DOI:
10.1039/C9RA09287K
(Review Article)
RSC Adv., 2019,
9, 41173-41191
Synthesis and biological activities of local anesthetics
Received
8th November 2019
, Accepted 27th November 2019
First published on 13th December 2019
Abstract
Local anesthetics are mainly used in stomatology, ophthalmology, gynecology and surgery to temporarily relieve pain. Local anesthetics act on nerve endings or around nerve trunks, and are combined with specific sodium ion (Na+) channel sites on the nerve membrane. They can affect the membrane potential by reducing Na+ passage through sodium ion channels, thus blocking the generation and conduction of nerve impulses, reversibly blocking the generation and conduction of sensory nerve impulses. Local anesthetics are used for convenience in local surgical operations and treatments. Herein, we mainly review the research progress on local anesthetics and discuss the important aspects of design, synthesis and biological activity of various new compounds.
 Shiyang Zhou | Shiyang Zhou is a Research Scholar at the College of Chemistry and Chemical Engineering, Hainan Normal University, China. He is pursuing PhD in chemistry from the Hainan Normal University. He has received his Master of Science degree in Chemical Biology from the Chongqing Normal University, China. He has over six years of experience in Industry and Academics. His research interests include the medicinal chemistry (drug design and synthesis, natural products semi-synthesis and full synthesis). |
1. Introduction
Anesthetics are a type of drug that act on the nervous system, causing it to be inhibited and resulting in the loss of the sense of pain.1–4 According to the different scopes of action of drugs, the department of anesthetics is divided into general anesthetics and local anesthetics.5–7 General anesthetics act on the central nervous system, causing it to be reversibly inhibited, thereby resulting in a loss of consciousness and sensation. Especially pain sensation and reflexes disappear and skeletal muscle relaxes, thus achieving the effect of general anesthesia, which is mainly used before surgery.8–11 In contrast, local anesthetics act on nerve endings or around nerve trunks, reversibly blocking the generation and conduction of sensory nerve impulses, and temporarily eliminating local sensation (mainly pain sensation) under the condition of consciousness. Moreover, it is convenient for local surgical operations and treatments.12–14 Local anesthetics are mainly used in stomatology, ophthalmology, gynecology and surgery to relieve pain temporarily.15 Local anesthetics combine with specific sodium ion (Na+) channel sites on the nerve membrane, and they can affect the membrane potential by reducing Na+ passage through the sodium ion channels, thus blocking the generation and conduction of nerve impulses.16 Local anesthetics decrease the excitability of nerve membranes, but have no effect on their resting potential.17–20 The application methods of local anesthesia can be divided into topical anesthesia, invasive anesthesia, conduction anesthesia, subarachnoid anesthesia and epidural anesthesia.21–24
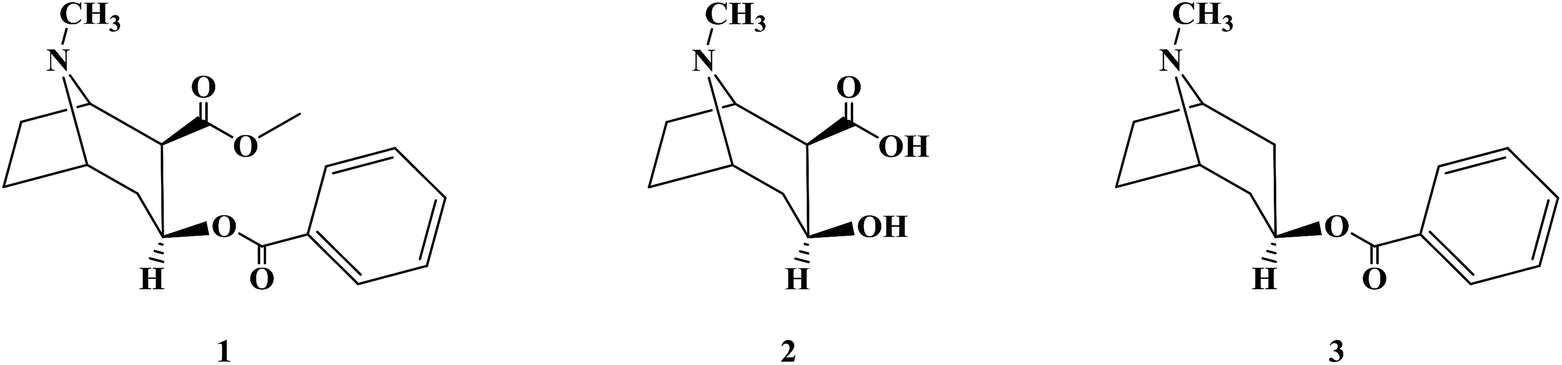
The first local anesthetic used was an active ingredient from coca leaves in South America (Erythroxylum coca Lam.). The Peruvians were known for chewing coca leaves to relieve pain as early as 1532. In 1860, Niemann extracted an alkaloid crystal from coca leaves and named it cocaine (1). Koller, an Austrian ophthalmologist, first formally used cocaine in clinical surgery in 1884. Also, he discovered a new local anesthesia technology, which led to the rapid development of local anesthetics.25–30 Cocaine has a local anesthesia effect and vasoconstriction effect, and it can reduce the bleeding of surgical wounds.31–34 Additionally, cocaine can be used in the nose, nasopharynx, mouth, throat and ear as surface anesthesia.35–39 However, cocaine is highly addictive, and easy to cause tissue irritation, allergic reactions and other toxic side effects. Also, its aqueous solution is not stable, and thus easily hydrolyzed during high-pressure disinfection; therefore, the clinical application of cocaine is greatly restricted, and it has long since been replaced by other local anesthetics.40–43 Because of the shortcomings of cocaine, researchers began to study and modify the structure of cocaine to find better local anesthetics. None of the three products from the hydrolysis of cocaine, ecgonine (2), benzoic acid and methanol, has the effect of local anesthesia.44 Substitution of benzoic acid with other carboxylic acids can form esters with ecgonine, resulting in reduced or complete loss of anesthetic effects, proving that benzoic acid esters are an important structure of cocaine with local anesthetic effects. The alkaloid tropacocaine (3) isolated from the leaves of coca in Java had only the structure of benzoate ester and no carboxylate methyl ester group in its molecular structure, which had the general effect of local anesthesia. Therefore, the local anesthesia effect of benzoate ester was further confirmed.45–47
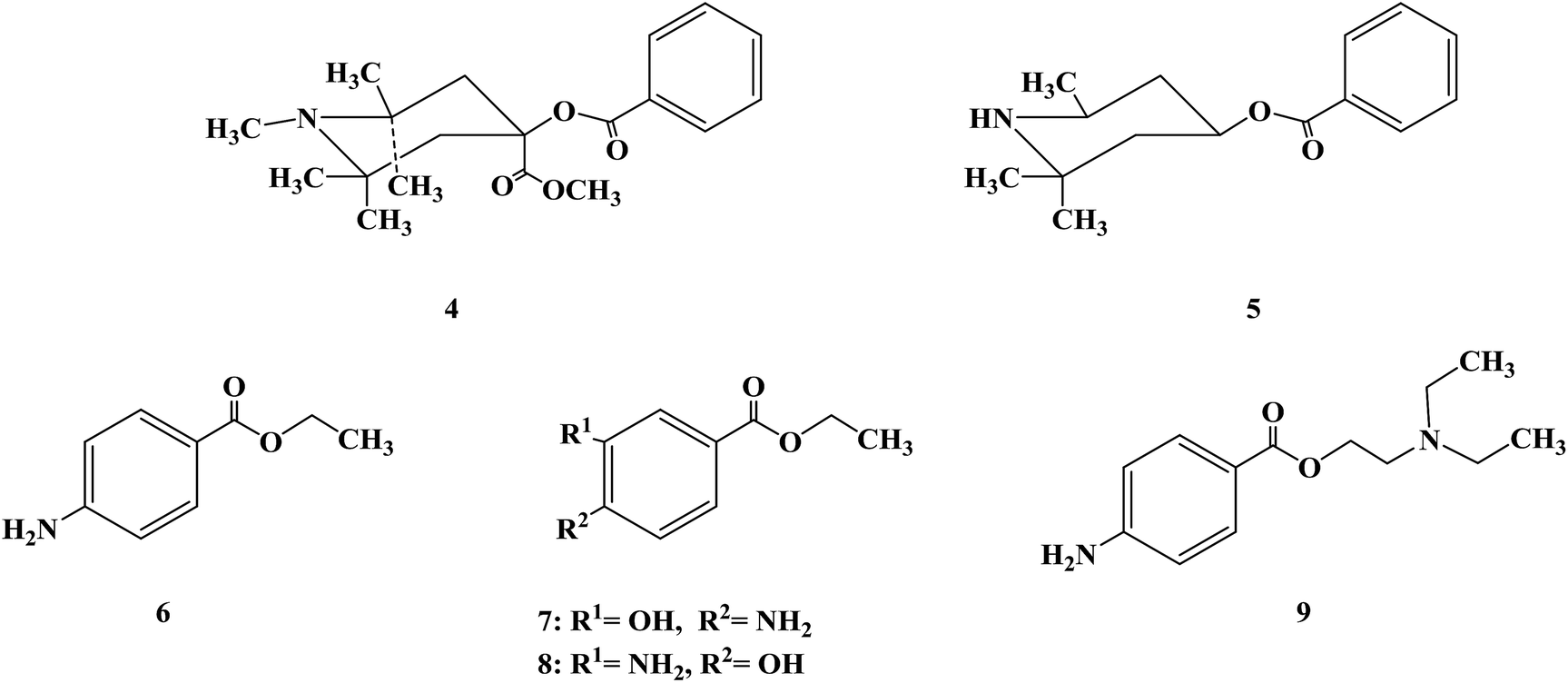
After determining the local anesthesia effect of benzoate ester, the structure of the parent nucleus of cocaine (ecgonine) was modified and simplified. When the N-methyl of ecgonine was removed, there was no significant effect on physiological activities, but the toxicity increased.48 If the nitrogen atom was quaternized, the activity was lost. For ecgonine, Corning further simplified its structure, and designed and synthesized two piperidol benzoate derivatives, α-eucaine (4) and β-eucaine (5). Both of them had a local anesthetic effect similar to cocaine, but their aqueous solution property was relatively stable, and their toxicity was also low, which indicate that the ecgonine double ring structure is not necessary for the local anesthesia effect.49–53 Simultaneously, this further proves the importance of the benzoate ester part from the side. Due to the significant research work on benzoate ester compounds, benzocaine (6) was first synthesized in 1890 and proven to have a local anesthesia effect. Subsequently, orthoform (7) and new orthoform (8) were successfully synthesized. Both of them exhibited strong local anesthesia effects, but they were not water-soluble and could not be injected.54–56 They could only be used for local anesthesia, while their salt was too acidic to be used. Because of their shortcomings, further structural transformation led to the successful development of the local anesthetic procaine (9) in 1904, which overcame these shortcomings.
The discovery of procaine made researchers realize the important role of the p-aminobenzoate structure in local anesthetics.57–59 Upon further structural modification of procaine, aromatic acid esters were obtained as local anesthetics. Based on the structure of aromatic acid esters, amide, aminoketone, aminoether and amino carbamate types of local anesthetics were developed (Table 1). In terms of drugs structure–activity relationship (SAR), the chemical structure of local anesthetics usually consists of three basic skeleton parts, a lipophilic aromatic ring, intermediate connecting functional group and hydrophilic amine group. In recent years, the local anesthetics used in clinical practice are mainly used to reduce toxicity and prolong the action time by changing the new dosage forms of existing local anesthetics.60,61 Herein, we mainly review the research progress of local anesthetics (2009–2019), including the design, synthesis and biological activity of various new compounds.
