
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccess to 1-amino-3,4-dihydroisoquinolines via palladium-catalyzed C–H bond aminoimidoylation reaction from functionalized isocyanides†
Zhuang Xiong*a,
Panyuan Caib,
Yingshuang Meia and
Jian Wang *b
*b
aSchool of Biotechnology and Sciences, Wuyi University, Jiangmen, Guangdong 529090, P. R. China. E-mail: wyuchemxz@wyu.edu.cn
bKey Laboratory of Functional Molecular Solids, Ministry of Education, Anhui Laboratory of Molecule-Based Materials, College of Chemistry and Materials Science, Anhui Normal University, Wuhu, Anhui 241000, P. R. China. E-mail: wang_jian989@163.com
First published on 18th December 2019
Abstract
Efficient access to 1-amino-3,4-dihydroisoquinolines, through palladium-catalyzed intramolecular C–H bond aminoimidoylation of α-benzyl-α-isocyanoacetates, has been developed. Consecutive isocyanide insertion and C–H bond activation were realized with C–N and C–C bonds formation in one step under redox neutral conditions, employing O-benzoyl hydroxylamines as electrophilic amino sources.
As a class of cyclic amidine compounds, 1-amino-3,4-dihydroisoquinolines are essential six-membered heterocycles which can be found in numerous natural products, functional chemicals and pharmaceutical agents (Scheme 1).1 For example, piperazine tethered dihydroisoquinoline and benzocyclic amide I was found to be an efficient cardiovascular agent.1a Phenylalanine-derived and arylamine or cyclic aliphatic-amine-containing 1-amino-3,4-dihydroisoquinolines (II–V) show outstanding BACE-1 inhibition, kinase VEGFR-2 inhibition, phosphodiesterase IV inhibition and hydrogen ion-sodium exchanger NHE-3 inhibition activities, respectively.1b–e Traditional preparation of such 1-amino-3,4-dihydroisoquinoline scaffolds is mainly based on nucleophilic substitution of readily prepared 3,4-dihydroisoquinolines2 and Bischler-Napieralski reaction starting from urea.3 Most of these methods suffer from multiple-step synthesis, harsh conditions or the use of toxic reagents. Thus, developing an efficient and practical method for the construction of 1-amino-3,4-dihydroisoquinoline derivatives remain an attractive synthetic task.
