
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe influences of electronic effect and isomerization of salalen titanium(IV) complexes on ethylene polymerization in the presence of methylaluminoxane†
Sihan Lia,
Yuqiong Zhua,
Huaqing Lianga,
Xiuli Xiea,
Yipeng Zhana,
Guodong Liang a and
Fangming Zhu*ab
a and
Fangming Zhu*ab
aPCFM and GDHPPC Lab, School of Chemistry, Sun Yat-Sen University, 510275, China. E-mail: ceszfm@mail.sysu.edu.cn; Fax: +86-20-84114033; Tel: +86-20-84113250
bPCFM and GDHPPC Lab, School of Materials Science and Engineering, Sun Yat-Sen University, Guangzhou, 510275, China
First published on 17th December 2019
Abstract
Herein, two salalen titanium(IV) complexes were synthesized and characterized. These complexes coexisted as two isomers in certain conditions and underwent isomerization, as evidenced by 1H NMR spectroscopy. Furthermore, the molar ratio of the two isomers ranged from 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15 at 30 °C to 100:34 at 120 °C, driven by thermal energy, based on variable temperature 1H NMR characterization. Both complexes were employed as catalysts for ethylene polymerization in the presence of methylaluminoxane (MAO). The influence of the electronic effects of different substituent groups at the ortho position of the phenolate on ethylene polymerization behaviors, molecular weight and molecular weight distributions of the resulting polyethylene was investigated. The fluorinated salalen titanium(IV) complex revealed relatively high catalytic activity and thermal stability owing to the electron-withdrawing inductive effect. Moreover, disentangled linear polyethylene with ultrahigh molecular weight (Mw up to 3000 kDa) and narrow molecular weight distribution (Mw/Mn ∼ 2) was obtained in the polymerization temperature range of 30 °C to 50 °C.
:15 at 30 °C to 100:34 at 120 °C, driven by thermal energy, based on variable temperature 1H NMR characterization. Both complexes were employed as catalysts for ethylene polymerization in the presence of methylaluminoxane (MAO). The influence of the electronic effects of different substituent groups at the ortho position of the phenolate on ethylene polymerization behaviors, molecular weight and molecular weight distributions of the resulting polyethylene was investigated. The fluorinated salalen titanium(IV) complex revealed relatively high catalytic activity and thermal stability owing to the electron-withdrawing inductive effect. Moreover, disentangled linear polyethylene with ultrahigh molecular weight (Mw up to 3000 kDa) and narrow molecular weight distribution (Mw/Mn ∼ 2) was obtained in the polymerization temperature range of 30 °C to 50 °C.
Introduction
Polyethylene is the most widely produced plastic (by volume) in the world, with 80 million metric tons produced annually.1,2 It has attracted considerable attention owing to a combination of its relatively low cost of production, large-scale production in factories, and extremely wide range of applications, as well as the variableness of its accomplishable physical performance.3 The physical properties of polyethylene are controlled by nearly limitless combinations of branched structures,4–6 molecular weights and molecular weight distributions.2,7,8 Polyethylene originates from ethylene addition polymerization, including radical polymerization3,9–11 and coordination polymerization.12–14 The strategy of radical polymerization is limited to high-pressure apparatus and forms low-density polyethylene as a result of the high degree of short and long chain branching, as well as hyperbranching.9 Ethylene coordination polymerization is the most widespread technology, which includes the heterogeneous Ziegler–Natta,14,15 homogeneous metallocene16–18 and post-metallocene19–22 catalysts. Advances in catalyst research with homogeneous catalysts have given rise to new classes of polyethylene,23–25 as these catalysts can provide control over branch structures,26–28 molecular weight and molecular weight distributions. Investigations of homogeneous catalysts have provided useful insights into the influences of the nature of the transition metal29,30 and ligand geometry31,32 on polymerization behaviour, branch structure and molecular weight of the polymerization product. In the last 30 years, considerable efforts have focused on post-metallocene coordination catalysts for ethylene polymerization in light of the vast array of different ligands and metal combinations for these catalysts.33 Our understanding of the influences of the metal and ligands on ethylene polymerization behaviour and its structures is still gradually developing. [OABO]-type bridged bis(phenolate) group 4 metal complexes have attracted considerable attention for the controlled coordination polymerization of olefins30,34–44 because of the abundant modifiable positions at the skeleton ligands and the formation of various stereoisomers as well as their interactive conversion.45,46 Such complexes typically include two labile monodentate groups of cis geometrical relationship and a chelating ligand that controls the electronic and steric environments, thereby affecting the activity of the catalyst and the molecular weights and molecular weight distributions of resulting polymers.42,47,48As reported by Kol and co-workers,45 titanium(IV) complexes bearing salalen-type ligands with bulky alkyl substituents at the ortho position of the imine-side phenol and electron-withdrawing halo groups at the ortho and para positions of the amine-side phenol were employed for propylene polymerization. The electronic effect of the halo phenolate substituent was found to play a less significant role not only in polymerization behaviour but also in the molecular weight of the resulting polypropylene. In order to investigate the influence of the electronic effects on ethylene polymerization behaviours, molecular weight, and the molecular weight distribution of polyethylene, the hydrogen atom at the ortho position of the imine-side phenol was replaced by a fluorine group. We predicted that a conjugated system consisting of a phenol ring and an imine donor could contribute to the strong electron-withdrawing inductive effect of the fluorine group on decreasing the electron cloud density on the titanium(IV) centre. It is obvious that the electronic effect of the substituent on the phenol ring at the amine side makes it difficult to modify the electron cloud density of the titanium(IV) centre due to the non-conjugated amine moiety.
Herein, we demonstrated the synthesis of two salalen titanium(IV) complexes and their performance in ethylene polymerization in the presence of methylaluminoxane (MAO). Complex [LigFTiCl2] showed exceptional thermal stability and catalytic activity in ethylene polymerization, yielding linear polyethylene with ultrahigh molecular weight. Most importantly, both complexes were found to reveal thermally-driven isomerization behaviour in toluene and promoted the formation of polyethylene with broad molecular weight distribution at higher polymerization temperature.
