
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective loading of platinum or silver cocatalyst onto a hydrogen-evolution photocatalyst in a silver-mediated all solid-state Z-scheme system for enhanced overall water splitting†
Junya Osakia,
Masaomi Yodaa,
Toshihiro Takashima ab and
Hiroshi Irie*ab
ab and
Hiroshi Irie*ab
aSpecial Doctoral Program for Green Energy Conversion Science and Technology, Integrated Graduate School of Medicine, Engineering, and Agricultural Sciences, University of Yamanashi, 4-3-11 Takeda, Kofu, Yamanashi 400-8511, Japan
bClean Energy Research Center, University of Yamanashi, 4-3-11 Takeda, Kofu, Yamanashi 400-8511, Japan. E-mail: hirie@yamanashi.ac.jp
First published on 17th December 2019
Abstract
We selectively loaded a hydrogen (H2)-evolution cocatalyst, either platinum (Pt) or silver (Ag), onto a H2-evolution photocatalyst, zinc rhodium oxide (ZnRh2O4), in a Ag-inserted ZnRh2O4 and bismuth vanadium oxide (Bi4V2O11) hetero-junction photocatalyst (ZnRh2O4/Ag/Bi4V2O11) by a photo-deposition method. The selective loading of Pt or Ag was achieved by taking advantage of the band-gap difference between ZnRh2O4 (1.2 eV) and Bi4V2O11 (1.7 eV) and increased the overall water-splitting activity of the photocatalyst.
Hydrogen (H2) is a versatile, non-polluting energy carrier because the chemical energy stored in the H–H bond is released when H2 combines with oxygen (O2) and yields only water as the reaction product. Accordingly, an energy infrastructure based on H2 represents an ideal solution to energy-related environmental issues for reducing carbon dioxide (CO2) emissions. Photocatalytic water-splitting using sunlight represents one of the cleanest methods to produce H2.1 Since the Honda–Fujishima effect was first reported in 1972,2 various water-splitting methods, including electrode and powder systems, have been extensively investigated for the possibility of simple, large scale production of H2.2,3 Following the demonstration by Domen and coworkers that a solid solution of gallium nitride (GaN)–zinc oxide (ZnO) is capable of splitting water at wavelengths up to ∼480 nm,4 numerous researchers have attempted to identify powdered photocatalysts that utilize longer wavelengths of visible light for efficient water-splitting using solar energy.5
Previously, we fabricated a hetero-junction photocatalyst consisting of zinc rhodium oxide (ZnRh2O4) and bismuth vanadium oxide (Bi4V2O11), which functioned as H2- and O2-evolution photocatalysts, respectively, and either silver (Ag) or gold (Au) as a conductive layer (ZnRh2O4/Ag/Bi4V2O11 or ZnRh2O4/Au/Bi4V2O11) after Tada and coworkers.6 Recently, similar hetero-junction photocatalysts were reported.7
In ZnRh2O4/Ag/Bi4V2O11 or ZnRh2O4/Au/Bi4V2O11, overall pure water-splitting proceeded under irradiation with red light (wavelengths larger than 700 nm)8–10 via Ag or Au, which mediated the transfer of photo-excited electrons from the conduction band (CB) of Bi4V2O11 to the valence band (VB) of ZnRh2O4. Thus, the photo-excited electrons in ZnRh2O4 and holes in Bi4V2O11 effectively reduced and oxidized water, respectively, to accomplish overall water splitting. The modification of the cocatalyst with a metal or metal oxide is essential to enhance the water-splitting activity. The selective loading of a H2-favorable cocatalyst onto ZnRh2O4 is expected to increase H2 evolution and thereby increase the overall water-splitting activity of this hetero-junction photocatalyst. Here, we used platinum (Pt) and Ag as H2-evolution cocatalysts and attempted to selectively deposit Pt or Ag onto ZnRh2O4 in ZnRh2O4/Ag/Bi4V2O11.
