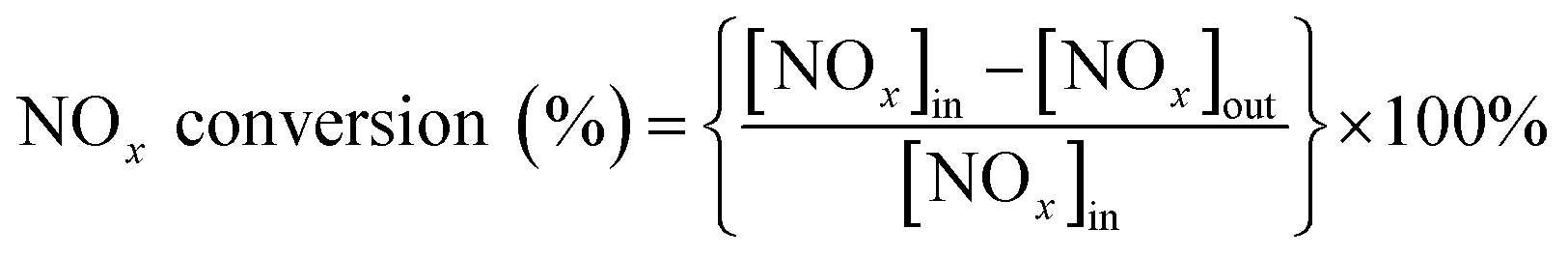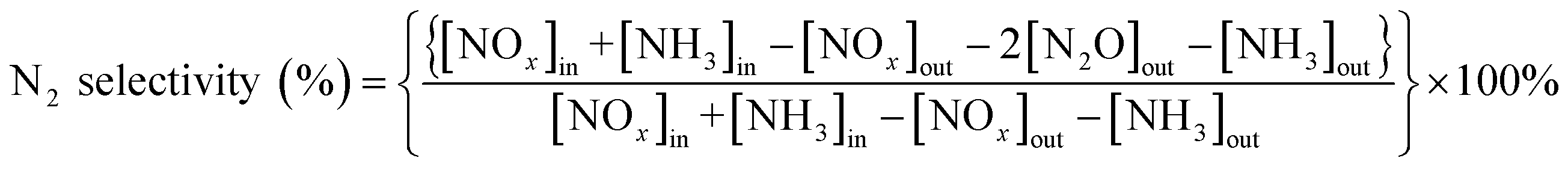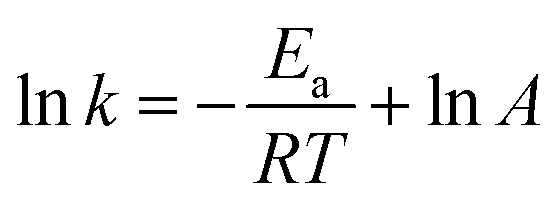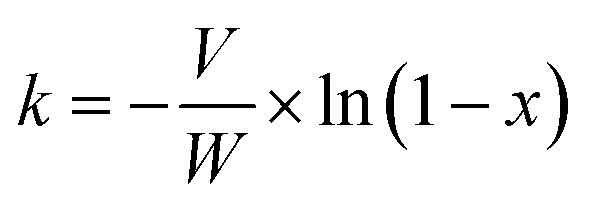DOI:
10.1039/C9RA08391J
(Paper)
RSC Adv., 2019,
9, 39699-39708
Enhancement of NH3-SCR performance of LDH-based MMnAl (M = Cu, Ni, Co) oxide catalyst: influence of dopant M†
Received
14th October 2019
, Accepted 14th November 2019
First published on 2nd December 2019
Abstract
Transition metal (Cu, Ni, Co) doped MnAl mixed oxide catalysts were prepared through a novel method involving the calcination of hydrotalcite precursors for the selective catalytic reduction of NOx with NH3 (NH3-SCR). The effects of transition metal modification were confirmed by means of XRD, BET, TEM, XPS, NH3-TPD, and H2-TPR measurements. Experimental results evidenced that CoMnAl-LDO presented the highest NOx removal efficiency of over 80% and a relatively high N2 selectivity of over 88% in a broad working temperature range (150–300 °C) among all the samples studied. Moreover, the CoMnAl-LDO sample possessed better stability and excellent resistance to H2O and SO2. The reasons for such results could be associated with the good dispersion of Co3O4 and MnOx, which could consequently provide optimum redox behavior, plentiful acid sites, and strong NOx adsorption ability. Furthermore, dynamics calculations verified the meaningful reduction in apparent activation energy (Ea) for the CoMnAl-LDO sample, which is in agreement with the DeNOx activity.
1. Introduction
Nitric oxide (NOx), which is mainly generated from stationary and automotive sources, has definitely given rise to many problems for the environment and has resulted in health concerns for humans.1–3 With gradually tightening regulations on NOx emission limits, post-treatment of nitrogen oxides has become one of the most important research challenges the world faces today in the environmental domain. Currently, selective catalytic reduction (SCR) has been put into wide commercial utilization to abate NOx.4–6 Most practical catalysts in the SCR system are vanadium-based catalysts that must be installed upstream of dedusting and desulfurization to satisfy their optimal operating temperature of 300–400 °C or the catalysts could be poisoned by high concentrations of dust particles and sulfur-containing ammonium salts.7–9 In this situation, it is necessary to develop low-temperature and environmentally friendly catalysts which can efficiently work in the downstream and avoid those problems mentioned above.
Mn-based DeNOx catalysts have been extensively explored in the low-temperature NH3-SCR field due to their unique redox properties, as they are sensitive to H2O and SO2.10,11 To overcome these issues, transition metals have been introduced to Mn-based catalysts to modulate their redox ability and acid sites.12–14 For example, Qiu successfully designed MnxCo3−xO4 nanoparticles through nanocasting, which presented superior catalytic performance to bulk MnCo2O4 synthesized through the co-precipitation method.15
In recent years, calcined layered double hydroxides (LDHs) have shown potential use as catalysts for several reasons, such as the possibility of tuning the composition in wide scope, their high surface areas and homogenous distribution of introduced cations.16–18 In addition, Al was proved to be of great necessity to enhance the self-stability of the LDH materials. From LDH precursors, Yan et al.19 prepared Cu0.5Mg1.5Mn0.5Al0.5Ox, which presented superior catalytic performance in a wide temperature range compared to Mn/γ-Al2O3 prepared by conventional methods. This method might enable good dispersion of the active metal species to provide strong internal interactions for the Cu0.5Mg1.5Mn0.5Al0.5Ox catalyst.
Based on the abovementioned points, using LDHs own innate advantages to accomplish the effective doping of transition metal into MnAl oxides could facilitate DeNOx performances. Herein, Cu-, Ni- or Co-doped MnAl mixed metal oxide catalysts were investigated for NH3-SCR at low temperatures, taking into account the characterization results of XRD (X-ray diffraction), BET (Brunauer–Emmett–Teller), XPS (X-ray photoelectron spectroscopy), NH3-TPD (NH3-temperature programmed desorption), H2-TPR (H2-temperature programmed reduction), NO + O2-TPD (NO + O2-temperature programmed desorption), etc. The addition of Co into MnAl oxide is proposed to dramatically increase its catalytic ability and behaviour, as CoMnAl-LDO presented preferable DeNOx activity (over 80% NO conversion) and N2 selectivity in a broad working temperature window (150–300 °C). In addition, the CoMnAl-LDO sample possessed better stability and excellent resistance to H2O and SO2 at 240 °C.
2. Experimental section
2.1 Preparation of MMnAl-LDO (M = Cu, Ni, Co)
The MMnAl-LDH (M = Cu, Ni, and Co) materials were prepared by co-precipitation method. 50% Mn(NO3)2, Al(NO3)3·9H2O and Cu(NO3)2·3H2O (or Ni(NO3)2·6H2O, Co(NO3)2·6H2O) were dissolved in deionized water, in which the molar ratios of M2+ to Mn2+ and M2+ to M3+ were 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 4:1 (M2+/Mn2+ = 3 and M2+/M3+ = 4:1), respectively. Then, the above aqueous solution was added into 1 mol l−1 NaOH solution with vigorous stirring. In order to avoid oxidation of M2+, N2 was injected during the whole aging process. The pH of the solution was kept at 9–10. The resulting slurry was transferred into a reaction vessel for about 6 h at 60 °C. Finally, MMnAl-LDH (M = Cu, Ni, and Co) materials were obtained by drying at 60 °C overnight in an oven. After being calcined at 500 °C for 5 h at a rate of 2 °C min−1 in air, diverse mixed metal oxides were prepared which were labelled CuMnAl-LDO, NiMnAl-LDO and CoMnAl-LDO. Meanwhile, the MnAl-LDH and corresponding MnAl-LDO were prepared by a similar method.
:1 and 4:1 (M2+/Mn2+ = 3 and M2+/M3+ = 4:1), respectively. Then, the above aqueous solution was added into 1 mol l−1 NaOH solution with vigorous stirring. In order to avoid oxidation of M2+, N2 was injected during the whole aging process. The pH of the solution was kept at 9–10. The resulting slurry was transferred into a reaction vessel for about 6 h at 60 °C. Finally, MMnAl-LDH (M = Cu, Ni, and Co) materials were obtained by drying at 60 °C overnight in an oven. After being calcined at 500 °C for 5 h at a rate of 2 °C min−1 in air, diverse mixed metal oxides were prepared which were labelled CuMnAl-LDO, NiMnAl-LDO and CoMnAl-LDO. Meanwhile, the MnAl-LDH and corresponding MnAl-LDO were prepared by a similar method.