Results and discussion
The salalen ligand precursors (L1 & L2) were synthesized by Schiff-base condensation of N-methylethylenediamine with salicylaldehyde and fluoro-substituted salicylaldehyde, followed by nucleophilic attack of the N-methyl secondary amine with the bromomethyl derivative of the corresponding phenol. Subsequently, L1 and L2 were reacted with 2 equivalents of n-BuLi to remove protons from the hydroxyl moieties, resulting in the formation of oxygen anions. Finally, the salalen titanium(IV) complexes were synthesized with tetradentate dianionic chelating ligands, including an imine and an amine internal donor bound to phenol arms, in high to quantitative yields, as described in Scheme 1.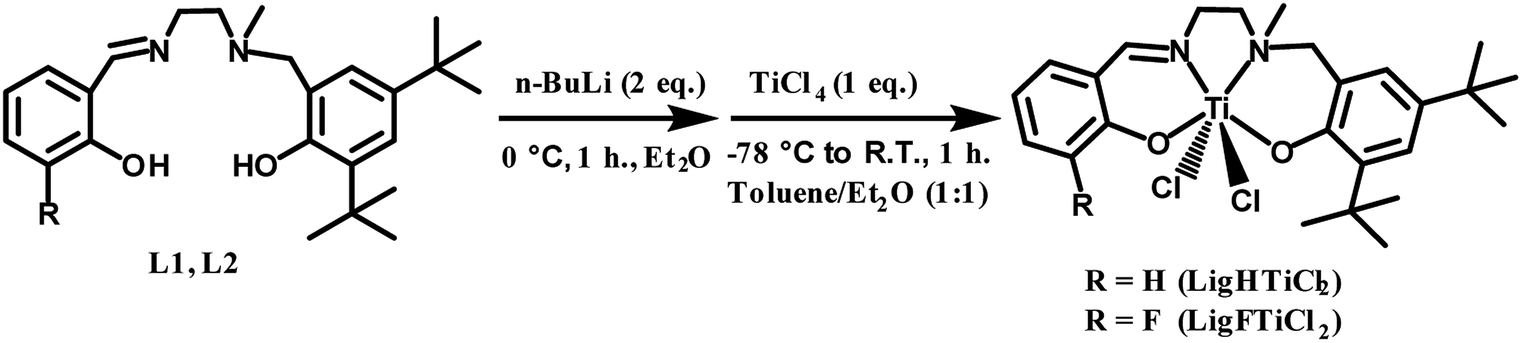 | ||
| Scheme 1 Synthesis route for salalen ligand precursors and their titanium(IV) complexes. | ||
The resulting complexes were labelled [LigHTiCl2] and [LigFTiCl2], and their single crystals were grown from toluene/n-hexane and dichloromethane/n-hexane at room temperature, respectively. As shown in Fig. 1, single-crystal X-ray diffraction analysis indicated that the titanium atom adopts distorted octahedral coordination, with one tetradentate ligand moiety and two chlorine atoms. The distortion is mainly in terms of the N1–Ti1–O2 angles of 159.76(17)° and 159.88(6)° deviating by 180°. The tetradentate salalen ligands form a fac (around the amine) – mer (around the imine) mode, wrapping around the titanium(IV) center for both complexes and leading to a cis-geometrical relationship between the two labile monodentate chlorine ligands. The structure parameters, including selected bond lengths and angles, are displayed in Fig. 1.
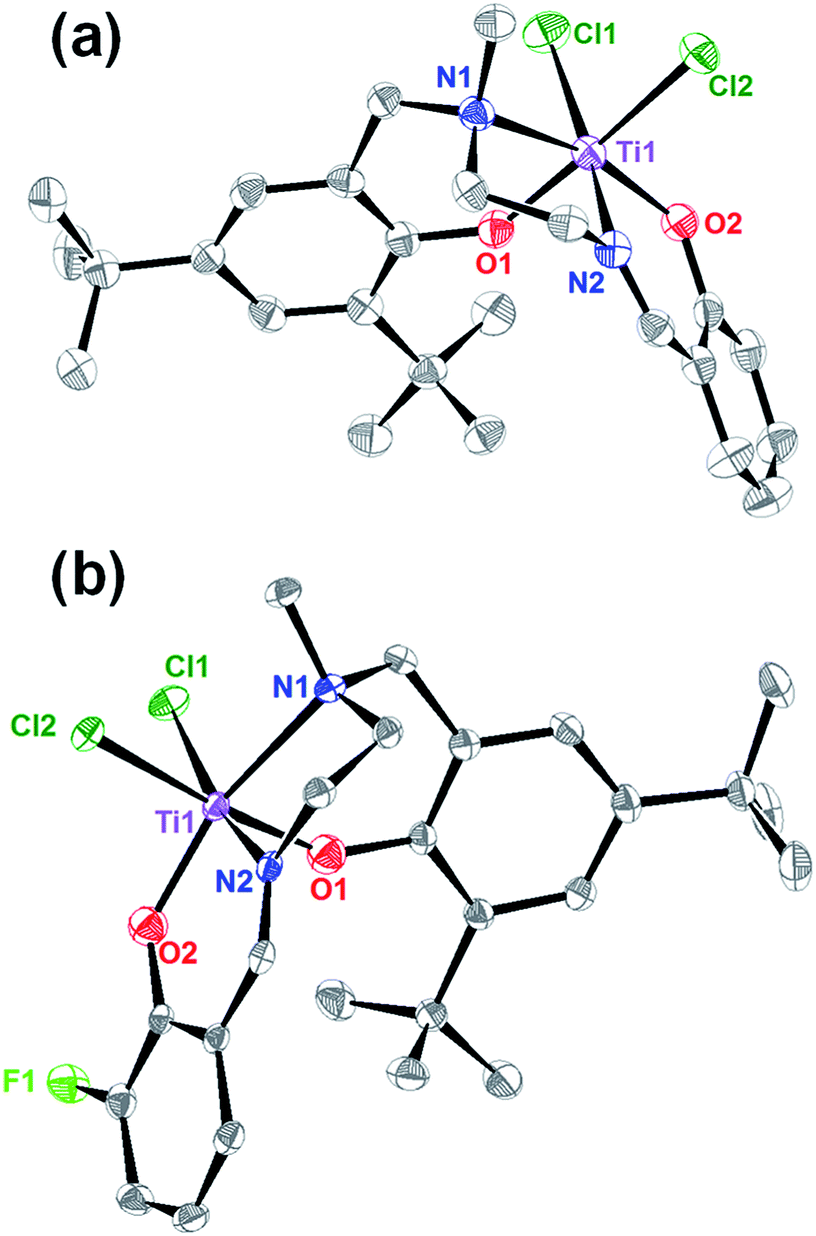 | ||
| Fig. 1 (a) Molecular structure of [LigHTiCl2]. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms and one toluene molecule have been omitted for clarity. Selected bond lengths (Å) and angles (deg): Ti(1)–O(1) 1.8246(48), Ti(1)–O(2) 1.8397(39), Ti(1)–N(1) 2.2926(46), Ti(1)–N(2) 2.1559(44), Ti(1)–Cl(1) 2.3246(17), Ti(1)–Cl(2) 2.3624(21); Cl(1)-Ti(1)-Cl(2) 89.69(6), Cl(2)–Ti(1)–N(2) 87.18(13), N(2)–Ti(1)–O(1) 89.56(18), O(1)–Ti(1)–Cl(1) 92.43(14), N(1)–Ti(1)–Cl(2) 91.88(13), Cl(2)–Ti(1)–O(2) 92.97(13), O(2)–Ti(1)–O(1) 92.25(17), O(1)–Ti(1)–N(1) 81.91(17), Cl(1)–Ti(1)–O(2) 107.16(13), O(2)–Ti(1)–N(2) 83.96(18), N(2)–Ti(1)–N(1) 76.66(18), N(1)–Ti(1)–Cl(1) 92.50(13), Cl(1)–Ti(1)–N(2) 168.60(14), Cl(2)–Ti(1)–O(1) 173.52(14), N(1)–Ti(1)–O(2) 159.76(17). (b) Molecular structure of [LigFTiCl2]. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and angles (deg): Ti(1)–O(1) 1.8141(12), Ti(1)–O(2) 1.8422(13), Ti(1)–N(1) 2.2975(16), Ti(1)–N(2) 2.1509(17), Ti(1)–Cl(1) 2.3032(6), Ti(1)–Cl(2) 2.3934(5); Cl(1)-Ti(1)-Cl(2) 90.09(2), Cl(2)–Ti(1)–N(2) 83.32(4), N(2)–Ti(1)–O(1) 90.95(6), O(1)–Ti(1)–Cl(1) 93.99(5), N(1)–Ti(1)–Cl(2) 90.74(4), Cl(2)–Ti(1)–O(2) 92.22(4), O(2)–Ti(1)–O(1) 93.53(6), O(1)–Ti(1)–N(1) 81.71(6), Cl(1)–Ti(1)–O(2) 109.14(4), O(2)–Ti(1)–N(2) 84.40(6), N(2)–Ti(1)–N(1) 76.18(6), N(1)–Ti(1)–Cl(1) 90.75(4), Cl(1)–Ti(1)–N(2) 165.23(5), Cl(2)–Ti(1)–O(1) 171.45(5), N(1)–Ti(1)–O(2) 159.88(6). | ||
Note that the introduction of a fluorine group at the ortho position of the imine-side phenol leads the Ti–Cl bond length in [LigFTiCl2] of 2.3934(5) Å to be much longer than that in [LigHTiCl2] of 2.3624(21) Å. Furthermore, the longer bond length corresponding to weaker binding suggests that the [LigFTiCl2] is more easily alkylated by MAO than [LigHTiCl2] and more in favour of the formation of active species as well as the insertion of ethylene into the titanium–carbon bond. Therefore, the [LigFTiCl2]/MAO could afford higher catalytic activity for ethylene polymerization than [LigHTiCl2]/MAO.