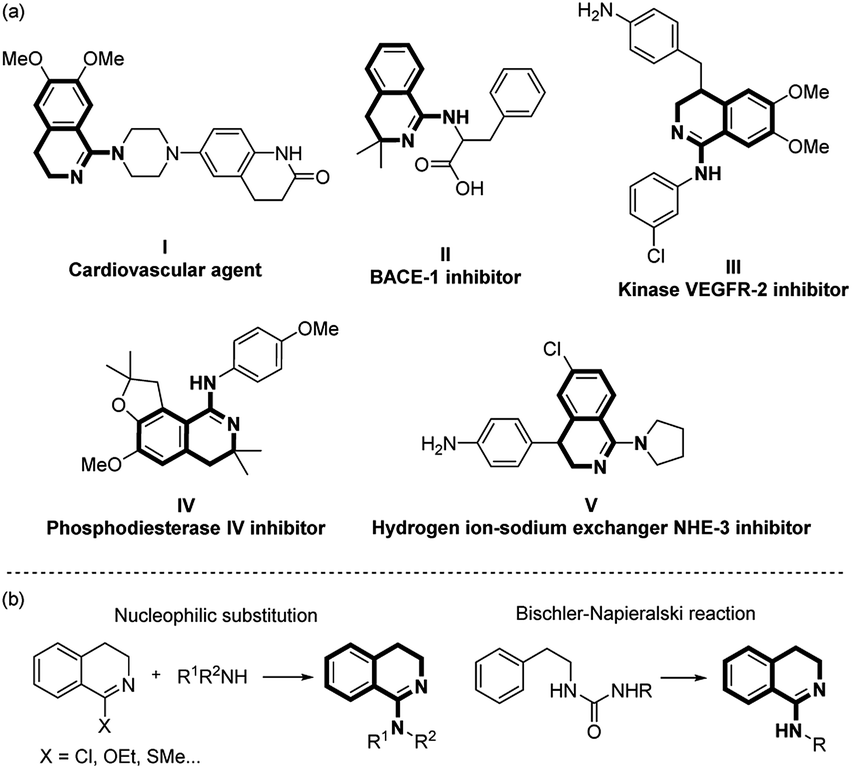 | ||
| Scheme 1 (a) Representative bioactive molecules containing the 1-amino-3,4-dihydroisoquinoline moiety. (b) Traditional synthetic methods. | ||
In recent decades, isocyanides have been extensively studied in multicomponent reactions,4 imidoyl radical-involved transformations5 and transition-metal-catalyzed insertion reactions6 due to their diverse and unique reactivities. A variety of acyclic imine derivatives were synthesized via palladium-catalyzed imidoylation reactions.7 For the construction of biorelevant nitrogen-containing heterocycles via Pd-catalyzed non-functionalized isocyanide insertion reactions, a nucleophilic group is usually preinstalled to the substrates containing C–X (C–H) bonds, followed by Pd-catalyzed oxidative addition (C–H bond activation)/isocyanide insertion/ligand substitution/reductive elimination sequence.8 For example, Maes and coworkers developed an efficient palladium-catalyzed synthesis of 4-aminoquinazolines from N-(2-bromoaryl)amidines using amidine as an internal nucleophile.8a Access to the same scaffold through palladium-catalyzed amidine-directed C–H bond imidoylation/cyclization reaction was reported by Zhu group.8c Isocyanide acted as a C1 synthon in these cyclization reactions, with the terminal carbon of isocyanide being used in the ring formation.9 Considering the diversity of R group on the nitrogen atom of isocyanide, the so-called functionalized isocyanide strategy was widely developed as well, with various N-heterocycles such as oxazoles,10 indoles,11 phenanthridines,12 9H-pyrido[3,4-b]indoles13 and nonaromatic azepines14 being synthesized efficiently. Zhu and coworkers developed the first