = 0.049 μM, p = 1.27 × 10−2). The mean concentration of Fe also decreased from 0.077 ± 0.020 to 0.054 ± 0.020 μM, however in our sample set, this was not statistically significant.
The effect of metal ion impurities in 68Ga radiolabelling reactions with THP
To identify the effect of the presence of trace/interfering metal ions on radiolabelling THP with 68Ga, THP was radiolabelled with 68Ga in a solution containing a spike of each metal ion. We included the metals identified as being present in generator eluate at concentrations >10 nM (Al, Fe, natGa, Pb, Ti and natZn), as well as Ni and Cr, which are present in steel needles often used for preparation of radiopharmaceuticals. In these experiments, we used only a small amount of 68Ga eluate (2–6 MBq, 70–90 μL) to minimise the amount of interfering metal ions from other sources, such as the generator itself. We have also assumed that the metal ions are present as Al3+, Fe3+, natGa3+, Pb2+, Ti4+, Zn2+, Ni2+ and Cr3+.The results of competition experiments with THP and 68Ga3+ in the presence of a single spiked metal ion (at metal ion concentrations of 50 nM to 500 μM, THP concentrations of 5 μM, pH 7 in carbonate solution) are shown in Fig. 4 and summarised in Table SI-2.† In the absence of a metal ion spike, the RCY of [68Ga(THP)] was consistently ≥97%. The presence of Ni2+, Zn2+, Pb2+ and Cr3+ had no effect on 68Ga radiolabelling efficiency, even at metal ion concentrations 100-fold greater than THP concentration.
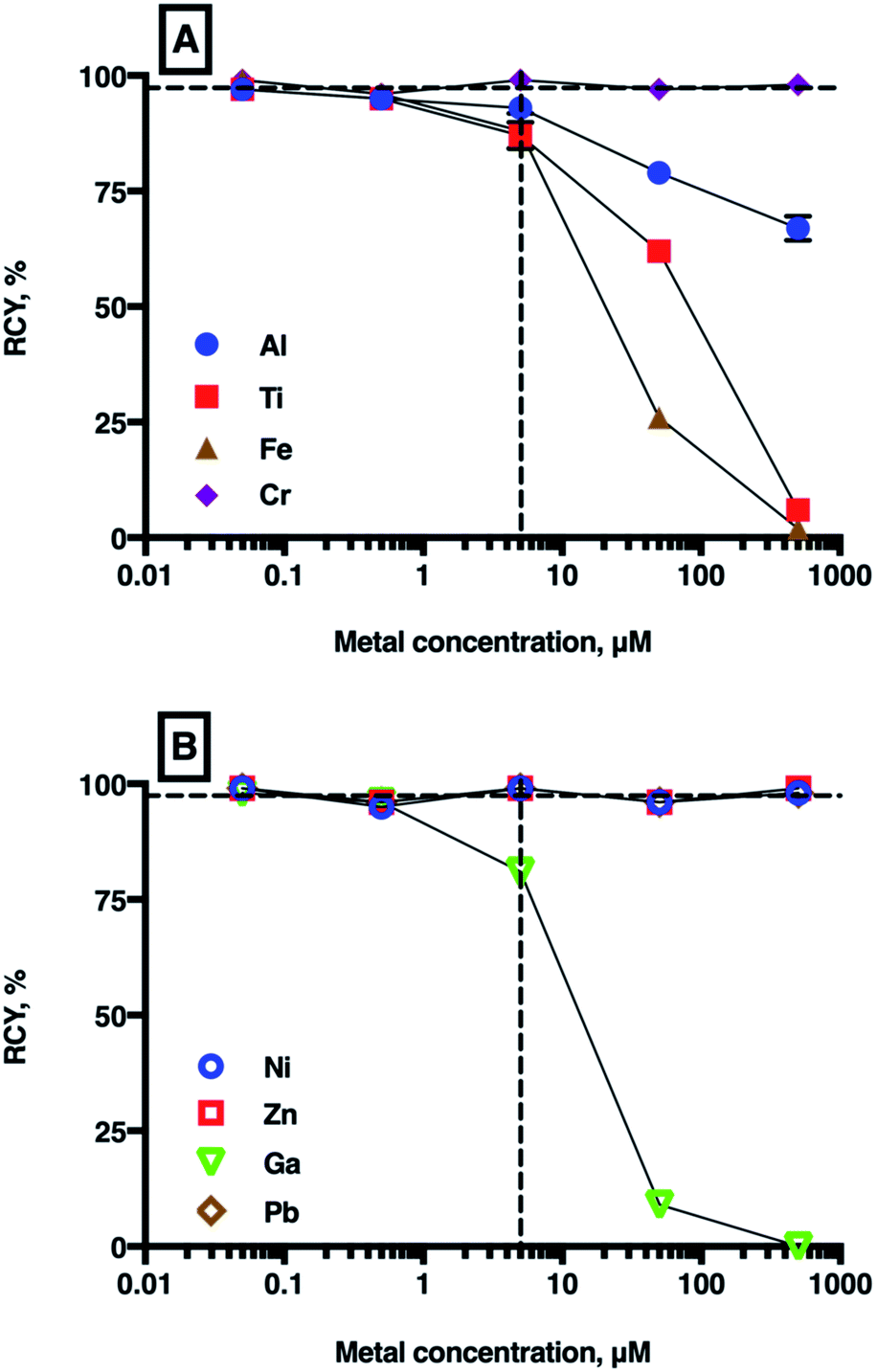 | ||
| Fig. 4 Mean RCYs of [68Ga(THP)] in the presence of increasing concentrations of metal ions ((A): Al3+, Ti4+, Cr3+, Fe3+ and (B): Ni2+, Zn2+, natGa3+, Pb2+). The vertical dashed line indicates equimolar concentrations of chelator and metal ion (5 μM), and the horizontal dashed guide indicates 97% (n = 7) RCY, which is achieved in the absence of added metal ions. For most values, error bars representing standard deviation are smaller than the symbols. For a full list of mean RCY (n = 3) and standard deviation values see Table SI-4.† | ||
