
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of hydroxyethyl tetrathiatriarylmethyl radicals OX063 and OX071†
Martin Ponceletab,
Justin L. Huffmanab,
Valery V. Khramtsovbc,
Ilirian Dhimitrukad and
Benoit Driesschaert*ab
aDepartment of Pharmaceutical Sciences, School of Pharmacy, West Virginia University, Morgantown, WV 26506, USA
bIn Vivo Multifunctional Magnetic Resonance center, Robert C. Byrd Health Sciences Center, West Virginia University, Morgantown, WV 26506, USA. E-mail: benoit.driesschaert@hsc.wvu.edu
cDepartment of Biochemistry, School of Medicine, West Virginia University, Morgantown, WV 26506, USA
dSchool of Health and Natural Sciences, Mercy College, Dobbs Ferry, NY 10522, USA
First published on 30th October 2019
Abstract
We report the synthesis of hydroxyethyl tetrathiatriarylmethyl radical OX063 and its deuterated analogue OX071 for biomedical EPR applications.
Soluble organic radicals such as tetrathiatriarylmethyls (TAM, trityl) or nitroxides have been used extensively for biomedical electron paramagnetic resonance (EPR) and dynamic nuclear polarization (DNP). The in vivo applications of nitroxide radicals are hampered by their fast bio-reduction, leading to an EPR-silent hydroxylamine. In addition, their broad linewidths and hyperfine couplings with the nitrogen nucleus (l = 1) of the nitroxide fragment decrease the analytical sensitivity and performance as polarizing agents. The second class of spin probes widely used, tetrathiatriarylmethyl radicals, was first reported by Nycomed Innovation in the late 90's. They chemically modified Gomberg's trityl radical (Fig. 1) with the aim to avoid hyperfine splitting, increase the stability and provide water solubility. The two most popular structures are the Finland trityl (FT) and its more hydrophilic analogue OX063.1 Those radicals exhibit unmatched properties, such as a single-line EPR spectrum, ultra-narrow linewidth (<200 mG) and water solubility.2,3 The publication in the scientific literature in 2002 by Reddy et al.2 of the synthesis of Finland trityl enabled the synthesis of a wide variety of Finland-based structures for biomedical EPR applications. FT-based trityls showing sensitivities to physiological parameters, such as pO2, pH, inorganic phosphate,4–7 thiol concentration8 or redox status9 have been reported. FT-based spin labels of biomacromolecules have allowed for distance measurements in DNA10 or proteins.11 Finally, high performance FT-nitroxide biradical polarizing agents have been developed.12 All of these structural modifications took place at the para position of the trityl scaffold, which is the only position that can be easily modified.
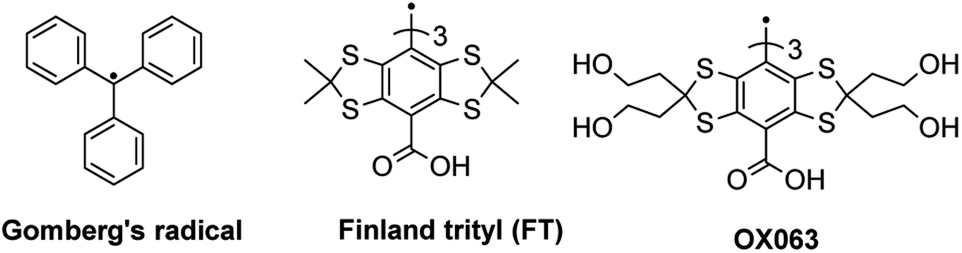 | ||
| Fig. 1 Structures of Gomberg's trityl, Finland trityl (FT) and OX063. | ||
Despite their widespread uses, the lipophilic core of FT-based molecules is responsible for their aggregation at low pH and hydrophobic interactions with plasma biomacromolecules (e.g. albumin13), resulting in a broadening of the EPR line. For this reason, in vivo applications are limited to intra-tissue deliveries only.6,7,14
On the other hand, OX063 shows a high hydrophilicity due to twelve additional alcohol functions, preventing interactions with biomacromolecules, allowing for a systemic delivery of the probe.15 Unfortunately, the synthesis of OX063 has not been reported in the scientific literature and its synthesis remained elusive16 although being commercially available at a very high cost (>$10,000 per g).17 In order to circumvent the limitations of FT-based structures, highly hydrophilic fragments such as PEGs,18,19 polypeptides,20,21 polyamidoamines,22 and dextrans,23 were conjugated. The high molecular weight of those probes decreases their spin density and tissue perfusion and none of them have been used beyond their initial proof of concept. Recently, a hybrid trityl radical possessing only one hydroxylated aryl group has been reported.16 However, to date, OX063 remains the sole spin probe used upon systemic delivery. Hereby we report the synthesis of OX063 and its partially deuterated analogue OX063-d24, also named OX071.