Interestingly, the Ti–N1 bond lengths in both complexes were longer than Ti–N2, indicating that the bond energy between the titanium atom and imine N-donor is stronger than that between the titanium atom and amine N-donor. In addition, the Ti–N1 bond length of 2.2926(46) Å in [LigHTiCl2] is slightly shorter than that in [LigFTiCl2], 2.2975(16) Å, implying the electron-withdrawing effect of the fluorine group at the ortho position of the imine side phenol.
The typical 1H NMR spectrum of [LigHTiCl2] strongly suggests that there are two isomers that coexist in toluene solution at 30 °C, [LigHTiCl2]-A and [LigHTiCl2]-B (Fig. 2). The major resonance signals of the corresponding hydrogen atoms in [LigHTiCl2] were attributed to [LigHTiCl2]-A, in accordance with the single-crystal structure. Meanwhile, the minor resonance signals were attributed to [LigHTiCl2]-B. The chemical shift of the hydrogen atoms of N-methyl displays an upfield shift from 3.21 to 2.70 ppm, indicating that the hydrogen atoms of the methyl group are far from the hydrogen atoms on the carbon bridge and on the methylene group, according to the van der Waals effect.
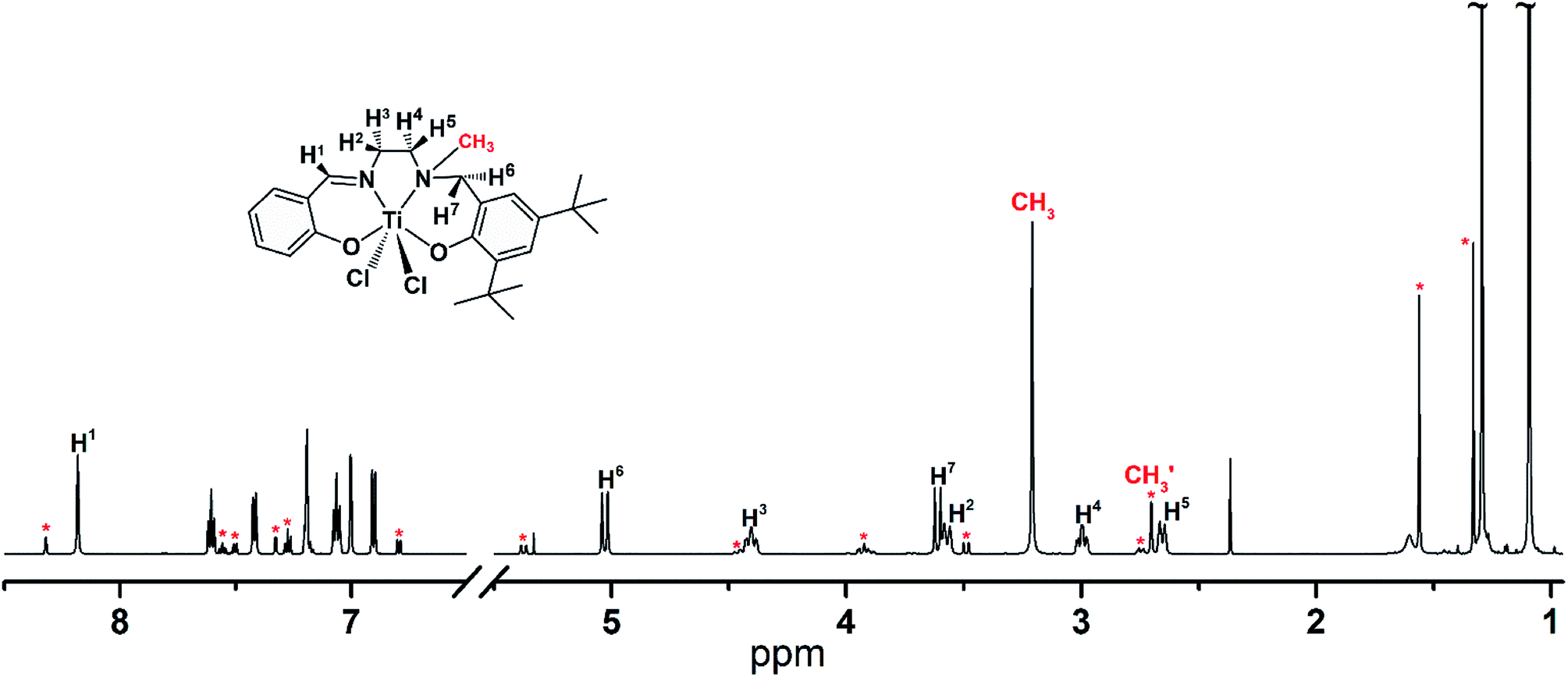 | ||
| Fig. 2 1H NMR of complex [LigHTiCl2]. Asterisks indicate some resonances of the minor component. | ||
As shown in Fig. 3, the content of the [LigHTiCl2]-B isomer increases from 15% to 34%, as calculated from variable temperature 1H NMR, with increasing temperature from 30 °C to 120 °C. Moreover, the corresponding chemical shifts of the hydrogen atoms in both conformational isomers can still be assigned though 1H NMR.
