
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMonitoring glycosidase activity for clustered sugar substrates, a study on β-glucuronidase†
Yoan Brissonneta,
Guillaume Compainb,
Brigitte Renouxb,
Eva-Maria Krammer c,
Franck Daligaultd,
David Deniauda,
Sébastien Papotb and
Sébastien G. Gouin*a
c,
Franck Daligaultd,
David Deniauda,
Sébastien Papotb and
Sébastien G. Gouin*a
aUniversité de Nantes, CEISAM, Chimie Et Interdisciplinarité, Synthèse, Analyse, Modélisation, UMR CNRS 6230, UFR des Sciences et des Techniques, 2, rue de la Houssinière, BP 92208, 44322 Nantes Cedex 3, France
bInstitut de Chimie des Milieux et des Matériaux de Poitiers, IC2MP, Université de Poitiers, UMR-CNRS 7285, 4 Rue Michel Brunet, 86022 Poitiers, France
cStructure et Fonction des Membranes Biologiques, Université Libre de Bruxelles (ULB), Brussels, Belgium
dUniversité de Nantes, UFIP, UMR CNRS 6286, UFR des Sciences et des Techniques, France
First published on 4th December 2019
Abstract
Determination of glycosidase hydrolysis kinetics for a monovalent sugar substrate is relatively straightforward and classically achieved by monitoring the fluorescence signal released from the sugar-conjugated probe after enzymatic hydrolysis. Naturally occuring sugar epitopes are, however, often clustered on biopolymers or at biological surfaces, and previous reports have shown that glycosidase hydrolytic rates can differ greatly with multivalent presentation of the sugar epitopes. New probes are needed to make it easier to interpret the importance of substrate clustering towards a specific enzyme activity. In this work, we developed multivalent glucuronide substrates attached to fluorescent amino-coumarines through self-immolative linkers to enable real time-monitoring of the hydrolysing activity of E.coli β-glucuronidases (GUS) towards clustered substrates. GUS are exoglycosidases of considerable therapeutic interest cleaving β-D-glucuronides and are found in the lysosomes, in the tumoral microenvironment, and are expressed by gut microbiota. GUS showed a much lower catalytic efficiency in hydrolysing clustered glucuronides due to a significantly lower enzymatic velocity and affinity for the substrates. GUS was 52-fold less efficient in hydrolysing GlcA substrates presented on an octameric silsequioxane (COSS) compared with a monovalent GlcA of similar chemical structure. Thus, kinetic and thermodynamic data of GUS hydrolysis towards multivalent glucuronides were easily obtained with these new types of enzymatically-triggered probes. More generally, adapting the substrate nature and valency of these new probes, should improve understanding of the impact of multivalency for a specific enzyme.
Introduction
Glycoclusters bearing several copies of a sugar ligand on a chemical or biological core have been extensively developed to target carbohydrate-binding-proteins (lectins).1–3 In numerous reports, the tethered ligands are shown to provide synergistic affinity enhancement compared with their corresponding monovalent references. Large multivalent effects with glycoclusters have been reported on a wide range of lectins, which are often multimeric in nature or multivalently displayed at a cell surface. The enhanced selectivity and avidity is generally explained by an increased local concentration (effective molarity) of the additional ligands close to the binding sites and in theoretical models by a statistical component (degeneracy prefactor) growing non-linearly with increased multivalency of both ligands and receptors.4Although, mostly exemplified on carbohydrate-binding proteins, multivalency effects have also been demonstrated on carbohydrate-processing enzymes (glycosidases and glycosyltransferases). Indeed, these enzymes were previously shown to display very different hydrolytic profiles towards multimerized substrates compared with monovalent analogues. N-acetylglucosamine-capped glycolipids can form microdomains in phospholipid bilayers. Webb and co-workers showed an accelerated galactosylation of these clustered sugars by bovine β(1,4)-galactosyltransferase. This was explained by a lower apparent KM attributed to multivalent binding.5 Boons and co-workers observed that bacterial sialidases flanked by a lectin domain hydrolyse polyvalent sialosides with much higher catalytic efficiency than their monovalent counterparts.6 In stark contrast, O-glucosides and O-mannosides multivalently displayed on nanodiamond particles were shown to be fully stable and even to inhibit their matching enzymes (glycosidases and mannosidases, respectively).7
These multivalent effects were also observed with clustered transition-state inhibitors of glycosidases.8–11 After the first evidence for a multivalent inhibitory effect with clusterized deoxynojirymicin iminosugars was shown on the jack bean mannosidase (jbMan) model,12 large synergistic effects were observed with this enzyme,13,14 and on biologically relevant GH targets such as golgi-mannosidase,15,16 hexosaminidases,17 sialidases,6,17 and the glycosyltransferases WaaC.18 More surprisingly, polyvalent iminosugars were also shown to improve the maximal velocity and catalytic efficiency of a galactosidase with potential application in the food industry.19,20 Thus multivalent analogues of substrate transition-states (TS) represent new tools to modulate GH activity.