asymmetric C–H bond imidoylation reaction from easily accessible α,α-dibenzyl-α-isocyanoacetates with chiral quaternary-carbon-containing 3,4-dihydroisoquinolines being generated (Scheme 2a).15 Recently, a catalytic system controlled regioselective C–H bond imidoylation was realized for the selective synthesis of 5 or 6-membered cyclic imines (Scheme 2b).16 Aryl halides were mostly used in these transformations while N–O compounds were rarely reported as electrophiles in Pd-catalyzed isocyanide insertion reactions.17 Zhu group reported the synthesis of phenanthridin-6-amine derivatives from O-benzoyl hydroxylamines and commonly used biaryl isocyanides.17c However, a methyl or methoxyl carbonyl group was needed at the ortho position of isocyano group, which critically limited the application of the reaction. Herein, we developed an efficient and practical synthesis of 1-amino-3,4-dihydroisoquinolines, through palladium-catalyzed intramolecular C–H bond aminoimidoylation reaction of α-benzyl-α-isocyanoacetates. Consecutive isocyanide insertion and C–H bond activation were realized with C–N and C–C bonds being constructed in one step, employing O-benzoyl hydroxylamines as electrophilic amino sources.
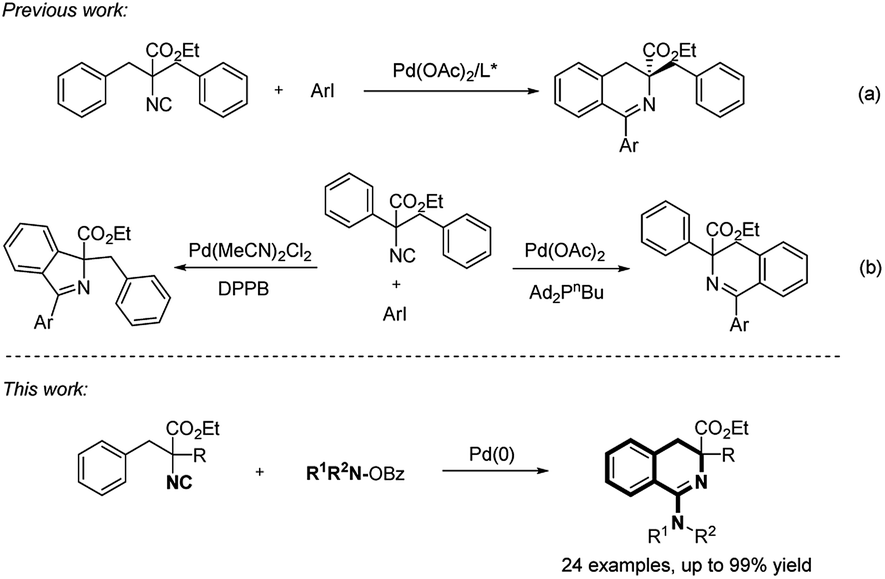 | ||
| Scheme 2 Palladium-catalyzed imidoylative cyclization of α-benzyl-ethylacetates. | ||
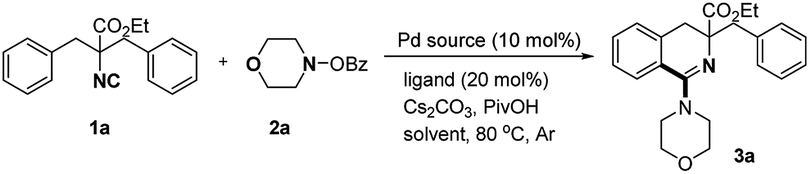
The reaction conditions were optimized with ethyl 2-benzyl-2-isocyano-3-phenylpropanoate (1a) and morpholino benzoate (2a) as model substrates, and ethyl 3-benzyl-1-morpholino-3,4-dihydroisoquinoline-3-carboxylate (3a) was obtained in 34% yield in the presence of Pd(OAc)2 (1.5 equiv.), PPh3 (20 mol%), Cs2CO3 (1.0 equiv.) and PivOH (0.6 equiv.) in toluene at 80 °C (entry 1, Table 1). Dioxane was proven to be the best choice after screening of solvents and the product was generated in 76% yield (entries 2–6). The reaction was totally suppressed in high polar