The synthesis starts with the construction of the protected aryl moiety 4 (Scheme 1). The condensation of dimethyl acetonedicarboxylate with the 1,2,4,5-tetrathiobenzene generated in situ leads to thioketal 2, recovered by a simple filtration. Next, the four methyl esters of 2 were reduced using 4.5 equivalents of LiAlH4 and the resulting alcohols were protected with tert-butyl groups using isobutene and triflic acid as a source of tert-butyl cation. The synthesis of 4 only requires one column chromatography purification and can be performed on multiple gram-scale.
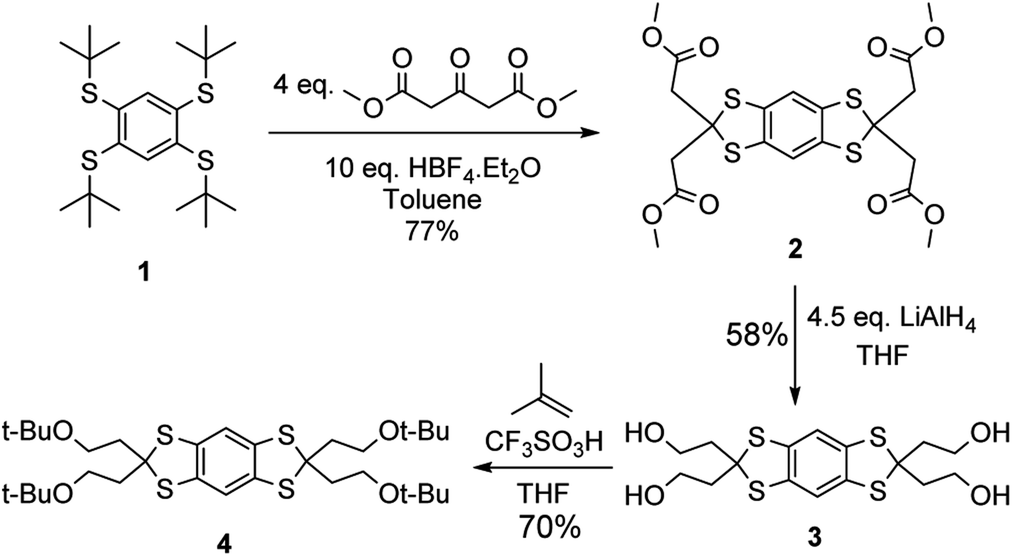 | ||
| Scheme 1 Synthesis of the protected key intermediate 4. | ||
Our initial attempt to generate the trityl alcohol 6 upon three successive additions of the aryllithium generated from the direct deprotonation of 4 using n-BuLi to diethyl carbonate, as classically performed for the synthesis of FT2,24 (Scheme 2, path A), failed to provide any amount of trityl alcohol. Unreacted material, mixed with unidentified compounds was recovered. The use of other alkyllithium reagents (sec-BuLi, tert-BuLi) or other solvents (THF, n-hexane) did not result in any improvement. We hypothesized that the incomplete lithiation of 4 was responsible for this result, as unreacted alkyllithium reagent could react with the diethyl carbonate or open the thioketal after nucleophilic attack on the sulfur.16,25 In order to quantitatively form the desired aryllithium of 4, we thought to use a halogen–metal exchange reaction and undertook the synthesis of the iodinated derivative 5. To avoid the possible attack of the base on the sulfur,16,25 we used the more sterically hindered LiTMP. The treatment of 4 with 2.5 equivalents of LiTMP at −78 °C, followed by the addition of iodine, resulted in the formation of the mono-iodide 5 in an excellent yield. Indeed, less than 5% of diiodinated derivative was formed (Scheme 2, path B).