2.2 Catalyst characterization
The X-ray diffraction (XRD) patterns for pristine hydrotalcites and corresponding mixed metal oxides were tested with a Rigaku D/max-2500 diffractometer with a Cu/Kα radiation source. The operating range for 2θ was from 5 to 85° and the scanning rate was 8° min−1. FT-IR experiments were carried out on an FTS 3000 MX FT-IR (Bruker Vertex70) spectrophotometer. The morphologies of the obtained hydrotalcites were measured by an SU8010 SEM apparatus with an accelerating voltage of 200 kV. TEM was carried out on a JEM 2100F transmission electron microscope. The TGA was performed on a Q50 TGA analyzer equipped with a quadrupole mass spectrometer (QMS). The range of testing temperature was from room temperature to 900 °C in flowing Ar (80 mL min−1) at the heating rate of 10 °C min−1. The surface areas for hydrotalcite-derived mixed oxides were obtained via the BET method. The operating instrument was a Micromeritics ASAP-2020 which needs −196 °C liquid nitrogen during operation. In addition, the t-plot approach was used to analyze the total pore volume and the pore size distributions were analyzed by BJH (Barrett–Joyner–Halenda) method. H2-TPR was performed in a TP-5080 instrument. A U-shaped quartz reactor is filled with 80 mg catalyst (40–60 mesh). Before operation, the samples were activated at 300 °C for 1 h under Ar flow. Next, the temperature of the catalyst was decreased to room temperature. Then the catalyst was heated to 800 °C with a ramping rate of 10 °C min−1 in 5% H2/Ar (30 mL min−1) and data were collected during the process. For NH3-TPD, the amount of catalyst and the initial activation process were the same as H2-TPR. Next, unlike the H2-TPR, the catalyst was exposed to NH3 at 50 °C for 1 h, followed by removing unstably adsorbed species by blowing He for 30 min. Then, the desorption process of NH3 was performed with the temperature of the reactor rising to 750 °C at a rate of 10 °C min−1. NO + O2-TPD experiments were operated on a fixed bed reactor self-assembled in the laboratory. First, the catalyst was pre-treated at 300 °C under N2 flow for 1 h. Then, adsorption of 5 vol% O2 and 500 ppm NO was implemented at 50 °C for 1 h, followed by physical removal of adsorbed NOx with N2 for 0.5 h. Finally, the temperature of samples was raised from 50 to 400 °C with an ascending rate of 10 °C min−1 under N2 flow. A Thermo Fisher IS10 FTIR was used to record the concentrations of NOx for the inlet and outlet gases. XPS analysis was carried out on an ESCALab 250 electron spectrometer (Thermo Scientific Corporation) in which the Al Kα radiation source (1486.6 eV) was adopted at 15 kW accelerating power. In order to compensate for the charge effect of catalysts, the C 1s peak of 284.6 eV was used to calibrate all binding energies (BE) of catalysts. The in situ DRIFT spectra were carried out on a Nicolet 6700 instrument produced by the Simefei company and equipped with a liquid nitrogen-cooled MCT detector with 4 cm−1 resolution. Before each measurement, the sample was pre-treated under N2 atmosphere for 30 min (300 °C) and then cooled to 50 °C while the background was recorded. Subsequently, 500 ppm NO + 5% vol. O2 was introduced into the cell and the DRIFT spectra were recorded. The final spectra were obtained after NO + O2 co-adsorption for 30 min and purging with N2 for another 10 min.
2.3 Catalytic activity test
The activity testing was performed in fixed-bed reactor. A quartz reactor was filled with 0.3–0.4 g catalysts (40–60 mesh). A model flue gas (500 ppm NH3, 500 ppm NO, 5 vol% O2, 0 or 100 ppm SO2, 0 or 10 vol% H2O, and N2 as equilibrium gas) was used in all experiments. During the process, the flow rate was 187 cm3 min−1 and the gas hourly space velocity (GHSV) was 45000 h−1. A Thermo Fisher IS10 FTIR was used to record the concentrations of NOx for the inlet and outlet gases every 10 min in the temperature range of 90–360 °C.
The NOx conversion and N2 selectivity were separately calculated basing on the following formulas:
where the inlet concentrations of NO, NO
2 and NH
3 were labelled [NO]
in, [NO
2]
in and [NH
3]
in, respectively, and the outlet concentrations of NO, NO
2 and N
2O were labelled [NO]
out, [NO
2]
out and [N
2O]
out, respectively.
2.4 Kinetic test
The feed-gas mixture is the same as in the above activity test. In order to eliminate the diffusion effect, experiments were performed with different particle sizes and different flow rates. For the NH3-SCR model reaction, the apparent activation energies (Ea) for these catalysts were acquired from the following formula:
where the reaction rate constant (cm3 g−1 s−1), the pre-exponential factor (cm3 g−1 s−1), the standard gas constant (J mol−1 K−1), and the reaction temperature (K) were respectively labelled as k, A, R, and T.
The reaction rate constant (k) as a function of NO conversion (%) can be calculated through the formula below:
in which the reaction rate coefficient (cm
3 g
−1 s
−1), the catalyst weight (g), the volumetric flow rate (cm
3 s
−1) and the NO conversion (%) were labelled as
k,
W,
V and
x, respectively.
3. Results and discussion
3.1 The structure characterization of catalysts
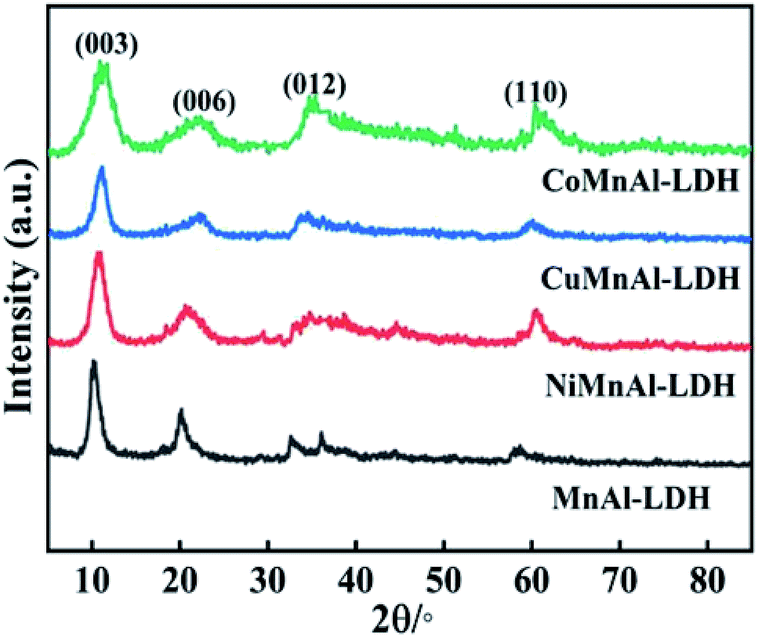
3.1.1 XRD analysis of pristine LDHs. XRD was used to investigate the crystalline micro-structures of the initial LDH precursors, as displayed in Fig. 1. All the prepared samples displayed distinctive diffraction peaks for the (003) and (006) planes characteristic of hydrotalcites.20 It is worth mentioning that the transition metal (Cu, Ni or Co) incorporation did cause distinct modification of the precursors in which their FWHM became significantly wider. Considering the Scherrer formula, this conveyed that the three doped precursors possessed relatively smaller crystal sizes. This phenomenon hinted that the transition metal (Cu, Ni or Co) doping changed the morphology of LDHs precursors, which should contribute to the dispersibility of the corresponding calcined products. This also can be certified by SEM and TEM characterization analysis.
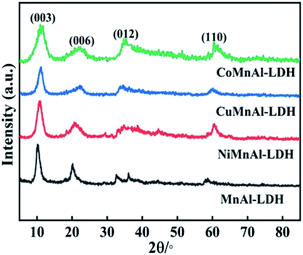 |
| | Fig. 1 XRD patterns of MnAl-LDH, CuMnAl-LDH, NiMnAl-LDH and CoMnAl-LDH. | |
3.1.2 SEM analysis. The morphologies of as-prepared LDHs were characterized using SEM. All samples presented brucite-like structure with well-defined platelets, as displayed in Fig. 2.
 |
| | Fig. 2 SEM images of (A) MnAl-LDH, (B) CuMnAl-LDH, (C) NiMnAl-LDH and (D) CoMnAl-LDH. | |
For unaltered MnAl-LDH, the agglomeration body was especially apparent, although there existed sheets on the surface of catalyst. After introducing the transition metal, the agglomeration was effectively improved. Surprisingly, the CoMnAl-LDH presented very thin and regular hexagonal platelets which were in favour of the gas–solid heterogeneous catalytic reactions.21 For NiMnAl-LDH, the nanoplatelets were the same as CoMnAl-LDH, but the stacking was a little disordered. CuMnAl-LDH had similar “flower-like” shapes, consisting of very thick nanoplatelets.