Table 1 The common types of local anesthetics
| Compound |
Structure |
Time to market |
Application methods |
| Procaine |
 |
1904 |
Infiltration anesthesia, conduction anesthesia, subarachnoid anesthesia and epidural anesthesia |
| Chloroprocaine |
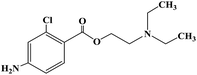 |
1952 |
Infiltration anesthesia, epidural anesthesia and conduction anesthesia |
| Hydroxyprocaine |
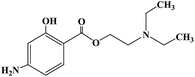 |
1960 |
Infiltration anesthesia |
| Tetracaine |
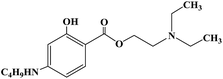 |
1988 |
Conduction anesthesia, subarachnoid anesthesia and epidural anesthesia |
| Oxybuprocaine |
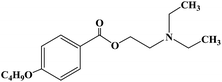 |
1975 |
Topical anesthesia |
| Tutocaine |
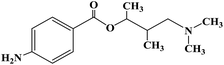 |
1976 |
Topical anesthesia and infiltration anesthesia |
| Butacaine |
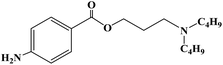 |
1976 |
Topical anesthesia and infiltration anesthesia |
| Dimethocaine |
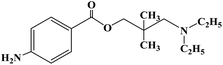 |
1938 |
Topical anesthesia and infiltration anesthesia |
| Thiocaine |
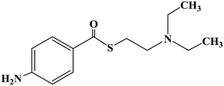 |
Halt sales |
Topical anesthesia and infiltration anesthesia |
| Lidocaine |
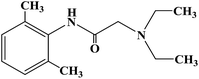 |
1948 |
Conduction anesthesia and epidural anesthesia |
| Mepivacaine |
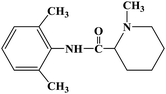 |
1986 |
Infiltration anesthesia, conduction anesthesia, epidural anesthesia and topical anesthesia |
| Bupivacaine |
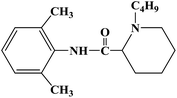 |
2000 |
Infiltration anesthesia, conduction anesthesia and epidural anesthesia |
| Ropivacaine |
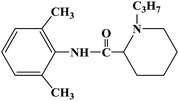 |
1996 |
Infiltration anesthesia, conduction anesthesia and epidural anesthesia |
| Trimecaine |
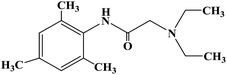 |
1965 |
Infiltration anesthesia, surface anesthesia and epidural anesthesia |
| Prilocaine |
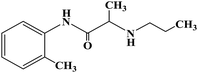 |
1993 |
Infiltration anesthesia, topical anesthesia and epidural anesthesia |
| Etidocaine |
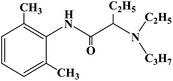 |
1976 |
Epidural anesthesia |
| Pyrrocaine |
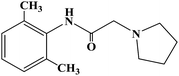 |
1964 |
Conduction anesthesia and epidural anesthesia |
| Butanilicaine |
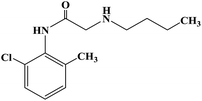 |
1982 |
Infiltration anesthesia and conduction anesthesia |
| Cinchocaine |
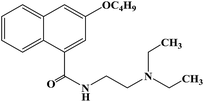 |
1985 |
Topical anesthesia, subarachnoid anesthesia and epidural anesthesia |
| Articaine |
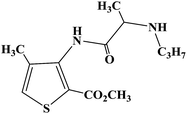 |
2002 |
Infiltration anesthesia and subarachnoid anesthesia |
| Dyclonine |
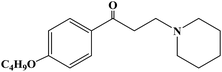 |
1956 |
Topical anesthesia |
| Falicaine |
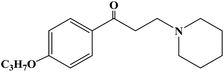 |
1957 |
Topical anesthesia |
| Quinisocaine |
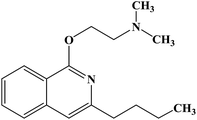 |
1957 |
Topical anesthesia |
| Pramocaine |
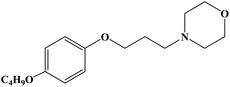 |
1977 |
Topical anesthesia |
| Diperodon |
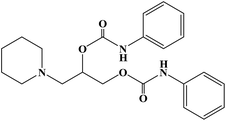 |
1980 |
Topical anesthesia |
| Heptacaine |
 |
1984 |
Infiltration anesthesia |
2. Synthesis and biological activities
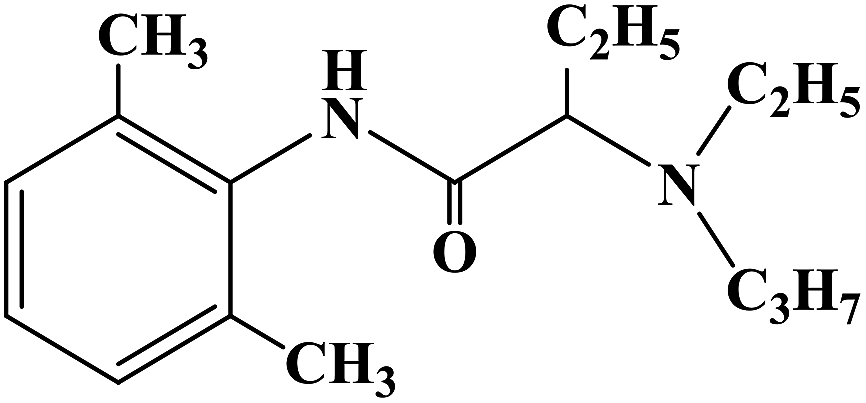
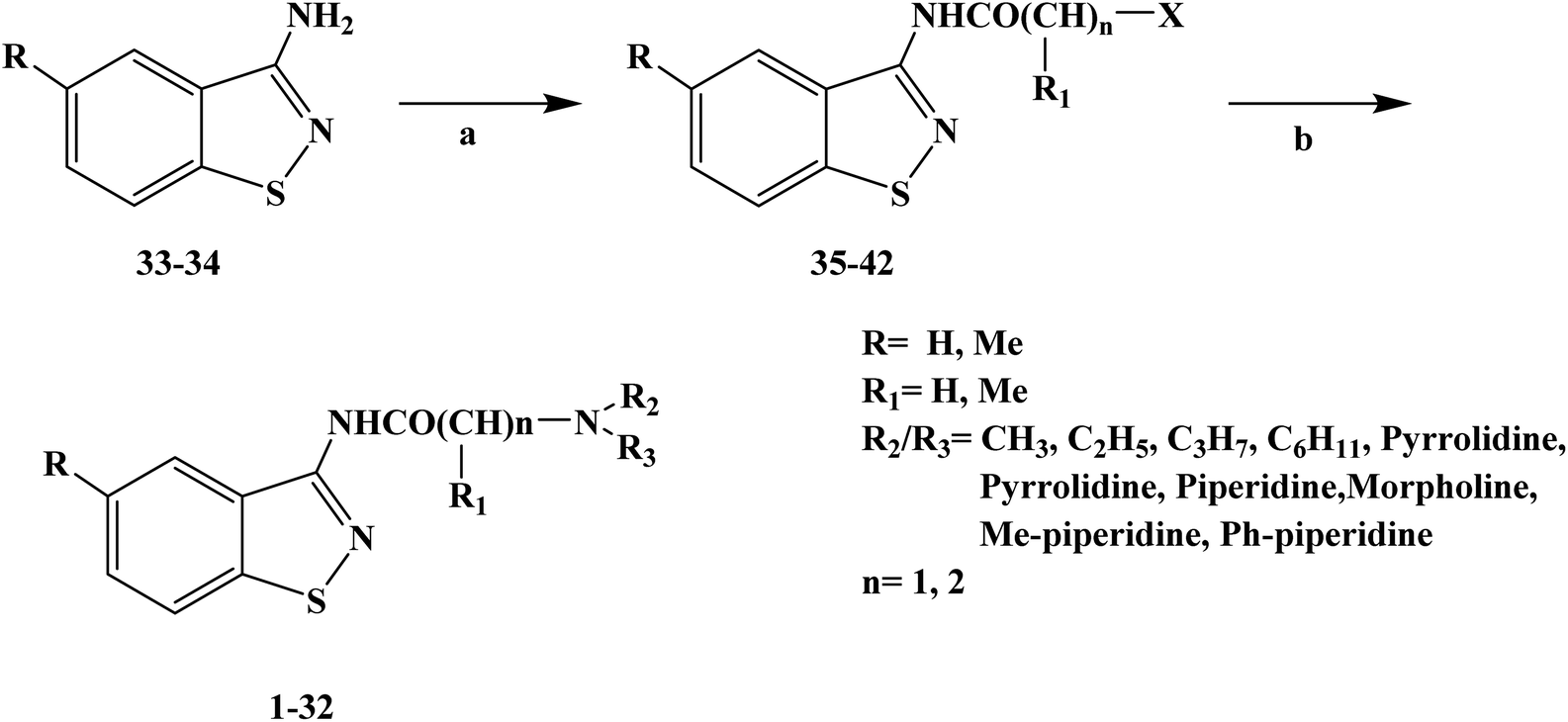
Local anesthesia is a compound that blocks the conduction of nerve fibers when applied locally to nerve tissue at appropriate concentrations. It blocks the voltage-sensitive sodium channels of each nerve fiber, eliminating their action potential. The greatest practical advantage of local anesthetics is that their effects can be reversed when they were removed from nerve tissue. In this case, nerve function and action potential are completely restored, with no evidence of damage to nerve fibers or cellular structures. Compounds with amino groups linked to heterocyclic nuclei, such as lipophilic molecules, were shown to be more active and less toxic than benzene analogues. It has been reported that 2-aminobenzothiazole, 2-aminothiazole derivatives and 3-aminobenzoazole-[d]-isothiazole derivatives have local anesthetic activity. In 2009, Carla F. et al.62 reported the design and synthesis of different thiazole and isothiazole derivatives for the study of local anesthesia. In the process of structural design of target compounds 1–32, they used the thiazole structure as the parent structure of the target compounds, and simultaneously retained the alkyl-acetamido or alkyl-propionamide groups that local anesthetic drugs have. In terms of structure–activity relationship (SAR), these target molecules were structurally similar to lidocaine, and thus they should be active in infiltration anesthesia and surface anesthesia. In the process of the synthesis of the target compounds, they used Scheme 1 as the synthetic route, and they chose the substituted thiazolamide as the starting material and dimethylformamide (DMF) as the reaction solvent to obtain intermediate compounds 35–42. After further reaction with imine at high temperature in anhydrous benzene, the crude products of the target compounds were obtained and then crystallized with EtOH/Et2O. All target compounds obtained good yields. The synthetic route had a few reaction steps, simple operation and easy reagents and raw materials. However, the synthetic route used benzene as the solvent, which will bring certain difficulties in the future industrial production. Thus, in the optimization of process conditions in the future, it is necessary to strengthen the in-depth study of solvents and try to choose low-toxic or non-toxic solvents. In the evaluation of local anesthetic activity, they chose the isolated sciatic nerve model to evaluate the local anesthetic effect of the target compounds. The results of local anesthesia screening in vitro showed that these compounds have a good local anesthesia effect, and the local anesthesia effect of some target compounds is better than that of the control product of lidocaine. To find the most promising target compounds, they also used the computer programs PASS and DEREK to predict the activity and toxicity of local anesthesia. The results of the QSAR study showed that the polarizability, polarity, hydrogen bond and molecular shape of the molecule had a positive and important effect on the local anesthetic activity of the target compounds. In addition, aromatic benzyl and single bond nitrogen also had a certain contribution to the local anesthetic activity of the target compounds. The data results of the local anesthesia model of frog and mouse showed that the calculated Pa of the local anesthesia effect of target compounds 5, 17, 27, 10 and 24 was in the range of 23–47% (the probability of passing was less than 50%), and thus these target compounds may become new local anesthetics, which need further research and development. Since these compounds are still in the process of in vitro screening, a follow-up study on their local anesthetic activity in vivo still needs to be carried out.
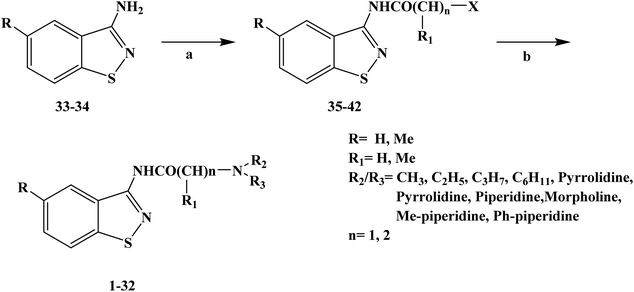 |
| | Scheme 1 Reagents and conditions: (a) ClCO(CHR)nX, DMF and (b) NN(R), benzene. | |
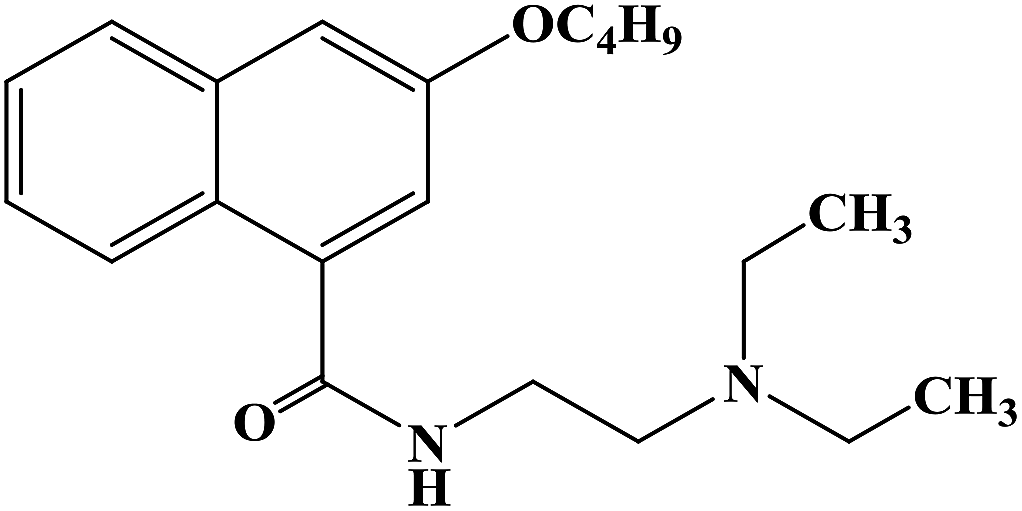
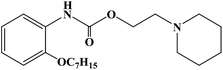
Two-thirds of the world's best-selling drugs are chiral compounds, and chiral drugs play an important role in drug discovery and drug therapy. Chiral drugs (enantiomers) differ in pharmacology, pharmacokinetics and toxicity, while single enantiomers have obvious advantages in the treatment of diseases. Therefore, the development of efficient methods for the synthesis of chiral compounds with high yield and enantiomer purity has become one of the important topics in the field of chemistry. Xanthone (9H-xanthon-9-one) derivatives are natural compounds with extensive biological and pharmacological activities, which have been proven important in the synthesis of new compounds. There are few reports on the synthesis of chiral derivatives despite their structural diversity and biological activity. Some chiral xanthone derivatives (CXDs) show good biological activity in the central nervous system (CNS). These compounds can both stimulate nerve activity and act as sedatives. Xanthone derivatives also have antiarrhythmic, anticonvulsive, antitumor, local anesthesia, antibacterial and antifungal effects. Some of these compounds have been shown to induce antihypertensive and vascular relaxant effects by inhibiting Ca2+ influx through receptor manipulation and/or voltage-sensitive channels. In 2012, Athina G. et al.63 designed and synthesized three chiral xanthone derivatives. In the process of the synthesis of target compounds 7a–7c, they chose an efficient and convenient synthesis route, as shown in Scheme 2. Under the catalysis of N,N-dimethyl-glycine, CuI and Cs2CO3, the Ullmann condensation reaction between intermediate compound 2 and m-methoxyphenol resulted in the formation of diaromatic ether compound 3. The coupling reaction was carried out at 90 °C for 14 h, and the yield of compound 3 was good (54%). Compound 4 was obtained by hydrolyzing methyl ester in a mixture of methanol/tetrahydrofuran and sodium hydroxide solution. Compound 5 was obtained by the molecular acylation of diaryl ether compound 4 with phosphorus pentoxide and methanesulfonic acid at room temperature. Finally, using TBTU and triethylamine as the catalyst, target compounds 7a–7c were obtained by reaction of tetrahydrofuran. The use of TBTU synthetic derivatives could be done at room temperature, resulting in high yield (97%), in a short reaction time (30 min to 2 h), with the absence of racemization (enantiomeric excess was more than 99%). This synthetic route used cheap raw materials to synthesize the target compounds, which has the characteristics of mild reaction temperature, relatively short reaction time and high overall yield. However, there are also many reaction steps, which bring some difficulties to the future process and need to be improved in the subsequent process conditions. The data from the local anesthesia activity study showed that when the concentration of CXDs (compounds 7a–7c) was high, they could block the sciatic nerve conduction in rats, and had no obvious protective effect on hypotonic hemolysis. It could be seen that the nerve conduction block induced by CXDs was mainly caused by the sodium ion (Na+) current. This effect could be isolated from their ability to stabilize the membrane, and became apparent only when the CXD (compounds 7a–7c) concentration increased to the high micromolar range. The chiral xanthone derivatives compounds 7a–7c are structurally similar to many local anesthetics, such as dibucaine, mepivacaine, lidocaine and bupivacaine, which may have the same molecular spatial structure and have the same pharmacophilia. The mechanism of nerve block induced by local anesthesia can be distinguished by its structure and by the way it crosses the cell membrane. Hydrophilic compounds diffuse to the axoplasm through open Na+ channel pores, while lipid-soluble drugs can easily enter the Na+ channel through the nerve membrane even if the (hydrophobic channel) is closed, achieving the effect of local anesthesia. The ability of compounds 7a–7c to prevent hypotonic hemolysis was also evaluated, and the results showed that there were no excited erythrocytes and no voltage-gated sodium ion channels. In general, these chiral derivatives show good activity in local anesthesia, which need further research and development.
