
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDegradation of tetrabromobisphenol A by a ferrate(VI)–ozone combination process: advantages, optimization, and mechanistic analysis†
Qi Hana,
Wenyi Dongac,
Hongjie Wang *ac,
Hang Mab,
Yurong Gud and
Yu Tiana
*ac,
Hang Mab,
Yurong Gud and
Yu Tiana
aSchool of Civil and Environmental Engineering, Harbin Institute of Technology (Shenzhen), Shenzhen, 518055, China. E-mail: hanhanqiqi1986@163.com; dwy1967@qq.com; whj1533qihan@163.com; Fax: +86 755 26033482; Tel: +86 755 26033482
bSchool of Architecture, Harbin Institute of Technology (Shenzhen), Shenzhen, 518055, China. E-mail: mahang@hit.edu.cn
cShenzhen Key Laboratory of Water Resource Utilization and Environmental Pollution Control, Shenzhen, 518055, China
dSchool of Construction and Environmental Engineering, Shenzhen Polytechnic, Shenzhen, 518055, China
First published on 17th December 2019
Abstract
This study systematically investigated the ferrate(VI)–ozone combination process for TBBPA degradation. Firstly, the advantages of a ferrate(VI)–ozone combination process were assessed as compared with a sole ozone and ferrate(VI) oxidation process. Then, the performance of the ferrate(VI)–ozone combination process was investigated under different experimental conditions, including the dosing orders of oxidants, dosing concentrations of oxidants, and the initial solution pH. At the same time, toxicity control (including the acute and chronic toxicity) and mineralization were analyzed after optimization. Finally, a mechanism was proposed about the synergetic effects of the ferrate(VI)–ozone combination process for decontamination. The ferrate(VI)–ozone combination process proved to be an efficient and promising technology for removing TBBPA from water. After being pre-oxidized by ferrate(VI) for 3 min and then co-oxidized by the two oxidants, TBBPA of 1.84 μmol L−1 could be completely degraded by dosing only 0.51 μmol L−1 of ferrate(VI) and 10.42 μmol L−1 of ozone within 10 min in wide ranges of pH (5.0–11.0). Up to 91.3% of debromination rate and 80.5% of mineralization rate were obtained, respectively. In addition, no bromate was detected and the acute and chronic toxicity were effectively controlled. The analysis of the proposed mechanism showed that there might exist a superposition effect of the oxidation pathways. In addition, the interactions between the two oxidants were beneficial for the oxidation efficiency of ferrate(VI) and ozone, including the catalytic effect of ferrate(VI) intermediates on ozone and the oxidation of low-valent iron compounds by ozone and the generated ·OH radical.
1. Introduction
As one of the most important brominated flame retardants (BFRs), tetrabromobisphenol A (TBBPA) has been widely used in the manufacturing of building materials, electronic products, plastics, textiles, et al.1 Owing to its extensive application and environmental persistency, TBBPA has frequently been detected in various environmental and biological matrices, such as water, sediments, air, aquatic organisms, animals and even human body.2,3 Moreover, toxicological researches have showed that TBBPA might induce severe damage to large fleas, fish, mice, and human transthyretin cells at a low concentration.4–6 It is imperative and significant to develop methods to remove TBBPA from the environment efficiently.At present, methods applied for the elimination of TBBPA mainly include biological degradation, adsorption, Fenton oxidation, photocatalytic oxidation,7–10 Nevertheless, the above technologies have some inherent drawbacks more or less, such as long periods, high costs, big sludge yields and difficulty in operations. In contrast, because of its reasonable cost performance and easy engineering implementation, ozonation has been considered as an efficient technology in practical application of bacteria sterilization, drinking water disinfection and removal of refractory organic pollutants11–13 have briefly investigated the degradation effect of ozonation on TBBPA and revealed that TBBPA could be quickly and effectively removed by ozonation, with the removal rate of TBBPA (50 mg L−1) reaching up to 99.3% under the ozone dosage of 52.3 mg h−1. However, due to the high mass ratio of bromine element (about 58.8%), the free bromide ion (Br−) produced by the debromination process might be further oxidized by ozone to form the by-product bromate (BrO3−), which is of genotoxic and carcinogenic properties.14 Moreover, many researches have showed that more toxic brominated intermediates might be generated during the degradation of TBBPA, such as tribromobisphenol A (Tri-BBPA), dibromobisphenol A (Di-BBPA), monobromobisphenol A, monobromophenol, dibromophenol, etc15–17, which might lead to the increase of the overall biological toxicity of the water samples. Thus, the sole degradation method cannot simultaneously solve the problems of efficiently degrading TBBPA and controlling the formation of organic or inorganic toxic products.
In recent years, some ozone combined technologies has attracted the attentions of scholars in terms of their good synergistic effect in decontamination and controlling by-products, such as O3-UV/VUV,18 O3–H2O2,19 KMnO4–O3.20 However, there still exist some irresistible defects of these combining methods. For example, the lamp used in the process of O3-UV/VUV was required below 200 nm; otherwise the inhibition of bromate might be not obvious. In addition, the VUV lamp is of high production cost and short service life, which limit the engineering application of this process. The required reaction conditions of the two other combined technologies were very harsh and needed to be strictly controlled. During the process of O3–H2O2, the value of H2O2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O3 and the dissolved ozone concentration was required to be bigger than 0.5 and less than 0.1 mg L−1, respectively, otherwise the bromate would increase. During the process of KMnO4–O3, only 26% of the formed bromate was decreased, which was not significant. In addition, the dosage of KMnO4 needs to be controlled less than 2.0 mg L−1, or the controlling effect of bromate would decrease and the concentration of heavy metal Mn in the effluent might exceed the standard (0.1 mg L−1).
:O3 and the dissolved ozone concentration was required to be bigger than 0.5 and less than 0.1 mg L−1, respectively, otherwise the bromate would increase. During the process of KMnO4–O3, only 26% of the formed bromate was decreased, which was not significant. In addition, the dosage of KMnO4 needs to be controlled less than 2.0 mg L−1, or the controlling effect of bromate would decrease and the concentration of heavy metal Mn in the effluent might exceed the standard (0.1 mg L−1).
In recent years, as a stronger oxidant than ozone, ferrate(VI) has been applied for degradation of various persistent organic compounds, such as personal care products (PCPS),21 endocrine disrupting chemicals (EDCs),22 pharmaceuticals23 and microcystins,23 et al. Data of the researches have indicated that ferrate(VI) oxidation was an efficient technique for pollution control.24,25 In addition, ferrate(VI) could avoid the formation of chlorinated DBPs and bromate, which are the by-products of chlorination and ozonation processes.26,27 Thus, the degradation of TBBPA by ferrate(VI) oxidation has been systematically investigated in our early studies.28 A 99.06% removal of TBBPA (1.84 μmol L−1) has been achieved via 30 min contacting reactions, with a ferrate(VI) dosage of 25.25 μmol L−1, initial pH of 7.0, and temperature of 25 °C. However, due to the high preparation cost and the instability in water of ferrate(VI), it was still not much of applying the sole ferrate(VI) oxidation in practical engineering. Recent, in order to reduce the cost, ferrate(VI) has been combined with other oxidants or methods, such as hypochlorite,29 hydrogen peroxide,30 ozone,31 photocatalytic oxidation,32,33 et al. The combination of ferrate(VI) and ozone has been certified to have a synergistic effect on sterilization. An ozone dose of 41.67 μmol L−1 should been required for inactivation of 99% enterobacterin; while only 20.83 μmol L−1 of ozone was necessary after pre-oxidation by ferrate(VI).34 According to the literature survey, the systematic study is still very few about the ferrate(VI)–ozone combination process, whose performance and relevant mechanism remains to be further studied.
In our previous studies, the systematic experiments have been carried out to investigate the controlling effect of bromate by ferrate(VI)–ozone combination process,35 which provided a basis for treating TBBPA contaminated water. The result indicated that bromate could be completely inhibited under wide conditions of ozone concentration (≤52.08 μmol L−1), initial bromide ion concentration (≤200 μg L−1), pH (3.0–9.0) and temperature (5–40 °C) with only 5.05 or 10.10 μmol L−1 ferrate(VI) being needed. Moreover, the controlling effect would be promoted with the increase of ferrate(VI) dosage. Thus, in the present study, the degradation of TBBPA by ferrate(VI)–ozone combination process was systematically investigated. Firstly, the advantages of ferrate(VI)–ozone combination process were concluded, such as the synergistic degradation of TBBPA, the high debromination level, the effective control of the toxicity and bromate. Then, the operating parameters were optimized, including the adding orders of oxidants, the adding concentrations of oxidants, and initial solution pH. Based on the optimization, the control of toxicities (acute and chronic toxicity) and the mineralization of TBBPA were further analyzed. At last, the possible mechanisms were proposed of the ferrate(VI)–ozone combination process.