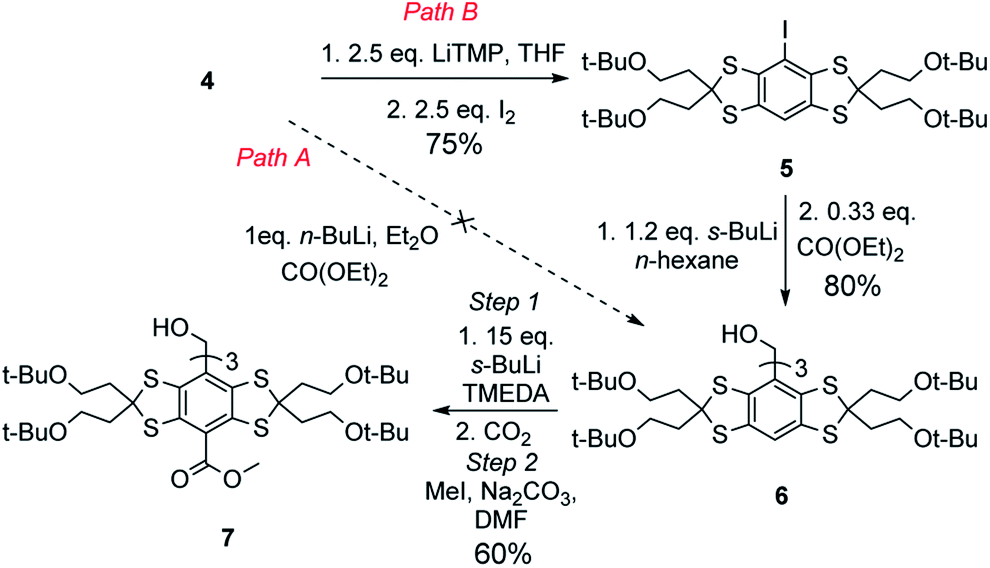 | ||
| Scheme 2 Synthesis of the trityl alcohol 7. | ||
The aryl iodide 5 was then treated at −78 °C with sec-BuLi in n-hexane to generate the corresponding aryllithium, followed by a slow addition of diethyl carbonate at room temperature to yield the trityl alcohol 6 in 80% yield. It is worth noting that the use of n-BuLi in diethyl ether did not result in the formation of the desired trityl 6 (Table 1, entry 1), as the deiodinated compound 4 was recovered together with unidentified compounds. The use of methyl chloroformate as an electrophile resulted in even more degradation (entry 2). A similar result was obtained in THF, with the exception of the formation of 40% of the butylated aryl analogue of 4 (entry 3).26 The use of sec-BuLi in THF prevented the formation of the butylated compound but did not result in the formation of the trityl alcohol 6 (entry 4). We found that only the use of the non-coordinating solvent n-hexane led to an efficient formation of trityl 6 in 80% yield (entry 5).