Powdered ZnRh2O4/Ag/Bi4V2O11 (molar ratio of ZnRh2O4 to Bi4V2O11 was 1.0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2) was prepared following procedures previously reported procedures (ESI-1†)8 and the photo-deposition of Pt or Ag was conducted under light irradiation at wavelength longer than 850 nm. Under these irradiation conditions, only ZnRh2O4 was photo-excited because the energy of irradiated light was higher than band-gap energy of ZnRh2O4 (1.2 eV) but smaller than that of Bi4V2O11 (1.7 eV). Due to the specific photoexcitation of ZnRh2O4, Pt or Ag was only photo-deposited onto this material, generating Pt/ZnRh2O4/Ag/Bi4V2O11 and Ag/ZnRh2O4/Ag/Bi4V2O11 (ESI-1†).
:1.2) was prepared following procedures previously reported procedures (ESI-1†)8 and the photo-deposition of Pt or Ag was conducted under light irradiation at wavelength longer than 850 nm. Under these irradiation conditions, only ZnRh2O4 was photo-excited because the energy of irradiated light was higher than band-gap energy of ZnRh2O4 (1.2 eV) but smaller than that of Bi4V2O11 (1.7 eV). Due to the specific photoexcitation of ZnRh2O4, Pt or Ag was only photo-deposited onto this material, generating Pt/ZnRh2O4/Ag/Bi4V2O11 and Ag/ZnRh2O4/Ag/Bi4V2O11 (ESI-1†).
In the powder XRD pattern of ZnRh2O4/Ag/Bi4V2O11 (Fig. S1, ESI-2†), the observed peaks mainly corresponded to two phases originating from ZnRh2O4 and Bi4V2O11, with the trace peaks ascribed to an unknown Ag oxide (likely AgVO3). Notably, no Ag peaks were observed in the XRD spectrum, which is plausible since the amount of Ag remaining in the composite was too small for detection. These observations are consistent with those reported previously.8 The valency of Ag in the photocatalyst was determined by XPS to be mainly zero (Ag0). The implications of this finding are discussed below.
UV-vis absorption spectra of ZnRh2O4/Ag/Bi4V2O11, Pt/ZnRh2O4/Ag/Bi4V2O11, and Ag/ZnRh2O4/Ag/Bi4V2O11 are shown in Fig. 1. In the spectra, Pt/ZnRh2O4/Ag/Bi4V2O11 and Ag/ZnRh2O4/Ag/Bi4V2O11 had greater absorption than that of ZnRh2O4/Ag/Bi4V2O11 in the longer wavelength region, a property that is derived from the deposited Pt and Ag.
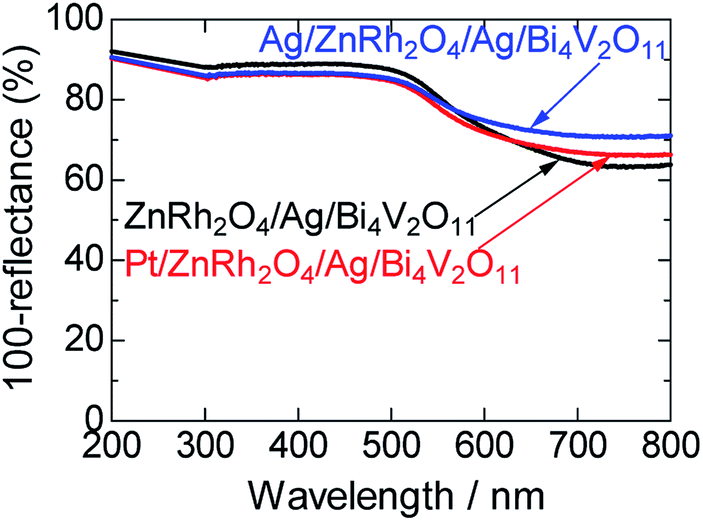 | ||
| Fig. 1 UV-visible absorption spectra of ZnRh2O4/Ag/Bi4V2O11, Pt/ZnRh2O4/Ag/Bi4V2O11, and Ag/ZnRh2O4/Ag/Bi4V2O11. | ||
Fig. 2a–f show XPS spectra of ZnRh2O4/Ag/Bi4V2O11, Pt/ZnRh2O4/Ag/Bi4V2O11, and Ag/ZnRh2O4/Ag/Bi4V2O11 for Bi 4f (Fig. 2a), V 2p (Fig. 2b), Zn 2p (Fig. 2c), Rh 3d (Fig. 2d), Ag 3d (Fig. 2e), and Pt 4f (Fig. 2f). As we assumed that the cocatalyst, Pt or Ag, was selectively deposited onto ZnRh2O4 and the Bi4V2O11 surface remained unaltered after the deposition of the cocatalyst, all of the XPS peaks were normalized using the peak areas of Bi 4f for the bare, Pt-deposited, and Ag-deposited ZnRh2O4/Ag/Bi4V2O11 photocatalysts (Fig. 2a). It was reasonable that the peak areas in the normalized V 2p3/2 spectra of the three photocatalysts were similar (Fig. 2b). Further, the normalized Zn 2p and Rh 3d peaks of Pt/ZnRh2O4/Ag/Bi4V2O11 and Ag/ZnRh2O4/Ag/Bi4V2O11 were smaller than those of ZnRh2O4/Ag/Bi4V2O11 (Fig. 2c and d). The results of the surface-sensitive XPS measurements indicate that Pt and Ag were selectively deposited onto ZnRh2O4.