2. Experimental section
2.1 Materials
TBBPA (98%, Aladdin) and ferrate(VI) (K2FeO4, purity ≥ 99%, Sigma-Aldrich, USA) were purchased and used without further purification. The solution of TBBPA and ferrate(VI) with desired concentrations were both prepared prior to experiments. The TBBPA powder was dissolved in 0.5% methanol solution, whose influence on TBBPA removal could be ignored. The ferrate(VI) solution was maintained at pH 9.0 with buffer (0.005 M Na2HPO4 and 0.001 M Na2B4O7·9H2O).36 Ozone dosed in this study was the saturated ozone water which was prepared by the method described in our earlier research.35 The other chemicals and reagents used in the experiments were of chromatographic or analytical grade. All reaction solutions were prepared with deionized and ultra pure water (Milli-Q Direct 8, USA).The acute toxicity was tested by using freeze-dried bacteria Vibro fischeri (V. fischeri), which was obtained from the manufacturers (DeltaTox, SDIX, USA; Moltox, USA) and stored at −20 °C. The chronic toxicity assessment (21 d) was carried out with Daphnia magna (D. magna), which was introduced from South China Institute of Environmental Sciences and was cultivated and domesticated for a long-term period in our laboratory. The young age fleas used for the experiments was cultured for three generations and with the age of 24 h, whose sensitivity determination was complied with the ISO standards.37
2.2 Experimental methods
Series of batch experiments for TBBPA degradation were conducted in 1000 mL conical beakers. The pH was adjusted by 1.2 M HCl or NaOH solution and the temperature was controlled by thermostatic water bath. The reaction system was started by adding calculated volume of oxidant into the reactor and stirred by magnetic stirrer (600 rpm). At certain intervals, 20 mL water samples were taken out and terminated by 0.5 mL 0.2 mM hydroxylamine hydrochloride solution. Then, the samples were centrifuged at 12000 rpm for 5 min prior to subsequent analysis. All the experiments were carried out in duplicate.
2.3 Analytical methods
The residual concentration of TBBPA was directly analyzed by a Waters Acquity H-class Ultra Performance Liquid Chromatography (UPLC) equipped with a Waters BEH C18 column (1.7 × 100 mm, 3.5 μm) and a TUV detector. The determination of ferrate(VI)'s concentration was based on the ABTS method.38 The principle was that ferrate(VI) could react with ABTS to form a stable green free radical ABTS˙+, which has specific absorption at 415 nm. The increase of the absorbance is linear with the increase of the concentration of ferrate(VI). The method and instrument used for analysis of formed Br−/BrO3− were the ion chromatography and a Dionex ICS-5000. The specific test methods have been reported in our previous studies.28,35A DeltaTox II luminometer (SDIX, USA) was applied for detecting the acute toxicities of the water samples. The method is accorded to the ISO standard and based on the inhibition of bioluminescence emitted by the luminescent bacteria V. fischeri.39 The inhibition of light emission was measured after a sample contact period of 15 min. Thereby the relative inhibitory rate (T%) was calculated based on the recorded normalized bioluminescence intensities (E). The chronic toxicities of samples were detected by the standard method of D. magna 21 d chronic toxicity test following OECD guidelines.40 The neonates (<24 h) of D. magna were exposed for 21 d to the reaction samples and the maximum non-observed effect concentration (NOEC) was obtained. Then, the chronic toxicity was converted to the toxic equivalent values by the formula (TU = 100%/NOEC), which was introduced by US Federal Environmental Protection Agency (USEPA) and expressed in terms of toxicity units (TU).41 The specific toxicity testing methods described above were detailed in ESI (Text S1).†
3. Results and discussion
3.1 Advantages of the ferrate(VI)–ozone combination process
The three oxidation systems (sole ozonation, sole ferrate(VI) oxidation and simultaneous oxidation) were compared from three aspects, including the degradation, mineralization and debromination of TBBPA, the formation and control of bromate, the control of toxicity. Based on the comparison, the advantages of the ferrate(VI)–ozone combination process were summarized, which could prepare for further optimization of the process.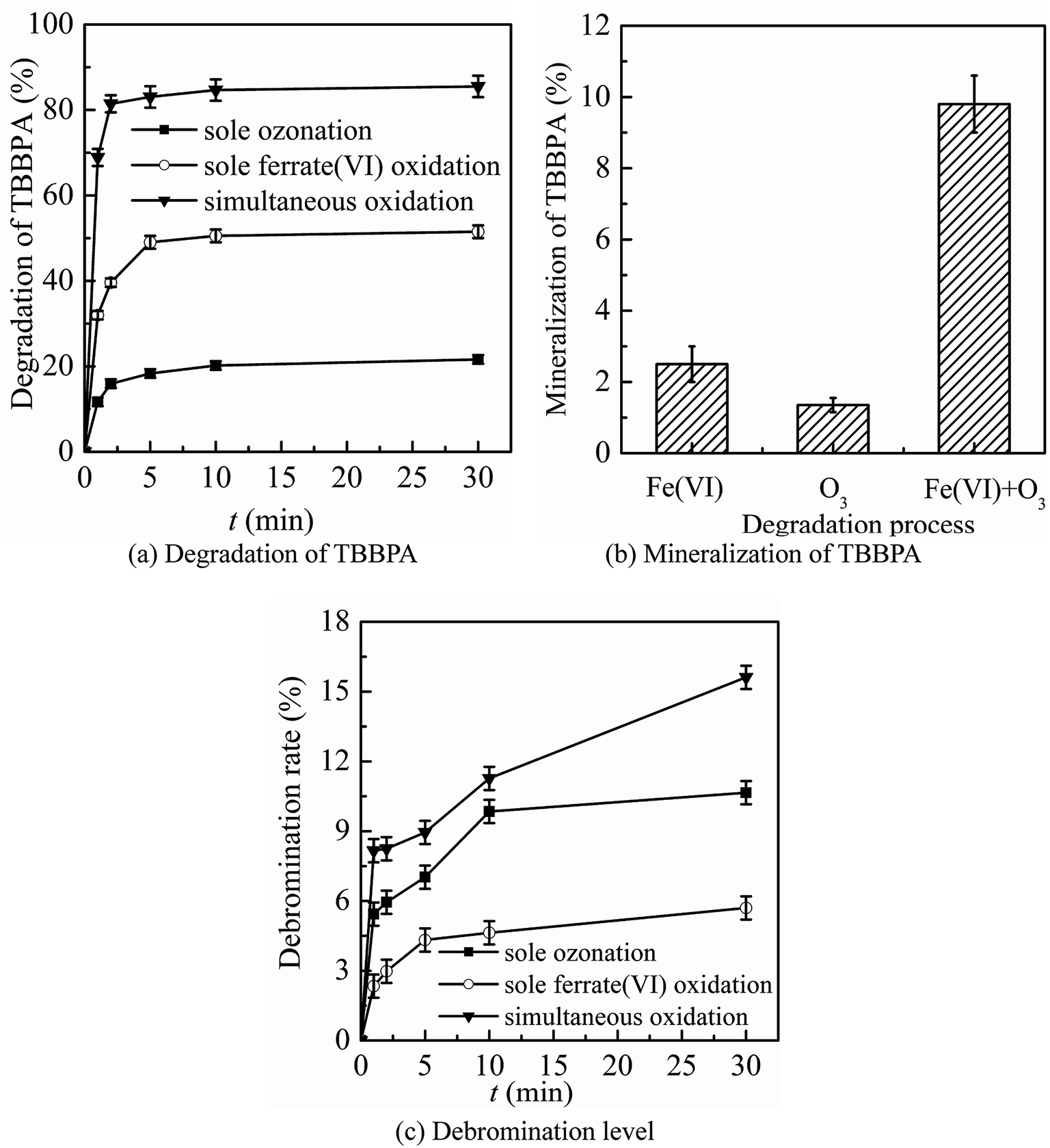 | ||
| Fig. 1 The synergistic effect of ferrate(VI) and ozone in the aspects of (a) degradation of TBBPA, (b) mineralization of TBBPA, (c) debromination level (experimental conditions: TBBPA concentration = 1.84 μmol L−1; ferrate(VI) concentration = 0.51 μmol L−1; ozone concentration = 0.51 μmol L−1; initial solution pH = 7.0; temperature = 25 ± 0.5 °C). | ||
It can be seen from Fig. 1(a) and (b) that the sole ferrate(VI) oxidation process had a stronger degradation effect on TBBPA than that in sole ozonation process under the same experimental conditions. Within 1 min reactions, the degradation rates of TBBPA by the sole ozonation and sole ferrate(VI) oxidation were 11.7% and 32.0%, respectively. After contacting for 30 min, the removals of TBBPA during the two oxidation systems reached to 21.6% and 51.5%, respectively. However, the degradation effect on TBBPA by simultaneous oxidation (68.9% and 85.5%) was much greater than the sum of the individual process effects (43.7% and 73.1%), which indicated the synergistic role of the two oxidants. In addition, this significant synergistic effect was also reflected in the mineralization of TBBPA. As seen from Fig. 1(b), the mineralization rate of TBBPA in simultaneous oxidation system was up to 9.8%, which was much higher than that of the sum of other two processes (3.9%).