As expected, the presence of natGa3+ reduced RCY of [68Ga(THP)] at natGa3+ concentrations ≥5 μM (to 81 ± 0.78% at 5 μM, 9 ± 0.87% at 50 μM, and 0% at 500 μM), but no significant changes were observed at concentrations <5 μM. Similarly, at concentrations ≥5 μM, Fe3+, Al3+ and Ti4+ all reduced RCY of [68Ga(THP)]. At metal ion concentrations equimolar with THP, RCY of [68Ga(THP)] was 88 ± 3.81% for Fe3+ solutions, 93 ± 2.30% for Al3+ solutions and 87 ± 2.86% for Ti4+ solutions. At 50 μM metal ion concentrations, RCY of [68Ga(THP)] was 26 ± 1.25% for Fe3+ solutions, 79 ± 0.47% for Al3+ solutions and 62 ± 1.53% for Ti4+ solutions.
Effect of ascorbate on radiolabelling THP in the presence of Fe3+
In many radiopharmaceutical formulations, ascorbic acid or sodium ascorbate is included to minimise radiolysis of the solution. The presence of sodium ascorbate (166 mM, THP concentrations of 5 μM, and 5–7 MBq 68Ga, pH 7 in bicarbonate solution) in THP radiolabelling reactions did not reduce radiolabelling efficiency of THP with 68Ga (RCY 96 ± 0.4%, n = 6). Ascorbate can reduce Fe3+ to Fe2+. To assess the practical implications of this, the RCY of [68Ga(THP)] in reactions containing 68Ga3+, THP, a spike of Fe3+ and ascorbate were compared with the RCY of reactions containing 68Ga, THP and a spike of Fe3+ only (Fig. 5). In reactions containing Fe3+ at concentrations of 5 μM, addition of ascorbate did not affect RCY of [68Ga(THP)]. In reactions in which Fe3+ was present at concentrations of 50 or 500 μM, the presence of ascorbate resulted in increased RCY of [68Ga(THP)]: at [Fe] = 50 μM, RCY increased from 26 ± 1.25% to 73 ± 4.17%, and at [Fe] = 500 μM Fe, RCY increased from 2 ± 1.99% to 31 ± 7.29%.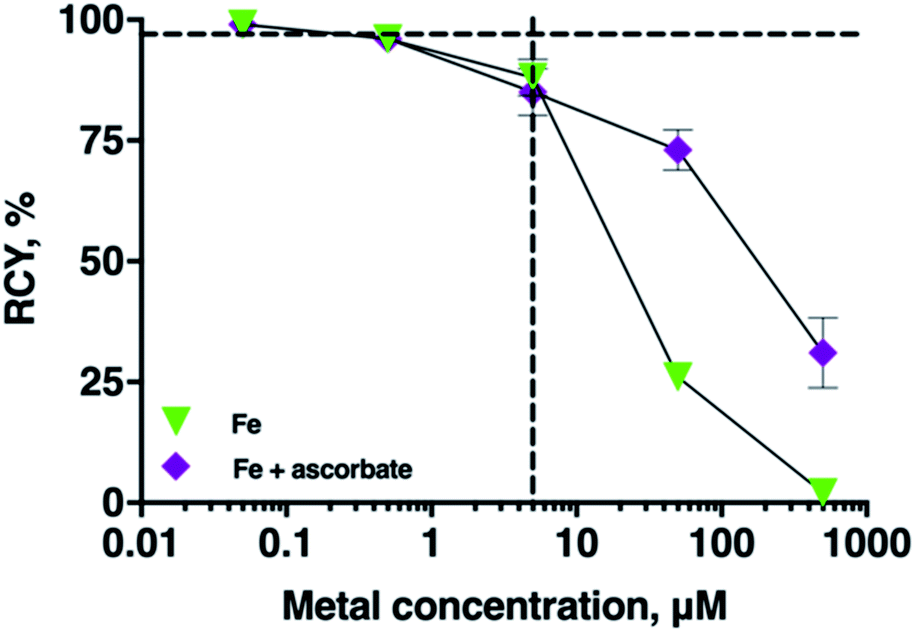 | ||