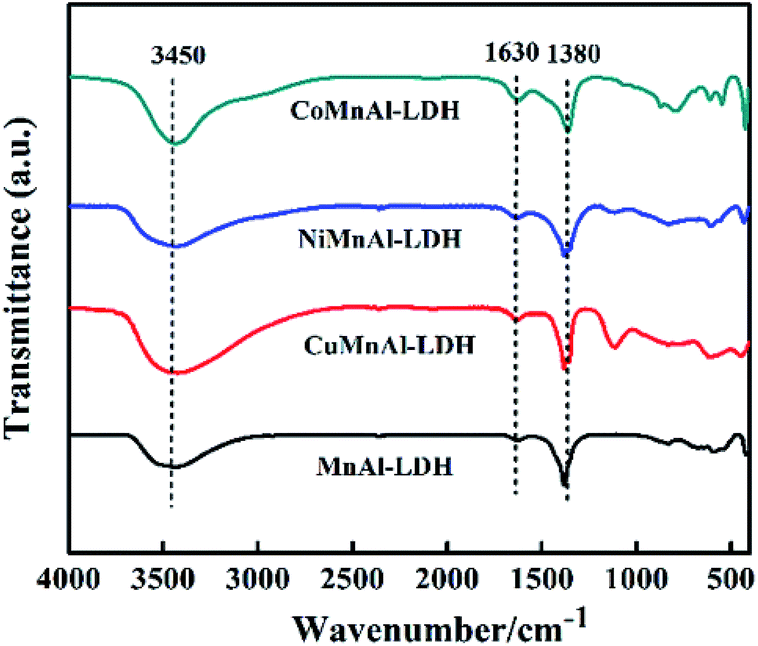
3.1.3 FT-IR. The FT-IR spectra of MMnAl-LDHs are shown in Fig. 3. According to previous literature, the FT-IR spectra of all pristine LDHs deliver typical adsorption peaks like Mg–Al hydrotalcite.22 It is clear that there exists a broad peak located at 3450 cm−1, which can be assigned to the stretching vibrations of the –OH groups of the brucite-like layers.23 All LDHs presented a similar peak located at 1630 cm−1 which can be assigned to the vibration of angular deformation of H2O molecules.24 The vibrations of CO32− are related to the absorption band located at 1380 cm−1.25,26 Finally, the absorption bands in the range of 800–500 cm−1 can be ascribed to the vibrations of M–O from M–OH, O–M–O, or M–O–M in the hydrotalcite structure.23–25 These data further suggest that LDH precursors were successfully synthesized.
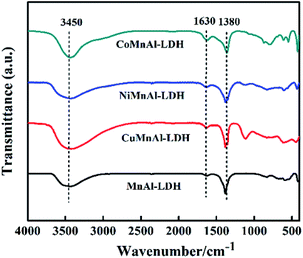 |
| | Fig. 3 FT-IR spectra of MnAl-LDH, CuMnAl-LDH, NiMnAl-LDH and CoMnAl-LDH. | |
3.2 SCR performance of the MMnAl mixed oxide catalysts
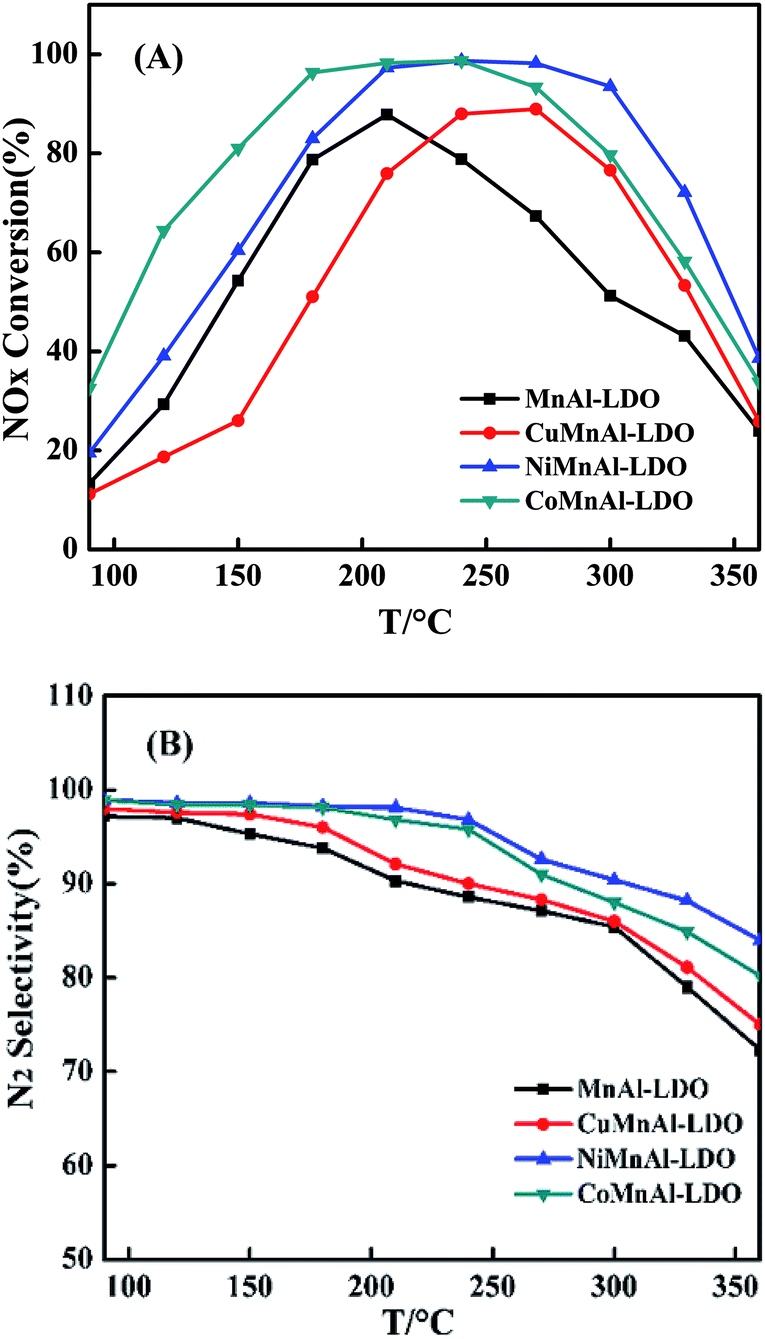
3.2.1 SCR activity. The Co-, Ni- or Cu-modified MnAl-LDO catalysts were obtained via calcination treatment at 500 °C to avoid the influence of residual organic fractions. The respective results of SCR activity and N2 selectivity of MnAl-LDO, CoMnAl-LDO, NiMnAl-LDO and CuMnAl-LDO catalysts are presented in Fig. 4(A) and (B). It is worth mentioning that the different doping transition elements (Co, Ni, Cu) in the MnAl mixed oxides significantly influenced the catalytic performance. Notably, although the activity over the Ni-doped MnAl-LDO had a slight improvement in the low temperature region (<250 °C), the activity over Co-doped catalyst sharply rose, achieving 81% conversion of NO at 150 °C and above 95% conversion in the range of 180 °C to 240 °C, much higher than the activity of the MnAl-LDO catalyst. However, another phenomenon can be observed during the process. After introduction of Cu into MnAl-LDO, the low-temperature activity (<250 °C) considerably decreased. Moreover, upon further increase of the temperature (>250 °C), the catalytic activity decreased dramatically for all catalysts. Fig. 4(B) shows the N2 selectivity over all catalysts. Note that the MnAl catalyst presented worse N2 selectivity than the Cu-, Co-, and Ni-doped catalysts in which the transition metals significantly enhanced the N2 selectivity. As can be seen from the diagram, the sequence of transition metal enhancement of N2 selectivity in the NH3-SCR reaction system was ranked as follows: Ni > Co > Cu.
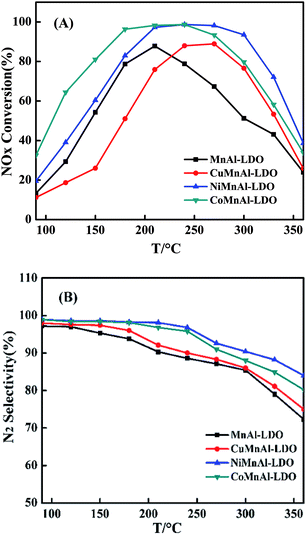 |
| | Fig. 4 (A) NOx conversion and (B) N2 selectivity of MnAl-LDO, CuMnAl-LDO, NiMnAl-LDO and CoMnAl-LDO. | |
3.3 Structure–property correlation
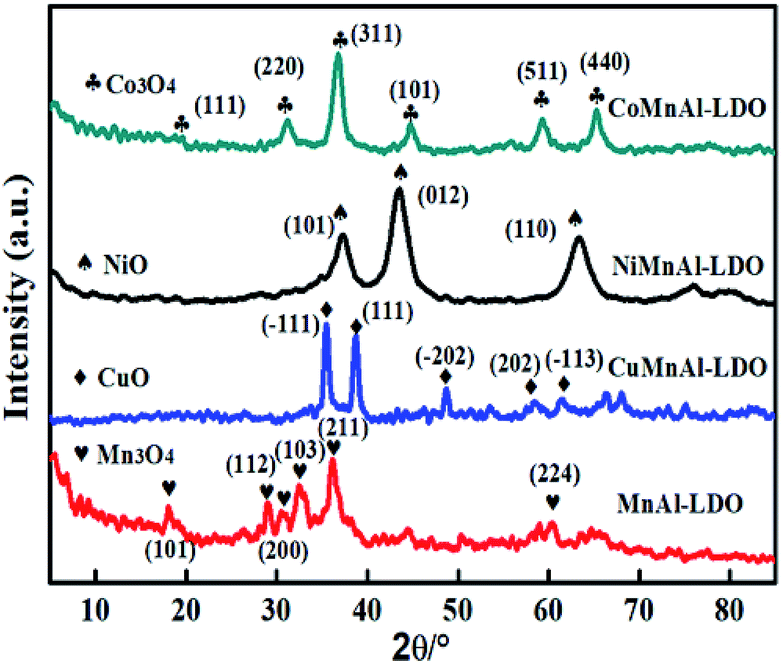
3.3.1 XRD analysis of MMnAl mixed metal oxides. In Fig. 5, the XRD result displays the crystal phases of the MnAl-LDO, CuMnAl-LDO, NiMnAl-LDO, CoMnAl-LDO catalysts calcined at 500 °C. For MnAl-LDO samples, the diffraction pattern showed peaks at 18.1°, 28.9°, 30.7°, 32.5°, and 36.1°, corresponding to Mn3O4.27 Meanwhile, the characteristic peaks assigned to CuO, NiO and Co3O4 were clearly observed in the respective CuMnAl-LDO, NiMnAl-LDO and CoMnAl-LDO catalysts.21,28,29 In addition, except for the MnAl-LDO, there is no noticeable diffraction peak for MnOx observed in the XRD patterns of the Cu-, Co- and Ni-doped catalysts, indicating the good dispersion of MnOx on the catalyst surface.