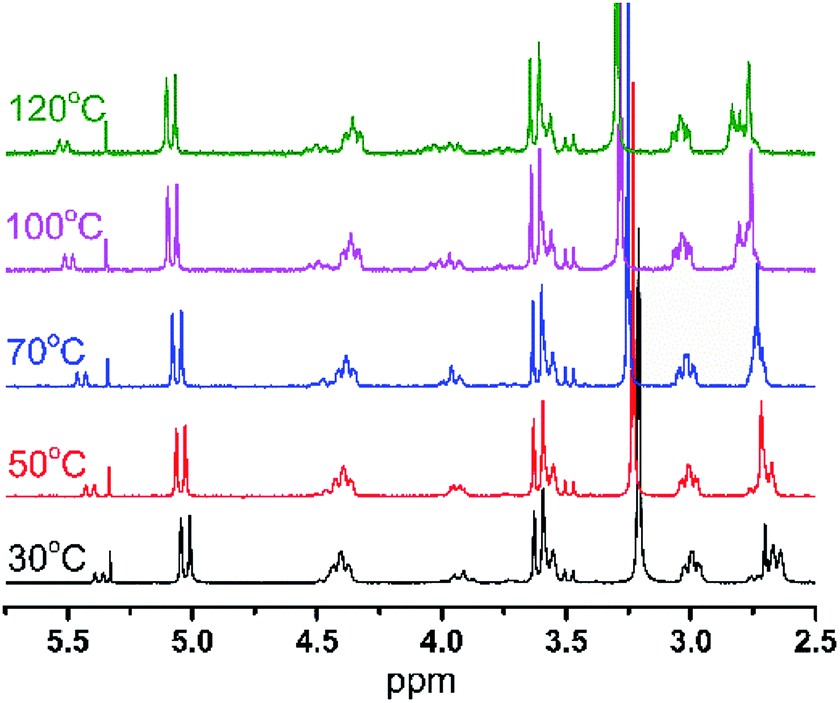 | ||
| Fig. 3 The 1H NMR spectra of [LigHTiCl2] at different temperatures. | ||
Upon activation by MAO, both complexes were tested for ethylene polymerization in toluene at temperatures ranging from 30 °C to 140 °C (Table 1). [LigHTiCl2] and [LigFTiCl2] showed moderate catalytic activities and produced linear polyethylene with ultrahigh molecular weight (Mw = 2950 and 2980 kDa, respectively) at room temperature (30–50 °C). However, [LigFTiCl2]/MAO displayed much higher catalytic activity than [LigHTiCl2]/MAO, supporting the structure analysis of both complexes. The formation of ultrahigh-molecular-weight polyethylene is mainly due to the combination of the steric bulk of the octahedral clathrate-like structure and the strong electron-donating ability of the two nitrogen-coordinated atoms relative to the two sulfur atoms in [OSSO]-type titanium complexes, which usually give rise to moderate-molecular-weight polyethylene (Mw ∼ 10 kDa).30,35,43 As shown in Fig. 4(a), the GPC traces for polyethylene obtained at 30 °C revealed a relatively narrow molecular weight distribution (Mw/Mn ∼ 2), as expected for an approximate single active species catalyst. As the polymerization temperature increases, the resulting polyethylene shows a gradually broadening molecular weight distribution, suggesting the presence of double or multiple active species as a result of the conformational isomerization of the complexes.42,49
| Entry | Precat. | T (°C) | Al/Ti | Yield (g) | Activityb | Mwc | Mw/Mnc | Tmd (°C) |
|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: Ti = 20 μmol; Al/Ti = 500; ethylene pressure = 220 psi, toluene = 70 mL; reaction time = 30 min.b In g[polymer] mmol−1 [Ti] h−1.c In 104 g mol−1, obtained from HT-GPC at 150 °C in 1,2,3-trichlorobenzene (TCB) against polystyrene standards.d Determined by differential scanning calorimetry, second heating cycle.e Reaction time = 15 min.f Reaction time = 45 min.g Reaction time = 60 min. | ||||||||
| 1 | LigHTiCl2 | 30 | 500 | 0.074 | 7.4 | 295 | 2.3 | 137.4 |
| 2 | LigHTiCl2 | 50 | 500 | 0.158 | 15.8 | 201 | 2.7 | 134.7 |
| 3 | LigHTiCl2 | 70 | 500 | 0.521 | 52.1 | 56.2 | 4.8 | 133.7 |
| 4 | LigHTiCl2 | 100 | 500 | 0.820 | 82.0 | 19.8 | 8.5 | 131.7 |
| 5 | LigHTiCl2 | 120 | 500 | 0.482 | 48.2 | 6.7 | 6.2 | 129.7 |
| 6 | LigHTiCl2 | 140 | 500 | 0.140 | 12.0 | 3.4 | 6.2 | 129.0 |
| 7 | LigFTiCl2 | 30 | 500 | 0.520 | 52.0 | 298 | 2.1 | 137.0 |
| 8 | LigFTiCl2 | 50 | 500 | 0.565 | 56.5 | 264 | 2.4 | 134.6 |
| 9 | LigFTiCl2 | 70 | 500 | 0.630 | 63.0 | 163 | 3.7 | 133.4 |
| 10 | LigFTiCl2 | 100 | 500 | 1.382 | 138.2 | 51.1 | 4.5 | 131.5 |
| 11 | LigFTiCl2 | 120 | 500 | 0.969 | 96.9 | 32.5 | 4.0 | 130.5 |
| 12 | LigFTiCl2 | 140 | 500 | 0.896 | 89.6 | 12.9 | 4.0 | 130.0 |
| 13 | LigFTiCl2 | 50 | 300 | 0.414 | 41.4 | 270 | 2.4 | 133.8 |
| 14 | LigFTiCl2 | 50 | 700 | 0.365 | 36.5 | 258 | 5.7 | 135.2 |
| 15 | LigFTiCl2 | 50 | 900 | 0.312 | 31.2 | 237 | 4.5 | 134.6 |
| 16e | LigFTiCl2 | 50 | 500 | 0.376 | 75.2 | 111 | 2.5 | 132.9 |
| 17f | LigFTiCl2 | 50 | 500 | 0.783 | 52.2 | 349 | 3.0 | 134.5 |
| 18g | LigFTiCl2 | 50 | 500 | 0.941 | 47.0 | 436 | 3.2 | 135.2 |
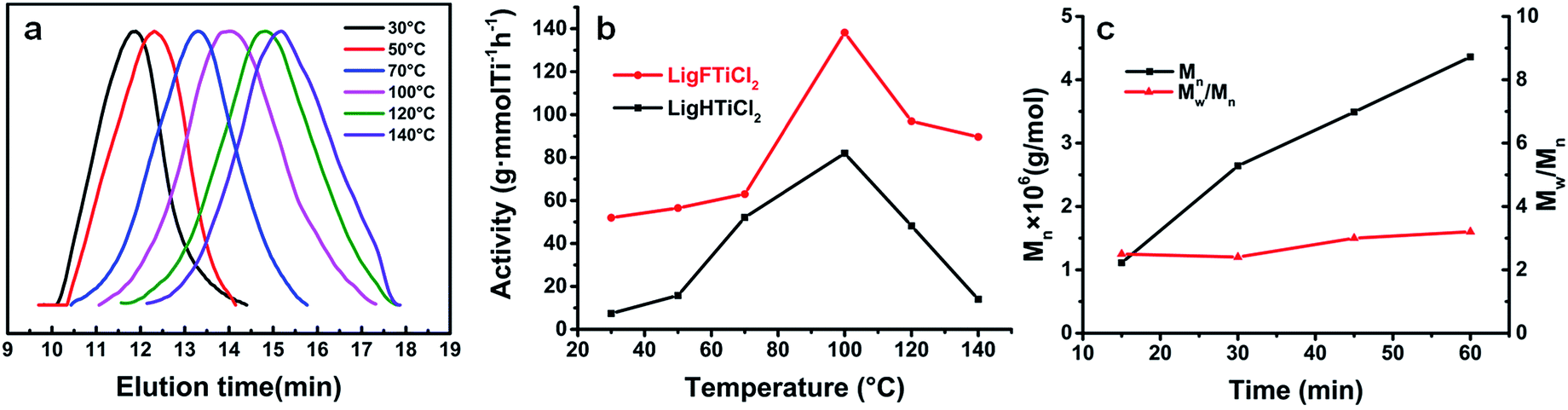 | ||
| Fig. 4 (a) GPC traces of polyethylene formed using LigHTiCl2/MAO at various temperatures. (b) Polymerization activity versus temperature. (c) Plots of Mn and Mw/Mn as a function of ethylene polymerization time. | ||
It should be noted that the polymerization temperature had a significant influence on the catalytic activities of the salalen titanium(IV) complexes/MAO and on the molecular weight as well as the molecular weight distribution of the resulting polyethylene.
Upon activation by MAO, [LigHTiCl2] and [LigFTiCl2] displayed their highest catalytic activities at 100 °C (Fig. 4(b)). The extraordinary thermal stability can be explained by the structure of these complexes, which displayed an octahedral spatial configuration with tetradentate dianionic chelating ligands.