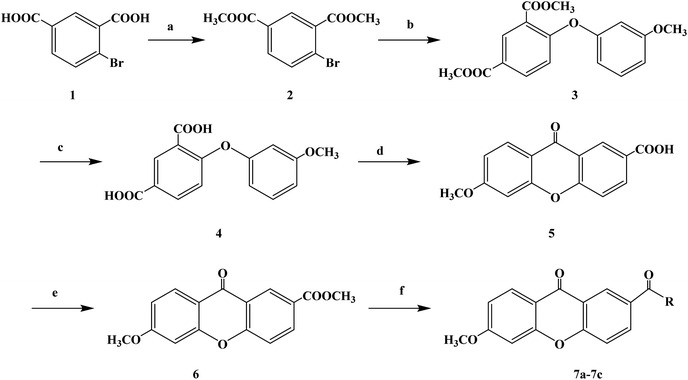 |
| | Scheme 2 Reagents and conditions: (a) methanol, H2SO4, reflux, 17 h; (b) m-methoxyphenol, CuI, Cs2CO3, N,N-dimethyl glycine, dioxane, N2, 90 °C, 14 h; (c) methanol/tetrahydrofuran, NaOH, r.t., 18 h; (d) P2O5, CH3SO3H, r.t., 22 h; (e) methanol, H2SO4, reflux, 19 h; and (f) substitute amine, TBTU, triethylamine, dry tetrahydrofuran, r.t., 30 min to 2 h. | |
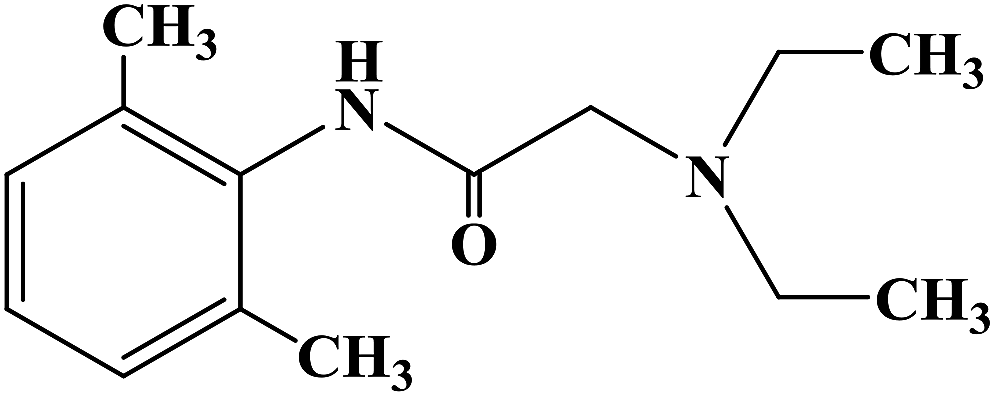
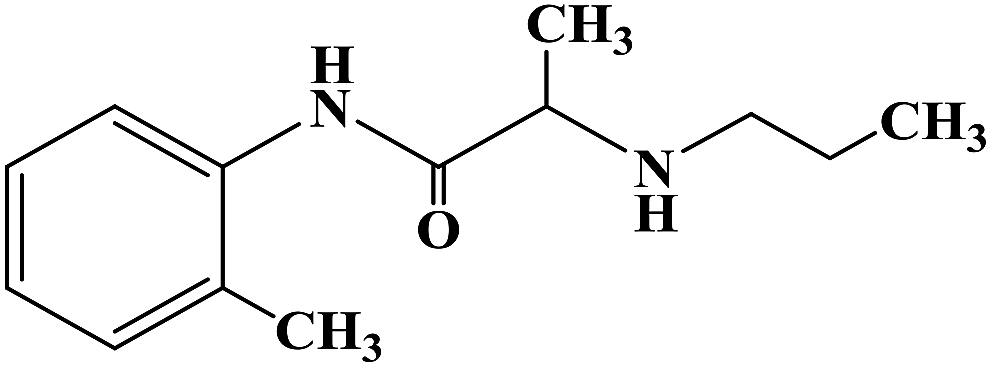
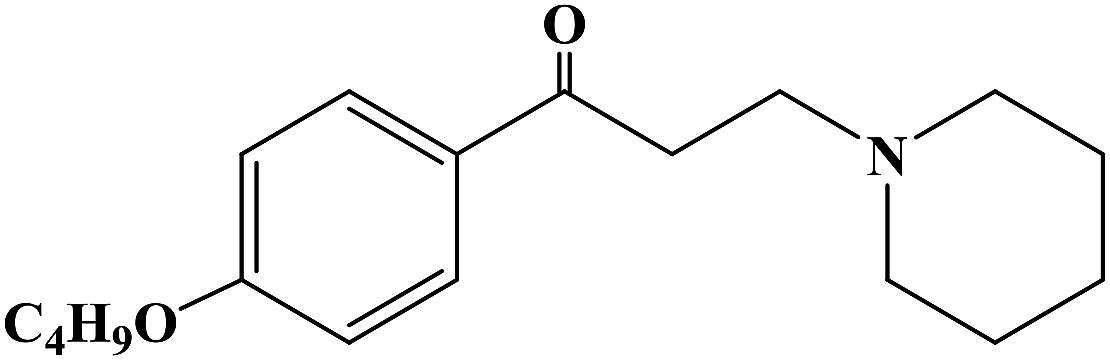
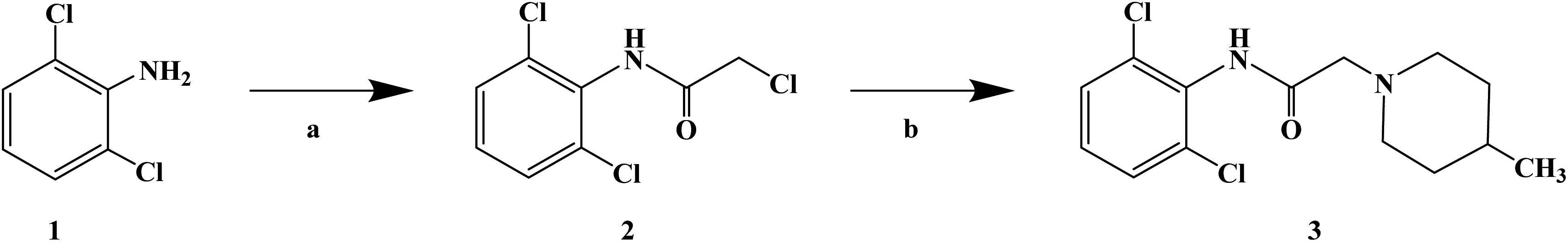
Sensory-neuron voltage-gated sodium (Na+) channels play an important role in chronic painful neuropathy caused by peripheral nerve injury. Local anesthetics can block sodium (Na+) channels and effectively treat neuropathic pain and diabetic neuropathy. Lidocaine, a local anesthetic and antiarrhythmic agent, reduces pain by blocking voltage-gated sodium (Na+) channels in sensory neurons around and at the central level of the spinal cord. Lidocaine is widely used clinically, but there is also a risk of nerve damage, particularly in patients with diabetic peripheral neuropathy, where neurotoxicity is more common. In 2013, Liliana G. et al.64 using lidocaine as the lead compound, designed and synthesized the lidocaine analogue N-(2,6-dichlorophenyl)-2-(4-methyl-1-piperidine) acetamide, which has less side effects, and evaluated its effect as local anesthesia. In the structural design of the target compound, lidocaine was modified through the bioisosteric principle, and the methyl group on the benzene ring in lidocaine was replaced by chlorine. This structural design improved the half-life and lipid solubility, and reduced the oxidation metabolism of the drug. The use of the cyclic analogue 4-methyl-piperidine instead of alkyl diethylamine generally did not change the pharmacokinetic effects such as lipophilicity and metabolism of the drug. The synthetic process shown in Scheme 3 was used to synthesize target compound 3. Initially, sodium bicarbonate (NaHCO3) was applied under the condition of acid and acetone as the solvent, and 2,6-dichloro (compound 1) and 2-chloro aniline acetyl chloride were used as the raw materials, which were reacted at 40 °C for 6 h to give the intermediate α-chlorine acetamide (compound 2). Finally, compound 2 and 4-methyl piperidine were reacted for 4 h at room temperature to synthesize target compound 3. The synthetic route was characterized by high yield (86.6%), simple operation and cheap reagents, thus reducing the synthesis cost. In the activity study, the formalin test was used to determine the safe activity of the compounds in the body of laboratory mice, which were used to evaluate the effect of local anesthesia. Two stages were used in the evaluation process. The first stage was acute pain response, and the second stage was the increase of primary afferent fiber input after tissue injury caused by chemical stimulation and local inflammatory reaction. The first stage of acute pain response began immediately after formalin management and rejection for more than 10 min. The second stage began about 15 min after formalin administration and lasted for about 1 h. A increased pain response in diabetic rats was observed with a lower dose of formalin than in the non-diabetic rats. Although 1% formalin had a higher harmful reaction to the non-diabetic rats, 0.5% formalin had no significant difference to the non-diabetic rats. This finding suggests that hyperalgesia in diabetic rats is caused by a sensitization process that changes both their peripheral and central levels. These results were consistent with previous observations using the same formalin concentration in the same model. The results showed that compound 3 has an anti-pain hypersensitivity effect because it could reduce the pain hypersensitivity behavior of diabetic rats. Although the mechanism of action of this compound is unknown, its mechanism may be similar to lidocaine due to its similar structure.
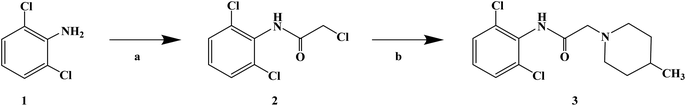 |
| | Scheme 3 Reagents and conditions: (a) 2-chloroacetyl chloride, NaHCO3, acetone, 40 °C, 6 h; and (b) 4-methylpiperidine, r.t., stirring, 4 h. | |
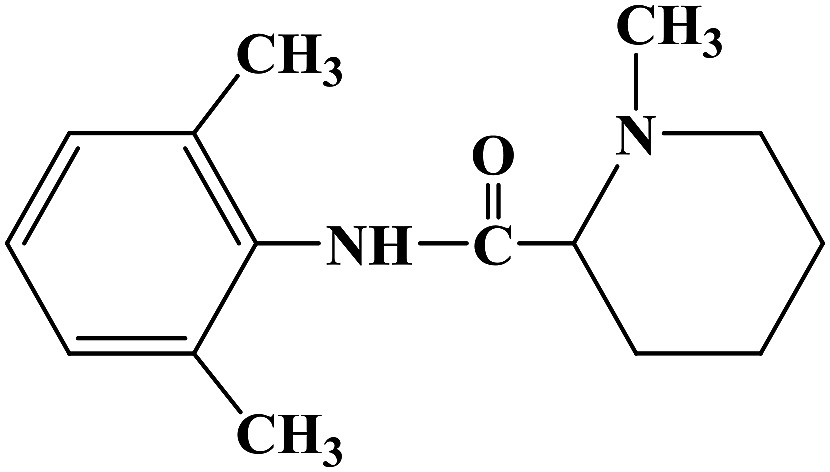
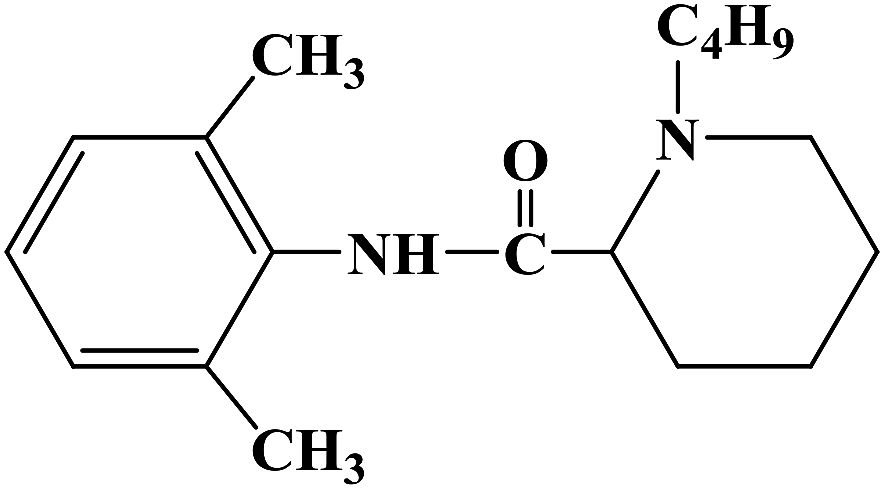
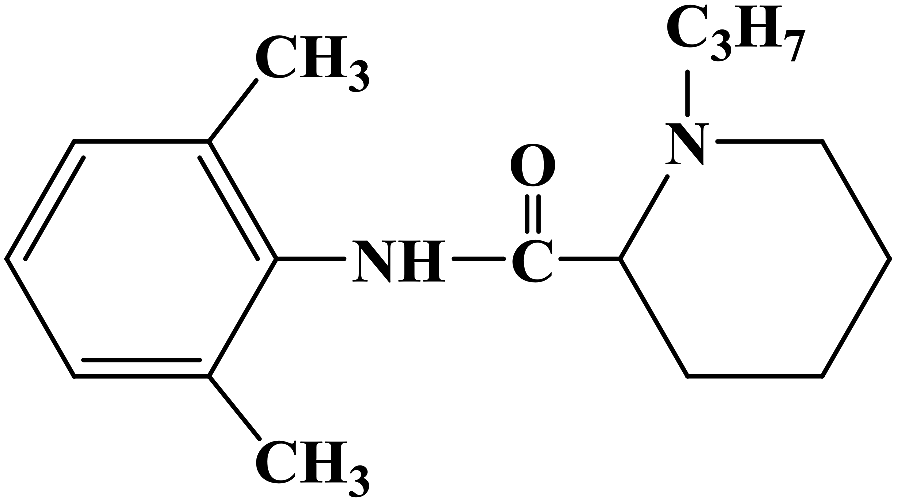
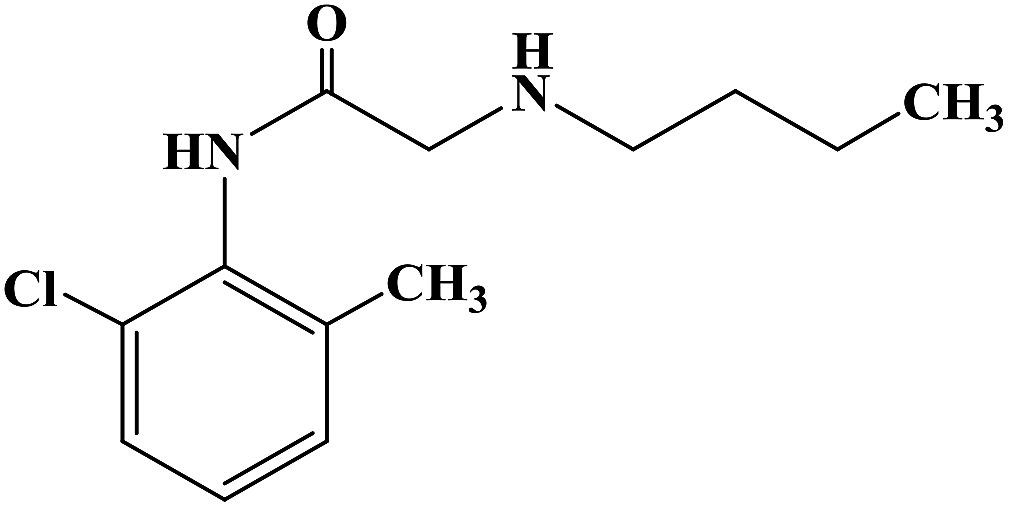
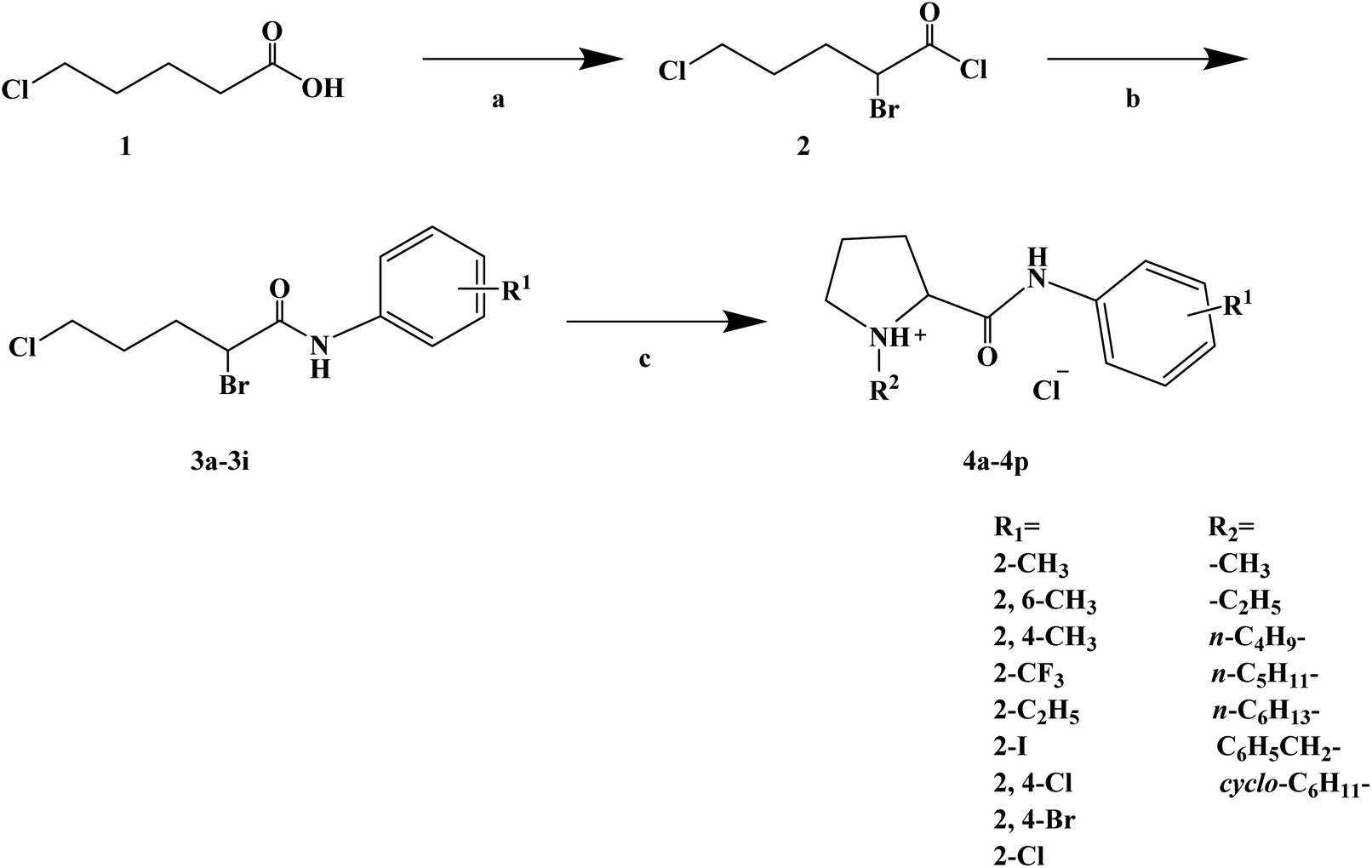
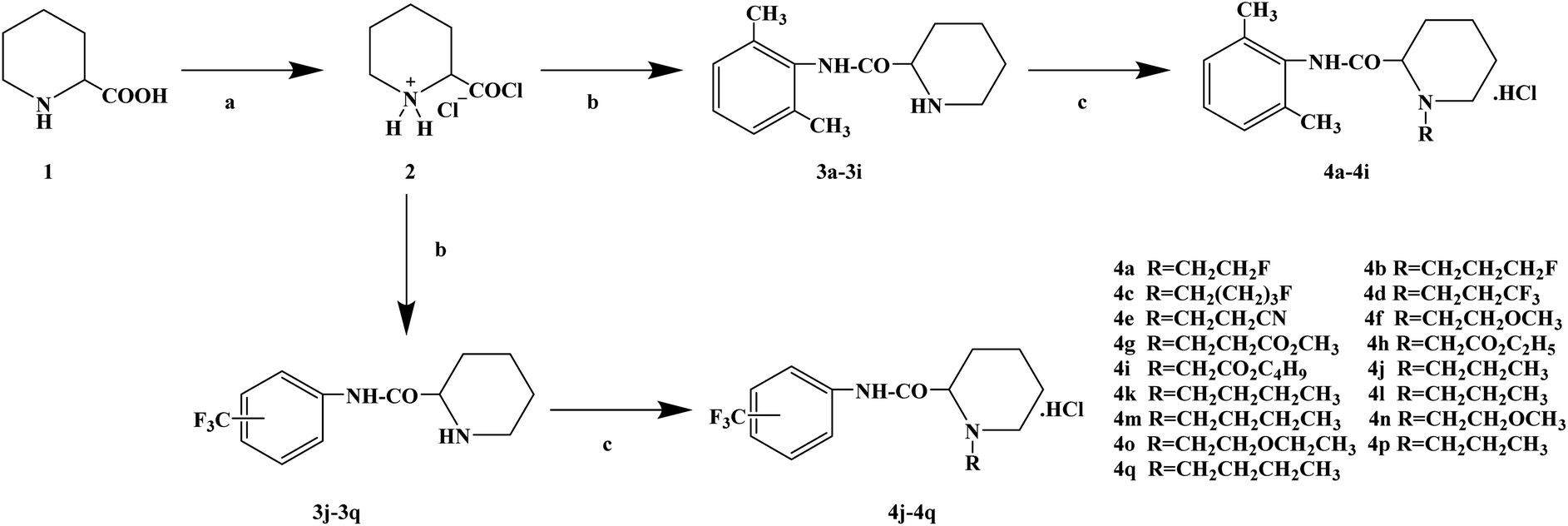
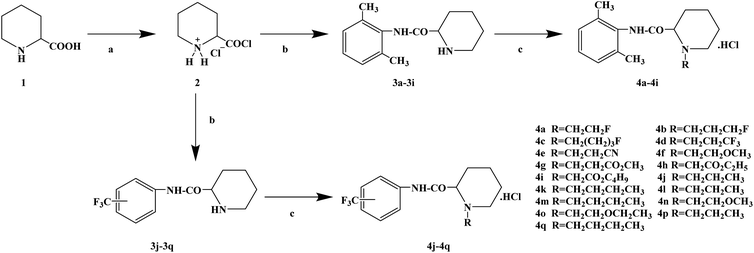
During the development of new effective local anesthetics, the N,N-dialkyl derivatives of glycine gradually evolved into N-alkyl substituted isoproline and proline. These new local anesthetics have the biological activity of arrhythmia in addition to the effect of local anesthesia. Studies have shown that the local anesthesia and antiarrhythmic action mechanism is the same, mainly by blocking voltage-gated sodium channels, calcium channels and potassium channels, resulting in membrane stability to prevent the initiation of action potential. In the process of drug development, the development of local anesthetics and antiarrhythmic drugs is usually studied and developed simultaneously. In 2013, Kalinin D. V. et al.65 reported the synthesis and biological activity of N-alkyl derivatives of proline aniline. In terms of synthesis, the designed synthetic route focused on two key biological activity substitutions of target compounds 4a–4p, and the substitutions of the tetrahydropyrrole ring and benzene ring (Scheme 4).The bromination of 5-chloropentanoic acid (compound 1) was accomplished by the action of bromine with phosphorus trichloride to produce 2-bromo-5-chloropentanoic acid. The chloropentanoic acid was then converted into the corresponding acyl chloride by SOCl2. Compound 2 was treated with anilines with different substituents under the condition of chloroform as the solvent to synthesize 2-bromo-5-chlorovalerate aryl amide (compound 3).The N-alkyl proline aryl amines underwent an intramolecular cyclization reaction after compound 3 was heated in toluene, which were further converted into the corresponding hydrochloride compounds 4a–4p under the condition of HCl. The synthetic route has mild reaction conditions, relatively simple operation, and the reagents used are cheap and easy to obtain. Simultaneously, the synthetic route does not use precious metal catalysts, and the total yield varies from 37% to 75%, but the total yield of most of the target compounds is not high, which needs to be optimized via the reaction conditions to improve the total yield, reduce the cost of synthesis, and provide favorable conditions for the realization of industrialization. In terms of biological activity, all the synthetic target compounds (4a–4p) were evaluated as local anesthesia in vitro and in vivo. The primary selection of the local anesthetic activity of the target compounds was to evaluate the biological activity of rabbit cornea by local anesthetic effect of a common surface anesthesia model. Further studies were conducted to evaluate the compounds with the best local anesthetic activity using a rat infiltration anesthesia model. The results of the structural–activity relationship (SAR) analysis showed that the substituents on the aromatic local anesthetics usually included one o-methyl group, sometimes two o-methyl groups or even p-methyl group. Among the synthesized compounds, the best biological activity was observed when the substituents on the aromatic ring were o-methyl. When the number of carbon atoms in alkyl chain R1 increased from methyl to ethyl, the