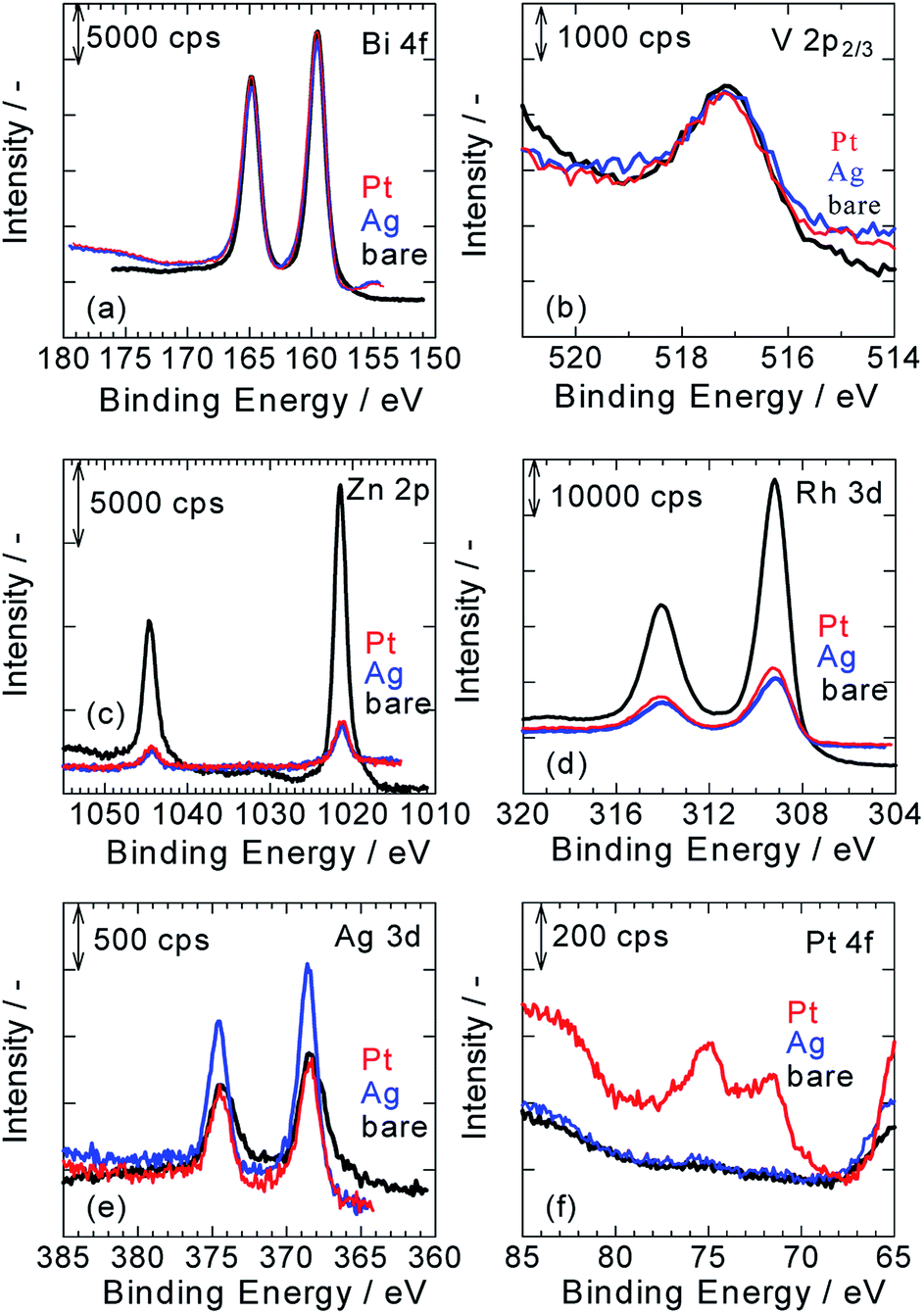 | ||
| Fig. 2 Normalized XPS spectra of ZnRh2O4/Ag/Bi4V2O11 (black lines), Pt/ZnRh2O4/Ag/Bi4V2O11 (red lines), and Ag/ZnRh2O4/Ag/Bi4V2O11 (blue lines) for Bi 4f (a), V 2p (b), Zn 2p (c), Rh 3d (d), Ag 3d (e), and Pt 4f (f). All spectra were calibrated with the C 1s peak, which was derived from a surface-contaminant hydrocarbon with a binding energy of 284.5 eV. | ||
The normalized Ag 3d peak area of Pt/ZnRh2O4/Ag/Bi4V2O11 was quite similar to that of ZnRh2O4/Ag/Bi4V2O11 (Fig. 2e). This result is reasonable because the Ag 3d spectra of the two photocatalysts are attributable to the Ag inserted between ZnRh2O4 and Bi4V2O11. In contrast, the Ag 3d spectrum of Ag/ZnRh2O4/Ag/Bi4V2O11 was larger than those of Pt/ZnRh2O4/Ag/Bi4V2O11 and ZnRh2O4/Ag/Bi4V2O11, indicating that the former spectrum originated from the photo-deposited Ag and Ag located between ZnRh2O4 and Bi4V2O11. In the normalized Pt 4f spectra, the Pt 4f peaks were only observed on Pt/ZnRh2O4/Ag/Bi4V2O11 and were not detected in the XPS spectra of Ag/ZnRh2O4/Ag/Bi4V2O11 and ZnRh2O4/Ag/Bi4V2O11 (Fig. 2f).