| Entrya | Base | Solvent | Electrophile | 6 (%) |
|---|---|---|---|---|
| a Base added at −78 °C, stirred for 15 min, warmed to room temperature, then the electrophile was added slowly over 3 h.b Butylated aryl analogue formed. | ||||
| 1 | n-BuLi | Et2O | CO(OEt)2 | 0 |
| 2 | n-BuLi | Et2O | ClCO2Me | 0 |
| 3 | n-BuLi | THF | CO(OEt)2 | 0b |
| 4 | s-BuLi | THF | CO(OEt)2 | 0 |
| 5 | s-BuLi | n-Hexane | CO(OEt)2 | 80% |
The introduction of carbonyl groups onto the trityl 6 was achieved by treatment with an excess of sec-BuLi (15 eq.) in anhydrous TMEDA at −30 °C, followed by bubbling of carbon dioxide. Interestingly, the treatment of 6 with 15 equivalents of tert-BuLi and TMEDA in benzene, followed by its addition to a solution of diethyl carbonate, as performed for the synthesis of FT,2,24 did not afford any esterified trityl, as the starting material was recovered (Table 2, entry 1). The same results were obtained in n-hexane, THF or diethyl ether (entries 2–4). When TMEDA was used as a solvent under similar conditions, a complex mixture of the starting material (8%) mono-(37%), di-(43%) and triester (7%) mixed with unidentified compounds (5%) was obtained, as determined by HPLC-MS, indicating that the deprotonation only occurred in TMEDA. Surprisingly, when diethyl carbonate was replaced by gaseous carbon dioxide, a clean mixture of triacid (70%) and diacid (30%) trityl alcohols was obtained. The carboxylic acids were then esterified from the mixture using iodomethane and sodium carbonate in DMF in order to allow a large-scale purification. 7 was obtained in 60% yield after purification on silica gel.
| Entry | Base | Solvent | Additive | Electrophile | 7 (%) |
|---|---|---|---|---|---|
| a Base (15 eq.) was added at room temperature, stirred for 2 h, then added to a solution of 30 eq. electrophile at room temperature and stirred for 1 h.b Base (15 eq.) was added at −30 °C, stirred for 2 h, then added to a solution of 30 eq. electrophile at room temperature and stirred for 1 h.c Base (15 eq.) was added at −30 °C, stirred for 2 h, then CO2 was bubbled for 30 min at −30 °C and 30 min at room temperature.d 15 eq.e Isolated yield after esterification. | |||||
| 1a | t-BuLi | C6H6 | TMEDAd | CO(OEt)2 | 0 |
| 2b | t-BuLi | n-Hexane | TMEDAd | CO(OEt)2 | 0 |
| 3b | t-BuLi | THF | TMEDAd | CO(OEt)2 | 0 |
| 4b | s-BuLi | Et2O | TMEDAd | CO(OEt)2 | 0 |
| 5b | s-BuLi | TMEDA | — | CO(OEt)2 | 7 |
| 6c | s-BuLi | TMEDA | — | CO2 | 60e |
The next step was the deprotection of the 12 alcohol groups (Scheme 3). The fully protected trityl alcohol 7 was heated at 45 °C for 90 minutes in formic acid, leading to a quantitative conversion of the tert-butyl ethers to formyl esters. Then, the tritylium cation was generated using triflic acid and subsequently reduced to radical by tin chloride(II). Finally, the esters were hydrolysed using sodium hydroxide, leading to OX063, isolated in 91% yield over the three steps.