As the mass ratio of bromine element is as high as 58.8%, it is an important part of TBBPA molecular structure. It has been confirmed in many studies28,42–44 that debromination process was one of the major degradation mechanisms of TBBPA, during which the free bromide ion (Br−) was formed. To some extent, the debromination rate indirectly reflected the degradation of TBBPA. As shown in Fig. 1(c), compared to the sole oxidation processes, the ferrate(VI)–ozone combination process also had higher level of debromination, which were 8.2% and 15.6% respectively at 1 min and 30 min. Thus, it further indicated the synergistic effect of ferrate(VI) and ozone. The ferrate(VI)–ozone combination process could maintain high debromination level as well as efficiently degrading TBBPA, which solved the problem of low debromination effect in sole ferrate(VI) oxidation process. However, compared to the high efficiency removal of TBBPA, there exists a hysteresis effect on the yield of free bromide, which was caused by the formation of a large amount of organic brominated intermediates.28,45,46 The generated free bromide might react with other organic products to form new brominated intermediates. As the low brominated organic intermediates being further degraded, the free bromide ions in the reaction system would gradually increase.
The preliminary analysis showed that the synergistic effect of ferrate(VI) and ozone could be attributed to the mutual chemical reactions between each other. On the one hand, it has been proved that the reduced intermediates of ferrate(VI) (such as hydrated iron ions, hydrated iron oxides and iron oxyhydroxide) had a catalyze role on ozone to generate more ·OH,47 which was beneficial to the degradation reactions. On the other hand, the reduction products of ferrate(VI) (Fe(III)) or (Fe(II)) might also be oxidized by the radical species of O2˙ to a high-valent iron-containing oxidant (Fe(V)),48 which could further degrade TBBPA. In summary, the corresponding oxidation efficiency of ferrate(VI) and ozone was improved in the combination system.
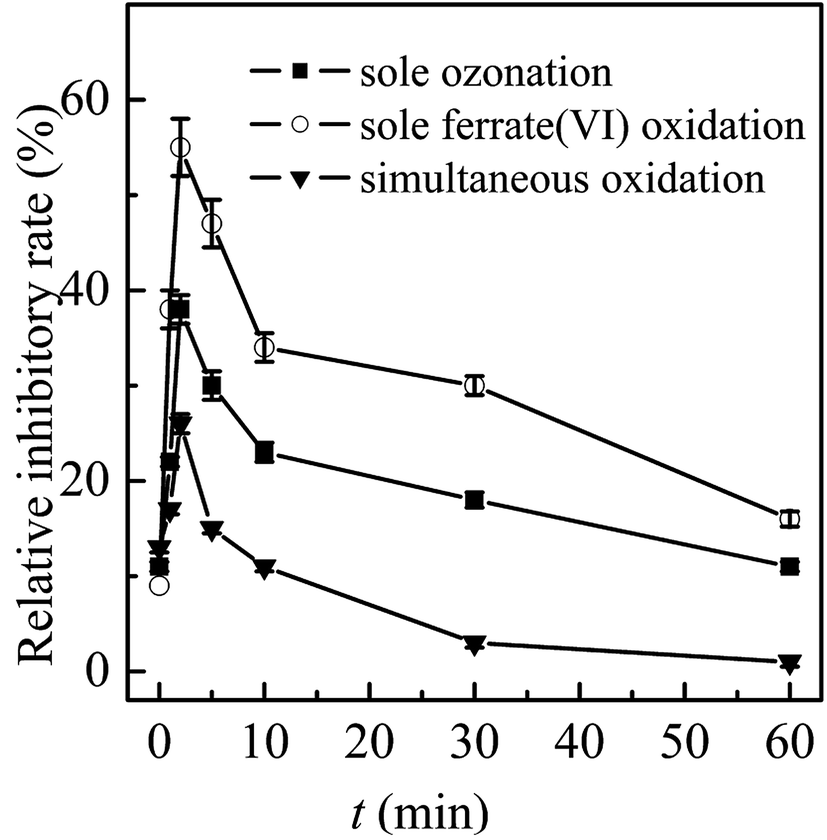 | ||
| Fig. 2 Toxicity control in the three oxidation systems. (Experimental conditions: TBBPA concentration = 1.84 μmol L−1; ferrate(VI) concentration = 0.51 μmol L−1; ozone concentration = 0.51 μmol L−1; initial solution pH = 7.0; temperature = 25 ± 0.5 °C). | ||
In summary, by comparing with the sole ozonation and sole ferrate(VI) oxidation process, it could be seen that the ferrate(VI)–ozone combination process has following advantages: the synergistic effect on degradation and mineralization of TBBPA, the high debromination level, the efficient control of by-product bromate and toxicity, which solved the problems of degrading TBBPA by the sole oxidation process.
3.2 Performance of the ferrate(VI)–ozone combination process
The main conditions of the ferrate(VI)–ozone combination process were further optimized, including the dosing order of oxidants, the dosing concentration of oxidants and the initial solution pH, with the degradation rate and debromination rate of TBBPA being selected as the indicators.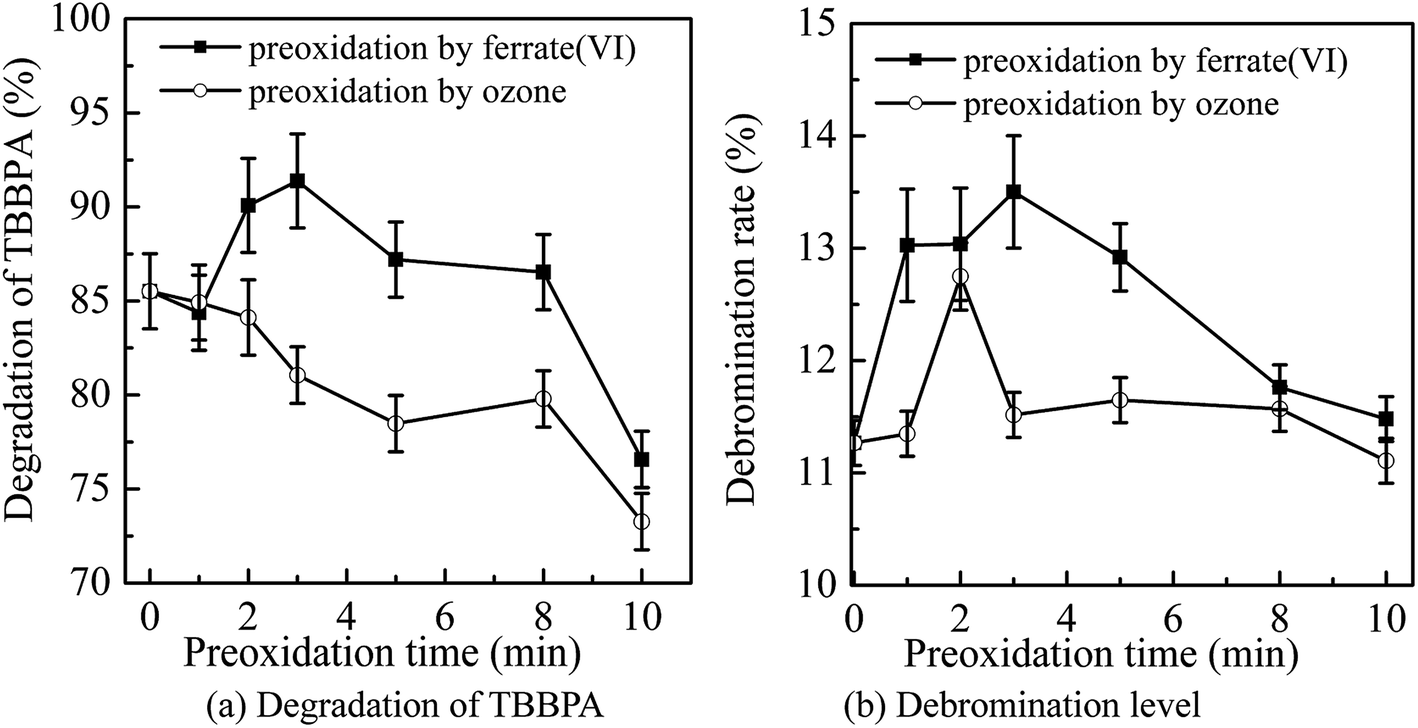 | ||
| Fig. 3 Optimization of dosing order of oxidants through detection of (a) degradation of TBBPA and (b) debromination level. (Experimental conditions: TBBPA concentration = 1.84 μmol L−1; ferrate(VI) concentration = 0.51 μmol L−1; ozone concentration = 0.51 μmol L−1; initial solution pH = 7.0; temperature = 25 ± 0.5 °C). | ||
It can been seen from Fig. 3 that the degradation and debromination rate of TBBPA in ferrate(VI) pre-oxidation system were both higher than that in ozone pre-oxidation one. In the ferrate(VI) pre-oxidation system, the degradation and debromination rate of TBBPA both increased first and then decreased, with the maximum of 91.4% and 13.5%, respectively. And the preferred pre-oxidation time was 3 min. While in the ozone pre-oxidation system, as the pre-oxidation time increased from 1 min to 10 min, the degradation rate of TBBPA gradually decreased from 84.9% to 73.2%, and the debromination rate fluctuated between 11.5%.