To quantitatively analyze the amounts and determine the valencies of Pt and Ag in the three photocatalysts, a peak deconvolution method was applied to the Bi 4f, Ag 3d, and Pt 4f spectra (Fig. S2 in ESI-3†). As we considered that the Bi4V2O11 surface remained unaltered after the Pt or Ag deposition, we estimated the amount of Pt or Ag in the photocatalysts based on the amount of Bi. First, peak deconvolution for Bi 4f was performed with the following parameters. The binding energies of Bi 4f7/2 and Bi 4f5/2 of Bi3+ were fixed at 159.5 ± 0.1 eV and 164.8 ± 0.1 eV, respectively.11 For the Pt- and Ag-deposited photocatalysts, an additional pair of small peaks was observed at 161.0 ± 0.1 eV and 166.3 ± 0.1 eV. The origin of the additional peaks was unclear; however, the peaks were likely derived from Bi3+–Cl− (e.g., BiOCl)12 or Bi3+–NO3− (Bi(NO3)3) remaining on the surface because Pt or Ag was photo-deposited in the H2PtCl6·6H2O or AgNO3 solution despite thorough washing with distilled water. The peak area ratio of Bi 4f5/2 to Bi 4f7/2 for Bi3+ was fixed at 0.75 (Fig. S2a, S2c, S2f and Table S1 in ESI-3†).