In this context there is strong interest in developing multivalent sugar probes that would allow direct measurement of glycosidase hydrolysis kinetics on clustered sugar substrates. It is more challenging to obtain a direct read out of a fluorescent signal from a multivalent system as the fluorophore needs to be released simultaneously from the sugar and the multivalent scaffold after glycosidase hydrolysis. To make this possible, we aimed to design multivalent sugars bearing a silent chromophore attached to the scaffold by a self immolative linker (Fig. 1).
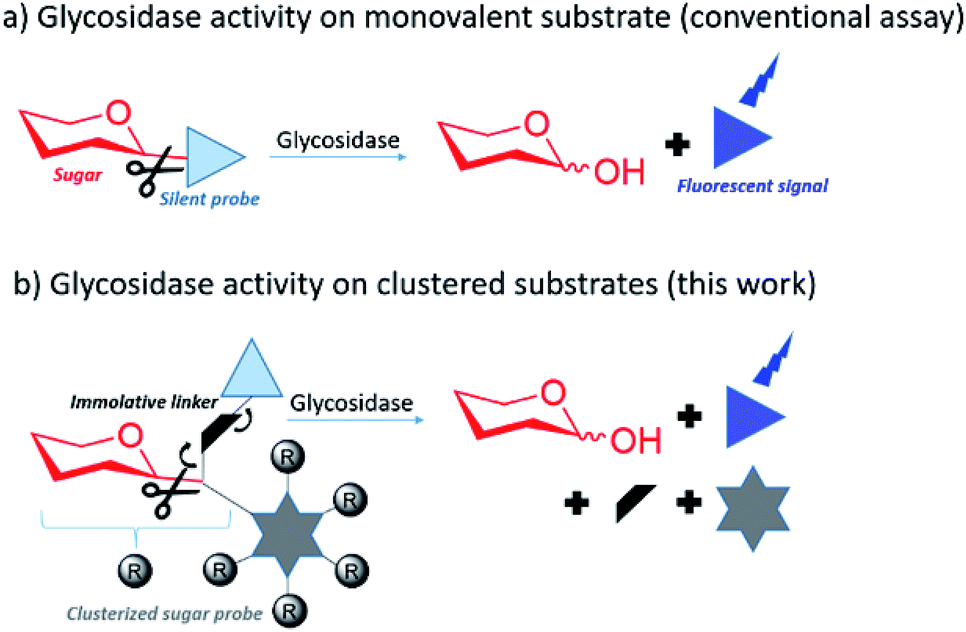 | ||
| Fig. 1 (a) The determination of glycosidase activity for a specific sugar substrate can be achieved by measuring a fluorescent signal after sugar cleavage from a glycoprobe. (b) In this work, we developed multivalent sugar substrates with silent chromophores attached through self-immolative linkers. The multivalent glycoprobes are designed to directly measure if a target glycosidase can hydrolyse clustered substrates with higher or lower catalytic efficiency than their monovalent counterparts. | ||
This concept was exemplified here with the synthesis of mono, di- and octavalent β-glucuronidase (GUS)-responsive pro-fluorophores (p-FluoGlcA, Fig. 2) to assess whether multivalency could be used to adjust GUS activity. More precisely, we compared the kinetic and catalytic efficiency of GUS when β-D-glucuronic acid (GlcA) substrates are mono- or multivalently displayed on a common core. Human β-glucuronidase (hGUS) is a homotetramer of 332 kDa with four active sites cleaving β-D-glucuronic acid (GlcA) from glycosaminoglycans.21 GUS is an exoglycosidase of considerable therapeutic interest found in lysosomes, in the tumor microenvironment, and is also expressed by symbiotic gut microbiota. Mutations in hGUS gene cause mucopolysaccharidosis type VII, a lysosomal storage disease caused by heparan sulfate and dermatan sulfate accumulation.22 Enzyme replacement therapy consisting in multiple injection of functional GUS proves efficient in preventing lysosomal storage in several tissues.23 The elevated GUS activity in specific tumours has also been successfully exploited for the selective delivery of cytotoxic agents.24 Potent anticancer drugs such as doxorubicin,25,26 or