surface anesthetic effect of the compounds was significantly reduced. Simultaneously, the number of substituents on the aromatic ring also affected the local anesthesia effect. With an increase in the number of methyl groups, the intensity and duration of the local anesthesia effect of the compound increased. The intensity and duration of the surface anesthetic effect of the compounds were significantly reduced by changing the substituent methyl group on the aromatic ring to trifluoromethyl or halogen. The N-alkyl substituents R2 in the pyrrolidone ring also play an important role in the surface local anesthetic activity. Accordingly, –CH3, –CH2CH3, n-C4H9, n-C5H11, n-C6H13, benzyl, and cyclohexyl were investigated. In the surface anesthesia experiments, when the substituents were methyl and ethyl, the compounds had no anesthetic effect. With an increase in the carbon chain length, the duration of the surface anesthesia was significantly prolonged, and the local anesthesia effect of the cyclohexyl substituted group was similar to that of the N-butyl substituted group. In local anesthesia tests in vivo, all the compounds worked quickly within 1–2 minutes of administration, blocking muscle contractions. The anesthesia time range of target compounds 4c, 4d, 4l and 4o was 35.0 ± 4.1 to 126.3 ± 24.3 min, which was similar to the control drugs. However, the total anesthesia time of compound 4c was shorter than that of the positive control substance (12.5 ± 2.9 min), and the complete prevention of stimulated muscle contraction was 102.5 ± 37.9 min and 58.8 ± 17.5 min, respectively. The local anesthesia time of compound 4d was longer, where the maximum block time was 20.0 ± 13.5 min, and the recovery was slow after anesthesia. Compounds 4l and 4o were basically the same in intensity and duration of surface local anesthesia, but compounds 4d, 4l and 4o showed obvious characteristics in the invasive anesthesia test. The duration of compound 4l anesthesia was 115.0 ± 12.2 min, while the duration of infiltration anesthesia induced by compound 4o was longer (126.3 ± 24.3 min). In general, compounds 4d, 4l and 4o were more active in inducing surface anesthesia than the positive controls. Meanwhile, the toxicity of compound 4o was significantly reduced, which make the compounds suitable for further research and development.
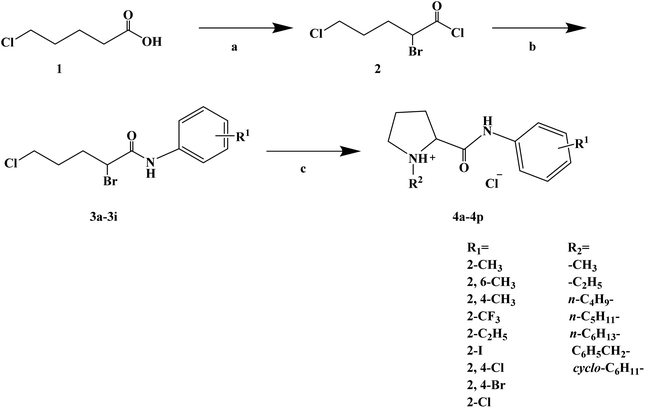 |
| | Scheme 4 Reagents and conditions: (a) (i) Br2,PCl3, 80 °C, 20 h; (ii) SOCl2, reflux, 1.5 h; (b) substituted anilines, CHCl3, Et3N, reflux, 1 h; and (c) (i) RNH2, KI, toluene, 6 h; (ii) HCl, CH3CH2OCH2CH3. | |
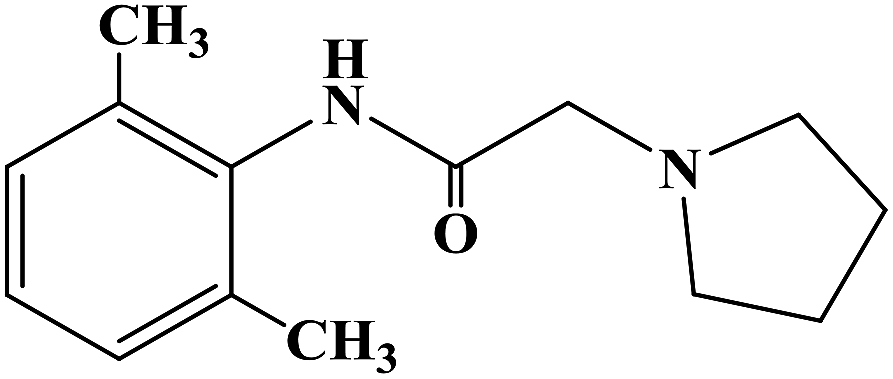
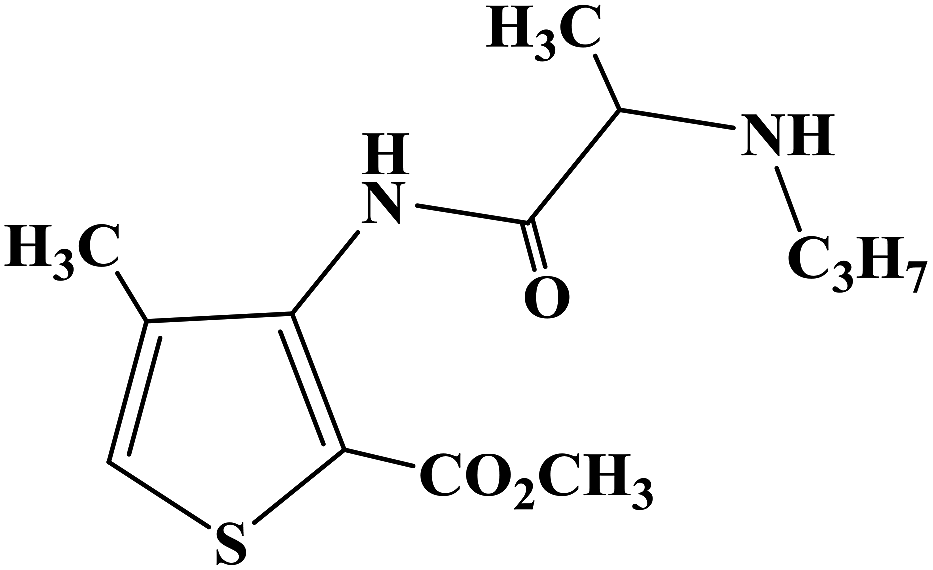
Heterocyclic compounds have the advantages of easy structural modification, structural diversity and good synthesis selectivity, and thus they are often used as preferred compounds in the research and development of new drugs. There are many types of heterocyclic compounds, among which those containing the imidazole structure are the most attractive to drug chemists. This is because compounds containing an imidazole structure have a wide range of medicinal value with different pharmacological effects, such as anti-malarial, anti-histamine, anti-bacterial, anti-inflammatory, anti-tuberculosis, analgesia, anti-insect drugs and anti-genic animal effects. In 2015, Yan R. et al.66 reported the β-cyclodextrin-propyl sulfonic acid (β-CD-PSA) catalysed one-pot synthesis of 1,2,4,5-tetrasubstituted imidazoles as local anesthetic drugs. They designed a total of 10 target compounds (compounds 5a–5j). They used R1 instead of a benzene ring and R2 instead of the imidazoles to get 1,2,4,5-tetrasubstituted imidazoles (compounds 5a–5j). Efficient synthetic methods for imidazole compounds have been widely studied. In the synthesis of 1,2,4,5-tetrasubstituted imidazoles compounds, usually with 1,2-diketone, hydroxy ketones or aldehydes ketomonoxime, the primary amine and ammonium acetate group are divided into four synthetic raw materials, with the use catalysts of HY zeolite, silicone/NaHSO4, BF3–SiO2, molecular iodine, HClO4–SiO2, InCl3·3H2O or potassium dodecatugstocobaltatetrihydrate (K5CoW12O40·3H2O) and functionalized silica (SiO2–Pr–SO3H). 1,2,4,5-Tetranomeric imidazole compounds are synthesized via a one-pot method with or without solvent. However, currently, the methods for synthesizing 1,2,4,5-tetrasubstituted imidazoles compounds have disadvantages such as long reaction time, low product yield, harsh reaction conditions, cumbersome processes and the use of toxic reagents. Yan R. et al. provided a green and economic synthetic method to obtain 1,2,4,5-tetrasubstituted imidazoles compounds, which used β-CD-PSA as a catalyst, as shown in the synthetic route in Scheme 5. In synthetic process, firstly the catalyst β-CD-PSA required for the reaction was synthesized under laboratory conditions, and then the conditions for the synthesis of the target compounds were optimized. The final selection for the one-pot synthetic method involved benzyl (compound 1), aniline or benzylamine (compound 2), aromatic aldehyde (compound 3) and ammonium acetate (compound 4) as raw materials, β-CD-PSA as the catalyst, reaction temperature of 100 °C under solvent-free conditions, and stirring for 15 min to 4 h, with the total yield of the final target compounds of 68–96%.The synthetic route has the advantages of simple operation, high total yield, no solvent and no pollution gas product, and it is environmentally friendly. Simultaneously, the catalyst β-CD-PSA used in the reaction process can be recycled and reused, and the recovery operation is simple and the recovery rate is high. In the study of the local anesthetic activity, they measured the local anesthetic activity of the target compounds by corneal anesthesia and mouse tail anesthesia. Firstly, two types of anesthesia models, surface anesthesia and infiltration anesthesia, were used to evaluate the local anesthesia activity of the target compounds in vitro. The results of local anesthetic activity in vitro showed that compounds 5g and 5i had a good local anesthetic effect (Table 2), while the other compounds had no significant local anesthetic effect. Among them, the local anesthetic effect of compound 5g was better than the positive control product lidocaine. From the analysis of the drug structural activity relationship (SAR), the presence of the benzylamine group has a significant effect on the local anesthetic effect of the target compounds, and aniline replaces benzylamine to eliminate the local anesthetic activity of the target compounds. Thus, the duration of the local anesthetic activity of compounds 5g, 5i and 5h was further determined by rat sciatic nerve block. The test results showed that the local anesthesia duration of compound 5g was similar to that of the positive control product lidocaine. In the study of local anesthetic activity in vivo, the acute toxicity and mouse therapeutic index test of compound 5g were investigated. The results showed that the acute toxicity of the compound 5g was less than that of the positive control product lidocaine. In terms of therapeutic index, that of compound 5g was significantly higher than that the positive control product lidocaine. The local anesthetic activities of the target compounds were screened via various methods and the results showed that these compounds are potential the local anesthetics. Combined with its effective synthetic method and obvious the local anesthetic activity, compound 5g may be a potential lead compound for further drug development.