In Fig. S2b, S2d, and S2g in ESI-3,† the Ag 3d peaks for the three photocatalysts were deconvoluted by a pair of Ag 3d peaks (Ag 3d5/2 and Ag 3d3/2, with binding energies of 368.6 ± 0.1 eV and 374.6 ± 0.1 eV, respectively), which is attributable to metallic Ag (Ag0).8,9,13 These results indicate that Ag was inserted as a metallic particle between ZnRh2O4 and Bi4V2O11 in ZnRh2O4/Ag/Bi4V2O11, Pt/ZnRh2O4/Ag/Bi4V2O11, and Ag/ZnRh2O4/Ag/Bi4V2O11.8,9 In addition, the Ag cocatalyst was also proved to be deposited as the metallic one. In contrast, the Pt 4f peaks for Pt/ZnRh2O4/Ag/Bi4V2O11 were deconvoluted by three pairs of Pt 4f peaks with binding energies of 71.3 eV (Pt 4f7/2) and 74.6 eV (Pt 4f5/2) for metallic Pt (Pt0), 72.3 eV (Pt 4f7/2) and 75.6 eV (Pt 4f5/2) for Pt2+, and 73.3 eV (Pt 4f7/2) and 76.6 eV (Pt 4f5/2) for Pt3+ (Fig. S2e in ESI-3†).14 The total Pt0, Pt2+, and Pt3+ areas were determined by summing the corresponding deconvolution areas of Pt 4f5/2 and Pt 4f7/2 (Table S1 in ESI-3†). The atomic ratios of Pt0, Pt2+, and Pt3+ vs. total Pt were calculated to be 0.63, 0.25, and 0.12, respectively, and by dividing the total Pt0, Pt2+, and Pt3+ areas by the total Pt area and represent the sum of the total Pt0, Pt2+, and Pt3+ areas. Thus, the major Pt species that functioned as the cocatalyst was Pt0; however, Pt4+ was not fully reduced to Pt0. The valence distribution was not observed for Ag, but observed for Pt. We are considering two possible explanations; one is a difference in the reduction potential between Ag and Pt deposition, Ag+ + e− → Ag0 (0.80 V vs. SHE), PtCl62− + 2e− → PtCl42− + 2Cl− (Pt4+ → Pt2+, 0.68 V vs. SHE), PtCl42− + 2e− → Pt0 + 4Cl− (Pt2+ → Pt0, 0.72 V vs. SHE) although that of PtCl62− + e− → PtCl52− + Cl− is unknown. Thus, forming Ag0 from the reduction of Ag+ is thermodynamically more favorable than forming Pt2+ from that of Pt4+ and Pt0 from that of Pt2+. The other reason is that the reduction product from Ag+ is only Ag0, but some reduction products are possible from Pt4+, such as Pt3+, Pt2+, Pt+, and Pt0, if the reduction proceeds. In fact, Pt4+ was not detected, and the reduction of Pt4+ proceeded.
The Ag and Bi areas were determined by summing the deconvolution areas of Ag 3d5/2 and Ag 3d3/2, and those of Bi 4f7/2 and Bi 4f5/2, respectively (Table S1 in ESI-3†). The atomic ratio of Ag to Bi was then calculated based on the Ag 3d (5.99) and Bi 4f sensitivity factors (9.14),15 and the mole percent of Ag to that of the sum of ZnRh2O4 and Bi4V2O11 (1.0 mole of ZnRh2O4 + 1.2 mole of Bi4V2O11) was determined. The weight percent (wt%) of Ag in the three photocatalysts was recalculated (Table S2 in ESI-3†). As expected, the amount of Ag in ZnRh2O4/Ag/Bi4V2O11 and Pt/ZnRh2O4/Ag/Bi4V2O11 was identical at 2.1 wt%. Thus, the Ag cocatalyst deposited in Ag/ZnRh2O4/Ag/Bi4V2O11 was estimated to be 1.1 wt% based on the assumption that the amount of Ag inserted in Ag/ZnRh2O4/Ag/Bi4V2O11 was 2.1 wt%. Similarly, the amount of Pt cocatalyst in Pt/ZnRh2O4/Ag/Bi4V2O11 was calculated to be 1.2 wt% (Table S2† in ESI-3†) based on the total Pt area and Pt 4f sensitivity factor (5.575).15
SEM images of ZnRh2O4/Ag/Bi4V2O11, Pt/ZnRh2O4/Ag/Bi4V2O11, and Ag/ZnRh2O4/Ag/Bi4V2O11 powders are shown (Figs. S3a–c in ESI-4†). All images were quite similar, and small ZnRh2O4 (∼200 nm) and large Bi4V2O11 (∼10 μm) particles can be clearly observed. However, the photo-deposited Pt and Ag were not observed.