monomethylauristatine E,27 have been successfully targeted to various tumours by the means of β-glucuronidase-responsive drug delivery systems, leading to impressive therapeutic efficacy in mice. Inhibition of GUS microbiota may also be required to alleviate drug and endobiotic toxicity. Indeed, cytotoxic compounds are glucuronidated by the liver for passivation but GUS from the microbiome cleaves the sugar in the gastrointestinal tract, restoring the initial drug toxicity.28
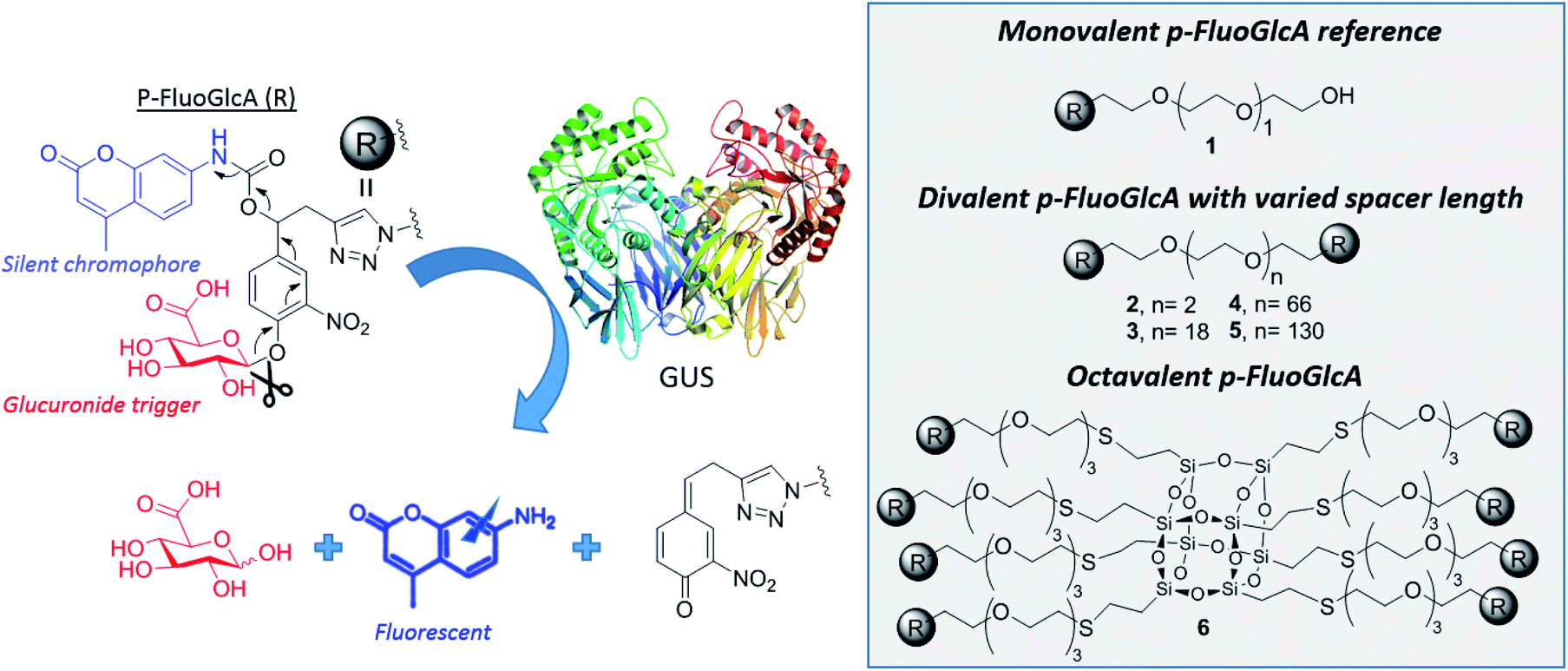 | ||
| Fig. 2 Measuring the impact of multivalency in GUS kinetics. A GUS-responsive pro-fluorophore p-FluoGlcA was developed for real-time monitoring of the enzymatic activity. GUS activity will hydrolyze the glucuronide trigger uncapping a self-immolative linker and releasing the fluorescent amino-coumarin. Detection of signal, reflecting GUS activity is compared for mono-, di- and octavalent p-FluoGlcA. | ||
We designed mono-, di- and octavalent FluoGlcA (Fig. 2) bearing a glucuronide trigger (in red) which, upon GUS cleavage uncap a self-immolative linker (in black) releasing an amino-coumarine dye (in blue, λex = 380 nm, λem = 450 nm). The self-immolative linker presented Fig. 2 has been selected due to its widespread use in the design of enzyme-responsive prodrugs.29–31 Such aromatic linkers bearing a nitro group allow a fast liberation kinetics of the drugs.32
Amino-coumarin functionalized by a propargyl-oxo-carbonyl group was previously shown to be virtually non-fluorescent but was converted into a highly fluorescent amino-coumarin after palladium-triggered depropargylation.33 Here, this responsive fluorescence is exploited for the kinetic measurement of GUS activity. Divalent p-FluoGlcA with varied PEG spacer arm length were designed to assess whether the spanning of two binding sites in GUS by the GlcA leads to a specific hydrolytic profile. Molecular dynamics experiments were performed to assess the average distance between the glucuronides ligands of compounds 2–5, and we found distances ranging from 8 to 59 Å (Chart 1). This is in accordance with previous calculations from polymer theory performed with a different set of compounds bearing PEG linkers of similar lengths.17,34 Compound 5 should therefore cover the distance of around 50 Å between two adjacent GUS binding sites, as determined from the crystal structure of E. coli GUS bound to an urea inhibitor (PDB code = 3LPG).35