solvents such as MeCN and DMSO (entries 5–6). The yield decreased sharply when the catalyst was changed to Pd(OPiv)2, Pd(TFA)2, PdCl2(MeCN)2 or Pd(PPh3)4 (entries 7–10). And no better results were detected by using other phosphine ligands (entries 12–17). Fortunately, the yield of 3a was increased to 84% at 110 °C (entry 18). Control experiments revealed that palladium catalyst and additives (Cs2CO3/PivOH) were essential and the reaction was much less efficient when using other bases such as CsOPiv, K2CO3, and Na2CO3 (entries 19–24). The yield decreased sharply to 61% when the catalyst loading was reduced to 5 mol% (entry 25).
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Ligand | Solvent | Yieldb |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.15 mmol), Pd-catalyst (0.01 mmol, 10 mol%), ligand (0.02 mmol, 20 mol%), Cs2CO3 (0.1 mmol, 1.0 equiv.), PivOH (0.06 mmol, 0.6 equiv.), 80 °C, Ar. A solution of 1a was added via a syringe pump within 1 h.b NMR yield with 1-iodo-4-methoxybenzene as an internal standard.c T = 110 °C.d Isolated yield.e Without Cs2CO3 and PivOH.f CsOPiv was used as the base.g K2CO3 was used as the base.h Na2CO3 was used as the base.i 5 mol% of catalyst was used. | ||||
| 1 | Pd(OAc)2 | PPh3 | Toluene | 34 |
| 2 | Pd(OAc)2 | PPh3 | THF | Trace |
| 3 | Pd(OAc)2 | PPh3 | DCE | 29 |
| 4 | Pd(OAc)2 | PPh3 | Dioxane | 76 |
| 5 | Pd(OAc)2 | PPh3 | MeCN | Trace |
| 6 | Pd(OAc)2 | PPh3 | DMSO | Trace |
| 7 | Pd(OPiv)2 | PPh3 | Dioxane | 31 |
| 8 | Pd(TFA)2 | PPh3 | Dioxane | 26 |
| 9 | PdCl2(MeCN)2 | PPh3 | Dioxane | 38 |
| 11 | Pd(PPh3)4 | Dioxane | 56 | |
| 12 | Pd(OAc)2 | BINAP | Dioxane | 30 |
| 13 | Pd(OAc)2 | dppb | Dioxane | 26 |
| 14 | Pd(OAc)2 | dppp | Dioxane | 28 |
| 15 | Pd(OAc)2 | XantPhos | Dioxane | 42 |
| 16 | Pd(OAc)2 | SPhos | Dioxane | 34 |
| 17 | Pd(OAc)2 | XPhos | Dioxane | 24 |
| 18c | Pd(OAc)2 | PPh3 | Dioxane | 84 (80)d |
| 19c | None | PPh3 | Dioxane | 0 |