As mentioned earlier, there existed a synergistic effect between the two oxidants in ferrate(VI)–ozone combination process, which was caused by the catalytic role of ferrate(VI) reduction products on ozone, and the oxidation of low-valent iron compounds by oxygen free radicals (O2˙). However, the catalytic effect was weakened by the way of ozone pre-oxidation. Moreover, ozone might be more activity to organic substances than inorganic iron compounds, which also resulted in the unsatisfactory degradation effect of TBBPA. As for the pre-oxidation by ferrate(VI), the synergistic effect could be enhanced via the sufficient interactions between the two oxidants. However, the catalytic effect would be weakened if the pre-oxidation time of ferrate(VI) was too long. Therefore, in the subsequent studies of ferrate(VI)–ozone combination process, TBBPA was firstly oxidized by ferrate(VI) for 3 min, and then co-degraded by ferrate(VI) and ozone.
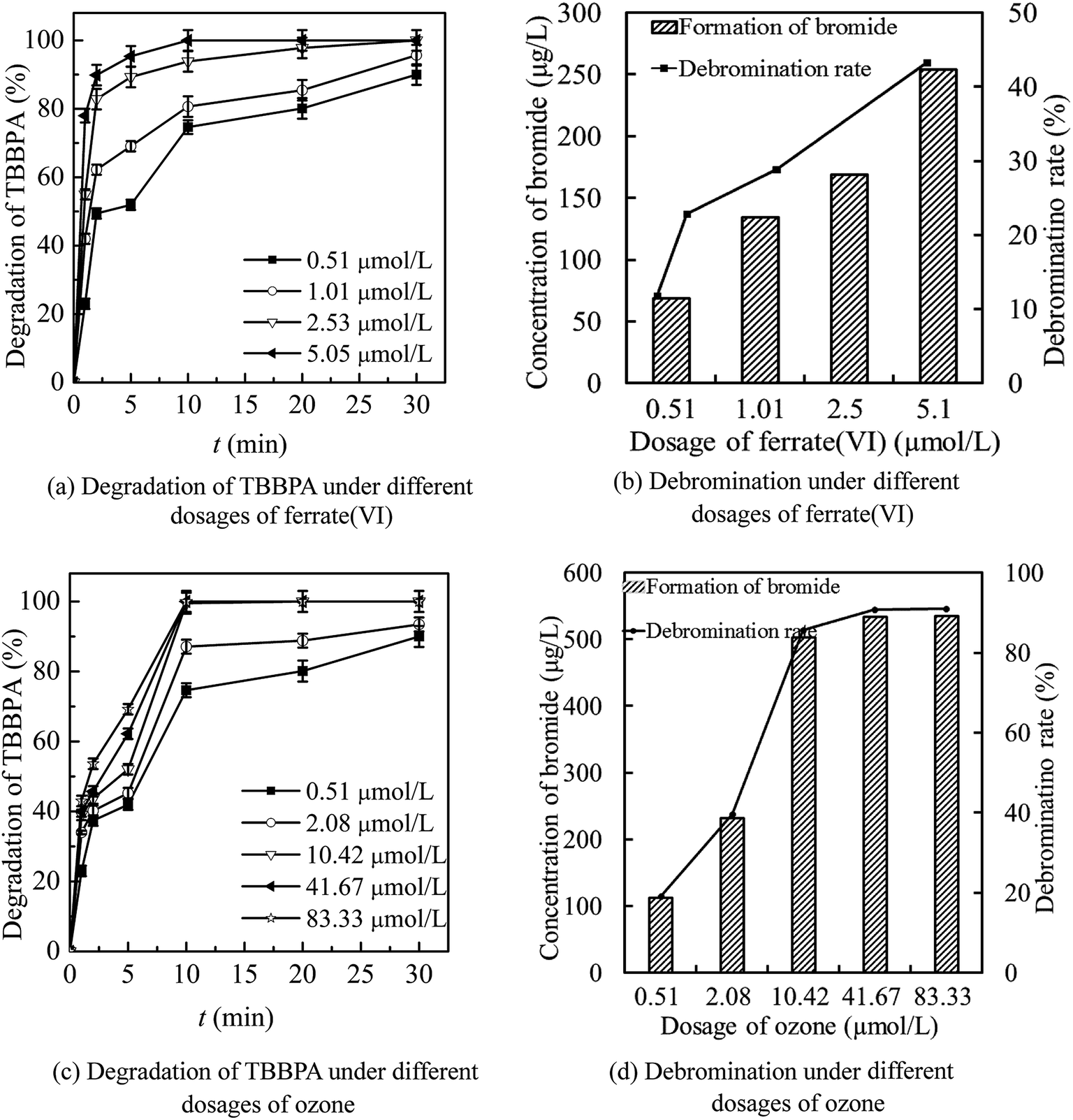 | ||
| Fig. 4 Optimization of dosing concentration of ferrate(VI) ((a) and (b)) and ozone ((c) and (d)) by detection the degradation of TBBPA ((a) and (c)) and the debromination level ((b) and (d)). (Experimental conditions: TBBPA concentration = 1.84 μmol L−1; initial solution pH = 7.0; temperature = 25 ± 0.5 °C; ferrate(VI) concentration = 0.51–5.05 μmol L−1; ozone concentration = 0.51–83.33 μmol L−1). | ||
As seen in Fig. 4(a) and (b), the increase of ferrate(VI) dosage was beneficial to the degradation of TBBPA by ferrate(VI)–ozone combination process. The degradation rate of TBBPA increased gradually from 90.1% to 100% as the ferrate(VI) dosage increased from 0.51 to 2.53 μmol L−1. The corresponding bromide concentration and debromination rate increased from 70.9 μg L−1 and 12.1% to 169.4 μg L−1 and 28.8%, respectively. In addition, the reaction time required for complete removal of TBBPA was reduced from 30 min to 10 min when the ferrate(VI) dosage increased to 5.05 μmol L−1. However, the debromination rate only was increased to 43.3% and still low, which indicated that only increasing the dosage of ferrate(VI) did not contribute much to the improvement of debromination effect. In view of the high economic cost of ferrate(VI) and the significant degradation effect of TBBPA at lower concentration of ferrate(VI) (0.51 μmol L−1), this dosage would be chosen for the following investigations of the ferrate(VI)–ozone combination process.
Data of Fig. 4(c) and (d) illustrated that increasing the dosage of ozone was more conducive to the increase of debromination rate. As the ozone dosage increased from 0.51 to 2.08 μmol L−1, the degradation rate of TBBPA increased from 90.1% to 93.5% after 30 min contacting reactions with ferrate(VI) and ozone. Correspondingly, the concentration of free bromide and the debromination rate increased from 70.9 μg L−1 and 12.1% to 233.0 μg L−1 and 39.6%, respectively. When the ozone concentration continued to increase to 10.42 μmol L−1, TBBPA could be completely removed within 10 min and the concentration of bromide was 504.1 μg L−1, with a high debromination rate of 85.7%. Moreover, the debromination rate could be further increased to 91.0% by increasing the ozone dosage to 83.33 μmol L−1. However, the by-product bromate as high as 25.22 μg L−1 was detected at the same time. Thus, after the comprehensive analysis of the degradation rate of TBBPA, the debromination rate and the formation risk of by-product bromate, 10.42 μmol L−1 was determined as the preferred dosage of ozone.