STEM imaging (high angle annular dark-field (HAADF), bright-field (BF)) and EDS-based elemental mapping of Pt/ZnRh2O4/Ag/Bi4V2O11 were performed (Fig. 3a–g). In the HAADF-STEM image (Fig. 3a), ZnRh2O4 and Bi4V2O11 particles were clearly distinguishable based on the SEM image (Fig. S3b in ESI-4†) and their previously reported sizes of ∼200 nm and ∼10 μm, respectively.8 Thus, the small aggregated particles in the HAADF-STEM image were ZnRh2O4 and were adjacent to a large particle of Bi4V2O11. The particle compositions were confirmed by the EDS-based elemental mapping of Bi and Rh in Fig. 3c and e, respectively. In addition, Ag particles were detected (Fig. 3d) between the areas of Bi (Fig. 3c) and Rh (Fig. 3e), indicating that Ag was inserted between the ZnRh2O4 and Bi4V2O11 particles. This finding was also confirmed by the line elemental analysis (Fig. 3g). Notably, the atomic percentages of Bi and V decreased at the boundary of Ag and ZnRh2O4, and those of Zn and Rh increased at the boundary of Ag and Bi4V2O11. Accordingly, the percentage of Ag increased and decreased at the boundary with Bi4V2O11 and ZnRh2O4, respectively, indicating that Ag was present at the Bi4V2O11 and ZnRh2O4 interface. As expected, Pt was only deposited onto ZnRh2O4 because Pt was only distributed in the vicinity of Rh (Fig. 3f). This finding was also confirmed by the line elemental analysis, as several sharp peaks derived from Pt were observed in the area of ZnRh2O4 (Fig. 3g). In addition, extremely small particles, corresponding to Pt, were observed only on ZnRh2O4 in the enlarged BF image of Pt/ZnRh2O4/Ag/Bi4V2O11 (Fig. 3b).
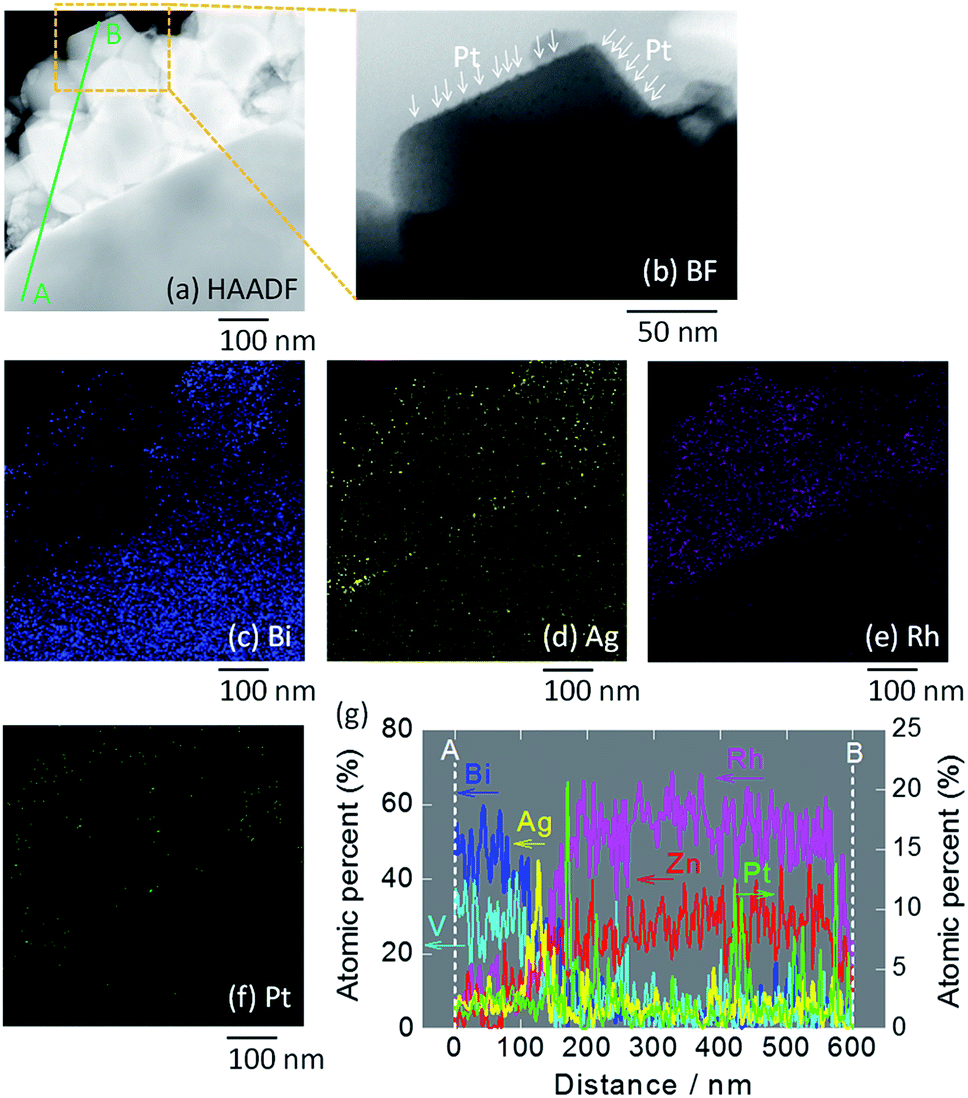 | ||