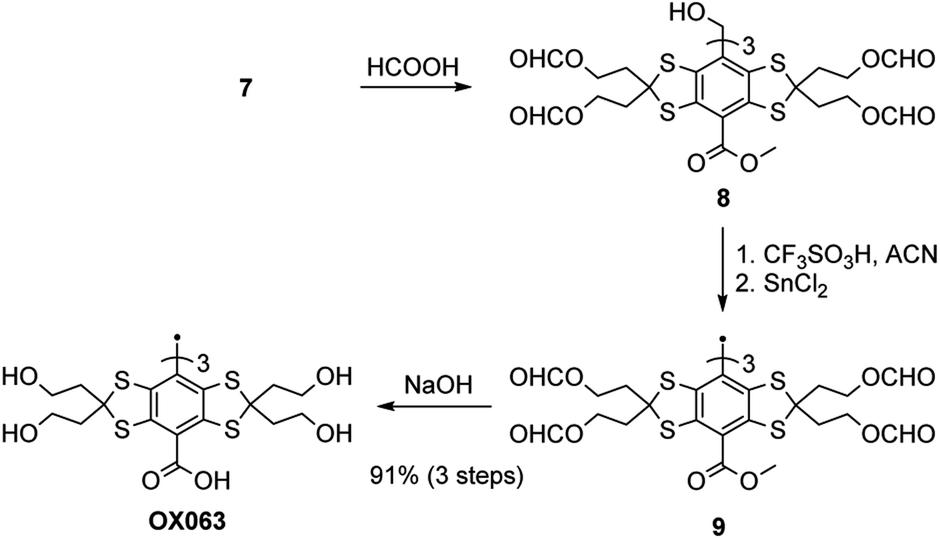 | ||
| Scheme 3 Conversion from 7 to OX063. | ||
OX063 EPR spectrum (50 μM) in PBS (10 mM, pH 7.4) recorded at X-band under nitrogen exhibits a single line pattern with a peak-to-peak linewidth of 160 mG (Fig. 2), which is consistent with the reported value.27
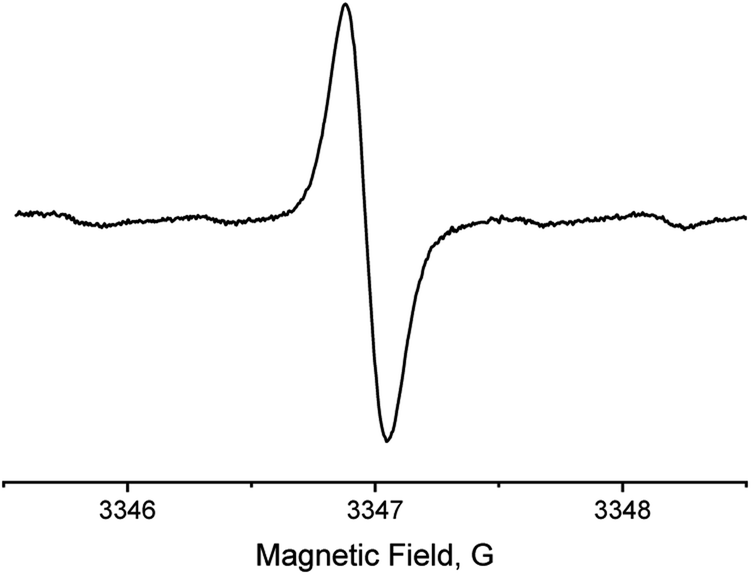 | ||
| Fig. 2 X-band EPR spectrum of OX063 (50 μM) in deoxygenated PBS (10 mM, pH = 7.4). | ||
The partial deuteration of the 12 methylene groups adjacent to the thioketals of OX063 leads to OX071 (Fig. 3), with a sharper linewidth.27 Indeed, unresolved splitting with hydrogen nuclei is responsible for an inhomogeneous broadening of the EPR line of OX063. The deuteration of OX063 decreases this inhomogeneous broadening due to the lower magnetic moment of the deuterium nucleus. A narrower linewidth increases the oxygen sensitivity and leads to a higher signal-to-noise ratio, which is of primary importance for in vivo applications.