| Fig. 5 Mean RCYs of [68Ga(THP)] in the presence of either increasing concentrations of Fe3+ (green triangles) or ascorbate (166 mM) and increasing concentrations of Fe3+ (n = 5). The vertical dashed line indicates equimolar concentrations of chelator and metal ion (5 μM), and the horizontal dashed guide indicates 96 ± 0.4% (n = 6) RCY in the absence of spiked Fe3+. For most values, error bars representing standard deviation are smaller than the symbols. | ||
Radiolabelling THP with cation-exchange processed 68Ga
Numerous clinical 68Ga-radiolabelling protocols include eluate pre-processing steps in which 68Ga is isolated on a cation exchange cartridge, followed by a washing step and subsequent elution of 68Ga from the cartridge. These methods concentrate 68Ga and isolate 68Ga from (i) non-radioactive metal ion impurities (such as Fe, Ti and Zn) and (ii) 68Ge that is often present, albeit in very low amounts, in generator eluate containing high concentrations of HCl. There are three well-used methods that employ solutions of different composition to wash and to release 68Ga from the cartridge: the “NaCl method”, in which aqueous 68Ga solutions contain 5 M NaCl and 0.13 M HCl,33,38 the “ethanol method”, in which 68Ga solutions contain 90% ethanol and 0.9 M HCl,9 and the “acetone method”, in which 68Ga solutions contain 97.6% acetone and 0.05 M HCl.10The reactivity and speciation of 68Ga in these mixtures with organic solvents could have effects on radiolabelling efficiency. To assess the tolerance of THP radiolabelling with pre-processed 68Ga in the presence of other components in solution such as NaCl, ethanol and acetone, we reacted THP with solutions of 68Ga processed according to these three methods. In these aqueous reaction solutions (final volume 300 μL) containing pre-processed 68Ga eluate (7–19 MBq, 50 μL, containing either ethanol, acetone or NaCl eluate solutions), the final THP concentration was 5 μM or 0.5 μM, and pH = 7.