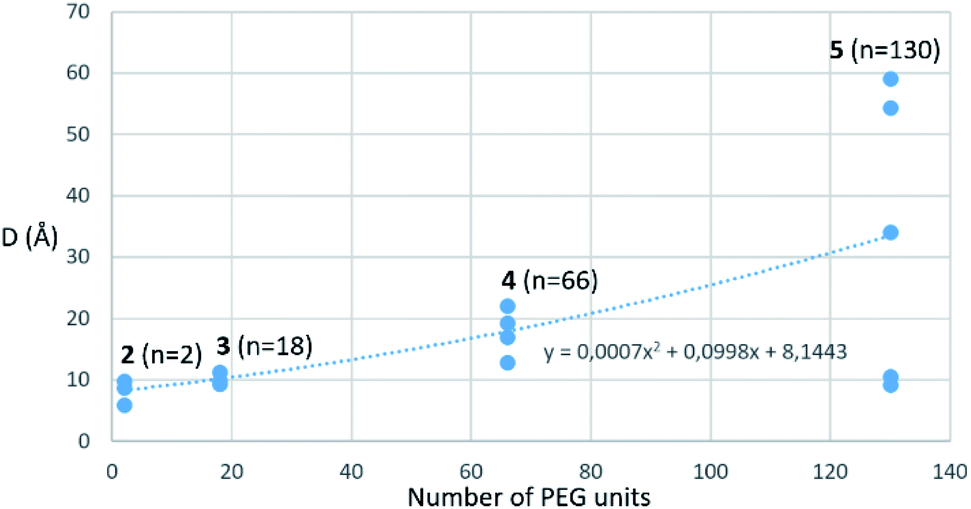 | ||
| Chart 1 Average distance d between the glucuronide ligands of compounds 2–5 plotted against the PEG number of the compounds. The distances were extracted from several 20 ns long, independent molecular dynamics simulations (for more details see Sup. Mat.) A nonlinear least squares regression using a power fitting function was used to fit the data points and is shown as a dotted line in the graph. | ||
Compound 6 was designed from a cubic octameric silsequioxane (COSS) scaffold. COSS are emerging as versatile core for the design of multivalent sugar ligands,36–38 the cubic geometry allowing an homogeneous spatial distribution of the grafted ligands.
Chemical synthesis
Compounds 1–6 (Scheme 1) were readily accessible in only three synthetic steps from the glucuronide 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 29,39 (Synthesis in ESI†). First, the 7-amino-4-methyl-2H-chromen-2-one 8 was introduced via nucleophilic substitution in the presence of hydroxybenzotriazole (HOBt) and (i-Pr)2NEt to give the carbamate 9 (38%). Full deprotection of the glucuronide moiety was then carried out using LiOH, giving the alkyne 10 with 58% yield after purification by reverse phase column chromatography. Finally, triazoles 1–6 were obtained by coupling 10 with the corresponding azide derivatives 11, using the well-known copper(I)-catalysed azide–alkyne 1,3-cycloaddition.40–43 After size-exclusion column purification, 1–6 were obtained with yields ranging from 58 to 95%.
29,39 (Synthesis in ESI†). First, the 7-amino-4-methyl-2H-chromen-2-one 8 was introduced via nucleophilic substitution in the presence of hydroxybenzotriazole (HOBt) and (i-Pr)2NEt to give the carbamate 9 (38%). Full deprotection of the glucuronide moiety was then carried out using LiOH, giving the alkyne 10 with 58% yield after purification by reverse phase column chromatography. Finally, triazoles 1–6 were obtained by coupling 10 with the corresponding azide derivatives 11, using the well-known copper(I)-catalysed azide–alkyne 1,3-cycloaddition.40–43 After size-exclusion column purification, 1–6 were obtained with yields ranging from 58 to 95%.