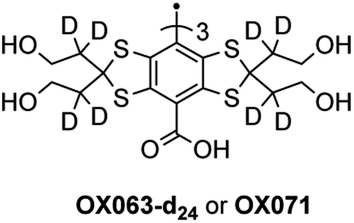 | ||
| Fig. 3 Structure of OX071. | ||
The synthesis of OX071 was achieved by exchange of the enolizable hydrogens of the intermediate 2 with CH3OD/CH3ONa in THF. The deuterated compound 2b was isolated in 85% yield without any purification (Scheme 4). OX071 was synthesized from 2b using the same procedures as OX063. The EPR spectrum of OX071 exhibits a single line pattern with a peak-to-peak linewidth of 80 mG (see ESI†), consistent with the value reported in the literature.27
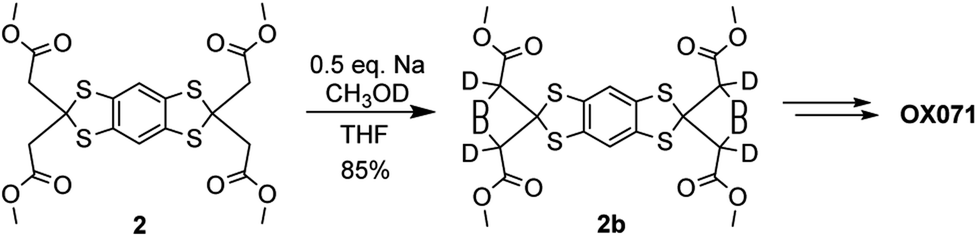 | ||
| Scheme 4 Deuteration of 2. | ||
Conclusions
We have developed an efficient synthesis of hydrophilic trityl radicals OX063 and its deuterated analogue OX071. Our synthetic protocol involves 7 steps and 4 chromatography columns and leads to OX063 with a total yield of 10%. This development will allow for the in vivo measurement of pO2 by EPRI and OMRI upon systemic delivery and for DNP applications. Moreover, our synthetic strategy will allow for the synthesis of new derivatives with extended functional sensitivity, such as phosphonated analogues for concurrent pO2, pH, and inorganic phosphate (Pi) measurement or new DNP agents and non-metallic contrast agents for MRI.28Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by the NIH grants (USA) EB023990, CA194013, CA192064, U54GM104942. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. West Virginia University Health Sciences Center is acknowledged for start-up fund to B. D. and WVU SURE program is acknowledged for funding support for J. L. H. We acknowledge use of the WVU Shared Research Facilities.Notes and references
- M. Thaning, PCT Int. Appl. WO9839277, 1998; S. Anderson, K. Golman, F. Rise, H. Wikström and L.-G. Wistrand, US Pat. 5530140, 1996.
- T. J. Reddy, T. Iwama, H. J. Halpern and V. H. Rawal, J. Org. Chem., 2002, 67, 4635–4639 CrossRef CAS PubMed.
- J. H. Ardenkjær-Larsen, I. Laursen, I. Leunbach, G. Ehnholm, L. G. Wistrand, J. S. Petersson and K. Golman, J. Magn. Reson., 1998, 133, 1–12 CrossRef.
- B. Driesschaert, V. Marchand, P. Levêque, B. Gallez and J. Marchand-Brynaert, Chem. Commun., 2012, 48, 4049–4051 RSC.
- V. Marchand, P. Levêque, B. Driesschaert, J. Marchand-Brynaert and B. Gallez, Magn. Reson. Med., 2017, 77, 2438–2443 CrossRef CAS.
- I. Dhimitruka, A. A. Bobko, T. D. Eubank, D. A. Komarov and V. V. Khramtsov, J. Am. Chem. Soc., 2013, 135, 5904–5910 CrossRef CAS.
- A. A. Bobko, T. D. Eubank, B. Driesschaert, I. Dhimitruka, J. Evans, R. Mohammad, E. E. Tchekneva, M. M. Dikov and V. V. Khramtsov, Sci. Rep., 2017, 7, 41233 CrossRef CAS PubMed.
- Y. Liu, Y. Song, A. Rockenbauer, J. Sun, C. Hemann, F. A. Villamena and J. L. Zweier, J. Org. Chem., 2011, 76, 3853–3860 CrossRef CAS.
- Y. Liu, F. A. Villamena, A. Rockenbauer and J. L. Zweier, Chem. Commun., 2010, 46, 628–630 RSC.
- G. Y. Shevelev, E. L. Gulyak, A. A. Lomzov, A. A. Kuzhelev, O. A. Krumkacheva, M. S. Kupryushkin, V. M. Tormyshev, M. V. Fedin, E. G. Bagryanskaya and D. V. Pyshnyi, J. Phys. Chem. B, 2018, 122, 137–143 CrossRef CAS PubMed.
- Z. Yang, Y. Liu, P. Borbat, J. L. Zweier, J. H. Freed and W. L. Hubbell, J. Am. Chem. Soc., 2012, 134, 9950–9952 CrossRef CAS.