We compared the RCY of these reactions with the RCY of reactions using unprocessed 68Ga (eluate straight from the generator). At 5 μM THP concentration, the RCY of [68Ga(THP)] was unaffected by using 68Ga eluent processed using either the NaCl method or the ethanol method, with RCY >96% (Table 1). At 0.5 μM THP concentration, the RCY was unaffected for reactions containing 68Ga eluent processed using the ethanol method, and slightly improved for reactions containing 68Ga eluent processed using the NaCl method (Table 1). In contrast, in reactions containing 68Ga eluent processed using the acetone method, the RCY of [68Ga(THP)] was significantly decreased at both THP concentrations of 5 μM and 0.5 μM.
| THP, μM | RCY ± SD (n = 3), % | |
|---|---|---|
| Unprocessed 68Ga | 5.0 | 96 ± 0.22 |
| 0.5 | 59 ± 3.16 | |
| NaCl method | 5.0 | 96 ± 0.58 |
| 0.5 | 70 ± 0.30 | |
| Ethanol method | 5.0 | 96 ± 0.35 |
| 0.5 | 58 ± 3.33 | |
| Acetone method | 5.0 | 92 ± 3.03 |
| 0.5 | 31 ± 10.26 |
Discussion
Previous studies12,13,27–29 have shown that THP and hydroxypyridinones have particularly high affinity for hard, oxiphilic cations such as Al3+, Fe3+ and Ga3+, with markedly lower affinity for divalent metal ions such as Zn2+ and Cu2+. Although THP has shown remarkably high affinity for Ga3+ (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K1 = 35.0, and pGa = 30.0 at pH 7.4) and quantitative RCYs when reacted with 68Ga3+ at chelator concentrations as low as 0.5 μM, trace metal impurities, particularly hard, oxiphilic metal ions, have a potential to compete with 68Ga3+ for complexation to THP.12,13,19 For example, for [Fe(THP)], logK1 = 34.2, and pM = 29.1 at pH 7.4.13
K1 = 35.0, and pGa = 30.0 at pH 7.4) and quantitative RCYs when reacted with 68Ga3+ at chelator concentrations as low as 0.5 μM, trace metal impurities, particularly hard, oxiphilic metal ions, have a potential to compete with 68Ga3+ for complexation to THP.12,13,19 For example, for [Fe(THP)], logK1 = 34.2, and pM = 29.1 at pH 7.4.13
Our competition experiments showed that the presence of Al3+, Fe3+, natGa3+ and Ti4+ reduces RCY of [68Ga(THP)] at concentrations equimolar with THP and higher. Analytical ICP-MS studies indicate that all of these metals are present in generator eluate, and that the amounts can vary significantly with (i) the time between generator elutions and (ii) the age of the generator. These findings have implications for design of kits that contain THP chelator bioconjugates. At present, the GalliProst™ kit contains 40 μg of THP-PSMA, and when reconstituted by simple addition of unprocessed 68Ga generator eluate, THP-PSMA is at a concentration of 5 μM. Here, the range of concentrations of Ti (1.17–1.51 μM), Fe (0.07–0.09 μM), natGa (0.04–0.21 μM) and Al (0.61–0.91 μM) observed in eluate are all <5 μM. Our experience to date5,16 shows that the presence of these metals does not affect 68Ga radiolabelling using GalliProst™ kits. However, if lower amounts/concentrations of THP bioconjugates were to be used, the combination of Al, Fe, Ga and Ti in (unprocessed) 68Ga eluate could decrease RCY of 68Ga radiolabelling in such kits.
68Ga radiolabelling of THP is less susceptible to the presence of some metal ions (Zn2+, Ni2+, Pb2+) compared to DOTA. Prior work has shown that the presence of a range of metal ions significantly decrease RCY of 68Ga-DOTA-based derivatives.22,36,37 For example, in 68Ga-radiolabelling reactions of DOTA-TATE, a 2:1 Pb2+:DOTA-TATE molar ratio decreases RCY of 68Ga-DOTA-TATE to less than 80% (compared to near quantitative RCY in the absence of spiked metal ion). For Zn2+, a 6:1 ratio decreases RCY to below 80%. The presence of Zn2+ in 68Ga-radiolabelling reactions of NOTA also decreases RCY of [68Ga(NOTA)].36 Similar to the case of THP, RCYs of 68Ga-labelled DOTA and NOTA decrease in the presence of a molar excess of Fe3+ (relative to chelator). In contrast to THP, RCY of 68Ga-labelled DOTA and NOTA are less susceptible to the presence of Al3+.36
The multi-sample ICP-MS analysis reported here is (to date) the most comprehensive and robust quantification of metal ion impurities in the widely used E&Z 68Ga generator, although we note that eluates in this study were obtained from 12–18 months old E&Z 68Ga generators. Previous studies have quantified trace metal impurities in 68Ga generator eluate, including eluate from a Cyclotron Co. Ltd. generator (0.1 M HCl mobile phase, titanium dioxide solid phase, from Obninsk, Russia), a generator developed by ANSTO (Australia's Nuclear Science and Technology Organisation) and single eluate samples from E&Z 68Ga generators (0.1 M HCl mobile phase, titanium dioxide solid phase).22,34,39 The amounts of metal ion impurities in our multi-sample ICP-MS analyses are of a similar magnitude to previously reported data for eluate deriving from generators with a titanium dioxide solid phase.14,22,34 Our data on Al, Ti, Fe and Ga concentrations in eluates obtained six months apart from each other suggest that the concentration of these metal ions decreases over time/number of elutions.