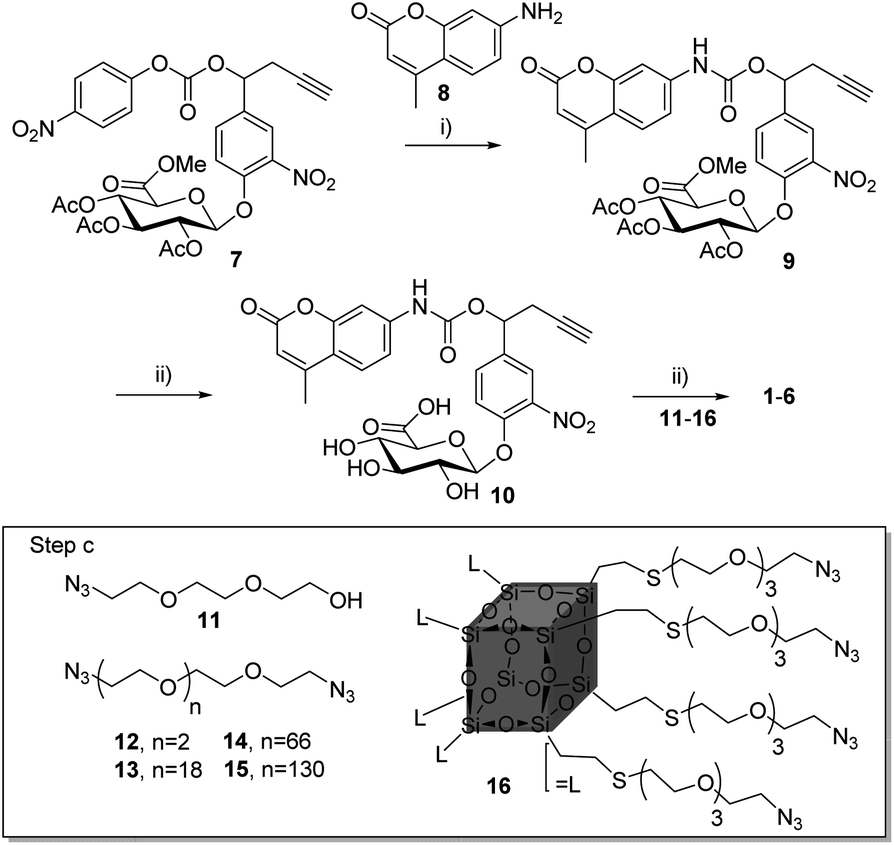 | ||
| Scheme 1 Synthesis of compounds 1–6. (i) 8, HOBt, DMF, (i-Pr)2NEt, 50 °C, 24 h, 38%; (ii) LiOH, H2O/MeOH, 0 °C, 2 h, 58%; (iii) CuSO4, NaAsc, dioxane–water, r.t., 12 h, with 11 for 1 (95%), 12 for 2 (78%), 13 for 3 (67%), 14 for 4 (61%), 15 for 5 (62%), 16 for 6 (58%). | ||
Enzymatic assays
Compounds 1–6 were assessed against the commercially available β-glucuronidase from E. coli. E.coli GUS shows a low sequence similarity with human GUS (45%) but both enzymes are tetravalent with a conserved three-dimensional fold.35 The fluorescence signal was directly monitored during the reaction and converted into concentration by a calibration curve. The Michaelis constant Km and the maximal velocity Vmax values were determined from the Michaelis–Menten equation at a GUS concentration of 9.6 nM. The Km, Vmax and catalytic efficiency (Ceff = kcat/Km) are presented in Fig. 3 together with the fitted curves of GUS hydrolysis velocity as a function of the molecule concentration of 1–6 (average from quadruplicates). To improve comparison of the enzymatic efficiency per mol of GlcA ligand (instead of molecule) we calculated the R(Ceff) representing the fold-decrease of GUS catalytic efficiency per GlcA ligand of a multivalent compound X of valency VX, compared with the GlcA ligand of monovalent reference 1. R(Ceff) was obtained from the equation R(Ceff) = VX × (kcat1/Km1)/(kcatX/KmX). GUS showed an apparent decreased affinity (higher KM) and lower hydrolysis rate (lower Vm or kcat) for the multivalent GlcA 2–6 than monovalent 1. It follows that the relative hydrolytic catalytic efficiency R(Ceff) for a GlcA ligand was reduced by 4 to 52-fold when multimerized on scaffolds 2–6. This significant decrease may not be explained by different interactions between 1 and 2–6 in close binding site proximity because the GlcA environment is the same independently of the substrate valency.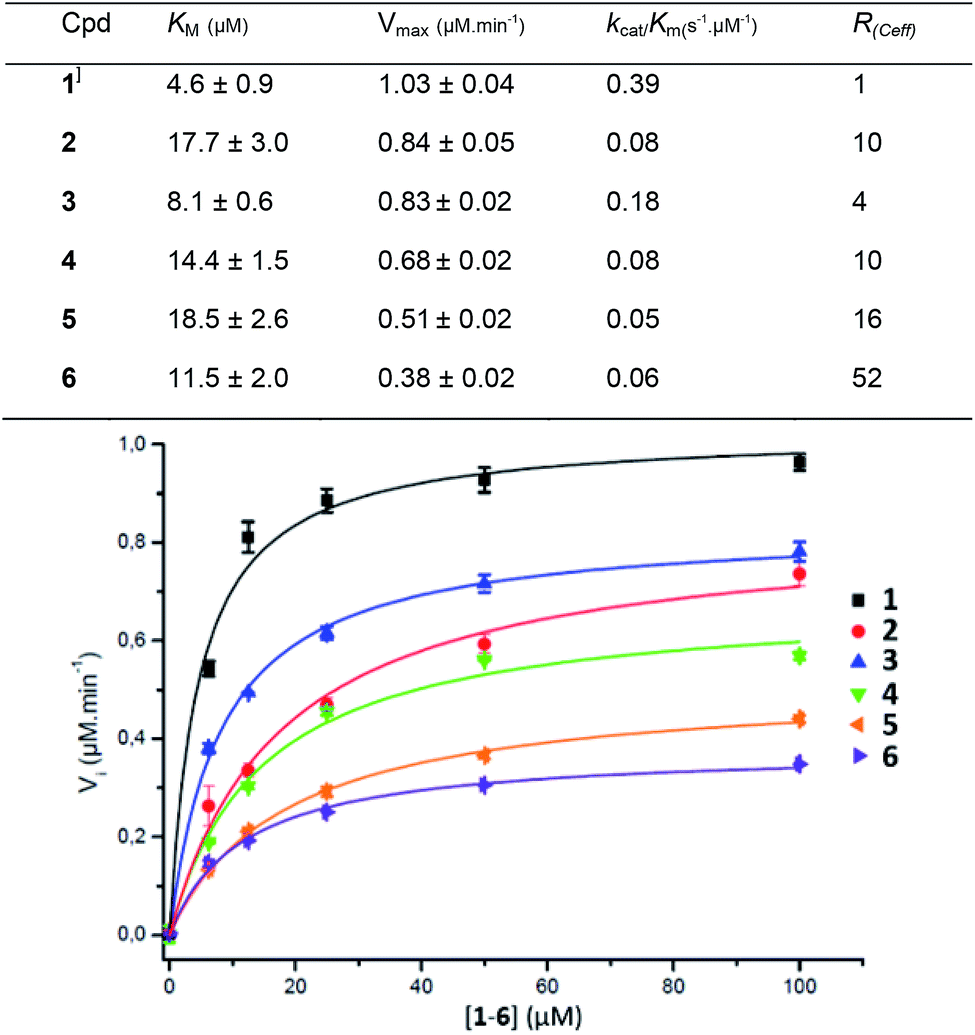 | ||
| Fig. 3 Kinetic parameters obtained from the Michaelis–Menten equation on GUS activity with compounds 1–6. | ||