Furthermore, the titanium(IV) atom was located in the centre of the octahedron, inhibiting the decomposition and associative chain transfer pathways.50 When the polymerization temperature reached 140 °C, the catalytic activity of [LigHTiCl2]/MAO decreased drastically, while [LigFTiCl2]/MAO retained a relatively high catalytic activity, which was 6-fold higher than that of [LigHTiCl2]/MAO. It is theorized that the fluorine atom may interact with the β-H of the growing polyethylene chain to reduce β-elimination reactions.51
As shown in Table 1, the influence of different Al/Ti molar ratios on ethylene polymerization was also investigated. The maximum catalytic activity of 5.7 × 104 g of PE (mol Ti)−1 × h−1 is observed at an Al/Ti molar ratio of 500 (Entry 8, Table 1). The lower Al/Ti molar ratios could not promote all the catalyst precursors to form active species. In contrast, the TMA coexisting in MAO increased with the increase of Al/Ti molar ratio. Therefore, high Al/Ti molar ratio would poison some catalyst precursors, causing the catalytic activity to decrease. In addition, the molecular weight of polyethylene remained at a relatively stable value, Mw ∼ 2500 kDa, indicating that the chain transfer to co-catalyst was not the main chain transfer mode.
The polymerization was carried out from 15 to 60 min, and the molecular weight increased with increasing polymerization time (Fig. 4(c)). However, the catalytic activity decreased significantly with increased reaction time, which can be attributed to the embedding of active species by polyethylene.
In order to investigate the disentangled state of the polyethylene, DSC experiments were performed using a thermal analysis protocol developed by Rastogi and co-workers (Scheme S1†).52 The presence of disentangled UHMWPE was confirmed by the appearance of two separate melting peaks in the DSC curve (Fig. 5), after annealing and isothermal crystallization for stipulated time, and our result is in good agreement with previous studies.52,53 A low-temperature melting peak at 130.7 °C and a higher-temperature melting peak at 139.2 °C indicate the crystallization from entangled and disentangled domains of the heterogeneous polymer melt, respectively.52 Furthermore, the ratio of the area under the low and high melting temperature peaks was found to change with the annealing time (at 160 °C), which also coincides with previous reports.52,53 Although this change in our experiments is not obvious as in previous reports due to the insufficient annealing time in our experiments, it still can prove the transformation of the heterogeneous distribution of entanglement density to a homogeneous state.52,53
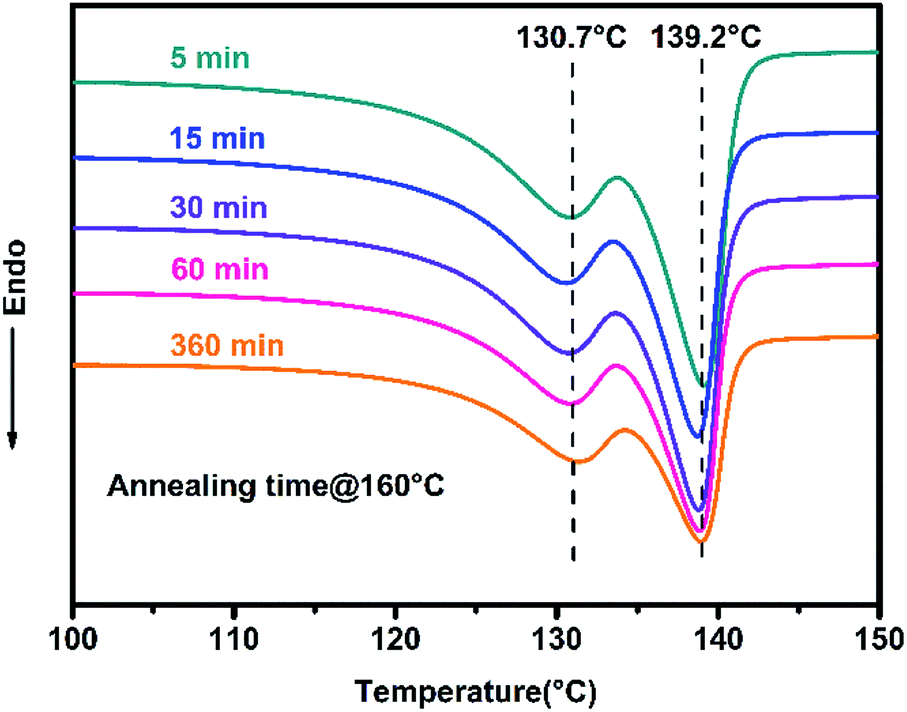 | ||
| Fig. 5 DSC plots of dis-UHMWPE sample (Table 1, Entry 7) obtained from the second heating cycle (Scheme S1† ramp G-H) with annealing time of 5, 15, 30, 60 and 360 min. | ||
Conclusions
In summary, two titanium(IV) complexes bearing salalen ligands were synthesized and characterized by single-crystal X-ray diffraction analysis and NMR. All the ligands bind to the titanium(IV) in a fac–mer mode with an approximate octahedral spatial configuration. It is worth noting that this type of complexation in solution yielded two coexisting isomers. It was discovered that [LigHTiCl2] and [LigFTiCl2] contain about 15% isomers at 30 °C, and the content of the isomers also increased with the increase of temperature. The result implies that the resulting polyethylene shows broader molecular weight distribution at relatively high polymerization temperatures. Both complexes were found to have the ability to catalyze the polymerization of ethylene in the presence of MAO. In addition, the complex containing a fluorine group at the ortho position of the imine-side phenol ([LigFTiCl2]) revealed higher catalytic activity and afforded disentangled linear polyethylene with ultrahigh molecular weight (Mw up to 3000 kDa) and narrow molecular weight distribution (Mw/Mn ∼ 2) at 30–50 °C. At high temperature (70–120 °C), these salalen titanium(IV) complexes gave rise to polyethylene with broad molecular weight distribution (Mw/Mn ∼ 6), resulting from the multiple active species present due to the isomerization of the complexes. Future investigations will focus on designing novel salalen titanium(IV) complexes with high catalytic activities for ethylene polymerization, affording ultrahigh-molecular-weight polyethylene with narrow molecular weight distribution.Experimental section
General considerations
All operations involving air- or moisture-sensitive compounds were performed under an atmosphere of dried and purified nitrogen using standard vacuum-line, Schlenk, or glovebox techniques. The salalen ligand precursors L1, L2 and their complexes [LigHTiCl2], [LigFTiCl2] were synthesized according to published literature procedures.48Materials
Toluene and n-hexane were distilled from Na/K alloy, and dichloromethane was distilled from CaH2. Methylaluminoxane (MAO) was purchased from Akzo-Nobel as 1.5 M toluene solution and used as received. Commercial ethylene was used directly for polymerization without further purification. All the other reagents were purchased and used as received.Physical and analytical measurements
NMR spectroscopy of the organic compounds was carried out on a Bruker 400 MHz instrument in CDCl3 using TMS as a reference, and characterization of the complexes was carried out on a Bruker 500 MHz in C2D2Cl4 (1,1,2,2-tetrachloroethane-d2). Chemical shifts were reported in ppm on the scale relative to CDCl3 (δ = 7.26 for 1H NMR, δ = 77.00 for 13C NMR) and C2D2Cl4 (δ = 6.00 for 1H NMR, δ = 75.00 for 13C NMR). NMR analysis of polymers was carried out on a Bruker 500 MHz at 120 °C. Sample solutions of the polymer were prepared in o-C6H6Cl2/o-C6D6Cl2 (50% v/v) in a 10 mm sample tube. DSC analyses were conducted with a PerkinElmer DSC-4000 system. The DSC curves were recorded as second heating curves from 30 °C to 200 °C at a heating rate of 10 °C min−1 and a cooling rate of 10 °C min−1. High-temperature gel permeation chromatography (GPC) was performed on a Polymer Laboratories Ltd. PL-220 equipped with a refractive index (RI) detector. GPC columns were eluted with 1,2,4-trichlorobenzene (TCB) at 150 °C with a run rate of 1.0 mL min−1.Crystal structure determination
The crystals of the titanium complex were mounted on a glass fiber and transferred to a Bruker CCD platform diffractometer. Data obtained under ω–2θ scan mode were collected on a Bruker SMART 1000 CCD diffractometer with graphite-monochromated Cu Kα radiation (λ = 1.54184 Å) or Mo Kα radiation (λ = 0.71073 Å). The structures were solved using direct methods, while further refinement with full-matrix least-squares on F2 was obtained with the SHELXTL program package. All the non-hydrogen atoms were refined anisotropically. Hydrogen atoms were introduced in calculated positions with the displacement factors of the host carbon atoms.Ethylene polymerization
A mechanically stirred 200 mL Parr reactor was heated to 150 °C for 3 h under vacuum and then cooled to reaction temperature. The autoclave was pressurized to 15 psi of ethylene and vented three times. The autoclave was then charged with toluene under 15 psi of ethylene at the initialization temperature. The system was maintained by continuously stirring for 5 min, and then methylaluminoxane (MAO) and 5 mL of a solution of the titanium complex in toluene were sequentially charged into the autoclave under 15 psi ethylene. The ethylene pressure was raised to the specified value. The polymerization temperature was controlled by means of a heater and error was found to be ±2 °C, as monitored by an internal thermocouple. The polymerization was carried out for a specified time before being terminated by the addition of 5 mL methanol into the autoclave, after releasing the ethylene pressure. The resulting solution was then transferred into 500 mL of acidic methanol (10:1 methanol/HCl) with stirring overnight. The resulting precipitated polymer was collected and treated by filtration, washed with methanol several times, and dried in vacuum at 40 °C to a constant weight.