We assessed whether the inclusion of other chemical components, such as ascorbate or organic solvents commonly used in 68Ga generator eluate pre-processing affect RCY of [68Ga(THP)]. Addition of ascorbate to 68Ga radiolabelling reactions did not result in decreased RCYs of [68Ga(THP)], and in this respect, it is a suitable radiolytic stabiliser for radiopharmaceuticals containing THP chelators. At Fe concentrations of 50 and 500 μM, the presence of ascorbate improved RCY of [68Ga(THP)], compared to reactions undertaken with a spike of Fe, but without ascorbate. We attribute this to Fe3+ reduction to Fe2+. Fe2+ has significantly lower affinity for hydroxypyridinones compared to Fe3+. Others' experimental data show that the rate of ascorbate reduction of Fe3+ to Fe2+ is pH dependent, and can decrease by an order of magnitude as the pH in aqueous media increases from 5 to 6.40 Over the timeframe of THP radiolabelling reactions (<5 min), and at Fe concentrations in generator eluate (<5 μM), ascorbate reduction of Fe3+ is unlikely to be of practical importance.
In some radiopharmacies, it has become standard practice to process 68Ga generator eluate, in order to separate 68Ga from 68Ge breakthrough. The presence of sodium chloride or ethanol in solutions of processed 68Ga eluate does not adversely affect RCY of [68Ga(THP)] compared to radiolabelling reactions in which unprocessed eluate is used, but the use of processed eluate containing acetone does result in a slight decrease in RCY and should be avoided in the context of THP-based radiopharmaceuticals.
Concluding remarks
Metal impurities commonly present in 68Ga generator eluate can deleteriously affect chelator 68Ga radiolabelling of various bifunctional chelator-derived radiopharmaceuticals.14,22,36,37 We have provided experimental evidence that 68Ga radiolabelling of THP is less susceptible to the presence of some of these metal ions (Zn2+, Cr3+, Ni2+, Pb2+) compared to other clinically used chelators such as DOTA.Other metal ions (Fe3+, Ti4+, Al3+, natGa3+) can interfere with THP radiolabelling if present at sufficient concentration. However, the low concentrations at which these metals occur in generator eluate, combined with the amount of THP-based conjugate currently used in prefabricated kits, results in quantitative RCY of these 68Ga–THP conjugates. If lower amounts/concentrations of THP bioconjugates were used, the combination of Al3+, Fe3+, natGa3+ and Ti4+ in unprocessed 68Ga eluate could decrease RCY of 68Ga radiolabelling in such kits, and kit design should take this into account.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by a Cancer Research UK Career Establishment Award (C63178/A24959), the Swiss National Science Foundation through an Advanced Postdoc Mobility Fellowship (P300P2-177855_1), the London Metallomics Facility funded by the Wellcome Trust Multi-user Equipment Grant (202902/Z/16/Z), the Centre for Medical Engineering funded by the Wellcome Trust and the Engineering and Physical Sciences Research Council (WT088641/Z/09/Z), the King's College London and University College London Comprehensive Cancer Imaging Centre funded by Cancer Research UK and EPSRC in association with the Medical Research Council and Department of Health (England), and the National Institute for Health Research Biomedical Research Centre based at Guy's and St. Thomas' NHS Foundation Trust and King's College London (KCL). The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.References
- M. Fani, H. R. Maecke and S. M. Okarvi, Theranostics, 2012, 2, 481–501 CrossRef CAS PubMed.