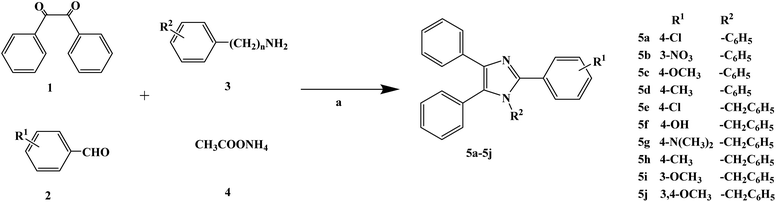 |
| | Scheme 5 Reagents and conditions: (a) solvent free, catalysed by β-CD-PSA, 100 °C, 15 min to 4 h. | |
Table 2 The local anesthetic activity of target compounds 5a–5j
| Compound |
Corneal anesthesia (%) |
Mouse tail anesthesia IC50 (mol L−1) |
| 5a |
20.3 ± 8.9 |
(11.3 ± 0.11) × 10−2 |
| 5b |
12.4 ± 9.3 |
(5.7 ± 0.42) × 10−2 |
| 5c |
15.5 ± 12.3 |
(6.5 ± 0.23) × 10−2 |
| 5d |
10.32 ± 0.12 |
(4.3 ± 0.54) × 10−2 |
| 5e |
21.65 ± 4.7 |
(3.4 ± 0.32) × 10−2 |
| 5f |
5.5 ± 3.5 |
(5.4 ± 0.14) × 10−2 |
| 5g |
121.23 ± 12.3 |
(1.6 ± 0.26) × 10−2 |
| 5h |
65.65 ± 5.8 |
(9.2 ± 0.16) × 10−2 |
| 5i |
95.6 ± 13.2 |
(7.4 ± 0.18) × 10−2 |
| 5j |
20.3 ± 10.3 |
(4.1 ± 0.45) × 10−2 |
| Lidocaine |
100 |
(2.1 ± 0.25) × 10−2 |
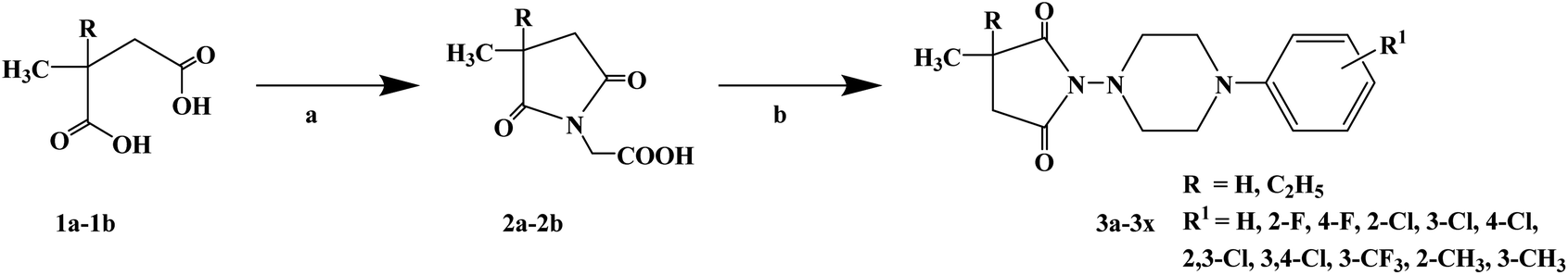
To determine the chemical structure characteristics of anticonvulsants, after decades of continuous research and numerous attempts, the basic chemical structure of anticonvulsants and nitrogen heterocycles containing an imine structure was finally determined, and it was found that there are usually carbonyl and phenyl or alkyl substituents in the structure of nitrogen heterocycles. In 2016, Jolanta O. et al.67 reported the design and synthesis of a series of 3-methyl-3-methyl-2,5-dioxo-pyrrolidin-1-yl-acetic acids and 3-ethyl-3-methyl-2,5-dioxo-pyrrolidin-1-yl-acetic acids as anticonvulsants. In the structural design, they introduced a methyl or ethyl and methyl substituent at the imine ring 3 site to evaluate the effect of the substituent on activity. These compounds were originally used as anticonvulsants, but some convulsants have analgesic effects in clinical practice. Therefore, some compounds have local anesthetic effects in subsequent studies. The synthetic process for the target compounds is shown in Scheme 6. Starting with 3-methyl succinic acid (compound 1a) and 3-ethyl-3-methyl succinic acid (compound 1b), the intermediates 3-methyl-3-methyl-succinic acid (compound 2a) and 3-ethyl-3-methyl-succinic acid (compound 2b) were synthesized by condensation reaction with glycine at 190–200 °C for 1.5 h. Compound 2a and compound 2b obtained from the first step were again reacted with arylpiperazine. Target compounds 3a–3x were obtained using dimethylformamide (DMF) as the solvent and N,N-carbonyldiimidazole (CID) as a dehydrator, with stirring at room temperature, giving yields ranging from 50% to 80%. This synthetic route has only a few reaction steps, the suitable solvent is cheap and easy to obtain, and the total yield is relatively high. However, the reaction temperature of the first step is relatively high, and a large amount of energy is required under high temperature conditions. Moreover, the reactor requirements are also relatively high, and the risk factor is relatively high. Local anesthetics block nerve conduction by altering the function of voltage-gated sodium channels and are used as adjunctive analgesics in the treatment of neuropathic pain. Compounds 3g, 3i, 3j, 3l, 3r and 3x were selected to test the effect of local anesthesia by the tail immersion test (Table 3). During the test, compounds 3j and 3r at a concentration of 2% significantly prolonged the response time of the animals to thermal stimulation (124% and 61%), while that of the positive control substance was 84%. Meanwhile, the binding ability of compounds 3g, 3i, 3j, 3l, 3r and 3x to sodium channels and calcium channels (Na+ and Ca2+ channels) was evaluated in vitro (Table 4). The test results showed that compound 3x had the best binding effect on Na+ and Ca2+ channels, with a binding capacity of 63.5 and 82.4 (positive control substances of 53.9 and 57.8), respectively. Thus, compound 3x may be a new local anesthetic, which needs to be further studied.
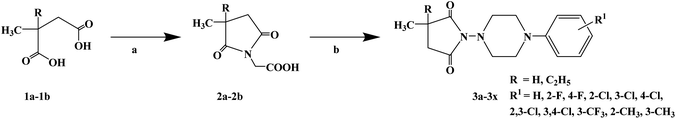 |
| | Scheme 6 Reagents and conditions: (a) glycine, 190–200 °C, 1.5 h; (b) arylpiperazine, CDI, DMF, r.t., 24 h. | |
Table 3 The local anesthetic activity in the tail immersion test
| Compound |
Concentration (%) |
Reaction time to painful stimulus (s) |
Prolongation of the time reaction (%) |
| 3g |
2 |
5.41 ± 0.53 |
12 |
| 3i |
2 |
5.66 ± 0.60 |
17 |
| 3j |
2 |
10.80 ± 1.74 |
124 |
| 3l |
2 |
5.53 ± 1.06 |
14 |
| 3r |
2 |
7.79 ± 0.95 |
61 |
| 3x |
2 |
7.06 ± 0.76 |
46 |
| Phenytoin |
2 |
8.90 ± 0.80 |
84 |
Table 4 In vitro binding assay results at a concentration 100 μM
| Compound |
Inhibition of control specific binding (%) |
| Voltage-sensitive Na+ channels |
Voltage-sensitive L-type Ca2+ channels |
| 3g |
36.6 |
13.5 |
| 3i |
28.5 |
17.2 |
| 3j |
20.9 |
4.2 |
| 3l |
33.4 |
71.0 |
| 3r |
32.5 |
30.8 |
| 3x |
63.5 |
82.4 |
| Phenytoin |
53.9 |
57.8 |
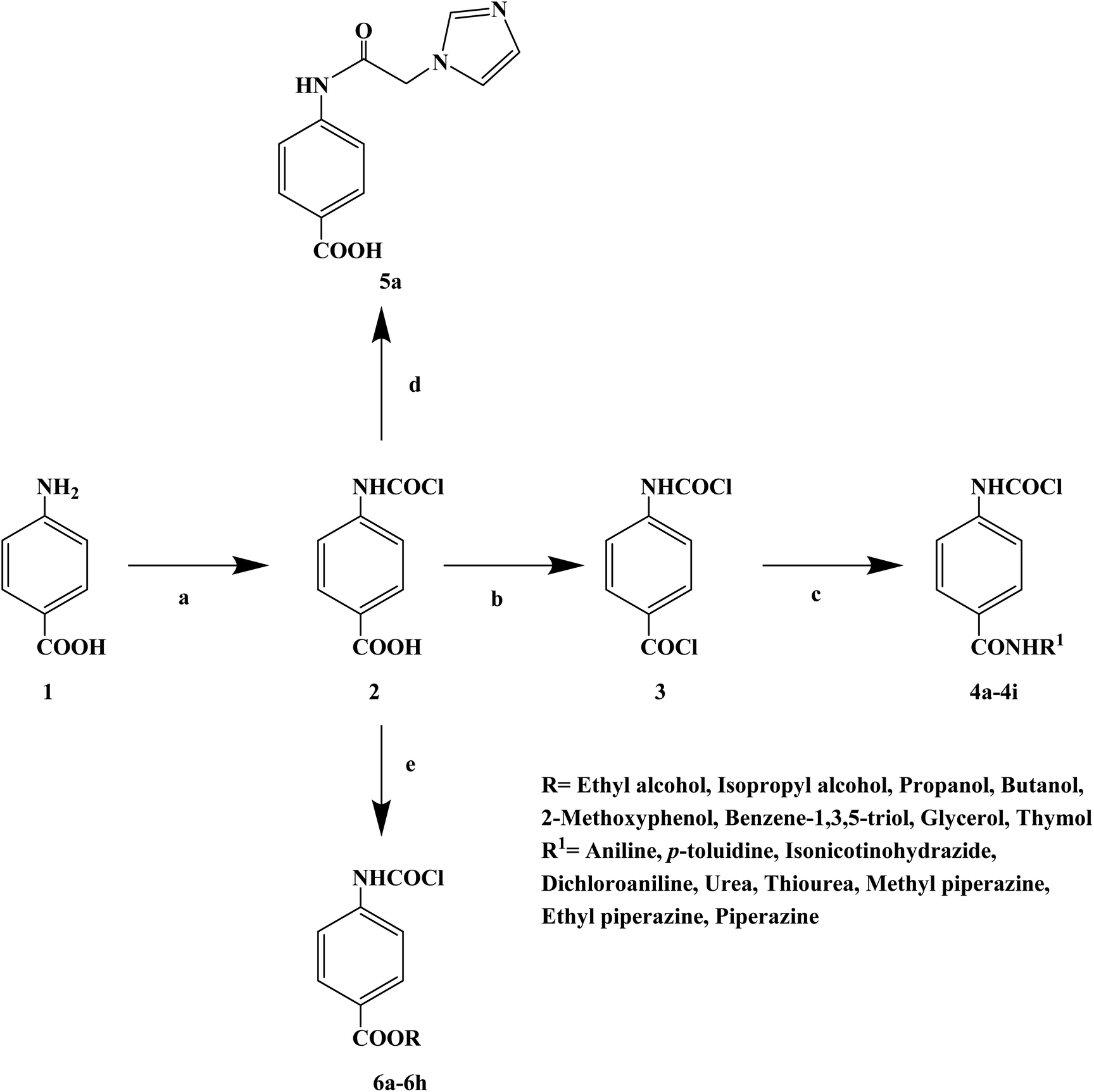
Local anesthetics can cause a reversible loss of sensation in a specific area of the body and control acute and chronic pain during surgery. However, the use of local anesthetics in medical practice also has serious side effects, leading to the need for safe and effective local anesthetics. The literature shows that benzimidazole, amide, benzotriazole, acetamide and other compounds can effectively inhibit sodium (Na+) channels and have obvious local anesthesia and antiarrhythmic activity. The chloroacetamido moiety also exists in many compounds and plays an important role in biological activities. In 2017, Ashish A. et al.68 the reported a series of 4-(2-chloroacetamido) benzoic acid derivatives, including 9 amide compounds, 1 imidazole compound and 8 ester compounds derivative (Scheme 7). A total of 19 target compounds were obtained (4a–4i, 5a, and 6a–h), and their local anesthetic activities were further studied. In the process of structurally designing the target compounds, they investigated relevant literature and found that many commercial local anesthetics contained the 4-aminobenzoic acid structure, in which the presence of an ester or amide bond is a key feature. Thus, they also chose 4-aminobenzoic acid as the parent structure. In the structural modification of the parent group, alkyl groups with strong ionization properties were replaced by amides, and carbonyl groups were placed next to the amides to enable them to enter the active pocket. A chloromethamphetamine was substituted on the carbonyl carbon of the amide. The presence of a chlorine atom will increase the ionization property of the amide through the induction effect, thus increasing its solubility in water. In the process of synthesizing the target compounds, the 4-aminobenzoic acid was synthesized by Fischer's esterification, Schotten–Baumann and substitution reactions. The 4-aminobenzoic acid (compound 1) and chloroacetyl chloride were treated with 35% sodium hydroxide solution for 10 min at a reaction temperature of 4–10 °C. The hydrochloric acid was then acidified to obtain the first intermediate 4-(2-chloroacetamido) benzoic acid (compound 2). Then compound 2 and thionyl chloride were continuously stirred in pyridine to form a second intermediate 4-(2-chloroacetamido)benzoyl chloride (compound 3). Then the corresponding amine dissolved in pyridine was added and vigorously mixed with compound 3, and after completion of the reaction with sulfuric acid treatment, 4-(2-chloroacetamido)-N-substituted benzamide derivatives were obtained (compounds 4a–4i). Compound 2 and imidazole were treated in dimethyl sulfoxide (DMSO). The mixture was heated at 60 °C for 5 h and substituted for chloride ions in the presence of sodium carbonate to form imidazole derivatives (compound 5a). Compound 1 and the corresponding alcohols were heated in the presence of hydrochloric or sulfuric acid and refluxed for 4–6 h to obtain the corresponding esters (compounds 6a–6h). The synthetic route has the advantages of short reaction time, cheap reagents and mild reaction conditions. However, the total yield of some target compounds in this synthetic route was low (13%), which needs to be optimized to improve the total yield. The activity of the invasive local anesthesia was determined by Bianchi's method. The results of infiltration local anesthesia showed that compounds 4b, 5a, 6a, 6b and 6d showed good local anesthetic activity, which was close to the local anesthetic effect of lidocaine hydrochloride. Analysis of the structure–activity relationship (SAR) results showed that compounds 4a–4i were amide derivatives of aromatic and aliphatic amines such as aniline, p-toluidine, dichloroaniline, urea, thiourea, methyl piperazine, ethyl piperazine and piperazine. The amides of aniline compound 4a and p-toluidine compound 4b showed significant local anesthetic effects. The other compounds 4c–4i exhibited a very low local anesthetic effect because both amide nitrogens were ionizable and easily dissolve in water, thus losing the local anesthetic effect. The imidazole derivative compound 5a exhibited significant local anesthetic activity, which has all the basic chemical structures of local anesthetics and its activity was 81%. Compounds 6a–6d were ester derivatives of ethyl, isopropyl, propyl and butanol, respectively, with a single ionizing group and their activity was between 60–80%. Compounds 6e–6h are phenolic ester compounds composed of 2-methoxyphenol, phloroglucinol, glycerol and thymol, respectively, with relatively low local anesthetic activity. In vivo, the rat sciatic nerve block method was used to further study the infiltration local anesthesia activities of compounds 4b, 5a, 6a, 6b and 6d (Table 5). The experimental results showed that compounds 4b, 5a, 6a, 6b and 6d were effective, among which compound 5a showed obvious infiltration local anesthetic activity. The effect of compound 5a on blocking the sciatic nerve in rats was better than that of lidocaine (53 min), and that of lidocaine was 27 min. The activity of compound 4b was lower than that of the control (11 min). In general, compound 5a is a new local anesthetic, which has the potential for further research and development.