Synthesis of 2-(bromomethyl)-4,6-di-tert-butylphenol
To a clear solution of 2,4-di-tert-butylphenol (17.4 g, 84.4 mmol) in glacial acetic acid (40 mL), paraformaldehyde (2.8 g, 93.24 mmol, 1.1 equiv.) was added at room temperature and stirred for 2 h. A HBr solution in acetic acid (90 mL, mmol, equiv.) was then added dropwise into the resulting solution. After 40 min of stirring at room temperature, the acetic acid was removed in vacuo, and an orange oil was obtained. Using n-hexane as a recrystallization solvent, a pale orange crystalline solid was obtained (20.7 g, 82%). 1H NMR (500 MHz, CDCl3) δ 1.29 (s, 9H, C(CH3)3), 1.43 (s, 9H, C(CH3)3), 4.58 (s, 2H, CH2Br), 5.28 (s, 1H, OH), 7.10 (d, 1H, ArH), 7.33 (d, 1H, ArH). 13C NMR (125 MHz, CDCl3) δ 32.23, 34.14, 35.46, 37.00, 37.59, 125.93, 127.55, 128.48, 139.20, 145.91, 154.48.Synthesis of 2-[((methylamino)ethylimino)methyl]-phenol
N-Methylethylenediamine (1.1 g, 15 mmol) was added to a solution of salicylaldehyde (1.8 g, 15 mmol) in toluene and refluxed for 2 h. The solvent was removed under vacuum, yielding a yellow oil (2.20 g, 82%). 1H NMR (500 MHz, CDCl3) δ 2.36 (s, 3H, CH3), 2.83 (t, 2H, CH2), 3.62 (t, 2H, CH2), 6.75–6.89 (m, 2H, ArH), 7.13–7.22 (m, 2H, ArH), 8.28 (s, 1H, N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH). 13C NMR (125 MHz, CDCl3) δ 36.3 (CH3), 51.9, 59.6 (CH2), 117.2, 118.5, 131.4, 132.4, (Ar–CH), 138.5 (Ar–C), 161.4 (Ar–O), 166.1 (CH).
CH). 13C NMR (125 MHz, CDCl3) δ 36.3 (CH3), 51.9, 59.6 (CH2), 117.2, 118.5, 131.4, 132.4, (Ar–CH), 138.5 (Ar–C), 161.4 (Ar–O), 166.1 (CH).
Synthesis of 2-[((methylamino)ethylimino)methyl]-6-fluorinephenol
N-Methylethylenediamine (1.1 g, 15 mmol) was added to a solution of 3-fluoro-2-hydroxybenzaldehyde (2.1 g, 15 mmol) in toluene and refluxed for 2 h. The solvent was removed under vacuum, yielding a yellow oil (2.75 g, 93%). 1H NMR (500 MHz, CDCl3) δ 2.46 (s, 3H, CH3), 2.93 (t, 2H, CH2), 3.74 (t, 2H, CH2), 6.86 (t, 1H, ArH), 6.94 (d, 1H, ArH), 7.28 (d, 1H, ArH), 8.39 (s, 1H, NCH). 13C NMR (125 MHz, CDCl3) δ 36.3 (CH3), 48.9, 51.5 (CH2), 117.5, 125.6, 128.8, 130.2 (Ar–C), 151.1 (Ar–O), 154.3 (Ar–F), 161.2 (CH).
Synthesis of L1
2-[((Methylamino)ethylimino)methyl]-phenol (0.87 g, 4.9 mmol) was dissolved in THF (40 mL), to which a solution of 2-(bromomethyl)-4,6-di-tert-butylphenol (1.4 g, 4.9 mmol) in THF (40 mL) was added. Triethylamine (5 mL) was then added, and the mixture was stirred at room temperature for 2 h. The white precipitate was then filtered and the solvent removed under vacuum. Using methanol as a recrystallization solvent, a yellow crystalline solid was obtained (1.1 g, 57%). 1H NMR (500 MHz, CDCl3) δ 1.27 (s, 9H, C(CH3)3), 1.36 (s, 9H, C(CH3)3), 2.39 (s, 3H, CH3), 2.84 (t, 2H, CH2), 3.74 (s, 2H, CH2), 3.78 (t, 2H, CH2), 6.82 (s, 1H, ArH), 6.88 (t, 1H, ArH), 6.96 (d, 1H, ArH), 7.17–7.35 (m, 3H, ArH), 8.37 (s, 1H, NCH). 13C NMR (125 MHz, CDCl3) δ 29.6, 31.7, 34.2, 34.8, 42.1, 57.0, 57.6, 62.3, 117.0, 118.7, 118.8, 121.0, 123.0, 131.4, 132.4, 135.9, 140.6, 154.3, 161.0, 166.3. HRMS (ESI) m/z: [M + H]+ calcd for C25H37N2O2, 397.2855, found, 397.2842.
Synthesis of L2
2-[((Methylamino)ethylimino)methyl]-6-fluorophenol (1.96 g, 10 mmol) was dissolved in THF (40 mL), to which a solution of 2-(bromomethyl)-4,6-di-tert-butylphenol (3.29 g, 11 mmol) in THF (40 mL) was added. Triethylamine (5 mL) was then added and the mixture stirred at room temperature for 2 h. The white precipitate was then filtered and the solvent removed under vacuum. Using methanol as a recrystallization solvent, a yellow crystalline solid was obtained (1.82 g, 44%).1H NMR (500 MHz, CDCl3) δ 1.27 (s, 9H, C(CH3)3), 1.35 (s, 9H, C(CH3)3), 2.39 (s, 3H, CH3), 2.85 (t, 2H, CH2), 3.74 (s, 2H, CH2), 3.80 (t, 2H, CH2), 6.74–6.84 (m, 2H), 7.03 (dd, 1H, ArH), 7.13 (td, 1H, ArH), 7.20 (dd, 1H, ArH), 8.37 (s, 1H, NCH). 13C NMR (125 MHz, CDCl3) δ 29.5, 31.7, 34.1, 34.8, 42.1, 56.8, 57.2, 62.1, 117.7, 118.5, 120.8, 123.0, 126.4, 135.6, 140.6, 149.9, 150.4, 152.3, 154.1, 167.0. 19F NMR (470 MHz, CDCl3) δ −138.1. HRMS (ESI) m/z: [M + H]+ calcd for C25H36FN2O2, 415.2761, found, 415.2755.