- M. Fani, P. K. Peitl and I. Velikyan, Pharmaceuticals, 2017, 10, 30 CrossRef PubMed.
- I. Velikyan, Theranostics, 2014, 4, 47–80 CrossRef CAS PubMed.
- D. Prince, D. Rossouw, C. Davids and S. Rubow, Mol. Imaging Biol., 2017, 19, 817–824 CrossRef CAS PubMed.
- J. D. Young, V. Abbate, C. Imberti, L. K. Meszaros, M. T. Ma, S. Y. A. Terry, R. C. Hider, G. E. Mullen and P. J. Blower, J. Nucl. Med., 2017, 58, 1270–1277 CrossRef CAS PubMed.
- A. Tirosh, G. Z. Papadakis, C. Millo, D. Hammoud, S. M. Sadowski, P. Herscovitch, K. Pacak, S. J. Marx, L. Yang, P. Nockel, J. Shell, P. Green, X. M. Keutgen, D. Patel, N. Nilubol and E. Kebebew, Gastroenterology, 2018, 154, 998–1008 CrossRef PubMed.
- M. McCarthy, T. Langton, D. Kumar and A. Campbell, Eur. J. Nucl. Med. Mol. Imaging, 2017, 44, 1455–1462 CrossRef CAS PubMed.
- M. Eder, O. Neels, M. Mueller, U. Bauder-Wuest, Y. Remde, M. Schaefer, U. Hennrich, M. Eisenhut, A. Afshar-Oromieh, U. Haberkorn and K. Kopka, Pharmaceuticals, 2014, 7, 779–796 CrossRef CAS PubMed.
- E. Eppard, M. Wuttke, P. L. Nicodemus and F. Rosch, J. Nucl. Med., 2014, 55, 1023–1028 CrossRef CAS PubMed.
- K. P. Zhernosekov, D. V. Filosofov, R. P. Baum, P. Aschoff, H. Bihl, A. A. Razbash, M. Jahn, M. Jennewein and F. Rosch, J. Nucl. Med., 2007, 48, 1741–1748 CrossRef CAS PubMed.
- M. T. Ma, C. Cullinane, K. Waldeck, P. Roselt, R. J. Hicks and P. J. Blower, EJNMMI Res., 2015, 5, 11 CrossRef PubMed.
- R. Cusnir, C. Imberti, R. C. Hider, P. J. Blower and M. T. Ma, Int. J. Mol. Sci., 2017, 18, 23 Search PubMed.
- C. Imberti, Y. L. Chen, C. A. Foley, M. T. Ma, B. M. Paterson, Y. F. Wang, J. D. Young, R. C. Hider and P. J. Blower, Dalton Trans., 2019, 48, 4299–4313 RSC.
- M. I. Tsionou, C. E. Knapp, C. A. Foley, C. R. Munteanu, A. Cakebread, C. Imberti, T. R. Eykyn, J. D. Young, B. M. Paterson, P. J. Blower and M. T. Ma, RSC Adv., 2017, 7, 49586–49599 RSC.
- D. Lobeek, G. M. Franssen, M. T. Ma, H. J. Wester, C. Decristoforo, W. J. G. Oyen, O. C. Boerman, S. Y. A. Terry and M. Rijpkema, J. Nucl. Med., 2018, 59, 1296–1301 CrossRef CAS PubMed.
- M. S. Hofman, P. Eu, P. Jackson, E. Hong, D. Binns, A. Iravani, D. Murphy, C. Mitchell, S. Siva, R. J. Hicks, J. D. Young, P. J. Blower and G. E. Mullen, J. Nucl. Med., 2018, 59, 625–631 CrossRef CAS PubMed.
- M. T. Ma, C. Cullinane, C. Imberti, J. Baguna Torres, S. Y. A. Terry, P. Roselt, R. J. Hicks and P. J. Blower, Bioconjugate Chem., 2016, 27, 309–318 CrossRef CAS PubMed.
- S. Nawaz, G. E. D. Mullen, K. Sunassee, J. Bordoloi, P. J. Blower and J. R. Ballinger, EJNMMI Res., 2017, 7, 9 CrossRef PubMed.