The PEG length elongation from n = 2 to n = 130 for the divalent serie 2–5 also affected R(Ceff), suggesting that the scaffold and the GlcA–GlcA average distance play a role in the reduced Ceff. This scenario was enhanced with COSS 6 where the GlcA are also tethered through EG spacer arms. Despite an eight fold higher GlcA concentration due to the octavalency, the GUS Ceff of 6 was only 0.06 μmol min−1, each of the GlcA being hydrolysed with 52-fold lower efficiency compared with 1. Although it is difficult at present to hypothesize about a specific binding mode, the data clearly show that a multivalent display of the sugar substrates strongly affects GUS activity.
Conclusions
In summary we developed multivalent glucuronides with self-immolative linkers releasing a fluorescent amino-coumarin after GUS activation. The multivalent probes allowed easy monitoring of the kinetic parameters for GUS enzymatic activity. GUS was shown to hydrolyze the clustered glucuronide probes with much less efficiency than the monovalent glucuronide analogues. The scope of the multivalent probes developed here could potentially be broadened to any glycosidase by adapting the sugar nature and valency. This may help to improve understanding the intriguing function of multivalency in carbohydrate processing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out with financial support from the Conseil Régional des Pays de La Loire (MAGGIC project), the Centre National de la Recherche Scientifique (CNRS) and the French Ministère de l’Enseignement Supérieur et de la Recherche. E. M. K. is a postdoctoral researcher of the Fonds de la Recherche Scientifique de Belgique (F.R.S.-F.R.N.S.), Belgium.References
- D. Deniaud, K. Julienne and S. G. Gouin, Org. Biomol. Chem., 2011, 9, 966–979 RSC.
- S. Cecioni, A. Imberty and S. Vidal, Chem. Rev., 2015, 115, 525–561 CrossRef CAS PubMed.
- A. Bernardi, J. Jimenez-Barbero, A. Casnati, C. De Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K.-E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. V. Murphy, C. Nativi, S. Oscarson, S. Penades, F. Peri, R. J. Pieters, O. Renaudet, J.-L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schaffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709–4727 RSC.
- K. C. Tjandra and P. Thordarson, Bioconjugate Chem., 2019, 30, 503–514 CrossRef CAS PubMed.
- G. T. Noble, F. L. Craven, J. Voglmeir, R. Šardzík, S. L. Flitsch and S. J. Webb, J. Am. Chem. Soc., 2012, 134, 13010–13017 CrossRef CAS PubMed.