| 20c | Pd(OAc)2 | None | Dioxane | 56 |
| 21c,e | Pd(OAc)2 | PPh3 | Dioxane | 0 |
| 22c,f | Pd(OAc)2 | PPh3 | Dioxane | 41 |
| 23c,g | Pd(OAc)2 | PPh3 | Dioxane | 24 |
| 24c,h | Pd(OAc)2 | PPh3 | Dioxane | 31 |
| 25c,i | Pd(OAc)2 | PPh3 | Dioxane | 61 |
With the optimized conditions in hand, the scope of functionalized isocyanide was investigated first, using morpholino benzoate 2a as the coupling partner (Scheme 3). Both electron rich and withdrawing substituents such as Me, t-Bu, F, Cl, CF3, CO2Me and NO2 at the para position of aromatic ring can tolerate the reaction conditions, and generate the corresponding products in good to excellent yields (3a–3h). Isocyanides bearing ortho substituents on the phenyl ring underwent the reaction smoothly, generating the products 3i and 3j in 58% and 82% yields respectively. Even substrates bearing two substituents were well suited to the reaction conditions, affording the aminoimidoylation products without loss of efficiency (3k–3l). Compound 3m was delivered with excellent regioselectivity in 63% yield when m-Cl substituted starting material was employed in the reaction, which indicated the hindrance sensitivity of this reaction. Modifying the ester moiety in 1 to a methyl ester or an amide did not impede the reaction, giving 3n and 3o with excellent yields. Changing one of the benzyl groups to alkyl substituents didn't decrease the reaction efficiency, generating products 3p and 3q in moderate yields.
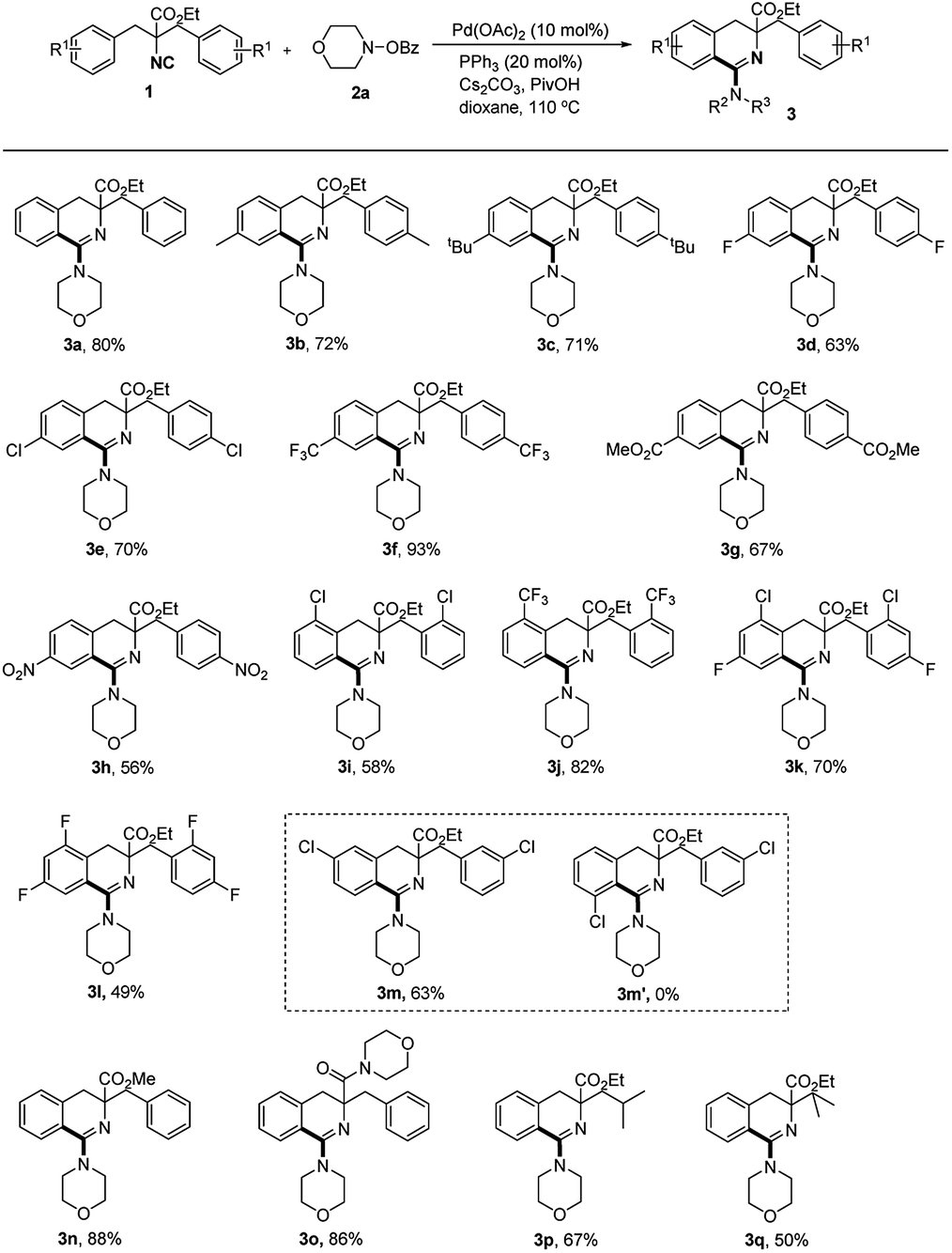 | ||
| Scheme 3 Scope of isocyanides. Reaction conditions: 1 (0.1 mmol), 2a (0.15 mmol), Pd(OAc)2 (0.01 mmol, 10 mol%), PPh3 (0.02 mmol, 20 mol%), Cs2CO3 (0.1 mmol, 1.0 equiv.), PivOH (0.06 mmol, 0.6 equiv.), 110 °C, Ar. A solution of 1 was added via a syringe pump within 1 h. Isolated yields. | ||