- C. Imberti, S. Y. A. Terry, C. Cullinane, F. Clarke, G. H. Cornish, N. K. Ramakrishnan, P. Roselt, A. P. Cope, R. J. Hicks, P. J. Blower and M. T. Ma, Bioconjugate Chem., 2017, 28, 481–495 CrossRef CAS PubMed.
- I. Velikyan, J. Labelled Compd. Radiopharm., 2015, 58, 99–121 CrossRef CAS PubMed.
- I. Velikyan, Molecules, 2015, 20, 12913–12943 CrossRef CAS PubMed.
- E. Oehlke, V. Le, N. Lengkeek, P. Pellegrini, T. Jackson, I. Greguric and R. Weiner, Appl. Radiat. Isot., 2013, 82, 232–238 CrossRef CAS PubMed.
- C. H. Taliaferro and A. Martell, Inorg. Chim. Acta, 1984, 85, 9–15 CrossRef CAS.
- J. Šimeček, M. Schulz, J. Notni, J. Plutnar, V. Kubíček, J. Havlíčková and P. Hermann, Inorg. Chem., 2012, 51, 577–590 CrossRef PubMed.
- R. Grazina, L. Gano, J. Sebestik and M. A. Santos, J. Inorg. Biochem., 2009, 103, 262–273 CrossRef CAS PubMed.
- Z. D. Liu and R. C. Hider, Med. Res. Rev., 2002, 22, 26–64 CrossRef CAS PubMed.
- E. T. Clarke and A. E. Martell, Inorg. Chim. Acta, 1992, 191, 57–63 CrossRef.
- S. Chaves, S. M. Marques, A. M. F. Matos, A. Nunes, L. Gano, T. Tuccinardi, A. Martinelli and M. A. Santos, Chem.–Eur. J., 2010, 16, 10535–10545 CrossRef CAS PubMed.
- M. A. Santos, M. Gil, L. Gano and S. Chaves, J. Biol. Inorg Chem., 2005, 10, 564–580 CrossRef CAS PubMed.
- M. Brom, L. Joosten, W. J. G. Oyen, M. Gotthardt and O. C. Boerman, EJNMMI Res., 2012, 2, 4 CrossRef PubMed.
- J. Notni, K. Pohle and H.-J. Wester, EJNMMI Res., 2012, 2(28), 25 Search PubMed.
- D. J. Berry, Y. M. Ma, J. R. Ballinger, R. Tavare, A. Koers, K. Sunassee, T. Zhou, S. Nawaz, G. E. D. Mullen, R. C. Hider and P. J. Blower, Chem. Commun., 2011, 47, 7068–7070 RSC.
- D. Mueller, I. Klette, R. P. Baum, M. Gottschaldt, M. K. Schutz and W. A. P. Breeman, Bioconjugate Chem., 2012, 23, 1712–1717 CrossRef CAS PubMed.
- I. Velikyan, G. J. Beyer and B. Langstrom, Bioconjugate Chem., 2004, 15, 554–560 CrossRef CAS PubMed.
- S. Chaves, R. Delgado and J. J. R. F. Da Silva, Talanta, 1992, 39, 249–254 CrossRef CAS PubMed.
- J. Šimeček, P. Hermann, H.-J. Wester and J. Notni, ChemMedChem, 2013, 8, 95–103 CrossRef PubMed.
- R. Chakravarty, S. Chakraborty, A. Dash and M. R. A. Pillai, Nucl. Med. Biol., 2013, 40, 197–205 CrossRef CAS PubMed.
- D. Mueller, W. A. P. Breeman, I. Klette, M. Gottschaldt, A. Odparlik, M. Baehre, I. Tworowska and M. K. Schultz, Nat. Protoc., 2016, 11, 1057–1066 CrossRef CAS PubMed.
- P. He, B. P. Burke, G. S. Clemente, N. Brown, N. Pamme and S. J. Archibald, React. Chem. Eng., 2016, 1, 361–365 RSC.
- Y. H. P. Hsieh and Y. P. Hsieh, J. Agric. Food Chem., 2000, 48, 1569–1573 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07723e |
| This journal is © The Royal Society of Chemistry 2019 |