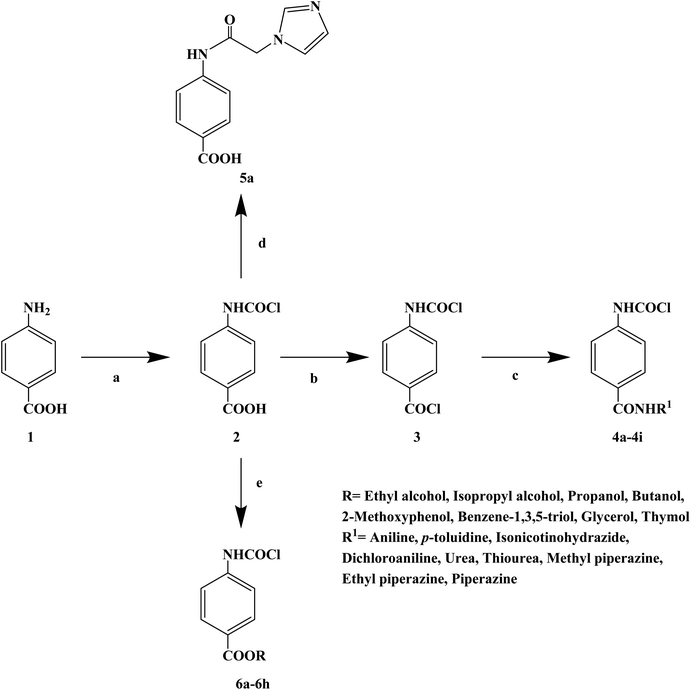 |
| | Scheme 7 Reagents and conditions: (a) chloroacetyl chloride, HCl, 4–10 °C, stirring, 10 min; (b) thionyl chloride, pyridine, 0–4 °C, stirring; (c) corresponding amide derivatives, conc. H2SO4, reflux; (d) imidazole, AlCl3, reflux; and (e) corresponding alcoholic derivative, conc. H2SO4, reflux. | |
Table 5 Infiltration local anesthetic activity (n = 6)
| Compound |
Enduring time at 15 min (s) |
Enduring time at 30 min (s) |
Enduring time at 45 min (s) |
Mean enduring time (s) |
% |
| 4b |
82.5 ± 1.66 |
75 ± 1.89 |
75 ± 2.08 |
77.5 |
61 |
| 5a |
109 ± 1.41 |
102 ± 1.18 |
94.5 ± 1.26 |
101.8 |
81.44 |
| 6a |
108.5 ± 0.04 |
100.5 ± 0.19 |
100.5 ± 0.17 |
103.2 |
81.87 |
| 6b |
90 ± 0.56 |
71.5 ± 0.81 |
71.5 ± 0.77 |
77.7 |
61.76 |
| 6d |
103 ± 1.47 |
96.5 ± 1.84 |
96.5 ± 1.66 |
98.7 |
78.45 |
| Lignocaine |
143.5 ± 2.11 |
117 ± 2.70 |
117 ± 2.91 |
125.8 |
100 |
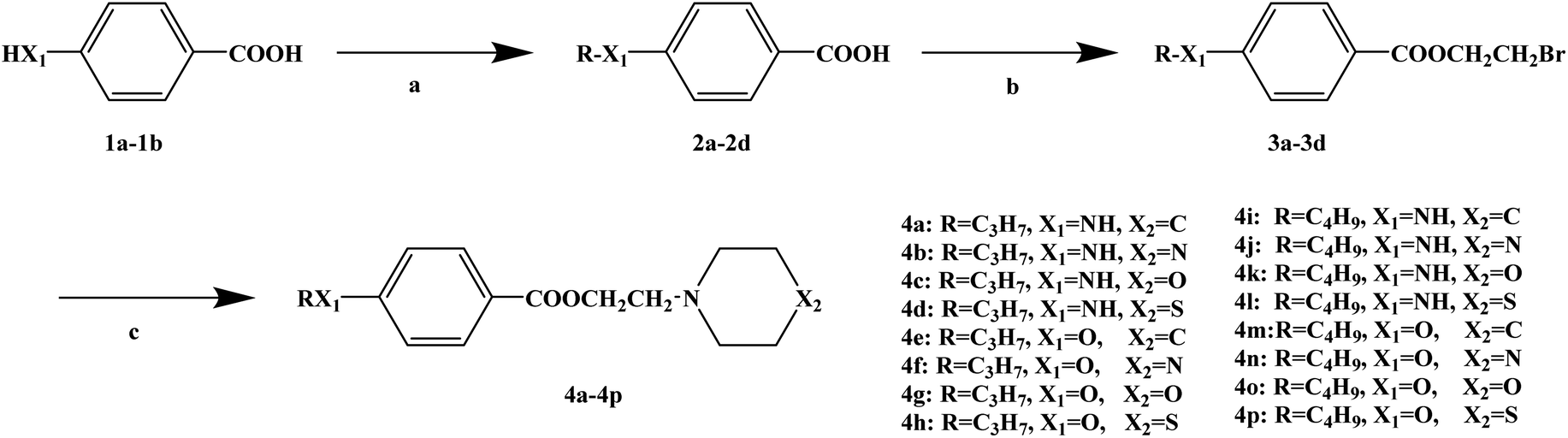
Ropivacaine is the S-enantiomer of an N-alkyl pipecoloxylidine derivative, which is the first local anesthetic with chiral activity, and is widely used in clinical infiltration anesthesia, conduction anesthesia and epidural anesthesia. It has a long of local anesthesia and analgesic effect. However, ropivacaine also has serious safety risks in clinical practice. When the concentration of ropivacaine in human blood is too high, it may cause toxicity to the cardiovascular and central nervous system, and even cause allergic reactions in some patients. Thus far, the mechanism of the effect of ropivacaine on local anesthesia is not clear. Ropivacaine is a multitarget drug that acts on the gamma-aminobutyric acid a receptor (GABAA-R) and N-methyl-D-aspartate acid receptor (NDMA-R). Sodium (Na+) channels are a key target of local anesthetics and these two receptors regulate sodium channels. Previous studies on the structural modification of ropivacaine mainly focused on the substitution of –CH3 on the phenyl group or the substitution of –CH2CH2CH3 on piperidine with different alkyl groups. In 2017, Wen L. et al.69 reported the design and synthesis of ropivacaine analogues for local anesthesia. In the process of structural design, they used ropivacaine as the lead compound to design two series of compounds, 4a–4q (17 new substituted imines). In the first series of compounds, 4a–4i, different substituents were selected to replace –CH2CH2CH3 on piperidine. In the second series of compounds, 4j–4q, the methyl groups were replaced by –CF3 at the o-positions, m-positions and p-positions. Meanwhile, the –CH2CH2CH3 on piperidine ring was also substituted and modified. The process for the synthesis of the target compounds is shown in Scheme 8. The synthetic route takes piperic acid (compound 1) as the starting material, hydrochloric acid and sulfoxide chloride as additives, and toluene as the reaction solvent to convert compound 1 into acyl chloride salt (compound 2). Compound 2 was then treated with substituted aniline and reacted at 58 °C for 5 h to form compounds 3a–3i and 3j–3q. Finally, bromoalkyl and hydrochloric acid were used to treat compounds 3a–3i and 3j–3q. Potassium carbonate (K2CO3) was used as an acid dressing agent and dimethylformamide (DMF) as the reaction solvent in N-alkylation reaction. The N-alkylation reaction lasted 10 h at 80 °C, and the salt reaction lasted 5 min at room temperature to obtain the final target compounds 4a–4q. The total yield of the target compounds ranged from 17.5% to 87.7%. The synthetic route has the advantages of mild reaction conditions, cheap reagents and simple operation. However, using this synthetic route, the total yield of some products is too low, and too low yield will bring great problems to the synthesis cost, which needs to be further optimized in follow-up work. In the evaluation of the local anesthesia effect, sciatic nerve block activity, infiltration anesthesia activity, corneal anesthesia activity and spinal cord anesthesia activity were used as evaluation indexes. Ropivacaine was used as a positive control substance to test the local anesthesia activity in vitro. Firstly, the local anesthesia effect of all the target compounds 4a–4q was screened by a sciatic nerve block test in toads in vitro (Table 6). The preliminary screening results in vitro showed that these compounds increased the blocking effect of the sciatic nerve on electrical stimulation, with ED50 values ranging from 0.012 to 0.64 (positive control for ropivacaine was 0.013), with the highest activity shown by compound 4b. In terms of latent period, that of target compounds 4a–4q ranged from 27.7 to 59.4 min. Based on the results of the preliminary in vitro screening, compounds 4a, 4b, 4c, 4j and 4l were selected to test the efficacy of invasive anesthesia in guinea pigs. The results of the infiltration anesthesia test showed that the local anesthetic effect of compounds 4c and 4l was similar to that of the positive control ropivacaine, and the local anesthetic activity of other compounds was lower than that of the positive control. Furthermore, compounds 4a, 4b, 4c, 4j and 4l were used to test the local surface anesthesia effect of these compounds (Table 7). The results of the surface anesthesia test showed that compound 4l had a similar local anesthetic effect as the positive control ropivacaine, while the effect of the other compounds was poor in comparison with the positive control. Finally, compounds 4a, 4b, 4c, 4j and 4l were tested for spinal anesthesia in order to further study their local surface anesthesia effect. The experimental results showed that the ED50 produced by compounds 4l and 4b was 5.02 and 7.87, respectively, while the effects of compounds 4a, 4c and 4j were poor. The evaluation of local anesthesia in vitro found that compound 4l had the best activity, and thus molecular docking of compound 4l and ropivacaine was conducted to further study its local anesthesia mechanism. The molecular docking results showed that compound 4l interacts with receptor proteins of VGSC, GABAA-R and NDMA-R, which may help optimize and predict the activity of these ropivacaine analogues as potential local anesthetics.
 |
| | Scheme 8 Reagents and conditions: (a) (i) HCl,PhCH3, r.t., 1 h; (ii) PhCH3, SOCl2, 55 °C, 1 h; (b) substituted anilines, 58 °C, 5 h; and (c) (i) RBr, K2CO3, DMF, 80 °C, 10 h; (ii) HCl, r.t., 5 min. | |
Table 6 The sciatic nerve block activities of compounds 4a–4q
| Compound |
ED50 (mg mL−1) |
Latent period |
| 4a |
0.014 |
35.1 |
| 4b |
0.012 |
28.9 |
| 4c |
0.021 |
35.1 |
| 4d |
0.021 |
34.6 |
| 4e |
0.023 |
37.8 |
| 4f |
0.021 |
37.5 |
| 4g |
0.20 |
38.6 |
| 4h |
0.64 |
39.3 |
| 4i |
0.42 |
59.4 |
| 4j |
0.026 |
32.8 |
| 4k |
0.027 |
28.6 |
| 4l |
0.021 |
33.7 |
| 4m |
0.022 |
32.8 |
| 4n |
0.026 |
29.0 |
| 4o |
0.046 |
27.7 |
| 4p |
0.32 |
28.9 |
| 4q |
0.37 |
34.8 |
| Ropivacaine |
0.013 |
35.5 |
Table 7 The infiltration anesthesia activities, corneal anesthesia activities and spinal anesthesia activities of selected compounds
| Compound |
ED50 (mg mL−1) |
| Infiltration anesthesia |
Corneal anesthesia |
Spinal anesthesia |
| 4q |
1.06 |
5.68 |
19.11 |
| 4b |
3.02 |
2.64 |
7.87 |
| 4c |
0.88 |
5.42 |
29.30 |
| 4j |
4.02 |
4.25 |
34.56 |
| 4l |
0.66 |
0.87 |
5.02 |
| Ropivacaine |
0.61 |
0.85 |
4.27 |
Most ester anesthetics produce p-aminobenzoate (PABA) metabolically after use, which among the PABA and sulfa drugs have anaphylaxis and antagonistic effects. However, amide local anesthetics rarely cause anaphylaxis after use, but these drugs have a certain relationship with cardiac toxicity. Hydroxamic acid is a biologically active compound, which is a potential source in future drug development. In 2019, Ekhlas S. et al.70 reported for the first time that benzohydroxamic acid with local anesthetic activity was converted into 6 benzohydroxamic acid analogues 3a–3f from p-substituted ethyl benzoate as a raw material (Scheme 9). The synthetic strategy was to react substituted benzoate with hydroxylamine hydrochloride in equimolar amount, and then treat intermediate products 2a–2f with potassium hydroxide. The intermediate products were in the form of potassium salt, and target compounds 3a–3f were finally acidified with acetic acid. The synthetic route only takes two steps to obtain the target compounds (3a–3f), which is simple and easy to operate, and the reagents used are cheap and easy to obtain, thus laying a solid foundation for future industrialization. The total yield of all the analogues was medium, ranging from 41% to 75%, and the total yield was improved by optimizing the reaction conditions. In the evaluation of local anesthesia activity, the frog foot withdrawal reflex method was used to test the activity of the benzohydroxamic acid analogues, and benzocaine was used as the positive control. The local anesthetic activity tests were conducted with 5% dimethyl sulfoxide (DMSO) and 0.65% sodium hydroxide (NaOH) solutions, respectively. Three different concentrations of 40 μg mL−1, 100 μg mL−1 and 200 μg mL−1 were selected for each test regimen. In 5% DMSO, most of the six benzohydroxamic acid analogues 3a–3f showed a good local anesthesia effect. In particular, compounds 3d and 3e exhibited basically the same local anesthetic effect as the positive control product benzocaine at the same dose level. The drug structure–activity relationship (SAR) studies suggested that the better local anesthesia effect of compounds 3d and 3e was due to the fact that both methyl (–CH3) and methoxy (–OCH3) groups was located in the contra-apoplectic region of the strong electron donor. The presence of electrically active substituents in the adjacent or opposite position may increase the anesthetic effect of local anesthetics. When the local anesthetic activity was tested in 0.65% NaOH aqueous solution (sodium salt), compounds 3d and 3e also had a good local anesthetic effect when used as a sodium salt (0.65% NaOH). Among the six benzohydroxamic acid analogues 3a–3f, compounds 3d and 3e showed a good local anesthesia effect, which was comparable with that of the positive control product benzocaine. These compounds have the advantages of good thermal stability, not easily hydrolyzed in water, no allergic phenomenon due to the non-release of PABA, and no antagonistic effect of clinical drugs. Thus, compounds 3d and 3e can be used as new local anesthetics for further research and development.