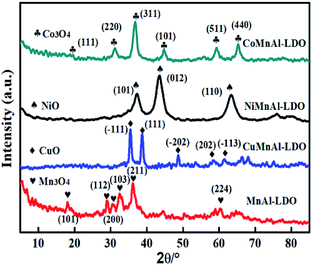 |
| | Fig. 5 The XRD patterns of MnAl-LDO, CuMnAl-LDO, NiMnAl-LDO and CoMnAl-LDO. | |
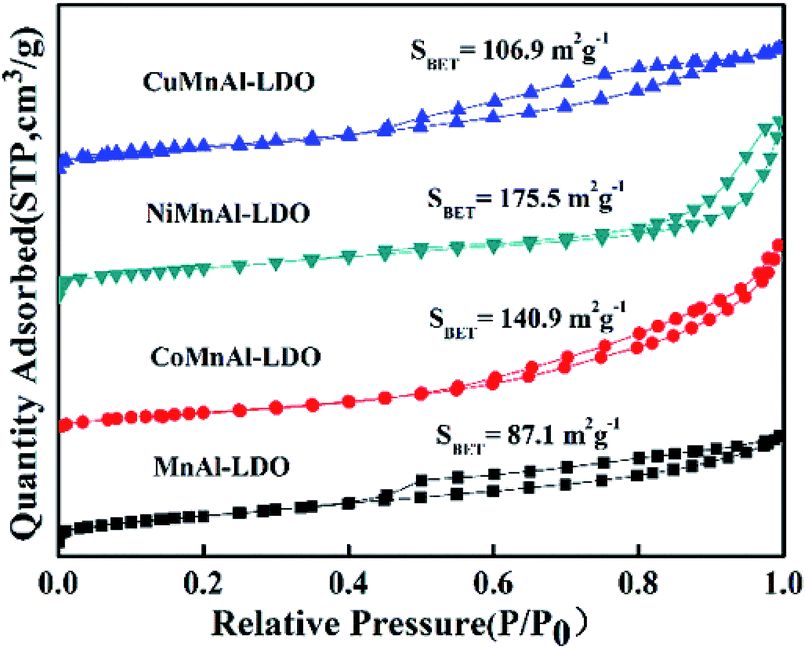
3.3.2 BET analysis. For all catalysts, the N2 adsorption–desorption isotherms are shown in Fig. 6. The isotherms presented a typical type IV isotherm accompanied by a type H2 hysteresis loop, indicating capillary condensation occurring in a definite mesoporous structure.30 It is clear that different doping transition metals affected the specific surface area to some degree, which may be related to the different calcined products from LDH precursors.8 From this diagram, it can be found that the specific surface area of CuMnAl-LDO was slightly improved. In addition, the NiMnAl-LDO and CoMnAl-LDO catalysts were found to have higher surface areas relative to the MnAl-LDO and CuMnAl-LDO catalysts, and the two former possessed higher DeNOx activity. The result suggests that the higher surface areas are in favour of DeNOx activity because they can offer more active sites for reactants.31,32 An interesting phenomenon was discovered, in that NiMnAl-LDO displayed the largest specific surface area among all the catalysts while showing inferior catalytic performance to CoMnAl-LDO, hinting that the specific surface area is not the single influencing factor.
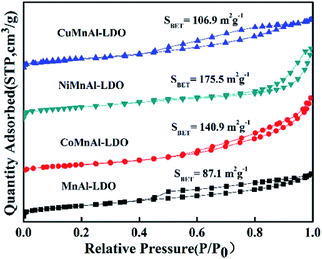 |
| | Fig. 6 The N2 adsorption–desorption isotherms of MnAl-LDO, CuMnAl-LDO, NiMnAl-LDO and CoMnAl-LDO. | |
3.3.3 TEM analysis. The TEM analysis further disclosed the microstructures of the different mixed oxides, as shown in Fig. 7. For MnAl-LDO, the particles were clearly nonuniform in size and distribution and displayed apparent agglomeration. After introducing Cu, the above shortcomings were not much improved. Inspiringly, the nanoparticles were very uniformly distributed over the surface of CoMnAl-LDO and NiMnAl-LDO catalysts in comparison to the other two catalysts. For the MnAl-LDO catalyst, the obvious (211) crystal plane of Mn3O4 can be referenced to the lattice fringe of 0.25 nm.33 The (−111) crystal plane of CuO and (012) crystal plane of NiO are found in Fig. 7(E) and (F) with lattice fringes of 0.25 nm and 0.35 nm,21,34 respectively. The lattice fringes with interplanar distances of 0.28 nm and 0.46 nm can be respectively ascribed to the (220) and (111) planes of Co3O4.35 These results further confirm the XRD results.
 |
| | Fig. 7 TEM images of (A and E) MnAl-LDO, (B and F) CuMnAl-LDO, (C and G) NiMnAl-LDO and (D and H) CoMnAl-LDO. | |
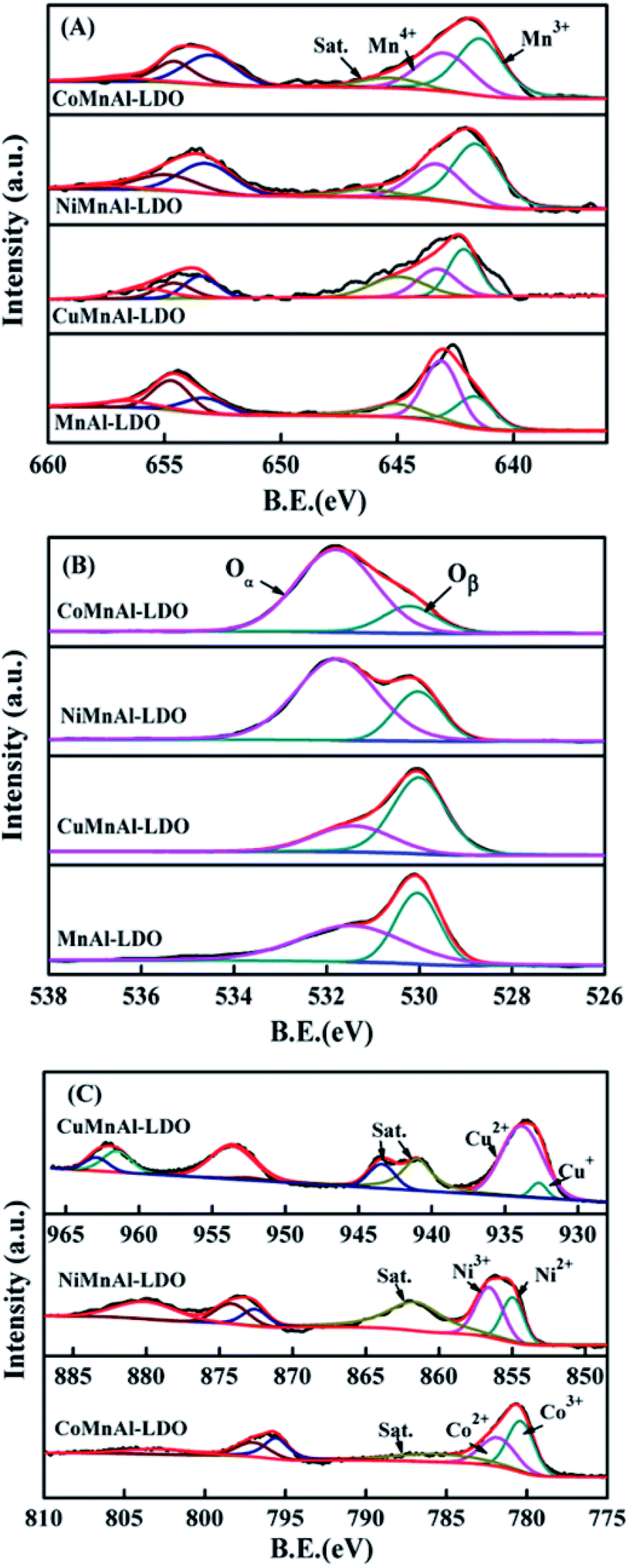
3.3.4 XPS analysis. In order to make clear the underlying causes, XPS spectra were acquired for MnAl-LDO and Cu-, Ni-, and Co-doped catalysts calcined at 500 °C, as illustrated in Fig. 8, with the relative concentration ratios summarized in Table 1. The XPS spectra of Mn 2p for all samples are shown in Fig. 8(A). After completing peak-fitting deconvolution for Mn 2p3/2, three distinct peaks at 642.0, 643.2, and 645.0 eV were observed, which can be correspondingly attributed to Mn3+, Mn4+ and a satellite peak. Apparently, the amount of surface Mn4+ over the MnAl-LDO catalyst was higher compared with the other catalysts, which can result in excessive NH3 oxidation at higher temperatures. Therefore, the MnAl-LDO catalyst afforded poor N2 selectivity, as shown in Fig. 4(B). However, the Co and Ni additions endowed the CoMnAl-LDO and NiMnAl-LDO catalysts with reasonable oxidation through the full operating temperature range, which can give rise to not only improved low-temperature activity but also enhanced N2 selectivity. Additionally, the Mn 2p3/2 peaks of the M-MnAl-LDO (M = Cu, Co, Ni) catalysts presented a minor shift toward lower binding energy compared to MnAl-LDO, indicating the existence of a strong interaction between MnOx and MOx, which would further make electron transfer more convenient.