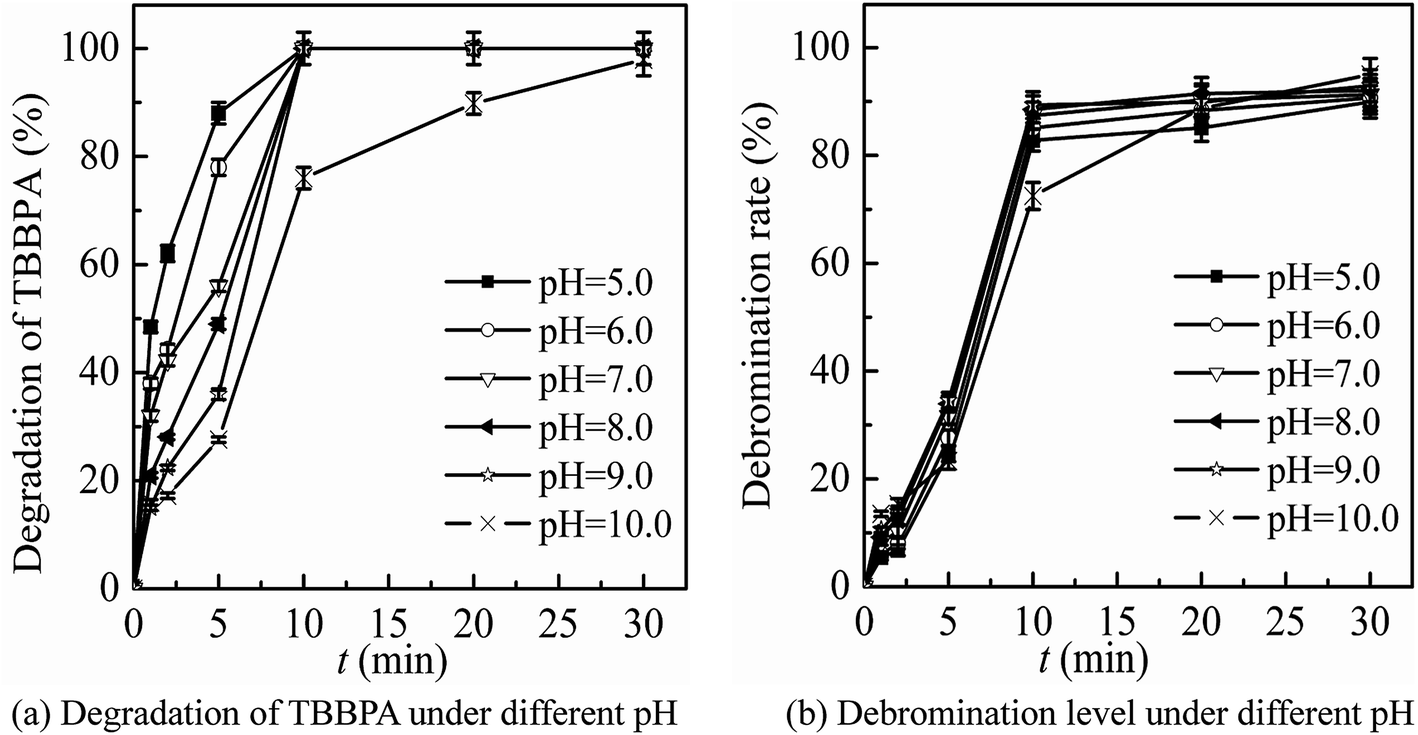 | ||
| Fig. 5 Optimization of the initial solution pH by detection of the degradation of TBBPA (a) and the debromination level (b). (Experimental conditions: TBBPA concentration = 1.84 μmol L−1; ferrate(VI) concentration = 0.51 μmol L−1; ozone concentration = 10.42 μmol L−1; initial solution pH = 5.0–10.0; temperature = 25 ± 0.5 °C). | ||
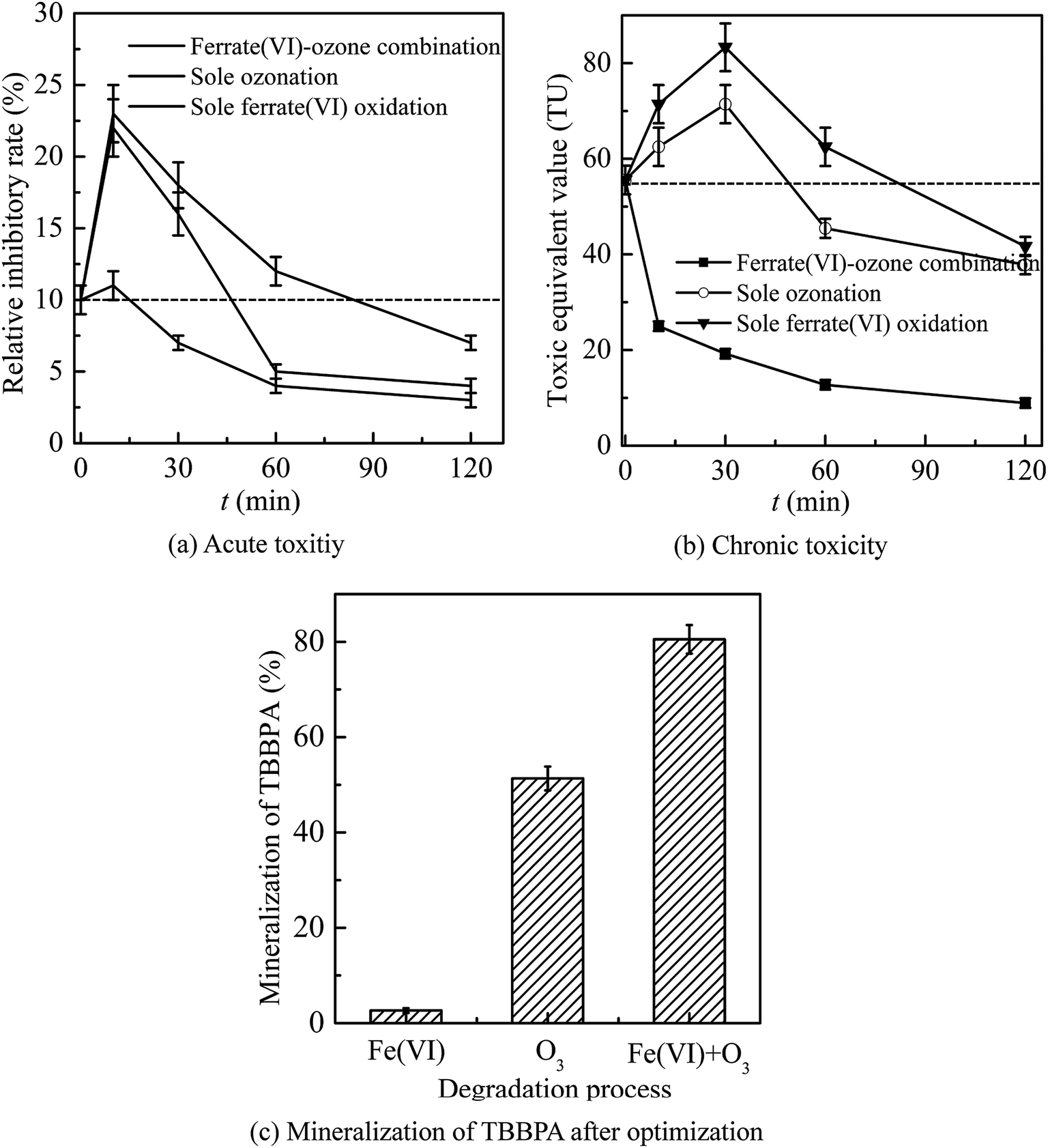 | ||
| Fig. 6 The control of acute toxicity (a) and the chronic toxicity (b) and mineralization of TBBPA (c) after optimization. (Experimental conditions: TBBPA concentration = 1.84 μmol L−1; ferrate(VI) concentration = 0.51 μmol L−1; ozone concentration = 10.42 μmol L−1; initial solution pH = 7.0; temperature = 25 ± 0.5 °C). | ||
In general, the ferrate(VI)–ozone combination process had stronger control ability of acute and chronic toxicity than the other two sole processes. As for the acute toxicity (as seen in Fig. 6(a)), after reaction for 10 min, the relative inhibitory rate of the luminescent bacteria in sole ferrate(VI) and ozone oxidation process were increased from 10% to 23% and 22%, respectively. While the value of T% was only 11% in the ferrate(VI)–ozone combination process, indicating the much lower acute toxicity. When the reaction was carried out for 30 min, the relative inhibitory rate had been reduced to 7% in the ferrate(VI)–ozone combination process, which was controlled below the initial toxicity of TBBPA. However, in the sole ferrate(VI) and ozone oxidation process, the values of T% were still as high as 18% and 16%, respectively. In addition, 60 and even 120 min were required for the sole oxidation process so as to control the toxicity below the initial value. As illustrated in Fig. 6(b), the chronic toxicity of TBBPA itself (1.84 μmol L−1) on D. magna was as high as 55.6 TU. In the sole ferrate(VI) and ozone oxidation process, after reaction for 30 min, the chronic toxicity increased to the maximum of 83.3 and 71.4 TU, respectively. Then, these toxic equivalent values gradually reduced to 41.7 and 37.9 TU at 120 min. The corresponding toxicity control rate in the sole ferrate(VI) and ozone oxidation process were 25.0% and 31.8%, respectively. In the ferrate(VI)–ozone combination process, the values of TU at reaction time 30 and 120 min were 19.2 and 8.9 TU, respectively, with 84.0% of the chronic toxicity being controlled. In summary, compared with the two sole oxidation process, the ferrate(VI)–ozone combination process exhibited a faster and stronger control effect on the acute and chronic toxicity.