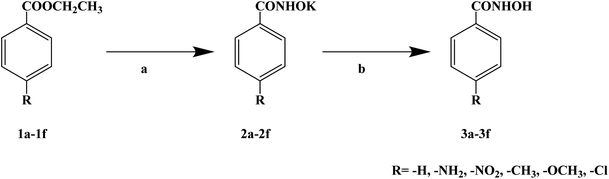 |
| | Scheme 9 Reagents and conditions: (a) NH2OH·HCl, KOH, CH3OH, ice bath, 5 min; and (b) CH3COOH, stirring. | |
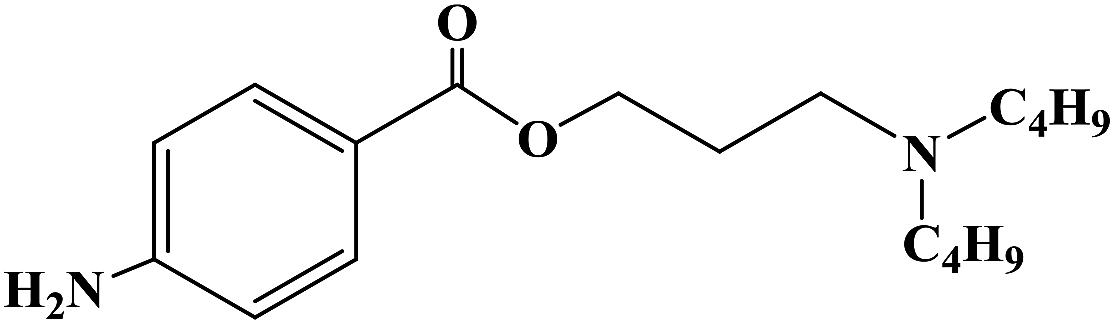
The action mechanism of local anesthetics is that the drug binds to the receptors of sodium (Na+) channels on the nerve membrane, preventing sodium ions from crossing the ion channels and blocking the conduction of nerve impulses to achieve the purpose of local anesthesia. Currently, there are many types of local anesthetic drugs used in clinic, such as tetracaine, dolicaine, dacronine and pramocaine. However, these local anesthetic drugs have many shortcomings, such as the toxicity of tetracaine is relatively high, and the drug dose is too prone to central nervous system and cardiovascular toxicity. However, procaine has some disadvantages such as short duration of anesthesia. It is expected that local anesthetics have the characteristics of fast acting time, long lasting acting time and low toxic and side effects. Huiying et al.71 reported that a series of novel benzoate compounds with local anesthetic effects were designed and synthesized with tetracaine and pramocaine as lead compounds. In the structural design of the target compounds, tetracaine and pramocaine were combined into a new structure by using the principle of splitting, and the structure was further optimized based on this new structure. A series of target compounds were obtained by optimizing the molecular structure after splicing based on the principle of bioisostere formation and modification with alkyl groups. From the structure activity relationship (SAR) of the drug, the designed target compounds also have three basic components, including a lipophilic section, a link section and a hydrophilic section. The synthetic route for the target compounds is shown in Scheme 10. The first alkylation step used 4-aminobenzoic acid (1a) or 4-hydroxybenzoic acid (1b) as the starting material, and R–Cl in dimethylformamide (DMF) to arrange it. In alkylation step, sodium carbonate anhydrous (Na2CO3) was used as the deacid reagent and dimethylaminopyridine (DMAP) as the catalyst, and the reflux reaction was completed in 12 h to obtain compounds 2a–2d. The esterification step used compounds 2a–2d and 2-bromoethanol in tetrahydrofuran (THF), and compounds 3a–3d were obtained. The esterification step used N,N′-carbonyldiimidazole (CDI) as the dehydrant, and the reflux reaction was completed in 8 h. Also, the second alkylation step used compounds 3a–3d and six-membered heterocyclic nitrogen in toluene, and compounds 4a–4p were obtained. In this step, potassium carbonate anhydrous (K2CO3) was used as the deacid reagent, and the reflux reaction was completed in 10 h. The synthetic route has a high total yield (76.3–90.4%), mild reaction conditions, and simple and safe operation. However, in the second step of this route, CDI was used as the dehydrating agent, and the price of CDI is relatively high, which increases the synthesis cost and is not conducive to the development of industrialization. Therefore, the selection of dehydrating agent needs to be improved due to the preferential treatment of subsequent process conditions. In the study of local anesthetic activity, rabbit corneal reflection was used to study the surface anesthetic effect of the target compound activity screening in vitro, and based on this, further mice abdomen acupuncture tests to study the target compounds infiltrating anesthesia effect, and with the rat tail nerve block test to study the effect of nerve block anesthesia using the target compounds. The studies on local anesthesia activity showed that these compounds had a good local anesthesia effect. From the analysis of the drug structure–activity relationship (SAR), it could be seen that substituents on benzene ring have a great influence on the local anesthesia effect. With an increase in substituent carbon chain length, the anesthetic effect time was prolonged. In the screening of the local anesthesia effect in vitro, the target compounds 4d, 4g, 4j, 4k, 4n and 4o showed a good surface anesthesia effect. In the further infiltration anesthesia test, the selected target compounds also showed good activity. The action time of compounds 4n and 4o was 4.5 h, which was better than the 2 h of the positive control substances tetracaine and pramocaine. When performing block anesthesia on compounds 4d, 4g, 4j, 4k, 4n and 4o, the experimental results showed that these compounds all showed the characteristics of quick analgesic effect and quick analgesic effect (Table 8). Among them, the analgesic initiation time of compound 4n was 6.4 ± 0.2 min, and the duration analgesic duration was 293.5 ± 3.4 min, which were better than that of the positive control. In addition, acute toxicity was evaluated by orally administering compounds 4d, 4g, 4j, 4k, 4n and 4o to mice (Table 4). The results of the acute toxicity test showed that the LD50 of compounds 4k, 4n and 4o was higher, among which compound 4n was 976.5 ± 4.5 mg kg−1. Furthermore, in the study on the mechanism of action of local anesthetics of the target compounds, they found that the target compounds enter sodium channels and bind to the receptor, thereby affecting the passage of sodium ions to achieve the effect of local anesthetics.
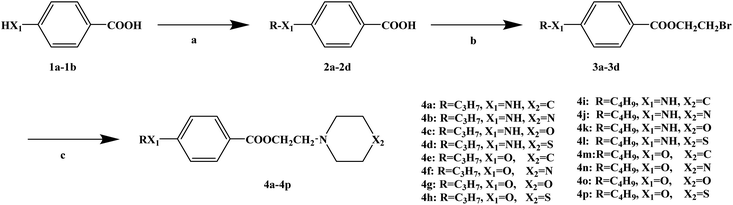 |
| | Scheme 10 Reagents and conditions: (a) R–Cl, Na2CO3, DMAP, reflux, 12 h; (b) 2-bromoethanol, CDI, THF, reflux, 8 h; and (c) six-membered heterocyclic nitrogen, K2CO3, toluene, reflux, 10 h. | |
Table 8 The target compounds of block anesthesia and acute toxicity test
| Compound |
Onset of analgesia (min) |
Duration of analgesia (min) |
LD50 (mg kg−1) |
| 4d |
8.8 ± 0.4 |
230.7 ± 3.2 |
543.7 ± 2.3 |
| 4g |
8.3 ± 0.4 |
254.6 ± 3.6 |
854.6 ± 3.7 |
| 4j |
12.5 ± 0.7 |
221,8 ± 3.0 |
735.1 ± 3.2 |
| 4k |
7.2 ± 0.3 |
279.9 ± 3.2 |
907.2 ± 4.3 |
| 4n |
6.4 ± 0.2 |
293.5 ± 3.4 |
976.5 ± 4.5 |
| 4o |
6.6 ± 0.2 |
286.4 ± 3.3 |
928.9 ± 4.9 |
| Tetracaine |
12.3 ± 0.9 |
172.3 ± 2.6 |
435.4 ± 3.3 |
| Pramocaine |
10.4 ± 0.8 |
166.5 ± 2.3 |
472.1 ± 3.6 |
In general, these newly designed local anesthetics include aromatic acid esters, amides, and imidazoles, which were mainly concentrated in amides (Table 9). Most of these target compounds were structurally modified based on commercial local anesthetics, which are traditional commercial local anesthetics such as lidocaine, ropivacaine and dyclonine. Imidazoles are novel local anesthetics used in local anesthesia. In terms of the synthesis of these compounds, cheap and easy starting materials are basically used, the conventional chemical synthetic method is used, and the ideal total yield is obtained, where the total yield of some target compounds can reach more than 90%. Simultaneously, the total yield of some target compounds is low, which needs to be optimized for the subsequent synthetic process to lay a solid foundation for the future industrialization. In terms of local anesthetic activity, most of these target compounds show good local anesthetic activity, and the local anesthetic effect of most of the target compounds is similar or better than the positive control substance. All of these compounds have potential for further research and development and are waiting to become the next generation of local anesthetics. However, there are still many follow-up studies, and most of the local anesthetic effects of these target compounds are still limited to in vitro screening or simple activity studies in vivo. Thus, there is still a long way to go before they become local anesthetics on the market. This field needs continuous efforts, and it is hoped that these compounds can become a new local anesthetics and serve human health.
Table 9 Synthesis and bioactivity summary of the target compounds
| Schemes |
Structure type |
Yields |
Biological activity |
Research phase |
References |
| 1 |
Amides |
45–60% |
Similar to lidocaine |
In vitro |
62 |
| 2 |
Aromatic acid esters |
97–99% |
Similar to lidocaine |
In vitro |
63 |
| 3 |
Amides |
86.6% |
Similar to lidocaine |
In vivo |
64 |
| 4 |
Aromatic acid esters |
37–75% |
The anesthesia time was 126.3 ± 24.3 min and infiltration anesthesia time was 126.3 ± 24.3 min |
In vitro and in vivo |
65 |
| 5 |
Imidazoles |
68–96% |
In vitro the mouse tail anesthesia IC50 was (1.6 ± 0.26) × 10−2 mol L−1 |
In vitro and in vivo |
66 |
| 6 |
Amides |
50–80% |
Better than that of lidocaine |
In vitro |
67 |
| 7 |
Amides |
13–79% |
In vitro, the response time of animals to thermal stimulation 61% to 124% |
In vitro and in vivo |
68 |
| 8 |
Amides |
17.5–87.7% |
In vitro ED50 values ranging from 0.012 to 0.64 and in vivo ED50 was 5.02 and 7.87 |
In vitro and in vivo |
69 |
| 9 |
Amides |
41–75% |
It's similar to benzocaine |
In vitro |
70 |
| 10 |
Aromatic acid esters |
76.3–90.4% |
The analgesic initiation time was 6.4 ± 0.2 min and the duration analgesic duration was 293.5 ± 3.4 min |
In vitro and in vivo |
71 |
3. Conclusions and perspective
Local anesthetics act on nerve endings or around nerve trunks, reversibly blocking the generation and conduction of sensory nerve impulses, and temporarily eliminating local sensation under the condition of consciousness, which are convenient for local surgical operation and treatment. Herein, we mainly reviewed the research progress of local anesthetics and discussed the important aspects of design, synthesis and biological activity of various new compounds. These target compounds are mainly concentrated in amides, which basically modify the structure of existing local anesthetics to obtain local anesthetics with better biological activity. However, imidazoles are novel compounds that have local anesthesia activity. In the synthetic process, these target compounds are generally synthesized by conventional reagents and conventional methods, and the final total yield is relatively ideal. In terms of local anesthetic activity of these target compounds, most of them show good activity in vitro or in vivo, and some of them have the same or better local anesthetic activity than the positive control products. However, these target compounds also have many shortcomings, which need to be continuously optimized in the follow-up work. In terms of the design of target compounds, the target compounds are mainly amide compounds, and there are few target compounds with new structures. Thus, more novel compounds with new structures should be designed in the future, and the diversity of compound structure should the focus. In terms of the synthesis of target compounds, the total yield of some compounds is low, which will increase the synthesis cost and hinder the development of industrialization. In terms of the activity of local anesthesia, some compounds are still in the stage of in vitro screening, and there is still a lot of work in the follow-up study of the activity in vivo. Meanwhile, most of these compounds lack of toxicological research. Thus, the latest research progress of local anesthetics can provide novel compounds for local anesthesia.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The project was sponsored by the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry (No. 2015-1098). The work was also supported by Chongqing Key Research Project of Basic Science & Frontier Technology (No. cstc2017jcyjBX0012), Foundation Project of Chongqing Normal University (No. 14XYY020), Chongqing General Research Program of Basic Research and Frontier Technology (No. cstc2015jcyjA10054), Chongqing Normal University Postgraduate's Research and Innovation Project (No. YKC17004), the National Natural Science Foundation (21662012, 41866005), and Postgraduate Research and Innovation Project of Hainan Normal University (Hsyx2018-8), and Open Foundation Project of Key Laboratory of Tropical Medicinal Resource Chemistry of Ministry of Education (RDZH2019002).
References
- A. Brown, R. Weiss and C. Greenberg, et al., Interscalene block for shoulder arthroscopy: comparison with general anesthesia, Arthroscopy, 1993, 9(3), 295–300 CrossRef CAS.
- G. Weinberg, T. VadeBoncouer and G. Ramaraju, et al., Pretreatment or resuscitation with a lipid infusion shifts the dose-response to bupivacaine-induced asystole in rats, Anesthesiology, 1998, 88(4), 1071–1075 CrossRef CAS PubMed.
- A. Zaggia and B. Ameduri, Recent advances on synthesis of potentially nonbioaccumulable fluorinated surfactants, Curr. Opin. Colloid Interface Sci., 2012, 17, 188–195 CrossRef CAS.