Then we moved our attention to the investigation of substrate scope of aminating partners (Scheme 4). It was found that various O-benzoyl hydroxylamines derived from piperidines could be smoothly employed in the reaction (4a–4e). Substituents such as Me, CO2Et, OMe and ketal group on the piperidines were well tolerated (4b–4e). Aminating reagent derived from Boc-protected piperazine was also compatible with this reaction, generating the product 4f in 99% yield. Fortunately, acyclic aminated product 4g was obtained as well in moderate yield.
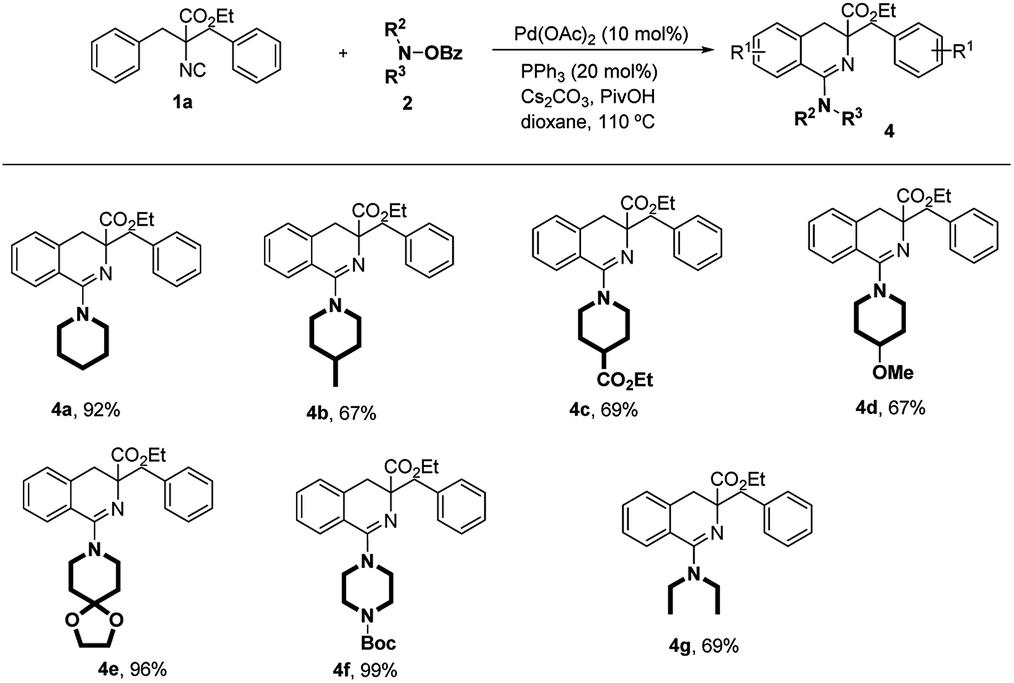 | ||
| Scheme 4 Substrate scope of aminating agents. Reaction conditions: 1a (0.1 mmol), 2 (0.15 mmol), Pd(OAc)2 (0.01 mmol, 10 mol%), PPh3 (0.02 mmol, 20 mol%), Cs2CO3 (0.1 mmol, 1.0 equiv.), PivOH (0.06 mmol, 0.6 equiv.), 110 °C, Ar. A solution of 1a was added via a syringe pump within 1 h. Isolated yields. | ||
When ethyl 2-isocyano-2,3-diphenylpropanoate was conducted in the reaction with 2a, the 3,4-dihydroisoquinoline product 6 was generated exclusively, albeit in only 37% yield (Scheme 5a). To examine if isoindoline could be afforded by this aminoimidoylation reaction, ethyl 2-isocyano-4-methyl-2-phenylpentanoate (7) was selected as the substrate with 2a (Scheme 5b). However, no desired product was generated under the standard conditions, showing different site-selectivity with the previous imidoylation reaction developed by Zhu and coworkers.16
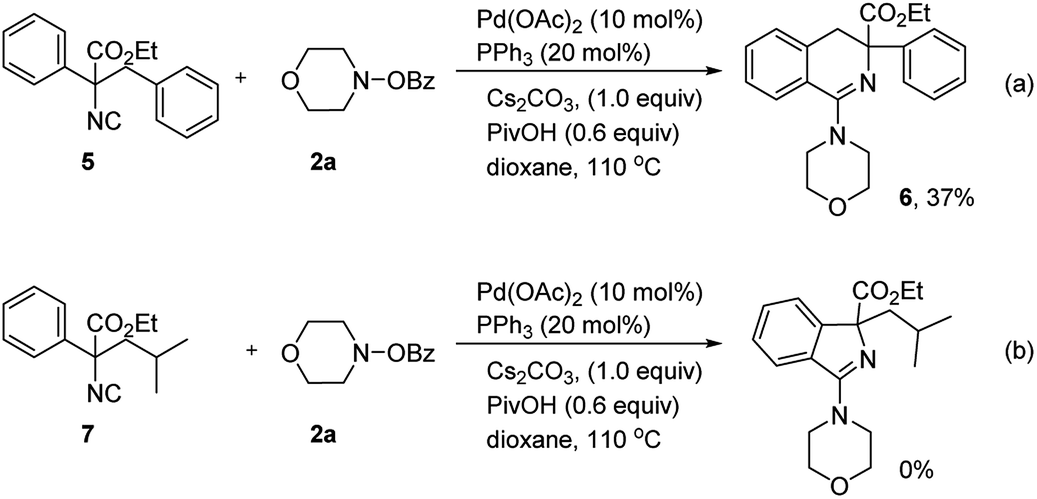 | ||
| Scheme 5 Reaction conditions: 5(7) (0.1 mmol), 2a (0.15 mmol), Pd(OAc)2 (0.01 mmol, 10 mol%), PPh3 (0.02 mmol, 20 mol%), Cs2CO3 (0.1 mmol, 1.0 equiv.), PivOH (0.06 mmol, 0.6 equiv.), 110 °C, Ar. A solution of 5(7) was added via a syringe pump within 1 h. Isolated yields. | ||
Gram-scale preparation of 3a was carried out smoothly under standard conditions (Scheme 6a). The reduction reaction of 3a was carried out using LiAlH4 and the corresponding product 8 was generated in 70% yield (Scheme 6b). The ester group was successfully transformed to tertiary alcohol (9) in moderate yield with Grignard reagent.