It also can be seen from Fig. 6(a) and (b) that the acute and chronic toxicity both increased to the highest at the initial stage of the reactions in the sole ferrate(VI) and ozone oxidation process. The increase of toxicity was caused by the accumulation of more toxic intermediates, such as TriBBPA, BPA, dibromo aromatics, et al.28 Toxicological data show that the values of LD50 (oral dose, mouse) for these products are approximately 2000, 2400, and 282 mg kg−1, respectively, which are much higher than that of TBBPA (LD50 3160 mg kg−1, oral dose, mouse) and indicating a higher toxicity than TBBPA.49 However, none of the above products were detected in the ferrate(VI)–ozone combination process. Thus, there was almost no increase in acute and chronic toxicity during the reactions of ferrate(VI)–ozone combination process (as shown in Fig. 6(a) and (b)), which was due to the synergetic effect of the two oxidants. The degradation and debromination rate of TBBPA were 100% and 91.3%, respectively. In addition, as shown in Fig. 6(c), the mineralization rate of TBBPA in the process of sole ferrate(VI) oxidation and sole ozonation were 2.7% and 51.3%, respectively. While up to 80.5% of the mineralization rate was obtained in the ferrate(VI)–ozone combination process, which was much larger than that of the sum of the two sole processes (54.0%) and showed the strong synergetic effect of the two oxidants.
3.3 Mechanism of the synergetic effect on decontamination
Since the suppression of bromate formation in ozonation process by using ferrate(VI) had been systematically investigated in our earlier studies,35 which had analyzed the mechanism in detail. Thus, this paper focus on the mechanism analysis of the synergetic effect on decontamination, which has been manifested in two aspects: one is the synergetic removal and mineralization of the target pollutants; the other is the efficient and rapid control of the toxicity.The mechanism of the synergetic effect in ferrate(VI)–ozone combination process would be analyzed from the oxidation pathways and the interactions between the two oxidants.
Firstly, the mechanisms of the sole ozone and ferrate(VI) oxidation process were learned again. During the ozonation process, the ozonolysis initiates a series of complex chain reactions and produces ·OH,50 as shown in Fig. 7(a). Therefore, the ozonation process on decontamination included the direct oxidation pathway by ozone molecular and the indirect oxidation pathway by ·OH. Similarly, the final products of ferrate(VI) in the aqueous solution reactions were found to be ferric hydroxides and oxygen, during which the atomic oxygen being generated and further producing ·OH. The main reactions were showed in Fig. 7(b).48 The pathways of ferrate(VI) oxidation process were also divided into the direct ferrate(VI) molecular oxidation and the indirect ·OH oxidation.51 Thus, in the direct oxidation pathway, ozone and ferrate(VI) are both strong oxidants, which have high redox potential (E0ferrate(VI) = 2.20 V; E0ozone = 2.076 V) and may have a superposition effect in the combined process. In the indirect oxidation pathway, ·OH generated in the combined process was much higher than the sole oxidation process, enhancing the decontamination efficiency.
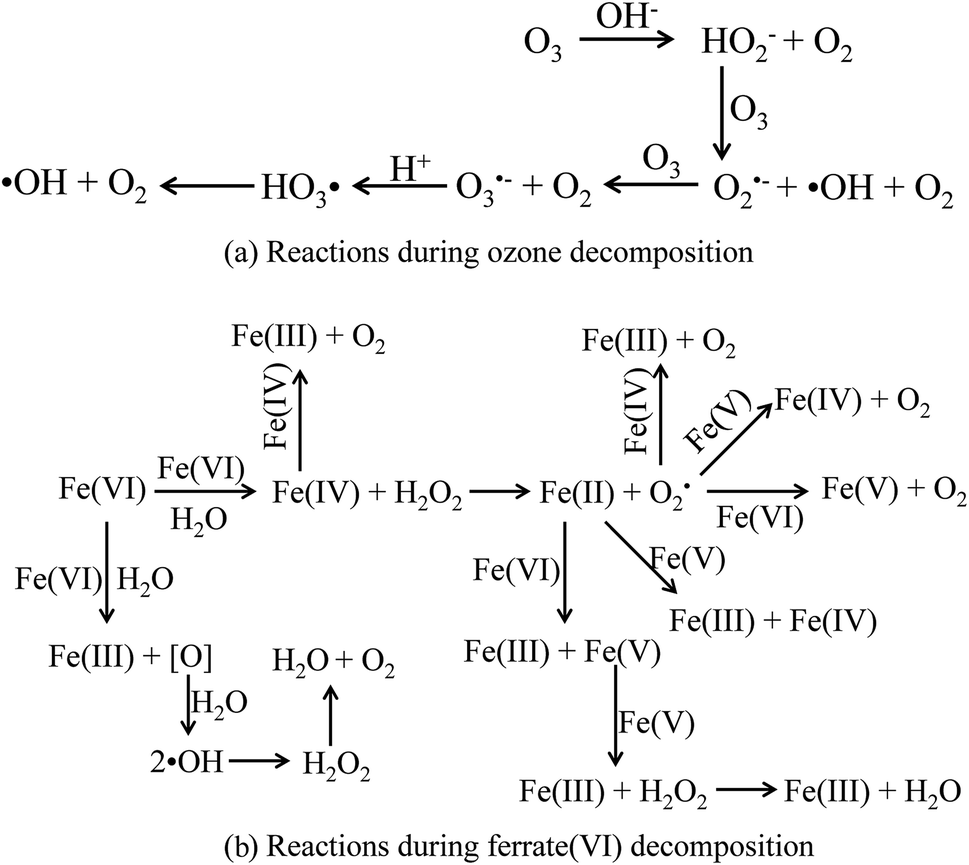 | ||
| Fig. 7 The decomposition process of ozone and ferrate(VI). (a) Reactions during ozone decomposition; (b) reactions during ferrate(VI) decomposition. | ||
Then, the mutual reactions between ozone and ferrate(VI) during the combination process were analyzed. As shown in Fig. 7(b), it depicted the self-decomposition process in the ferrate(VI) oxidation system, whose intermediates were complex and mainly included a variety of valence irons (such as Fe(V), Fe(IV), Fe(III), Fe(II)), free radicals (O2−, ·OH),24 H2O2 and other compounds.48 It is more complicated of ferrate(VI) oxidation reactions, including: the degradation of the target pollutants by Fe(VI) and ·OH; or by the reduced high-valence irons (Fe(V) and Fe(IV)), which were also of strong oxidizing properties and formed via 1-e and 2-e electron transfer of Fe(VI); the lower valence irons (Fe(III) and Fe(II)) could be oxidized by the free radicals (O2−·OH) or higher valence irons (eg Fe(VI), Fe(V) and Fe(IV)) to form Fe(III), Fe(IV) or Fe(V), which could also remove the contaminants. Thus, in the ferrate(VI)–ozone combination process, the interactions of the two oxidants mainly existed between the intermediates of ferrate(VI) and ozone. On the one hand, the ozone molecular and the generated ·OH could again oxidize the lower valence irons to form the higher valence irons so that improved the oxidation efficiency of ferrate(VI). On the other hand, the reduced iron intermediates of ferrate(VI) might have a catalytic role on ozone, thereby producing more ·OH, enhancing the decontamination efficiency in the indirect oxidation pathway and improving the oxidation efficacy of ozone. The later might have a more important impact on the synergetic decontamination efficiency of the combined process. In order to verify the catalytic effect of ferrate(VI) on ozone, the experiments were carried out under the following conditions: initial ozone concentrations of 20.83 μmol L−1, ferrate(VI) dosages of 0–10.10 μmol L−1, Fe3+ dosages of 0–89.29 μmol L−1. The variation of ozone concentration (expressed by the UV absorbance) and the degradation of TBBPA were investigated, as shown in Fig. 8.