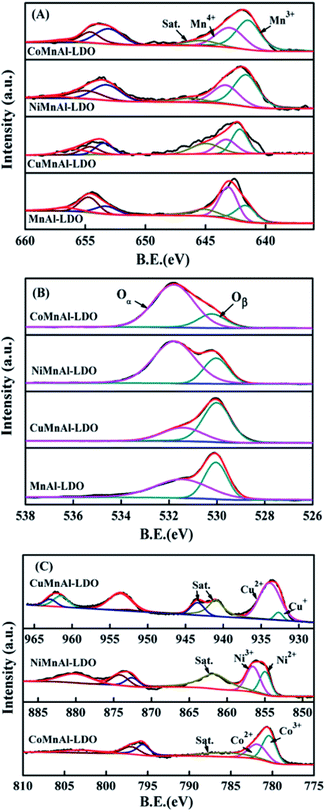 |
| | Fig. 8 XPS results of (A) Mn 2p, (B) O 1s, and (C) Cu 2p, Ni 2p, and Co 2p for MnAl-LDO, CuMnAl-LDO, NiMnAl-LDO and CoMnAl-LDO. | |
Table 1 Surface oxidation states calculated from XPS
| Catalysts |
Mn 2p |
O 1s |
Cu 2p |
Ni 2p |
Co 2p |
| Mn4+/Mn3+ |
Oα/Oβ |
Cu2+/Cu+ |
Ni3+/Ni2+ |
Co3+/Co2+ |
| MnAl-LDO |
1.66 |
1.10 |
— |
— |
— |
| CuMnAl-LDO |
0.62 |
0.68 |
12.04 |
— |
— |
| NiMnAl-LDO |
0.67 |
2.77 |
— |
1.38 |
— |
| CoMnAl-LDO |
0.75 |
3.70 |
— |
— |
1.47 |
Fig. 8(B) shows the XPS spectra of O 1s, which can be fitted into two characteristic peaks at 530.1 eV and 531.7 eV respectively belonging to lattice oxygen and chemisorbed oxygen, hereafter termed Oβ and Oα. From Table 1, it can be clearly observed that the Oα/Oβ ratio on the surface of the CoMnAl-LDO catalyst was higher than those of the other catalysts. The Oα, with higher mobility, is more conductive to oxidation reactions,36 which can enhance the DeNOx activity of CoMnAl-LDO catalyst to some extent. Further XPS data processing revealed that Co3+/Co2+, Ni3+/Ni2+ and Cu2+/Cu+ exist in the separate systems of CoMnAl-LDO, NiMnAl-LDO, CuMnAl-LDO. In general, more Co3+, Ni3+ and Cu2+ species were preferable for redox ability, while the existence of redox circles like Co3+ + Mn3+ ↔ Co2+ + Mn4+, Ni3+ + Mn3+ ↔ Ni2+ + Mn4+ and Cu2+ + Mn3+ ↔ Cu+ + Mn4+ also played a major role in improving the catalytic activity.37,38
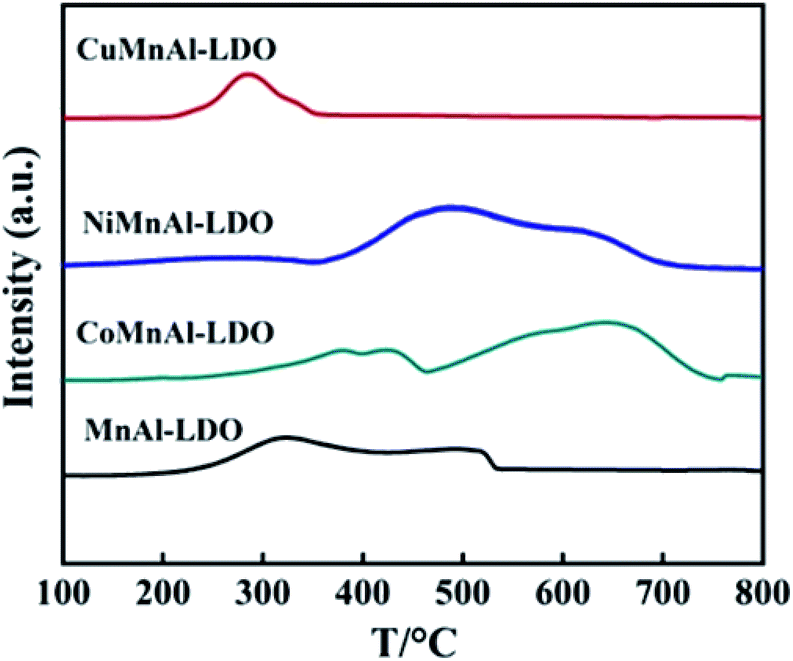
3.3.5 H2-TPR analysis. The H2-TPR technique was used to evaluate the surface redox behavior of the catalysts and the H2-TPR profiles are shown in Fig. 9. For the pure MnAl-LDO catalyst, two reduction peaks observed at roughly 324 and 502 °C were assigned to the continuous reduction of MnO2/Mn2O3/Mn3O4 → MnO.27 The TPR curve of CuMnAl-LDO catalyst shows a reduction peak at 284 °C which can be ascribed to the stepwise reduction of CuO, along with the reduction of MnOx.39 With regard to CoMnAl-LDO, there was a broad medium-temperature peak in the temperature range of 300–400 °C in which Co3O4 and MnOx were gradually reduced. Additionally, the peak in the high-temperature region (500–900 °C) could be assigned to the reduction of CoO or cobalt aluminates.40 For NiMnAl-LDO catalyst, the reduction peak located at 489 °C corresponded to the reduction of Ni3+/Ni2+ to NiO coupled with the reduction of amorphous MnOx.41 This result indicates that doping transition metals can change the redox ability of the MnAl-LDO catalyst. Among these metals, a more suitable interaction between CuOx species and MnOx may be responsible for the lower reducing temperature. As the number of reducible species on the surface of catalyst is also an important factor affecting the DeNOx activity, the hydrogen consumption for each catalyst was calculated and is shown in Table 2. Obviously, the CoMnAl-LDO (10.1 mmol g−1) and NiMnAl-LDO (11.3 mmol g−1) catalysts consumed more hydrogen than MnAl-LDO (6.4 mmol g−1), indicating there were more reducible species over the surfaces of CoMnAl-LDO and NiMnAl-LDO which provide more redox sites and enable sufficient redox reactions. For the CuMnAl-LDO catalyst, the presence of a reduction peak at lower temperature can be an indication of its good reducibility. However, the limited redox sites (hydrogen consumption: 2.6 mmol g−1) are a disadvantage which might account for its lower catalytic activity. The above results are, to some extent, in accordance with the catalytic performances of the catalysts.
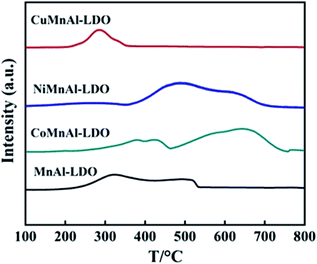 |
| | Fig. 9 H2-TPR results of MnAl-LDO, CuMnAl-LDO, NiMnAl-LDO and CoMnAl-LDO. | |
Table 2 H2 consumption and NOx desorption of the four catalysts
| Catalyst |
Reduction temperature (°C) |
H2 consumption (mmol g−1) |
NOx desorption (%) |
Total NOx desorption (a.u.) |
| T1 |
T2 |
100–150 °C |
150–200 °C |
>200 °C |
| CoMnAl-LDO |
395 |
646 |
10.1 |
38.9 |
31.7 |
29.4 |
2.5 |
| NiMnAl-LDO |
489 |
— |
11.3 |
59.5 |
40.5 |
— |
1.1 |
| CuMnAl-LDO |
284 |
— |
2.6 |
— |
42.9 |
57.1 |
1.3 |
| MnAl-LDO |
324 |
502 |
6.4 |
— |
56.3 |
43.7 |
1 |
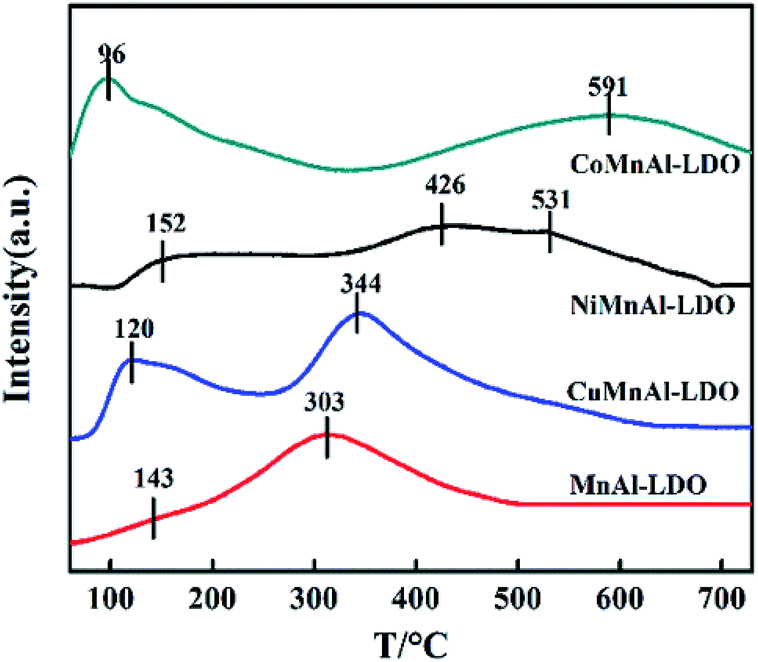
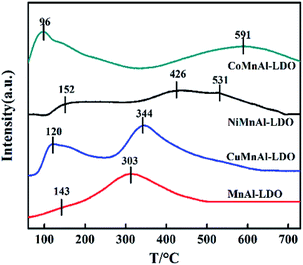
3.3.6 NH3-TPD analysis. NH3-TPD measurements were used to study the number and strength of the acid sites over the surface of the catalysts. As is well known, surface acidity is in favour of NH3 adsorption, which is an essential step for the reaction in the NH3-SCR system.42 The NH3-TPD profiles for all the catalysts are depicted in Fig. 10. The MnAl-LDO catalyst presented a small desorption peak at 143 °C belonging to the weak acid sites (<200 °C) and a large desorption peak at 303 °C belonging to the medium acid sites (200–450 °C). In contrast to MnAl-LDO catalyst, the weak acid sites of all transition-metal-doped catalysts increased significantly, suggesting that the addition of transition metals afforded more weak acid sites. Notably, although the weak acid sites of CuMnAl-LDO were improved, its ability for NOx adsorption was weak, as confirmed in the NO + O2-TPD analysis. The poor activity of CuMnAl in the low-temperature region can be further explained. It can also be seen that the strong acid sites, to a different extent, depend on the identity of the transition metal. Both NiMnAl-LDO and CoMnAl-LDO catalysts have a broad NH3 desorption peak above 400 °C, which can be attributed to continuous NH3 desorption on medium strong acid sites, while there were no strong acid sites on the surface of CuMnAl-LDO catalyst. These results demonstrated that strong acid sites were beneficial to NH3 adsorption, proportional to the wide temperature windows of NiMnAl-LDO and CoMnAl-LDO catalysts.