Synthesis of [LigHTiCl2]
To a solution of L1 (0.597 g, 1.51 mmol) in Et2O (30 mL) at 0 °C was added n-BuLi (2.5 M in hexane, 1.28 mL, 3.2 mmol, 2.1 equiv.) within 5 min. The solution was stirred for 1 h at this temperature. Then, TiCl4 (1 M in toluene, 1.2 mL, 1.2 mmol) and toluene (20 mL) was added to the resulting solution of L1-Li2 at −78 °C, and the mixture was stirred for 1 h. The solution was then gradually allowed to warm to room temperature. The insoluble inorganic salts were then filtered under nitrogen atmosphere. The filtration solvent was removed in vacuo, and the residual solid was washed with hexane three times and dried in vacuo to give a dark red powder. Recrystallization in cold toluene gave deep red crystals (537.5 mg, 70%). The complex [LigHTiCl2] is composed of a mixture of two isomers, A and B, in the ratio of 85:15 at 30 °C, respectively (from the 1H NMR spectra). For the major isomer, the amine atom is not detached from the titanium(IV) center and is represented by A, whereas B represents the amine atom detached from the titanium(IV) center in the minor isomer. Owing to the low content of the minor isomer, only the major isomer (A) was observed in the 13C NMR. 1H NMR (500 MHz, C2D2Cl4) δ 1.09 (s, 9H, C(CH3)A3), 1.29 (s, 9H, C(CH3)A3), 1.33 (s, 9H, C(CH3)B3), 1.56 (s, 9H, C(CH3)B3), 2.67 (d, 1H, CHA2), 2.70 (s, 3H, CHB3), 2.75 (d, 1H, CHB2), 3.00 (t, 1H, CHA2), 3.05 (s, 3H, CHA3), 3.50 (d, 1H, CHB2), 3.58 (d, 1H, CHA2), 3.62 (d, 1H, CHA2), 3.86–3.98 (m, 2H, CHB2), 4.42 (t, 1H, CHA2), 4.45 (t, 1H, CHB2), 5.04 (d, 1H, CHA2), 5.36 (d, 1H, CHB2), 6.91 (d, 1H, Ar-HA), 7.00 (s, 1H, Ar-HA), 7.06 (t, 1H, Ar-HA), 7.19 (s, 1H, Ar-HA), 7.27 (t, 1H, Ar-HB), 7.33 (m, 1H, Ar-HB), 7.41 (d, 1H, Ar-HA), 7.48–7.53 (m, 1H, Ar-HB), 7.56 (t, 1H, Ar-HB), 7.61 (t, 1H, Ar-HA), 8.18 (s, 1H, CHA), 8.32 (s, 1H, CHB). 13C NMR (125 MHz, C2D2Cl4) δ 30.6, 31.6, 32.7, 35.6, 35.7, 54.0, 57.7, 59.6, 67.5, 116.9, 123.1, 124.4, 124.7, 125.2, 127.3, 129.4, 130.2, 134.9, 136.9, 137.6, 136.9, 160.1, 163.2, 163.4.
Synthesis of [LigFTiCl2]
To a solution of L2 (0.482 g, 1.17 mmol) in Et2O (30 mL) at 0 °C was added n-BuLi (2.5 M in hexane, 1.0 mL, 2.5 mmol, 2.1 equiv.) within 5 min. The solution was stirred for 1 h at this temperature. TiCl4 (1 M in toluene, 1.2 mL, 1.2 mmol) and toluene (20 mL) were added to the resulting solution of L2-Li2 at −78 °C, and the mixture was stirred for 1 h. The solution was then gradually allowed to warm to room temperature. The insoluble inorganic salts were then filtered under nitrogen atmosphere. The filtration solvent was removed in vacuo, and the residual solid was washed three times with n-hexane and dried in vacuo to give a dark red powder. Recrystallization in cold toluene gave deep red crystals (443.1 mg, 71.2%). The complex [LigFTiCl2] is composed of a mixture of two isomers, A and B, in the ratio of 75:25 at 30 °C, respectively (from the 1H NMR spectra). For the major isomer, the amine atom is not detached from the titanium(IV) center and is represented by A, whereas B represents the amine atom detached from the titanium(IV) center in the minor isomer. Owing to the low content of the minor isomer, only the major isomer (A) was observed in the 13C NMR. 1H NMR (500 MHz, C2D2Cl4) δ 1.08 (s, 9H, C(CH3)A3), 1.29 (s, 9H, C(CH3)A3), 1.33 (s, 9H, C(CH3)B3), 1.55 (s, 9H, C(CH3)B3), 2.69 (d, 1H, CHA2), 2.71 (s, 3H, CHB3), 2.80 (dd, 1H, CHB3), 3.00 (t, 1H, CHA2), 3.23 (s, 3H, CHA3), 3.51 (d, 1H, CHB2), 3.57 (d, 1H, CHA2), 3.65 (d, 1H, CHA2), 3.85–4.01 (m, 2H, CHB2), 4.43 (t, 1H, CHA2), 4.47 (t, 1H, CHB2), 5.02 (d, 1H, CHA2), 5.34 (d, 1H, CHB2), 6.95–7.04 (m, 2H, Ar-HA), 7.06 (s, 1H, Ar-HB), 7.16–7.23 (m, 2H, Ar-HA), 7.24–7.36 (m, 4H, Ar-HB), 7.39 (t, 1H, Ar-HA), 8.21 (s, 1H, CHA), 8.35 (s, 1H, CHB). 13C NMR (125 MHz, C2D2Cl4) δ 30.6, 31.6, 32.6, 35.6, 35.7, 54.3, 57.8, 59.7, 67.7, 122.5, 123.3, 123.5, 124.7, 124.7, 125.3, 126.2, 127.4, 129.8, 136.9, 147.3, 148.6, 160.1, 162.7, 162.8. 19F NMR (470 MHz, C2D2Cl4) δ −129.9 (Ar-FA), −129.0 (Ar-FB).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21174167, 51573212) and the NSF of Guangdong Province (S2013030013474, 2014A030313178).Notes and references
- M. C. Baier, M. A. Zuideveld and S. Mecking, Angew. Chem., Int. Ed., 2014, 53, 9722–9744 CrossRef CAS PubMed.
- M. Stürzel, S. Mihan and R. Mülhaupt, Chem. Rev., 2016, 116, 1398–1433 CrossRef PubMed.
- S. L. Aggarwal and O. J. Sweeting, Chem. Rev., 1957, 57, 665–742 CrossRef CAS.
- D. J. Lohse, S. T. Milner, L. J. Fetters, M. Xenidou, N. Hadjichristidis, R. A. Mendelson, C. A. García-Franco and M. K. Lyon, Macromolecules, 2002, 35, 3066–3075 CrossRef CAS.
- S. Costeux, P. Wood-Adams and D. Beigzadeh, Macromolecules, 2002, 35, 2514–2528 CrossRef CAS.
- B. H. Bersted, J. D. Slee and C. A. Richter, J. Appl. Polym. Sci., 1981, 26, 1001–1014 CrossRef CAS.