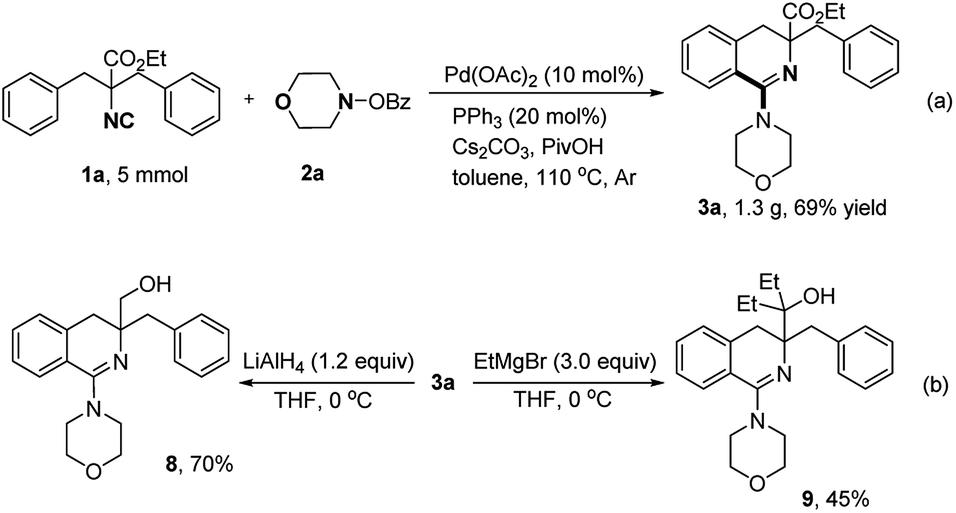 | ||
| Scheme 6 Gram-scale preparation and diversifications of 3a. | ||
In order to get further insight into the reaction mechanism, a radical trapping reaction with 0.1 equivalents of TEMPO was carried out (Scheme 7a). The product 3a was generated without loss of efficiency, which indicated that the radical-involved pathway could be ruled out. A plausible mechanism was then proposed in Scheme 7b. The reaction was initiated with oxidative addition of O-benzoyl hydroxylamine 2a to the in situ generated Pd(0) species. Migratory isocyanide insertion to intermediate B afforded aminoimidoyl Pd(II) intermediate C. The following intramolecular C–H bond activation and reductive elimination via E yielded the final product 3a and regenerated Pd(0) to complete the catalytic cycle.
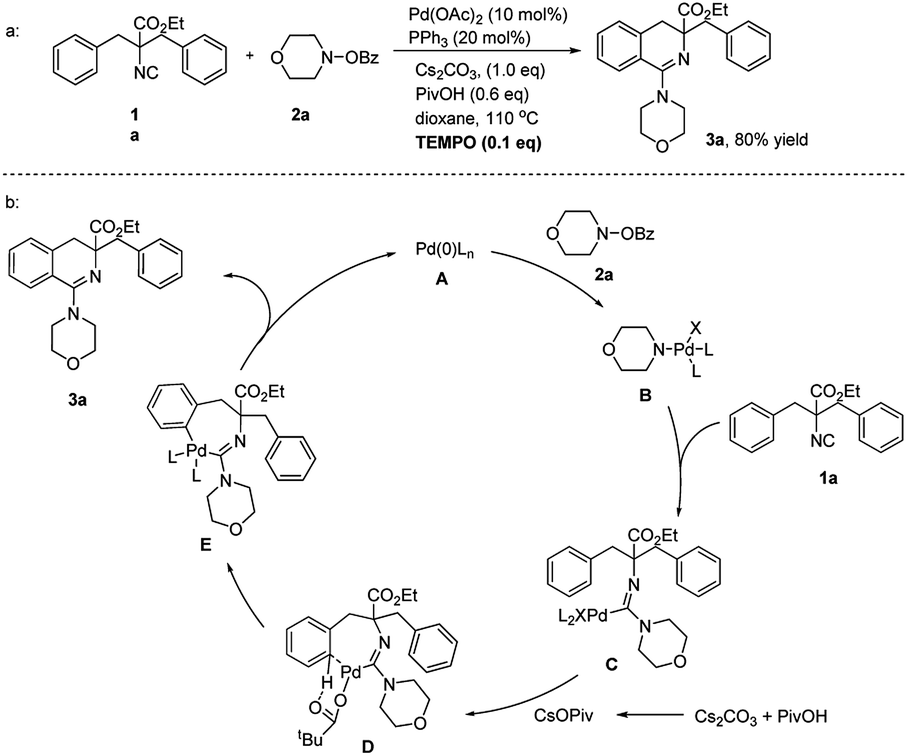 | ||
| Scheme 7 Proposed mechanism. | ||
In conclusion, we have developed an efficient and practical method to prepare 1-amino-3,4-dihydroisoquinolines via palladium-catalyzed intramolecular C–H bond aminoimidoylation of α-benzyl-α-isocyanoacetates under redox-neutral conditions. Consecutive isocyanide insertion and C–H bond activation were realized with C–N and C–C bonds formation in one step, employing O-benzoyl hydroxylamines as electrophilic amino sources. A broad range of substrates were applicable to the reaction in good to excellent yields.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Natural Science Foundation of China (21702003), Natural Science Foundation of Anhui Province (1808085QB31), Wuyi University and Anhui Normal University for financial support.Notes and references
-
(a) K. Masayuki, N. Shigetaka, I. Kiyotaka and T. Hisashi, Eur. Pat. Appl., EP370381, 1990
; (b) S. Bowers, Y.-Z. Xu, S. Yuan, G. D. Probst, R. K. Hom, W. Chan, A. W. Konradi, H. L. Sham, Y.-L. Zhu, P. Beroza, H. Pan, E. Brecht, N. Yao, J. Lougheed, D. Tam, Z. Ren, L. Ruslim, M. P. Bova and D. R. Artis, Bioorg. Med. Chem. Lett., 2013, 23, 2181 CrossRef CAS PubMed