 |
| | Fig. 10 NH3-TPD results of MnAl-LDO, CuMnAl-LDO, NiMnAl-LDO and CoMnAl-LDO. | |
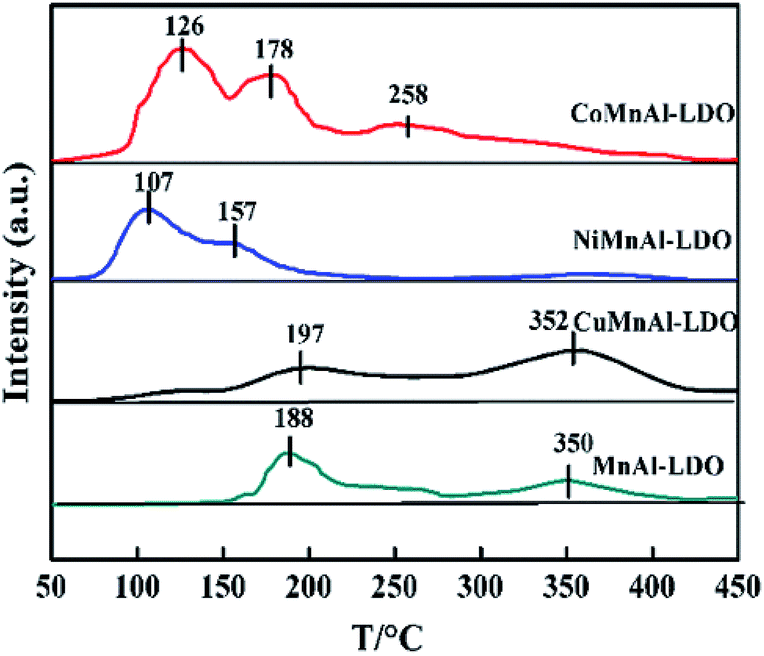
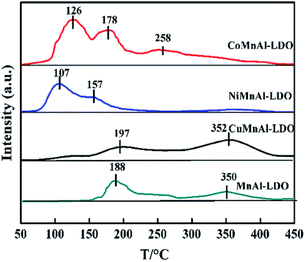
3.3.7 NO + O2-TPD analysis. As with the acid sites, the catalysts' adsorption behavior of the other major reactant (NOx) and its further activation also play an important role in the SCR reaction. Hence, NO + O2-TPD for each catalyst was investigated and the results are shown in Fig. 11, with the in situ DRIFT spectra of NO + O2 co-adsorption displayed in Fig. 12. The MnAl-LDO catalyst exhibits two desorption peaks located at 188 and 350 °C. After Cu modification, the number of NOx adsorption sites slightly increased from 1 to 1.3 in CuMnAl-LDO, as shown in Table 2. However, it can be seen that the intensity of the desorption peak at low temperature (197 °C) decreased, while that of the high temperature desorption peak (352 °C) increased, implying the formation of more stable (normally more inactive) adsorbed NOx species. This result can explain to a certain degree the inferior low temperature DeNOx activity of the CuMnAl-LDO catalyst. For the CoMnAl-LDO and NiMnAl-LDO catalysts, the desorption peaks were shifted to lower temperature region, indicating the Co- or Ni-doping can remarkably reduce the stability of the adsorbed NOx species, which is more likely to provide reactive intermediates. Meanwhile, as can be seen in Table 2, doping with Co and Ni caused a new desorption peak below 150 °C, in which CoMnAl-LDO has a larger total desorption amount of NOx than NiMnAl-LDO. These characteristics of CoMnAl-LDO could be associated with its superior low temperature DeNOx activity.
 |
| | Fig. 11 NO + O2-TPD results of MnAl-LDO, CuMnAl-LDO, NiMnAl-LDO and CoMnAl-LDO. | |
 |
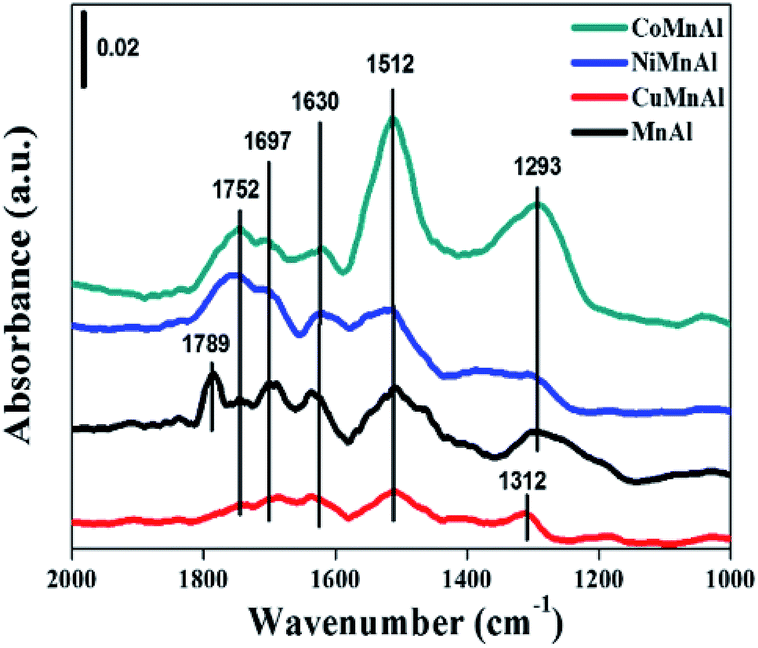
| | Fig. 12 In situ DRIFT spectra of NO + O2 co-adsorption at 50 °C over MnAl-LDO, CuMnAl-LDO, NiMnAl-LDO and CoMnAl-LDO catalysts. | |
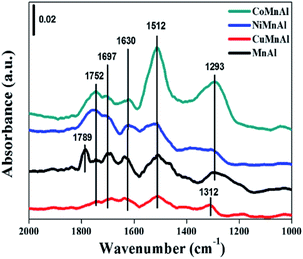
3.3.8 In situ DRIFT spectra. From the in situ DRIFT spectra (Fig. 12), the IR bands between 1620–1800 cm−1 can be ascribed to physisorbed NxOy molecules,43 corresponding to the desorption peaks below 150 °C in Fig. 11. Generally speaking, weakly adsorbed NOx species can be easily activated to participate in the NH3-SCR reaction and are beneficial to the improvement of DeNOx performance at low temperatures. In addition, the bands located at around 1512 cm−1 can be assigned to monodentate nitrate with relatively higher stability and can be related to the desorption peak between 150–200 °C. The desorption peaks at higher temperatures (>200 °C) can be attributed to more stable bidentate or bridged species, which are normally not as active as the monodentate species.44 In summary, doping transition metals (Cu, Ni, Co) can affect the state of adsorption sites for NOx on the surface of MnAl-LDO catalysts to some extent. In comparison to CuMnAl-LDO and MnAl-LDO catalysts, CoMnAl-LDO and NiMnAl-LDO catalysts presented better low temperature catalytic activity based on their suitable NOx adsorption.