- S. Thobhani, B. Ember, A. Siriwardena and G.-J. Boons, J. Am. Chem. Soc., 2003, 125, 7154–7155 CrossRef CAS PubMed.
- A. Siriwardena, M. Khanal, A. Barras, O. Bande, T. Mena-Barragán, C. O. Mellet, J. M. G. Fernández, R. Boukherroub and S. Szunerits, RSC Adv., 2015, 5, 100568–100578 RSC.
- S. G. Gouin, Chem.–Eur. J., 2014, 20, 11616–11628 CrossRef CAS PubMed.
- P. Compain and A. Bodlenner, ChemBioChem, 2014, 15, 1239–1251 CrossRef CAS PubMed.
- C. Matassini, C. Parmeggiani, F. Cardona and A. Goti, Tetrahedron Lett., 2016, 57, 5407–5415 CrossRef CAS.
- N. Kanfar, E. Bartolami, R. Zelli, A. Marra, J.-Y. Winum, S. Ulrich and P. Dumy, Org. Biomol. Chem., 2015, 13, 9894–9906 RSC.
- J. Diot, M. I. García-Moreno, S. G. Gouin, C. O. Mellet, K. Haupt and J. Kovensky, Org. Biomol. Chem., 2008, 7, 357–363 RSC.
- P. Compain, C. Decroocq, J. Iehl, M. Holler, D. Hazelard, T. Mena Barragán, C. Ortiz Mellet and J.-F. Nierengarten, Angew. Chem., Int. Ed., 2010, 49, 5753–5756 CrossRef CAS PubMed.
- M. L. Lepage, J. P. Schneider, A. Bodlenner, A. Meli, F. De Riccardis, M. Schmitt, C. Tarnus, N.-T. Nguyen-Huynh, Y.-N. Francois, E. Leize-Wagner, C. Birck, A. Cousido-Siah, A. Podjarny, I. Izzo and P. Compain, Chem.–Eur. J., 2016, 22, 5151–5155 CrossRef CAS PubMed.
- Y. Brissonnet, C. Ortiz Mellet, S. Morandat, M. I. Garcia Moreno, D. Deniaud, S. E. Matthews, S. Vidal, S. Šesták, K. El Kirat and S. G. Gouin, J. Am. Chem. Soc., 2013, 135, 18427–18435 CrossRef CAS PubMed.
- S. Mirabella, G. D'Adamio, C. Matassini, A. Goti, S. Delgado, A. Gimeno, I. Robina, A. J. Moreno-Vargas, S. Šesták, J. Jiménez-Barbero and F. Cardona, Chem.–Eur. J., 2017, 23, 14585–14596 CrossRef CAS PubMed.
- Y. Brissonnet, C. Assailly, A. Saumonneau, J. Bouckaert, M. Maillasson, C. Petitot, B. Roubinet, B. Didak, L. Landemarre, C. Bridot, R. Blossey, D. Deniaud, X. Yan, J. Bernard, C. Tellier, C. Grandjean, F. Daligault and S. G. Gouin, Chem.–Eur. J., 2019, 25, 2358–2365 CrossRef CAS PubMed.
- M. Durka, K. Buffet, J. Iehl, M. Holler, J.-F. Nierengarten and S. P. Vincent, Chem.–Eur. J., 2012, 18, 641–651 CrossRef CAS PubMed.
- Y. Brissonnet, S. Ladevèze, D. Tezé, E. Fabre, D. Deniaud, F. Daligault, C. Tellier, S. Šesták, M. Remaud-Simeon, G. Potocki-Veronese and S. G. Gouin, Bioconjugate Chem., 2015, 26, 766–772 CrossRef CAS PubMed.
- D. Alvarez-Dorta, Y. Brissonnet, A. Saumonneau, D. Deniaud, J. Bernard, X. Yan, C. Tellier, F. Daligault and S. G. Gouin, ChemistrySelect, 2017, 2, 9552–9556 CrossRef CAS.
- S. Jain, W. B. Drendel, Z. Chen, F. S. Mathews, W. S. Sly and J. H. Grubb, Nat. Struct. Mol. Biol., 1996, 3, 375–381 CrossRef CAS PubMed.
- S. Tomatsu, S. Fukuda, K. Sukegawa, Y. Ikedo, S. Yamada, Y. Yamada, T. Sasaki, H. Okamoto, T. Kuwahara, S. Yamaguchi and T. K. H. Shintaku, Am. J. Hum. Genet., 1991, 48, 89–96 CAS.
- H. Naz, A. Islam, A. Waheed, W. S. Sly, F. Ahmad and Md. I. Hassan, Rejuvenation Res., 2013, 16, 352–363 CrossRef CAS PubMed.
- I. Tranoy-Opalinski, T. Legigan, R. Barat, J. Clarhaut, M. Thomas, B. Renoux and S. Papot, Eur. J. Med. Chem., 2014, 74, 302–313 CrossRef CAS PubMed.