- G. Mathies, M. A. Caporini, V. K. Michaelis, Y. Liu, K.-N. Hu, D. Mance, J. L. Zweier, M. Rosay, M. Baldus and R. G. Griffin, Angew. Chem., Int. Ed., 2015, 54, 11770–11774 CrossRef CAS.
- Y. Song, Y. Liu, W. Liu, F. A. Villamena and J. L. Zweier, RSC Adv., 2014, 4, 47649–47656 RSC.
- A. A. Gorodetskii, T. D. Eubank, B. Driesschaert, M. Poncelet, E. Ellis, V. V. Khramtsov and A. A. Bobko, Sci. Rep., 2019, 9, 12093 CrossRef.
- B. Epel, M. C. Maggio, E. D. Barth, R. C. Miller, C. A. Pelizzari, M. Krzykawska-Serda, S. V. Sundramoorthy, B. Aydogan, R. R. Weichselbaum, V. M. Tormyshev and H. J. Halpern, Int. J. Radiat. Oncol., Biol., Phys., 2019, 103, 977–984 CrossRef.
- Y. Qu, Y. Li, X. Tan, W. Zhai, G. Han, J. Hou, G. Liu, Y. Song and Y. Liu, Chem.–Eur. J., 2019, 25, 7888–7895 CrossRef CAS.
- M. Serda, Y.-K. Wu, E. D. Barth, H. J. Halpern and V. H. Rawal, Chem. Res. Toxicol., 2016, 29, 2153–2156 Search PubMed.
- Y. Song, Y. Liu, C. Hemann, F. A. Villamena and J. L. Zweier, J. Org. Chem., 2013, 78, 1371–1376 CrossRef CAS.
- W. Liu, J. Nie, X. Tan, H. Liu, N. Yu, G. Han, Y. Zhu, F. A. Villamena, Y. Song, J. L. Zweier and Y. Liu, J. Org. Chem., 2017, 82, 588–596 CrossRef CAS.
- B. Driesschaert, A. A. Bobko, T. D. Eubank, A. Samouilov, V. V. Khramtsov and J. L. Zweier, Bioorg. Med. Chem. Lett., 2016, 26, 1742–1744 CrossRef CAS.
- B. Driesschaert, P. Levêque, B. Gallez and J. Marchand-Brynaert, Tetrahedron Lett., 2013, 54, 5924–5926 CrossRef CAS.
- Y. Liu, F. A. Villamena and J. L. Zweier, Chem. Commun., 2008, 4336–4338 RSC.
- M. Poncelet, B. Driesschaert, O. Tseytlin, M. Tseytlin, T. D. Eubank and V. V. Khramtsov, Bioorg. Med. Chem. Lett., 2019, 29, 1756–1760 CrossRef CAS PubMed.
- I. Dhimitruka, M. Velayutham, A. A. Bobko, V. V. Khramtsov, F. A. Villamena, C. M. Hadad and J. L. Zweier, Bioorg. Med. Chem. Lett., 2007, 17, 6801–6805 CrossRef CAS PubMed.
- H. Hintz, A. Vanas, D. Klose, G. Jeschke and A. Godt, J. Org. Chem., 2019, 84, 3304–3320 CrossRef CAS.
- R. E. Merrill and E. Negishi, J. Org. Chem., 1974, 39, 3452–3453 CrossRef CAS.
- B. Epel and H. J. Halpern, in Methods in Enzymology, ed. P. Z. Qin and K. Warncke, Academic Press, 2015, vol. 564, pp. 501–527 Search PubMed.
- H. V. T. Nguyen, A. Detappe, N. M. Gallagher, H. Zhang, P. Harvey, C. Yan, C. Mathieu, M. R. Golder, Y. Jiang, M. F. Ottaviani, A. Jasanoff, A. Rajca, I. Ghobrial, P. P. Ghoroghchian and J. A. Johnson, ACS Nano, 2018, 12, 11343–11354 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08633a |
| This journal is © The Royal Society of Chemistry 2019 |