3.4 Kinetic analysis
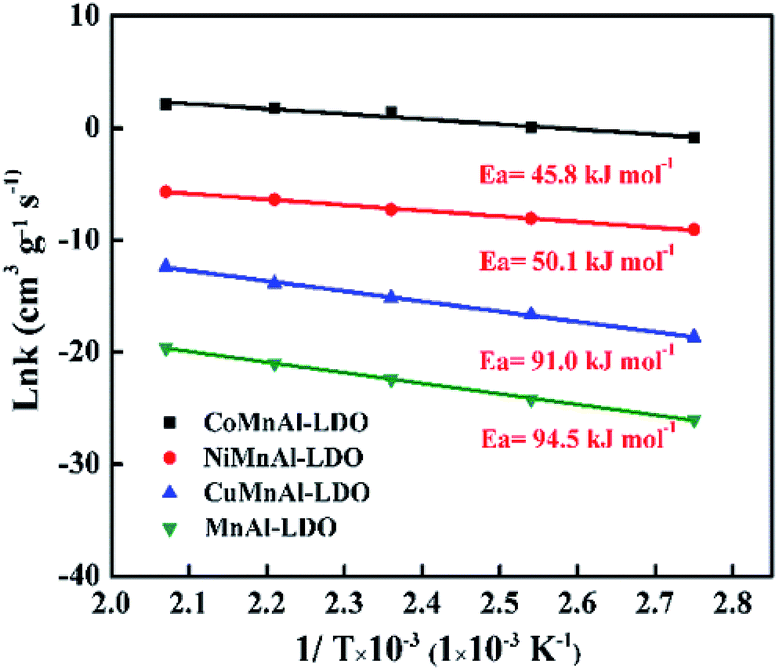
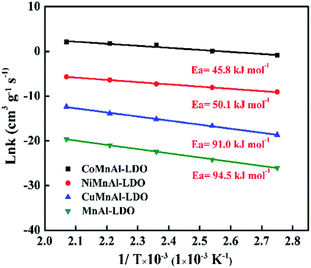
3.4.1 Apparent activation energy. The calculation of apparent activation energy (Ea) can provide a valuable reference for evaluating the different catalytic activities. In the conventional NH3-SCR reaction, the concentration of O2 in the reactants is greatly excessive and NH3 has a strong adsorption capacity. Thus, it is usually regarded as a quasi-first-order reaction (the reaction rate is first-order with respect to NO and zero-order with respect to NH3) for kinetic evaluation.45–49 Although this assumption may be imperfect, it can still provide a relatively valuable reference as a general way of calculating Ea in this study and the relevant formula for Ea has been presented in Section 2.4. It should be pointed out that the influences of internal and external diffusion have been excluded before calculating the apparent activation energy; corresponding experimental data are exhibited in Table S1, Fig. S1 and S2.† Fig. 13 shows the activation energy curves of the four catalysts at 90–210 °C; the Ea for MnAl-LDO, CuMnAl-LDO, NiMnAl-LDO, and CoMnAl-LDO were about 94.5 kJ mol−1, 91.0 kJ mol−1, 50.1 kJ mol−1, and 45.8 kJ mol−1, respectively. The results suggest that the removal of NO was accessible for NiMnAl-LDO and CoMnAl-LDO and difficult for MnAl-LDO and CuMnAl-LDO catalysts, in agreement with the results of DeNOx activity. This result also indicates that the doping of MnAl-LDO by a transition metal (Co, Ni, Cu) can influence the activation energy of the reaction process to a certain extent, thereby promoting or inhibiting the NH3-SCR reaction.
 |
| | Fig. 13 Arrhenius plots of SCR reaction rates over MnAl-LDO, CuMnAl-LDO, NiMnAl-LDO and CoMnAl-LDO. | |
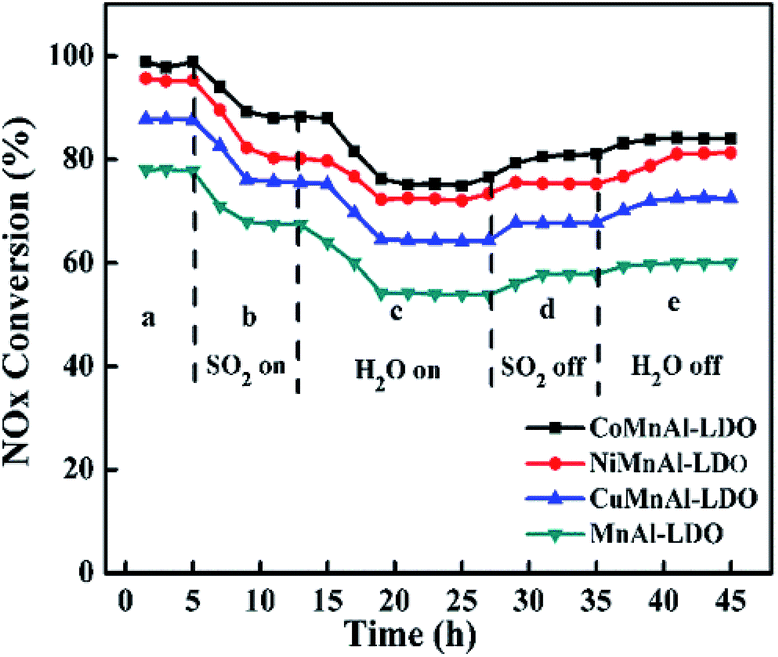
3.5 Stability test and H2O/SO2 resistance test
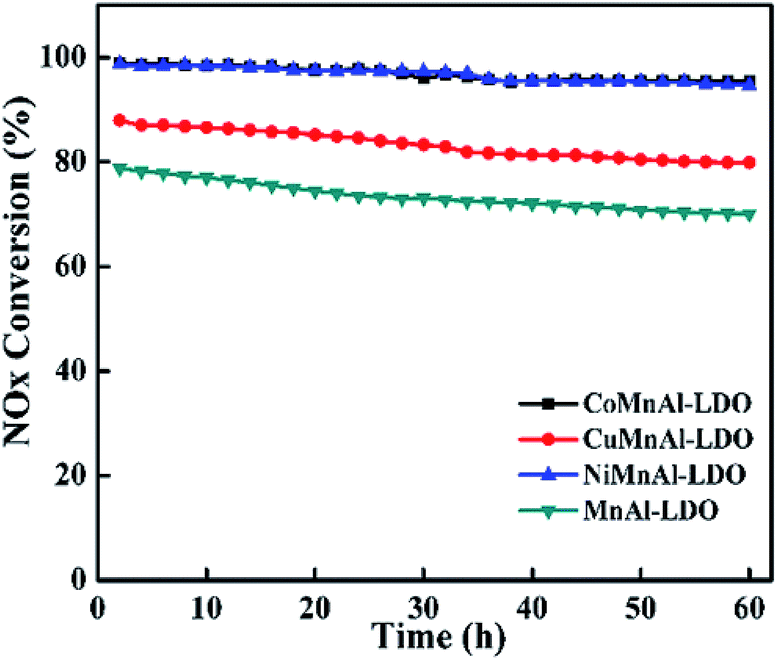
3.5.1 Stability test. To measure the duration of DeNOx activity for all the catalysts, a stability experiment was performed at 240 °C for 60 h under the same flue gas concentration adopted above, as shown in Fig. 14. After the test, the fluctuation value of DeNOx activity was 3.2% for the CoMnAl-LDO, which is slightly lower than that for NiMnAl-LDO (4%). For the CuMnAl-LDO and NiMnAl-LDO catalysts, the fluctuation values for the DeNOx activity were approximately 80%, much higher than that of CoMnAl-LDO. It is evident that the CoMnAl-LDO possesses a better stability.
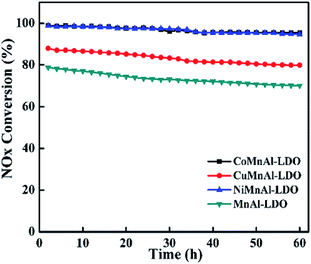 |
| | Fig. 14 The results of stability tests over MnAl-LDO, CuMnAl-LDO, NiMnAl-LDO and CoMnAl-LDO. | |
3.5.2 H2O/SO2 resistance test. In practical use, residual fractions of SO2 and H2O will have some unpleasant side effects on the elimination of NOx for all the catalysts. Therefore, the SO2/H2O durability as a function of time for all catalysts was tested in the NH3-SCR reaction at 240 °C, as shown in Fig. 15. Initially, the NOx conversions were at 98.7%, 98.7%, 88.0%, and 78.8% for CoMnAl-LDO, NiMnAl-LDO, CuMnAl-LDO, and MnAl-LDO, respectively. When SO2 was forced into the reaction system, all catalysts possessed lower DeNOx activity, declining to 88.3%, 80.2%, 75.6%, and 67.4% for CoMnAl-LDO, NiMnAl-LDO, CuMnAl-LDO, and MnAl-LDO, respectively. When both SO2 and H2O were inleted to the reaction system, a dramatic decrease occurred for all catalysts, especially for CuMnAl-LDO and MnAl-LDO with deceases as high as 23% and 25%. This suggests that there existed a synergistic inhibition effect on the NH3-SCR reaction process due to the coexistence of SO2 and H2O. Previous reports have proved that the phenomenon is a result of the formation of sulfate particles that block pores and cover the catalyst surface.50 When SO2 was cut off, there were varied recoveries of DeNOx activity. When the supplies of both SO2 and H2O were closed, it was observed that the DeNOx abilities of CoMnAl-LDO and NiMnAl-LDO catalysts were restored to about 84% and 81.3%, while the DeNOx abilities for MnAl-LDO and CuMnAl-LDO were only restored to 60.1% and 72.5%, respectively. Thus, it can be deduced that the CoMnAl-LDO catalyst displayed excellent SO2 and H2O tolerance.
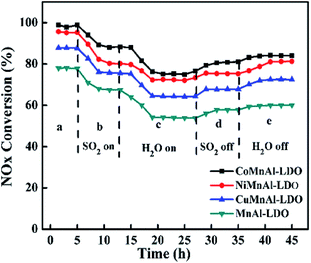 |
| | Fig. 15 The results of SO2/H2O durability over MnAl-LDO, CuMnAl-LDO, NiMnAl-LDO and CoMnAl-LDO. | |
4. Conclusions
In this study, a series of transition metal (Cu, Ni, Co) modified MnAl catalysts were obtained via calcining the corresponding LDH precursors and evaluated for the NH3-SCR reaction. The obtained information suggested that the catalytic activities of the catalysts were strongly dependent on the identity of their transition metal, for which the NH3-SCR reactivity order was CoMnAl-LDO > NiMnAl-LDO > CuMnAl-LDO. The fine catalytic performance in the broadest working temperature window over the CoMnAl-LDO catalyst should be attributed to its good dispersion of active species over the CoMnAl-LDO surface, which could contribute to its optimum redox behavior, plentiful acid sites and NOx adsorption sites. These findings might open the future potential for LDHs and provide a new mentality for heterogeneous catalysis.