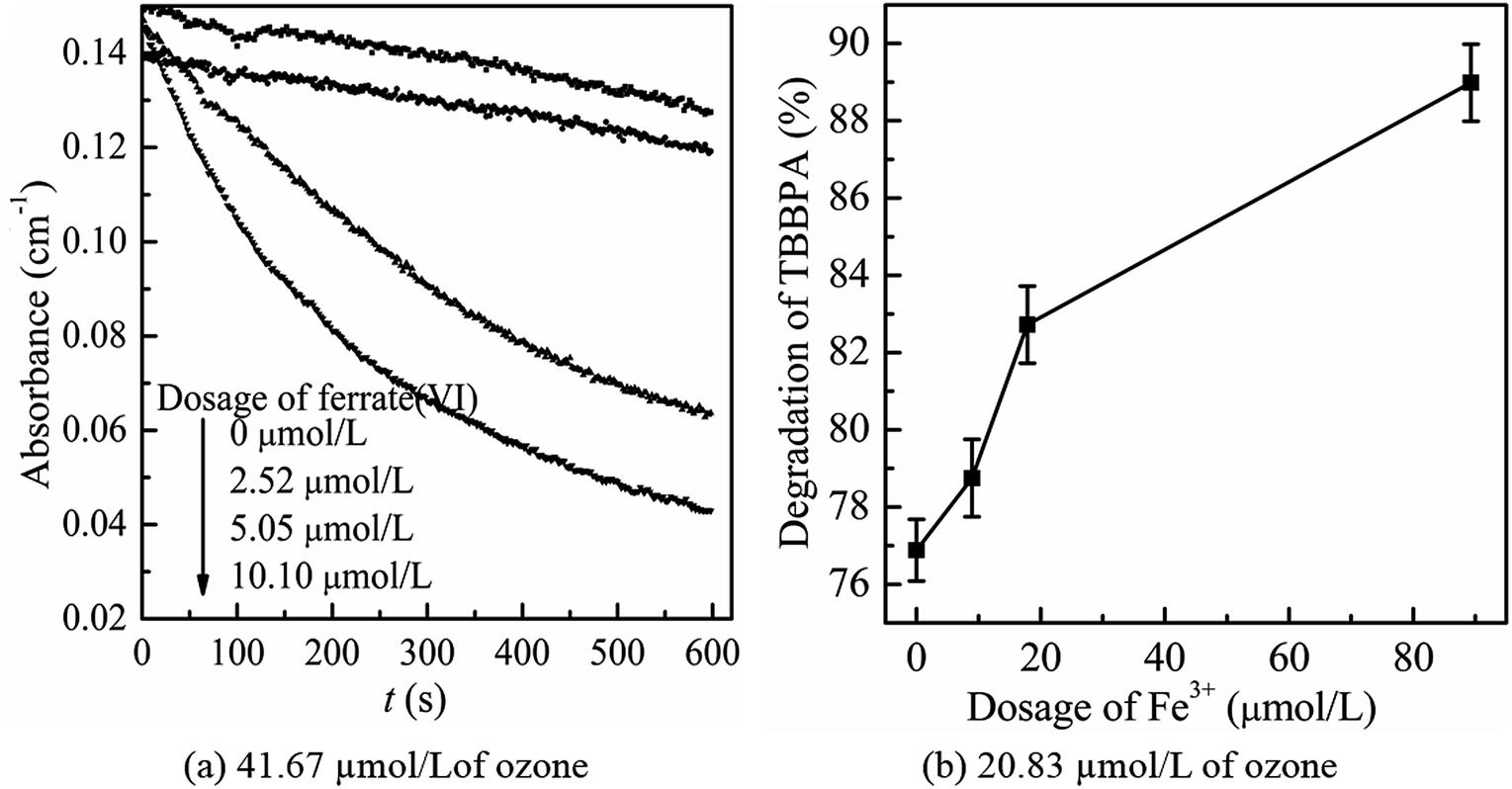 | ||
| Fig. 8 The catalytic effect of ferrate(VI) on ozone decomposition under the ozone concentration of 41.67 μmol L−1 (a) and 20.83 μmol L−1 (b). (Experimental conditions: ozone concentration = 41.67 μmol L−1; TBBPA concentration = 1.84 μmol L−1; ferrate(VI) dosage = 2.52, 5.05, 10.10 μmol L−1; Fe3+ dosage = 8.93, 17.86, 89.29 μmol L−1; initial solution pH = 7.0; temperature = 25 ± 0.5 °C). | ||
It can be seen from Fig. 8(a) that the decomposition of ozone was promoted with the increase of ferrate(VI) dosage, indicating a certain catalytic effect of ferrate(VI) on ozone. In addition, the main reduction products Fe(III), such as hydrated iron ions, hydrated iron oxides and iron oxyhydroxide, have been speculated could catalyze ozone to produce more ·OH,47 which was described in eqn (1)–(4). This conclusion was also obtained in Fig. 8(b), which illustrated that the degradation rate of TBBPA significantly increased from 76.88% to 88.99% as the dosage of Fe3+ increased from 0 to 89.29 μmol L−1.
| Fe3+ + O3 + H2O → FeO2+ + H+ +·OH + O2 | (1) |
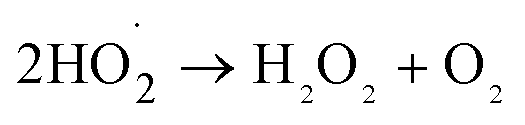 | (2) |
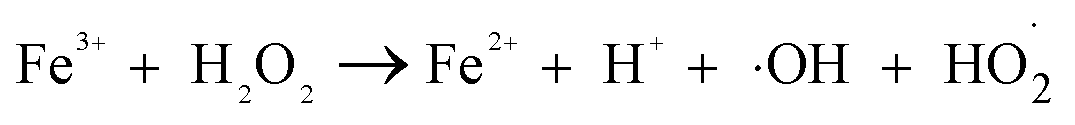 | (3) |
| Fe2+ + H2O2 → Fe3+ + H+ + ·OH + OH− | (4) |
In summary, the mechanism of the synergetic effect in ferrate(VI)–ozone combination process could be summarized as follows:
(1) In terms of oxidation pathway, both of the direct and indirect oxidation pathways might have the superposition effect, thereby enhancing the decontamination efficiency.
(2) Ozone and the generated ·OH could oxidize the low valence iron reduction products of ferrate(VI), and the intermediates of ferrate(VI) also could catalyze ozone to generate more ·OH. The interactions of the two oxidants could improve the oxidation efficiency of ferrate(VI) and ozone.
It was the superposition effect of the oxidation pathways and the interactions between the two oxidants that ensured the strong synergetic degradation efficiency of ferrate(VI)–ozone combination process.
4. Conclusion
The degradation of TBBPA by ferrate(VI)–ozone combination process was systematically investigated in this study, and the major conclusions are summarized as follows:(1) Compared to the sole ferrate(VI) and ozone oxidation process, the ferrate(VI)–ozone combination process had some advantages, including the synergetic effect on degradation and mineralization of TBBPA, the high level of debromination rate, the effective control of the by-product bromate and toxicities.
(2) In the ferrate(VI)–ozone combination process, after being pre-oxidized by ferrate(VI) for 3 min and then co-degraded by the two oxidants, TBBPA (1.84 μmol L−1) could be completely removed by low dosages of ferrate(VI) (0.51 μmol L−1) and ozone (10.42 μmol L−1), with the debromination and mineralization rate as high as 91.3% and 80.5%, respectively. There was no risk of forming the by-product bromate. Moreover, the acute and chronic toxicity could be significantly controlled below the initial one within 10 min.
(3) The mechanism analysis showed that the synergetic effect in ferrate(VI)–ozone combination process might be caused by the superposition effect of the oxidation pathways and the interactions between the two oxidants.
(4) The interactions between the two oxidants mainly included the catalytic effect of ferrate(VI) intermediates on ozone, such as hydrated iron ions, hydrated iron oxides and iron oxyhydroxide; and the oxidation of the low-valent iron compounds by ozone and the generated ·OH.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to appreciate the supports of Study on the Ozone Degradation Mechanism and Toxicity Control of Tetrabromobisphenol A based on Quantum Chemistry (China Postdoctoral Science Foundation: 2018M641831).References
- S. Meriç, H. Selçuk and V. Belgiorno, Water Res., 2005, 39, 1147–1153 CrossRef PubMed.
- I. A. Lang, T. S. Galloway, A. Scarlett, W. E. Henley, M. Depledge, R. B. Wallace and D. Melzer, J. Am. Med. Assoc., 2008, 300, 1303–1310 CrossRef CAS PubMed.