-
(a) G. D. Diana, W. B. Hinshaw and H. E. Lape, J. Med. Chem., 1977, 20, 449 CrossRef CAS PubMed
-
(a) W. M. Whaley and T. R. Govindachari, Organic Reactions, ed. R. Adams, John Wiley and Sons, New York, 1951, vol. 6, pp. 74–150 Search PubMed
- For reviews of multicomponent reactions, see:
(a) V. Nair, C. Rajesh, A. U. Vinod, S. Bindu, A. R. Sreekanth, J. S. Mathen and L. Balagopal, Acc. Chem. Res., 2003, 36, 899 CrossRef CAS PubMed
- For reviews of imidoyl radical-involved reactions, see:
(a) J. Lei, J. Huang and Q. Zhu, Org. Biomol. Chem., 2016, 14, 2593 RSC
- For reviews of isocyanide insertion reactions, see:
(a) A. V. Lygin and A. Meijere de, Angew. Chem., Int. Ed., 2010, 49, 9094 CrossRef CAS PubMed
-
(a) M. Kosugi, T. Ogata, H. Tamura, H. Sano and T. Migita, Chem. Lett., 1986, 15, 1197 CrossRef
-
(a) G. Baelen, S. Kuijer, L. Rýček, S. Sergeyev, E. Janssen, F. De Kanter, B. Maes, E. Ruijter and R. Orru, Chem.–Eur. J., 2011, 17, 15039 CrossRef PubMed
- M. Giustiniano, A. Basso, V. Mercalli, A. Massarotti, E. Novellino, G. C. Tron and J. Zhu, Chem. Soc. Rev., 2017, 46, 1295 RSC
-
(a) J. Wang, S. Luo, J. Huang, T. Mao and Q. Zhu, Chem.–Eur. J., 2014, 20, 11220 CrossRef CAS
-
(a) T. Nanjo, C. Tsukano and Y. Takemoto, Org. Lett., 2012, 14, 4270 CrossRef CAS
- For selected examples, see:
(a) M. Tobisu, K. Koh, T. Furukawa and N. Chatani, Angew. Chem., Int. Ed., 2012, 51, 11363 CrossRef CAS
- S. Tang, J. Wang, Z. Xiong, Z. Xie, D. Li, J. Huang and Q. Zhu, Org. Lett., 2017, 19, 5577 CrossRef CAS
- J. Wang, S. Tang and Q. Zhu, Org. Lett., 2016, 18, 3074 CrossRef CAS
- J. Wang, D.-W. Gao, J. Huang, S. Tang, Z. Xiong, H. Hu, S.-L. You and Q. Zhu, ACS Catal., 2017, 7, 3832 CrossRef CAS
- S. Tang, S.-W. Yang, H. Sun, Y. Zhou, J. Li and Q. Zhu, Org. Lett., 2018, 20, 1832 CrossRef CAS
-
(a) W. Hu, J. Li, Y. Xu, J. Li, W. Wu, H. Liu and H. Jiang, Org. Lett., 2017, 19, 678 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09139d |
| This journal is © The Royal Society of Chemistry 2019 |