- P. H. J. Houba, E. Boven, I. H. van der Meulen-Muileman, R. G. G. Leenders, J. W. Scheeren, H. M. Pinedo and H. J. Haisma, Br. J. Cancer, 2001, 84, 550–557 CrossRef CAS PubMed.
- T. Legigan, J. Clarhaut, B. Renoux, I. Tranoy-Opalinski, A. Monvoisin, J.-M. Berjeaud, F. Guilhot and S. Papot, J. Med. Chem., 2012, 55, 4516–4520 CrossRef CAS PubMed.
- T. Legigan, J. Clarhaut, B. Renoux, I. Tranoy-Opalinski, A. Monvoisin, C. Jayle, J. Alsarraf, M. Thomas and S. Papot, Eur. J. Med. Chem., 2013, 67, 75–80 CrossRef CAS PubMed.
- B. D. Wallace, A. B. Roberts, R. M. Pollet, J. D. Ingle, K. A. Biernat, S. J. Pellock, M. K. Venkatesh, L. Guthrie, S. K. O'Neal, S. J. Robinson, M. Dollinger, E. Figueroa, S. R. McShane, R. D. Cohen, J. Jin, S. V. Frye, W. C. Zamboni, C. Pepe-Ranney, S. Mani, L. Kelly and M. R. Redinbo, Chem. Biol., 2015, 22, 1238–1249 CrossRef CAS PubMed.
- B. Renoux, L. Fangous, C. Hötten, E. Péraudeau, B. Eddhif, P. Poinot, J. Clarhaut and S. Papot, MedChemComm, 2018, 9, 2068–2071 RSC.
- W. Viricel, G. Fournet, S. Beaumel, E. Perrial, S. Papot, C. Dumontet and B. Joseph, Chem. Sci., 2019, 10, 4048–4053 RSC.
- T. Legigan, J. Clarhaut, I. Tranoy-Opalinski, A. Monvoisin, B. Renoux, M. Thomas, A. Le Pape, S. Lerondel and S. Papot, Angew. Chem., Int. Ed., 2012, 51, 11606–11610 CrossRef CAS PubMed.
- E. Bouvier, S. Thirot, F. Schmidt and C. Monneret, Bioorg. Med. Chem., 2004, 12, 969–977 CrossRef CAS PubMed.
- J. Wang, B. Cheng, J. Li, Z. Zhang, W. Hong, X. Chen and P. R. Chen, Angew. Chem., Int. Ed., 2015, 54, 5364–5368 CrossRef CAS.
- R. H. Kramer and J. W. Karpen, Nature, 1998, 395, 710–713 CrossRef CAS PubMed.
- B. D. Wallace, H. Wang, K. T. Lane, J. E. Scott, J. Orans, J. S. Koo, M. Venkatesh, C. Jobin, L.-A. Yeh, S. Mani and M. R. Redinbo, Science, 2010, 330, 831–835 CrossRef CAS PubMed.
- Y. Gao, A. Eguchi, K. Kakehi and Y. C. Lee, Org. Lett., 2004, 6, 3457–3460 CrossRef CAS PubMed.
- N. Kanfar, A. Mehdi, P. Dumy, S. Ulrich and J.-Y. Winum, Chem.–Eur. J., 2017, 23, 17867–17869 CrossRef CAS PubMed.
- B. Trastoy, D. A. Bonsor, M. E. Pérez-Ojeda, M. L. Jimeno, A. Méndez-Ardoy, J. M. García Fernández, E. J. Sundberg and J. L. Chiara, Adv. Funct. Mater., 2012, 22, 3191–3201 CrossRef CAS.
- B. Renoux, T. Legigan, S. Bensalma, C. Chadéneau, J.-M. Muller and S. Papot, Org. Biomol. Chem., 2011, 9, 8459–8464 RSC.
- P. V. Babu, S. Mukherjee, D. R. Gorja, S. Yellanki, R. Medisetti, P. Kulkarni, K. Mukkanti and M. Pal, RSC Adv., 2014, 4, 4878 RSC.
- Y. Yu, J. Zou, L. Yu, W. Ji, Y. Li, W.-C. Law and C. Cheng, Macromolecules, 2011, 44, 4793–4800 CrossRef CAS.
- K. Zhang, M. A. Lackey, Y. Wu and G. N. Tew, J. Am. Chem. Soc., 2011, 133, 6906–6909 CrossRef CAS PubMed.
- K. S. S. Praveena, E. V. V. Shivaji Ramarao, N. Y. S. Murthy, S. Akkenapally, C. Ganesh Kumar, R. Kapavarapu and S. Pal, Bioorg. Med. Chem. Lett., 2015, 25, 1057–1063 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra08847d |
| This journal is © The Royal Society of Chemistry 2019 |