- J. Lee, B. C. Lee, J. S. Ra, J. Cho, I. S. Kim, N. I. Chang, H. K. Kim and S. D. Kim, Chemosphere, 2008, 71, 1572–1592 Search PubMed.
- G. A. K. Anquandah, V. K. Sharma, V. R. Panditi, P. R. Gardinali, H. Kim and M. A. Oturan, Chemosphere, 2013, 91, 105–109 CrossRef CAS PubMed.
- R. J. West and P. A. Goodwin, Dow Company Report, 1997 Search PubMed.
- A. Karci, I. Arslan-Alaton and M. Bekbolet, J. Hazard. Mater., 2013, 263, 275–282 CrossRef CAS PubMed.
- B. Uhnáková, R. Ludwig, J. Pěknicová, L. Homolka, L. Lisá, M. Šulc, A. Petříčková, F. Elzeinová, H. Pelantová, D. Monti, V. Křen, D. Haltrich and L. Martínková, Bioresour. Technol., 2011, 102, 9409–9415 CrossRef PubMed.
- X. Peng, Z. Wang, J. Huang, B. R. Pittendrigh, S. Liu, X. Jia and P. K. Wong, Water Res., 2017, 122, 471–480 CrossRef CAS PubMed.
- Y. H. Zhong, X. L. Liang, Y. Zhong, J. X. Zhu, S. Y. Zhu, P. Yuan, H. P. He and J. Zhang, Water Res., 2012, 46, 4633–4644 CrossRef CAS PubMed.
- N. H. El Najjar, M. Deborde, R. Journel and N. K. Vel Leitner, Water Res., 2013, 47, 121–129 CrossRef CAS PubMed.
- F. R. M. Umar, L. H. Fan and H. A. Aziz, Chemosphere, 2013, 90, 2197–2207 CrossRef PubMed.
- S. L. H. J. Zhang, H. X. Ren, L. P. Wang and L. J. Tian, Procedia Earth Planet. Sci., 2009, 1, 1–5 CrossRef.
- S. G. Zimmermann, A. Schmukat, M. Schulz, J. Benner, U. von Gunten and T. A. Ternes, Environ. Sci. Technol., 2012, 46, 876–884 CrossRef CAS PubMed.
- R. F. Dantas, M. Canterino, R. Marotta, C. Sans, S. Esplugas and R. Andreozzi, Water Res., 2007, 41, 2525–2532 CrossRef CAS PubMed.
- T. Debenest, F. Gagné, A. N. Petit, C. André, M. Kohli and C. Blaise, Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol., 2010, 152, 407–412 CAS.
- B. Uhnáková, R. Ludwig, J. Pěknicová, L. Homolka, L. Lisá, M. Šulc, A. Petříčková, F. Elzeinová, H. Pelantová, D. Monti, V. Křen, D. Haltrich and L. Martínková, Bioresour. Technol., 2011, 102, 9409–9415 CrossRef PubMed.
- N. Ortuño, J. Moltó, J. A. Conesa and R. Font, Environ. Pollut., 2014, 191, 31–37 CrossRef PubMed.
- J. Reungoat, B. I. Escher, M. Macova and J. Keller, Water Res., 2011, 45, 2751–2762 CrossRef CAS PubMed.
- H. S. Kim, H. Yamada and H. Tsuno, Water Sci. Technol., 2006, 53, 169 CrossRef CAS PubMed.
- W. Y. Dong, Z. J. Dong, F. OuYang and Y. Dong, Adv. Mater. Res., 2010, 113–116, 1490–1495 CAS.
- B. Yang and G. G. Ying, Water Res., 2013, 47, 2458–2466 CrossRef CAS PubMed.
- K. Machalová Šišková, D. Jančula, B. Drahoš, L. Machala, P. Babica, P. G. Alonso, Z. Trávníček, J. Tuček, B. Maršálek, V. K. Sharma and R. Zbořil, Phys. Chem. Chem. Phys., 2016, 18, 18802–18810 RSC.
- V. K. Sharma, L. Chen and R. Zboril, ACS Sustainable Chem. Eng., 2016, 4, 18–34 CrossRef CAS.
- J. Chen, Y. Qi, X. Pan, N. Wu, J. Zuo, C. Li, R. Qu, Z. Wang and Z. Chen, Water Res., 2019, 158, 338–349 CrossRef CAS PubMed.
- J. Chen, N. Wu, X. Xu, R. Qu, C. Li, X. Pan, Z. Wei and Z. Wang, Environ. Sci. Technol., 2018, 52, 12592–12601 CrossRef CAS PubMed.
- Y. Y. Eng, V. K. Sharma and A. K. Ray, Chemosphere, 2006, 63, 1785–1790 CrossRef CAS PubMed.
- V. K. Sharma, Environ. Sci. Technol., 2010, 44, 5148–5152 CrossRef CAS PubMed.
- Q. Han, W. Dong, H. Wang, T. Liu, Y. Tian and X. Song, Chemosphere, 2018, 198, 92–102 CrossRef CAS PubMed.
- G. T. Li, N. G. Wang, B. T. Liu and X. W. Zhang, Desalination, 2009, 249, 936–941 CrossRef CAS.
- J. D. Rush, Z. W. Zhao and B. H. J. Bielski, Free Radical Res., 1995, 24(3), 187–198 CrossRef PubMed.
- Y. Ma, N. Y. Gao, W. H. Chu and C. Li, Technol. Water Treat., 2010, 36, 10–15 CAS.
- K. Winkelmann, V. K. Sharma, Y. Lin, K. A. Shreve, C. Winkelmann, L. J. Hoisington and R. A. Yngard, Chemosphere, 2008, 72, 1694–1699 CrossRef CAS PubMed.
- B. L. Yuan, X. Z. Li and N. Graham, Water Res., 2008, 42, 1413–1420 CrossRef CAS PubMed.
- V. k. Sharma and B. H. J. Bielski, Inorg. Chem., 1991, 30, 4306–4310 CrossRef CAS.
- Q. Han, H. Wang, W. Dong, T. Liu and Y. Yin, Sep. Purif. Technol., 2013, 118, 653–658 CrossRef CAS.
- J. Chen, X. Xu, X. Zeng, M. Feng, R. Qu, Z. Wang, N. Nesnas and V. K. Sharma, Water Res., 2018, 143, 1–9 CrossRef CAS PubMed.
- T. Yamamoto, A. Yasuhara, H. Shiraishi and O. Nakasugi, Chemosphere, 2001, 42, 415–418 CrossRef CAS PubMed.
- Y. Lee, J. Yoon and U. von Gunten, Water Res., 2005, 39, 1946–1953 CrossRef CAS PubMed.
- S. C. Laws, S. A. Carey, J. M. Ferrell, G. J. Bodman and R. L. Cooper, Toxicol. Sci., 2000, 54, 154–167 CrossRef CAS PubMed.
- V. Bindhumol, K. C. Chitra and P. P. Mathur, Toxicology, 2003, 188, 117–124 CrossRef CAS PubMed.
- B.-V. Chang, S.-Y. Yuan and Y.-L. Ren, Ecol. Eng., 2012, 49, 73–76 CrossRef.
- J. An, L. Zhu, N. Wang, Z. Song, Z. Yang, D. Du and H. Tang, Chem. Eng. J., 2013, 219, 225–237 CrossRef CAS.
- S. Y. Pang, J. Q. Jiang, Y. Gao, Y. Zhou, X. L. Huangfu, Y. Z. Liu and J. Ma, Environ. Sci. Technol., 2014, 48, 615–623 CrossRef CAS PubMed.
- Y. Zhong, P. A. Peng and W. L. Huang, Chemosphere, 2012, 87, 1141–1148 CrossRef CAS PubMed.
- Q. Han, W. Dong, H. Wang, H. Ma, P. Liu, Y. Gu, H. Fan and X. Song, Chemosphere, 2019, 235, 701–712 CrossRef CAS PubMed.
- K. Zhang, J. Huang, W. Zhang, Y. Yu, S. Deng and G. Yu, J. Hazard. Mater., 2012, 243, 278–285 CrossRef CAS PubMed.
- T. Zhang, J. Lu, J. Ma and Z. Qiang, Water Res., 2008, 42, 1563–1570 CrossRef CAS PubMed.
- V. K. Sharma, R. Zboril and R. S. Varma, Acc. Chem. Res., 2015, 48, 182–191 CrossRef CAS PubMed.
- ChemIDplus Adv.
- P. Yang, S. Luo, H. Liu, W. Jiao and Y. Liu, J. Taiwan Inst. Chem. Eng., 2019, 96, 11–17 CrossRef CAS.
- Y. Lee, R. Kissner and U. von Gunten, Environ. Sci. Technol., 2014, 48, 5154–5162 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra07774j |
| This journal is © The Royal Society of Chemistry 2019 |