- S. Talebi, R. Duchateau, S. Rastogi, J. Kaschta, G. W. M. Peters and P. J. Lemstra, Macromolecules, 2010, 43, 2780–2788 CrossRef CAS.
- D. Romano, N. Tops, E. Andablo-Reyes, S. Ronca and S. Rastogi, Macromolecules, 2014, 47, 4750–4760 CrossRef CAS.
- P. Ehrlich and G. A. Mortimer, Adv. Polym. Sci., 1970, 7, 386–448 CrossRef CAS.
- E. Grau, J.-P. Broyer, C. Boisson, R. Spitz and V. Monteil, Macromolecules, 2009, 42, 7279–7281 CrossRef CAS.
- G. Billuart, E. Bourgeat-Lami, M. Lansalot and V. Monteil, Macromolecules, 2014, 47, 6591–6600 CrossRef CAS.
- U. Giannini, Die Makromolekulare Chemie, 1981, 5, 216–229 CrossRef CAS.
- E. Y.-X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391–1434 CrossRef CAS PubMed.
- P. Cossee, J. Catal., 1964, 3, 80–88 CrossRef CAS.
- J. J. Eisch, Organometallics, 2012, 31, 4917–4932 CrossRef CAS.
- P. J. Chirik, Organometallics, 2010, 29, 1500–1517 CrossRef CAS.
- P. T. Wolczanski and P. J. Chirik, ACS Catal., 2015, 5, 1747–1757 CrossRef CAS.
- M. Bochmann, Organometallics, 2010, 29, 4711–4740 CrossRef CAS.
- H. Mu, L. Pan, D. Song and Y. Li, Chem. Rev., 2015, 115, 12091–12137 CrossRef CAS PubMed.
- K. P. Bryliakov, Russ. Chem. Rev., 2007, 76, 253–277 CrossRef CAS.
- H. Makio, H. Terao, A. Iwashita and T. Fujita, Chem. Rev., 2011, 111, 2363–2449 CrossRef CAS PubMed.
- G. J. P. Britovsek, V. C. Gibson and D. F. Wass, Angew. Chem., Int. Ed., 1999, 38, 428–447 CrossRef CAS PubMed.
- K. H. Theopold, Eur. J. Inorg. Chem., 1998, 15–24 CrossRef CAS.
- G. Erker, Acc. Chem. Res., 2001, 34, 309–317 CrossRef CAS PubMed.
- G. Bhandari, Y. Kim, J. M. McFarland, A. L. Rheingold and K. H. Theopold, Organometallics, 1995, 14, 738–745 CrossRef CAS.
- R. Quijada, R. Rojas, G. Bazan, Z. J. A. Komon, R. S. Mauler and G. B. Galland, Macromolecules, 2001, 34, 2411–2417 CrossRef CAS.
- F. Zhu, Y. Fang, H. Chen and S. Lin, Macromolecules, 2000, 33, 5006–5010 CrossRef CAS.
- D. C. Aluthge, A. Sattler, M. A. Al-Harthi, J. A. Labinger and J. E. Bercaw, ACS Catal., 2016, 6, 6581–6584 CrossRef CAS.
- S. Segal, I. Goldberg and M. Kol, Organometallics, 2005, 24, 200–202 CrossRef CAS.
- M. Mella, L. Izzo and C. Capacchione, ACS Catal., 2011, 1, 1460–1468 CrossRef CAS.
- A. Stopper, J. Okuda and M. Kol, Macromolecules, 2012, 45, 698–704 CrossRef CAS.
- K. A. Frazier, R. D. Froese, Y. He, J. Klosin, C. N. Theriault, P. C. Vosejpka, Z. Zhou and K. A. Abboud, Organometallics, 2011, 30, 3318–3329 CrossRef CAS.
- S. Budagumpi, K.-H. Kim and I. Kim, Coord. Chem. Rev., 2011, 255, 2785–2809 CrossRef CAS.
- P. D. Knight, A. J. Clarke, B. S. Kimberley, R. A. Jacksonc and P. Scott, Chem. Commun., 2002, 352–353 RSC.
- C. Capacchione, A. Proto and J. Okuda, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 2815–2822 CrossRef CAS.
- C. Cuomo, M. Strianese, T. Cuenca, M. Sanz and A. Grassi, Macromolcules, 2004, 37, 7469–7476 CrossRef CAS.
- H. Zhu, M. Wang, C. Ma, B. Li, C. Chen and L. Sun, J. Organomet. Chem., 2005, 690, 3929–3936 CrossRef CAS.
- G. J. Clarkson, V. C. Gibson, P. K. Goh, M. L. Hammond, P. D. Knight, P. Scott, T. M. Smit, A. J. White and D. J. Williams, Dalton Trans., 2006, 5484–5491 RSC.
- M. Strianese, M. Lamberti, M. Mazzeo, C. Tedesco and C. Pellecchia, J. Mol. Catal. A: Chem., 2006, 258, 284–291 CrossRef CAS.
- S. Gendler, A. L. Zelikoff, J. Kopilov, I. Goldberg and M. Koi, J. Am. Chem. Soc., 2008, 130, 2144–2145 CrossRef CAS PubMed.
- C. Capacchione, D. Saviello, A. Avagliano and A. Proto, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4200–4206 CrossRef CAS.
- K. Huang, S. Zhou, D. Zhang, X. Gao, Q. Wang and Y. Lin, J. Organomet. Chem., 2013, 741–742, 83–90 CrossRef CAS.
- M. Białek, M. Pochwała and G. Spaleniak, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2111–2123 CrossRef.
- M. Mandal, D. Chakraborty and V. Ramkumar, RSC Adv., 2015, 5, 28536–28553 RSC.
- K. Press, A. Cohen, I. Goldberg, V. Venditto, M. Mazzeo and M. Kol, Angew. Chem., Int. Ed., 2011, 50, 3529–3532 CrossRef CAS PubMed.
- G.-J. M. Meppelder, H.-T. Fan, T. P. Spaniol and J. Okuda, Organometallics, 2009, 28, 5159–5165 CrossRef CAS.
- R. García-Zarracino, J. s. Ramos-Quiñones and H. Höpfl, J. Organomet. Chem., 2002, 664, 188–200 CrossRef.
- A. Yeori, S. Gendler, S. Groysman, I. Goldberg and M. Kol, Inorg. Chem. Commun., 2004, 7, 280–282 CrossRef CAS.
- G.-J. M. Meppelder, H.-T. Fan, T. P. Spaniol and J. Okuda, Inorg. Chem., 2009, 48, 7378–7388 CrossRef CAS PubMed.
- S. Matsui, T. P. Spaniol, Y. Takagi, Y. Yoshida and J. Okuda, Dalton Trans., 2002, 4529–4531 RSC.
- M. Mitani, J.-i. Mohri, Y. Yoshida, J. Saito, S. Ishii, K. Tsuru, S. Matsui, R. Furuyama, T. Nakano, H. Tanaka, S.-i. Kojoh, T. Matsugi, N. Kashiwa and T. Fujita, J. Am. Chem. Soc., 2001, 124, 3327 CrossRef PubMed.
- K. Liu, E. L. de Boer, Y. Yao, D. Romano, S. Ronca and S. Rastogi, Macromolecules, 2016, 49, 7497–7509 CrossRef CAS.
- R. P. Gote, D. Mandal, K. Patel, K. Chaudhuri, C. P. Vinod, A. K. Lele and S. H. Chikkali, Macromolecules, 2018, 51, 4541–4552 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CCDC 1921379 for [LigHTiCl2] and 1921380 for [LigFTiCl2]. NMR analyses of titanium complex, characterization of polymers. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ra08899g |
| This journal is © The Royal Society of Chemistry 2019 |