- J. Conder, R. Hoke and W. De Wolf, et al., Are PFCAs bioaccumulative? A critical review and comparison with regulatory criteria and persistent lipophilic compounds, Environ. Sci. Technol., 2007, 42(4), 995–1003 CrossRef PubMed.
- G. Weinberg, R. Ripper and D. Feinstein, et al., Lipid emulsion infusion rescues dogs from bupivacaine-induced cardiac toxicity, Reg. Anesth. Pain Med., 2003, 28(3), 198–199 CrossRef CAS.
- J. Picard and T. Meek, Lipid emulsion to treat overdose of local anaesthetic: the gift of the glob, Anaesthesia, 2006, 61(2), 107–109 CrossRef PubMed.
- M. Dinglasan, E. Edwards and S. Mabury, Fluorotelomer alcohol biodegradation yields poly and perfluorinated acids, Environ. Sci. Technol., 2004, 38, 2857–2864 CrossRef CAS.
- C. Wilson, D. Wilson and A. Feiring, et al., Disassembly via an environmentally friendly and efficient fluorous phase constructed with dendritic architectures, J. Polym. Sci., Part A: Polym. Chem., 2010, 48(11), 2498–2508 CrossRef CAS.
- C. Kausch, J. Leising and R. Medsker, et al., Thomas, Synthesis,characterization and unusual surface activity of a series of novel architecture,water-dispersible poly(fluorooxetane)s, Langmuir, 2002, 8(15), 5933–5938 CrossRef.
- M. Rosenblatt, M. Abel and G. Fischer, et al., Successful use of a 20% lipid emulsion to resuscitate a patient after a presumed bupivacaine-related cardiac arrest, Anesthesiology, 2006, 105(1), 217–225 CrossRef PubMed.
- R. Litz, M. Popp and S. Stehr, et al., Successful resuscitation of a patient with ropivacaine-induced asystole after axillary plexus block using lipid infusion, Anaesthesia, 2006, 61(8), 800–801 CrossRef CAS PubMed.
- G. Cave and M. Harvey, Intravenous lipid emulsion as antidote beyond local anesthetic toxicity: a systematic review, Acad. Emerg. Med., 2009, 16(9), 815–824 CrossRef PubMed.
- C. Fleck, E. Karge and K. Wennek, et al., Local anaesthetic effects and toxicity of seven new diethanolamine and morpholine derivatives of fomocaine. Testing in rats compared with procaine and tetracaine, Arzneimittelforschung, 2003, 53, 221–228 CAS.
- H. Fozzard, M. Sheets and D. Hanck, et al., The sodium channel as a target for local anesthetic drugs, Front. Pharmacol., 2011, 2, 1–6 Search PubMed.
- C. Gronwald, V. Vegh and M. Hollmann, The inhibitory potency of local anesthetics on NMDA receptor signalling depends on their structural features, Eur. J. Pharmacol., 2012, 674, 13–19 CrossRef CAS PubMed.
- A. Sirianni, K. Osterhoudt and D. Calello, et al., Use of lipid emulsion in the resuscitation of a patient with prolonged cardiovascular collapse after overdose of bupropion and lamotrigine, Ann. Emerg. Med., 2008, 51(4), 412–415 CrossRef PubMed.
- M. Harvey and G. Cave, Intralipid outperforms sodium bicarbonate in a rabbit model of clomipramine toxicity, Ann. Emerg. Med., 2007, 49(2), 178–185 CrossRef PubMed.
- M. Harvey, G. Cave and K. Hoggett, et al., Correlation of plasma and peritoneal diasylate clomipramine concentration with hemodynamic recovery after intralipid infusion in rabbits, Acad. Emerg. Med., 2009, 16(2), 151–156 CrossRef PubMed.
- G. Lipkind and H. Fozzard, Molecular model of anticonvulsant drug binding to the voltage-gated sodium channel inner pore, Mol. Pharmacol., 2010, 78, 631–638 CrossRef CAS PubMed.
- W. Li, B. Xiao and Y. Zhou, et al., Synthesis and analgetic activity of Imipramine Analoges, Chin. J. Org. Chem., 2010, 30, 898–903 CAS.
- W. Li and Q. You, Synthesis and local anesthetic activity of fluoro-substituted imipramine and its analogues, Bioorg. Med. Chem. Lett., 2007, 17, 3733–3735 CrossRef CAS.
- P. Paoletti, C. Bellone and Q. Zhou, NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease, Nat. Rev. Neurosci., 2013, 14, 383–400 CrossRef CAS PubMed.
- D. Crandell and G. Weinberg, Moxidectin toxicosis in a puppy successfully treated with intravenous lipids, J. Vet. Emerg. Crit. Care, 2009, 19(2), 181–186 CrossRef PubMed.
- S. Kampe, M. Warm and S. Kasper, et al., Concept for postoperative analgesia after pedicled TRAM flaps: Continuous wound instillation with 0.2% ropivacaine via multilumen catheters. A report of two cases, Br. J. Plast. Surg., 2003, 56(5), 478–483 CrossRef CAS.
- F. Tomoko, M. Umit and J. Kenneth, et al., Synthesis and characterization of novel amphiphilic telechelic polyoxetanes, Macromolecules, 2003, 36, 9383–9389 CrossRef.
- C. Moody and J. Field, Perfluorinated surfactants and the environmental implications of their use in fire-Fighting foams, Development, 2000, 18, 3864–3870 Search PubMed.
- L. Nielsen, P. Lumholt and L. Halmich, Local anaesthesia with vasoconstrictor is safe to use in areas with end-arteries in fingers, toes, noses and ears, Ugeskrift for Lægerer, 2014, 176(44), 44 Search PubMed.
- S. Györke and V. Lukyanenko, Dual effects of tetracaine on spontaneous sodium release in rat ventricular myocytes, J. Physiol., 1997, 500(2), 297–309 CrossRef.
- V. Silva, R. Silva and S. Damasceno, et al., Anti-inflammatory and antinociceptive activity of epiisopiloturine, an imidazole alkaloid isolated from Pilocarpus microphyllus, J. Nat. Prod., 2013, 76, 1071–1077 CrossRef CAS.
- S. Sondhi, S. Jain and M. Dinodia, et al., Synthesis of some thiophene, imidazole and pyridine derivatives exhibiting good anti-inflammatory and analgesic activities, Med. Chem., 2008, 4, 146–154 CrossRef CAS.
- J. Pandey, V. Tiwari and S. Verma, et al., Synthesis and antitubercular screening of imidazole derivatives, Eur. J. Med. Chem., 2009, 44, 3350–3355 CrossRef CAS PubMed.
- S. Derry, P. Wiffen and R. Moore, et al., Topical lidocaine for neuropathic pain in adults, Cochrane Database Syst. Rev., 2014, 7, CD010958 Search PubMed.
- A. Martí, D. Simancas and V. Anand, et al., Prophylactic lidocaine for myocardial infarction, Cochrane Database Syst. Rev., 2015, 8, CD008553 Search PubMed.
- H. Kubinyi, Strategies and Recent Technologies in Drug Discovery, Pharmazie, 1995, 50, 647–662 CAS.
- V. Poroikov and D. Filimonov, How to acquire new biological activities in old compounds by computer prediction, J. Comput.-Aided Mol. Des., 2002, 16, 819–824 CrossRef CAS.
- K. Hahnenkamp, M. Durieux and A. Hahnenkamp, Local anaesthetics inhibit signalling of human NMDA receptors recombinantly expressed in Xenopus laevis oocytes: role of protein kinase C, Br. J. Anaesth., 2006, 96, 77–87 CrossRef CAS.
- L. Slaughter, A. Patel and J. Slaughter, Pharmacological treatment of neonatal seizures: a systematic review, J. Child Neurol., 2013, 28(3), 351–364 CrossRef.
- R. Morabito, A. Marino and S. Dossena, et al., Nematocyst discharge in Pelagia noctiluca (Cnidaria, Scyphozoa) oral arms can be affected by lidocaine, ethanol, ammonia and acetic acid, Toxicon, 2014, 83, 52–58 CrossRef CAS.
- R. Dua, S. Shrivastava and S. Sonwane, et al., Pharmacological significance of synthetic heterocycles scaffold: A Review, Adv. Biol. Res., 2011, 5, 120–144 CAS.
- L. Zhang, X. Peng and G. Damu, et al., Comprehensive review in current developments of imidazole-based medicinal chemistry, Med. Res. Rev., 2014, 34, 340–437 CrossRef CAS.
- T. Wu, A. Nagle and K. Kuhen, et al., Imidazolopiperazines: Hit to lead optimization of new antimalarial agents, J. Med. Chem., 2011, 54, 5116–5130 CrossRef CAS.
- S. Saravanan, P. Selvan and N. Gopal, et al., Synthesis and antibacterial activity of some imidazole-5-(4H)one derivatives, Arch. Pharm., 2005, 338, 488–492 CrossRef CAS.
- M. Segal, G. Rogers and H. Needleman, et al., Hypokalemic sensory overstimulation, J. Child Neurol., 2007, 22(12), 1408–1410 CrossRef.
- D. Jackson, A. Chen and C. Bennett, Identifying true lidocaine allergy, J. Am. Dent. Assoc., JADA, 1994, 125(10), 1362–1366 CrossRef CAS.
- J. Picard, S. Ward and R. Zumpe, et al., Guidelines and the adoption of 'lipid rescue' therapy for local anaesthetic toxicity, Anaesthesia, 2009, 64(2), 122–125 CrossRef CAS.
- L. Wang, S. Zellmer and D. Printzenhoff, et al., Addition of a single methyl group to a small molecule sodium channel inhibitor introduces a new mode of gating modulation, Br. J. Pharmacol., 2015, 172, 4905–4918 CrossRef CAS.
- Y. Yang, J. Q. Si and C. Fan, et al., Effects of ropivacaine on GABAactivated currents in isolated dorsal root ganglion neurons in rats, Chin. J. Appl. Physiol., 2013, 29, 263–266 CAS.
- L. Zhang, K. Tanabe and F. Yanagidate, et al., Different effects of local anesthetics on extracellular signal-regulated kinase phosphorylation in rat dorsal horn neurons, Eur. J. Pharmacol., 2014, 3, 132–136 CrossRef PubMed.
- A. Gulihar, S. Robati and H. Twaij, et al., Articular cartilage and local anaesthetic: A systematic review of the current literature, J. Orthop., 2015, 12, S200–S210 CrossRef PubMed.
- W. A. Carterall, Molecular mechanisms of gating and drug block of sodium channels, Sodium Channels and Neuronal Hyperexcitability: Novartis Foundation Symposia, 2001, vol. 241, pp. 206–225 Search PubMed.
- A. Bender and R. Glen, Molecular similarity: a key technique in molecular informatics, Org. Biomol. Chem., 2004, 2(22), 3204–3218 RSC.
- A. Khanna, R. Saxena and A. Dutta, et al., Comparison of ropivacaine with and without fentanyl vs. bupivacaine with fentanyl for postoperative epidural analgesia in bilateral total knee replacement surgery, J. Clin. Anesth., 2017, 37, 7–13 CrossRef CAS.
- S. Sheu and W. Lederer, Lidocaine's negative inotropic and antiarrhythmic actions. Dependence on shortening of action potential duration and reduction of intracellular sodium activity, Circ. Res., 1985, 57(4), 578–590 CrossRef CAS.
- K. Collinsworth, S. Kalman and D. Harrison, The clinical pharmacology of lidocaine as an antiarrhythymic drug, Circulation, 1974, 50(6), 1217–1230 CrossRef CAS.
- A. Pupka, J. Sikora and J. Mauricz, et al., The usage of synthol in the body building, Polim. Med., 2009, 39(1), 63–67 Search PubMed.
- G. Lipkind and H. Fozzard, Molecular modeling of local anesthetic drug binding by voltage-gated sodium channels, Mol. Pharmacol., 2005, 68, 1611–1622 CrossRef CAS PubMed.
- M. Sugimoto, I. Uchida and S. Fukami, et al., The alpha and gamma subunit-dependent effects of local anesthetics on recombinantGABA(A) receptors, Eur. J. Pharmacol., 2000, 401, 329–337 CrossRef CAS.
- M. Volgraf, B. Sellers and Y. Jiang, et al., Discovery of GluN2A Selective NMDA receptor positive allosteric modulators (PAMs):tuning deactivation kinetics via structure-based design, J. Med. Chem., 2016, 59, 2760–2779 CrossRef CAS PubMed.
- N. Bernardo, M. Siqueira and M. DePaiva, et al., Caffeine and other adulterants in seizures of street cocaine in Brazil, Int. J. Drug Policy, 2003, 14(4), 331–334 CrossRef.
- A. Gargiulo, G. Burns and C. Huck, Dyclonine hydrochloride-a topical agent for managing pain, Ill. Dent. J., 1992, 61(4), 303–304 CAS.
- J. Schmidt, L. Blockus and R. Richards, The Pharmacology of Pramoxine Hydrochloride: A New Topical Local Anesthetic, Curr. Res. Anesth. Analg., 1953, 32(61), 418–425 CAS.
- F. Carla, O. Laura and E. Maria, et al., Synthesis of new chiral xanthone derivatives acting as nerve conduction blockers in the rat sciatic nerve, Eur. J. Med. Chem., 2012, 55, 1–11 Search PubMed.
- G. Athina, V. Paola and D. Nikos, et al., Evaluation of the local anaesthetic activity of 3-aminobenzo [d] isothiazole derivatives using the rat sciatic nerve model, Eur. J. Med. Chem., 2009, 44, 473–481 CrossRef.
- G. Liliana, N. Gabriel and E. María, et al., Antihyperalgesic activity of a novel synthesized analogue of lidocaine in diabetic rats, J. Pharm. Pharmacol., 2013, 65, 689–696 CrossRef.
- D. Kalinin, V. Pantsurkin and B. Syropyatov, et al., Synthesis, local anaesthetic and antiarrhythmic activities of N-alkyl derivatives of proline anilides, Eur. J. Med. Chem., 2013, 63, 144–150 CrossRef CAS PubMed.
- R. Yan, L. Ming and Z. Zong, et al., β-Cyclodextrin-propyl sulfonic acid catalysed one-pot synthesis of 1,2,4,5-tetrasubstituted imidazoles as local anesthetic agents, Molecules, 2015, 20, 20286–20296 CrossRef PubMed.
- J. Obniska, A. Rapacz and S. Rybka, et al., Synthesis, and anticonvulsant activity of new amides derived from 3-methyl- or 3-ethyl-3-methyl-2,5-dioxo-pyrrolidin-1-yl-acetic acids, Bioorg. Med. Chem., 2016, 24(8), 1598–1607 CrossRef CAS PubMed.
- A. Ashish, P. Ashok and R. Kavita, et al., 4-(2-Chloroacetamido) benzoic acid derivatives as local anesthetic agents: design, synthesis, and characterization, J. Pharm. Biosci., 2017, 4(6), 35–44 Search PubMed.
- L. Wen, D. Lina and M. Hong, et al., Synthesis, biological evaluation, and molecular docking of ropivacaine analogs as local anesthetic agents, Med. Chem. Res., 2017, 27(3), 954–965 Search PubMed.
- S. Ekhlas, E. Tilal and S. Malik, et al., Synthesis, characterization and assessment of local anesthetic activity of some benzohydroxamic acids, Asian J. Chem., 2019, 31(1), 181–185 CrossRef.
- Z. Huiying, C. Guangying and Z. Shiyang, Design, synthesis and biological activity evaluation of benzoate compounds as local anesthetic, RSC Adv., 2019, 9(12), 6627–6635 RSC.
|
| This journal is © The Royal Society of Chemistry 2019 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
Guangying Chen*a
*b and
Guangying Chen*a