Conflicts of interest
There are no conflicts of interest to declare.
Acknowledgements
The project was supported by National Natural Science Foundation of the China (No. 51978436), Scientific and Technological Innovation Programs of Higher Education Institutions in Shanxi (2019L0885) and the Doctoral Startup and Research Fund of Jinzhong University.
Notes and references
- Z. Y. Fan, Z. Y. Wang, J. W. Shi, C. Gao, G. Gao, B. R. Wang, Y. Wang, X. Chen, C. He and C. M. Niu, J. Catal., 2019, 370, 30–37 CrossRef CAS.
- C. Fang, D. S. Zhang, L. Y. Shi, R. H. Gao, H. R. Li, L. P. Ye and J. P. Zhang, Catal. Sci. Technol., 2013, 3, 803–811 RSC.
- U. Bentrup, A. Brückner, M. Richter and R. Fricke, Appl. Catal., B, 2001, 32, 229–241 CrossRef CAS.
- J. Muñiz, G. Marbán and A. B. Fuertes, Appl. Catal., B, 2000, 27, 27–36 CrossRef.
- Z. Y. Fan, J. W. Shi, C. Gao, G. Gao, B. R. Wang, Y. Wang, C. He and C. M. Niu, Chem. Eng. J., 2018, 348, 820–830 CrossRef CAS.
- B. Thirupathi and P. G. Smirniotis, J. Catal., 2012, 28, 874–883 Search PubMed.
- S. Djerad, M. Crocoll, S. Kureti, L. Tifouti and W. Weisweiler, Catal. Today, 2006, 113, 208–214 CrossRef CAS.
- B. H. Hou, Y. L. Du, X. Z. Liu, C. Ci, X. Wu and X. M. Xie, RSC Adv., 2019, 9, 24377–24385 RSC.
- J. P. Dunn, P. R. Koppula, H. G. Stenger and I. E. Wachs, Appl. Catal., B, 1998, 19, 103–117 CrossRef CAS.
- S. Andreoli, F. A. Deorsola, C. Galletti and R. Pirone, Chem. Eng. J., 2015, 278, 174–182 CrossRef CAS.
- X. L. Tang, J. M. Hao, W. G. Xu and J. H. Li, Catal. Commun., 2007, 8, 329–334 CrossRef CAS.
- F. Y. Gao, X. L. Tang, H. H. Yi, J. Y. Li, S. Z. Zhao, J. G. Wang, C. Chu and C. L. Li, Chem. Eng. J., 2017, 317, 20–31 CrossRef CAS.
- Z. H. Chen, Q. Yang, H. Li, X. H. Li, L. F. Wang and S. C. Tsang, J. Catal., 2010, 276, 56–65 CrossRef CAS.
- D. Fang, J. L. Xie, D. Mei, Y. M. Zhang, F. He, X. Q. Liu and Y. M. Li, RSC Adv., 2014, 4, 25540–25551 RSC.
- M. Y. Qiu, S. H. Zhan, H. B. Yu, D. D. Zhu and S. Q. Wang, Nanoscale, 2015, 7, 2568–2577 RSC.
- H. Meng, J. N. Liu, Y. L. Du, B. H. Hou, X. Wu and X. M. Xie, Catal. Commun., 2019, 119, 101–105 CrossRef CAS.
- Y. K. Shao, J. H. Li, H. Z. Chang, Y. Peng and Y. X. Deng, Catal. Sci. Technol., 2015, 5, 3536–3544 RSC.
- L. Chmielarz, P. Kuśtrowski, A. Rafalska-Łasocha, D. Majda and R. Dziembaj, Appl. Catal., B, 2002, 35, 195–210 CrossRef CAS.
- Q. H. Yan, S. N. Chen, C. Zhang, D. O'Hare and Q. Wang, J. Colloid Interface Sci., 2018, 526, 63–74 CrossRef CAS PubMed.
- X. Wu, Y. L. Feng, X. Z. Liu, L. L. Liu, Y. L. Du and Z. Li, Appl. Surf. Sci., 2019, 495, 143513 CrossRef CAS.
- Q. H. Yan, Y. Nie, R. Y. Yang, Y. H. Cui, D. O'Hare and Q. Wang, Appl. Catal., A, 2017, 538, 37–50 CrossRef CAS.
- C. Gennequin, T. Barakat, H. L. Tidahy, R. Cousin, J. F. Lamonier, A. Aboukaïs and S. Siffert, Catal. Today, 2010, 157, 191–197 CrossRef CAS.
- J. M. Gatica and H. Vidal, J. Hazard. Mater., 2010, 181, 9–18 CrossRef CAS PubMed.
- C. J. Wang and D. O'Hare, J. Mater. Chem., 2012, 22, 23064–23070 RSC.
- S. Kannan and V. Rives, J. Solid State Chem., 2004, 177, 319–331 CrossRef CAS.
- C. X. Yang, L. B. Liao, G. C. Lv, L. M. Wu, L. F. Mei and Z. H. Li, J. Colloid Interface Sci., 2016, 479, 115–120 CrossRef CAS PubMed.
- D. Zhao, C. Wang, F. Yu, Y. L. Shi, P. Cao, J. M. Dan, K. Chen, Y. Lv, X. H. Guo and B. Dai, Nanomaterials, 2018, 8, 620 CrossRef PubMed.
- X. J. Li, Y. L. Du, X. M. Guo, R. N. Wang, B. H. Hou and X. Wu, Catal. Lett., 2018, 149, 456–464 CrossRef.
- X. Wu, Y. L. Feng, Y. L. Du, X. Z. Liu, C. L. Zou and Z. Li, Appl. Surf. Sci., 2019, 495, 14353 Search PubMed.
- S. P. Mo, S. D. Li, W. H. Li, J. Q. Li, J. Y. Chen and Y. F. Chen, J. Mater. Chem. A, 2016, 4, 8113–8122 RSC.
- Q. M. Jia, S. Y. Shan, L. H. Jiang, Y. M. Wang and D. Li, J. Appl. Polym. Sci., 2012, 125, 3560–3566 CrossRef CAS.
- F. D. Liu and H. He, J. Phys. Chem. C, 2010, 114, 16929–16936 CrossRef CAS.
- S. N. Chen, Q. H. Yan, C. Zhang and Q. Wang, Catal. Today, 2019, 327, 81–89 CrossRef CAS.
- Y. Nie, Q. H. Yan, S. Chen, D. O'Hare and Q. Wang, Catal. Commun., 2017, 97, 47–50 CrossRef CAS.
- B. Meng, Z. B. Zhao, X. Z. Wang, J. J. Liang and J. S. Qiu, Appl. Catal., B, 2013, 129, 491–500 CrossRef CAS.
- X. Wu, Y. L. Feng, Y. L. Du, X. Z. Liu, C. L. Zou and Z. Li, Appl. Surf. Sci., 2019, 467, 802–810 CrossRef.
- F. Y. Gao, X. L. Tang, H. H. Yi, S. Z. Zhao, J. G. Wang, Y. R. Shi and X. M. Meng, Appl. Surf. Sci., 2018, 443, 103–113 CrossRef CAS.
- H. Meng, J. N. Liu, Y. L. Du, B. H. Hou, X. Wu and X. M. Xie, Catal. Commun., 2019, 119, 101–105 CrossRef CAS.
- C. Z. Sun, J. Zhu, Y. Y. Lv, L. Qi, B. Liu, F. Gao, K. Q. Sun, L. Dong and Y. Chen, Appl. Catal., B, 2011, 103, 206–220 CrossRef CAS.
- J. F. Lamonier, A. B. Boutoundou, C. Gennequin, M. J. Pérez-Zurita and S. Siffert, Catal. Lett., 2007, 118, 165–172 CrossRef CAS.
- S. X. Cai, D. S. Zhang, L. Y. Shi, J. Xu, L. Zhang, L. Huang, H. R. Li and J. P. Zhang, Nanoscale, 2014, 6, 7346–7353 RSC.
- X. Wu, H. Meng, Y. L. Du, J. N. Liu, B. H. Hou and X. M. Xie, ACS Appl. Mater. Interfaces, 2019, 11, 32917–32927 CrossRef CAS PubMed.
- K. I. Hadjiivanov, Catal. Rev., 2000, 42, 71–144 CrossRef CAS.
- F. D. Liu, H. He, Y. Ding and C. B. Zhang, Appl. Catal., B, 2009, 93, 194–204 CrossRef CAS.
- L. J. Yan, Y. Y. Liu, H. Hu, H. R. Li, L. Y. Shi and D. S. Zhang, ChemCatChem, 2016, 8, 2267–2278 CrossRef CAS.
- S. C. Xiong, Y. Liao, X. Xiao, H. Dang and S. J. Yang, J. Phys. Chem. C, 2015, 119, 4180–4187 CrossRef CAS.
- G. S. Qi and R. T. Yang, J. Catal., 2003, 217, 434–441 CrossRef CAS.
- G. S. Qi, R. T. Yang and R. Chang, Appl. Catal., B, 2004, 51, 93–106 CrossRef CAS.
- R. H. Gao, D. S. Zhang, P. Maitarad, L. Y. Shi, T. Rungrotmongkol, H. R. Li, J. P. Zhang and W. G. Cao, J. Phys. Chem. C, 2013, 117, 10502–10511 CrossRef CAS.
- X. N. Lu, C. Y. Song, S. H. Jia, Z. S. Tong, X. L. Tang and Y. X. Teng, Chem. Eng. J., 2015, 260, 776–784 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08391j |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Xu Wu
b and
Xu Wu