| Fig. 3 STEM images of Pt/ZnRh2O4/Ag/Bi4V2O11. HAADF-STEM image (a) and the BF enlargement (b) in which Pt is indicated by arrows, and EDS elemental maps (c–f), in which blue (c), yellow (d), pink (e), and green (f) colors correspond to Bi, Ag, Rh, and Pt, respectively. In (a), the line along which the elemental analysis was performed is shown (A to B). Atomic percentages of Bi, V, Zn, Rh, Ag, and Pt measured from the area of Bi4V2O11 (A) to that of ZnRh2O4 (B) (g). Enlarged images of (a)–(f) are shown in ESI-5.† | ||
The time courses of H2 and O2 evolution from Pt/ZnRh2O4/Ag/Bi4V2O11 and Ag/ZnRh2O4/Ag/Bi4V2O11 in comparison with ZnRh2O4/Ag/Bi4V2O11 under monochromic light irradiation at a wavelength of 700 nm were measured (Fig. 4a and b). The three photocatalysts evolved H2 and O2 from water at a molar ratio of ∼2 to 1 (2.1 to 1, 2.3 to 1, and 2.2 to 1 for ZnRh2O4/Ag/Bi4V2O11, Pt/ZnRh2O4/Ag/Bi4V2O11, and Ag/ZnRh2O4/Ag/Bi4V2O11, respectively), indicating that the overall water-splitting reaction proceeded efficiently. The band alignments of ZnRh2O4, Ag, and Bi4V2O11, and the charge transfer process are shown in Scheme S1 (ESI-6†). The photo-excited electrons in ZnRh2O4 and holes in Bi4V2O11 can effectively reduce and oxidize water, respectively. Specifically, Ag acted as a solid-state electron mediator for the electron transfer from the CB of Bi4V2O11 to the VB of ZnRh2O4. The deposition of Pt or Ag enhanced the H2 and O2 evolution rates, demonstrating that Pt and Ag functioned as cocatalysts for the overall water-splitting reaction. The apparent quantum efficiency (AQE) values for the reaction over Pt/ZnRh2O4/Ag/Bi4V2O11 (4.1 × 10−2%) and Ag/ZnRh2O4/Ag/Bi4V2O11 (4.6 × 10−2%) were ∼3-fold higher than that over ZnRh2O4/Ag/Bi4V2O11 (1.5 × 10−2%) (Tables 1 and S3 in ESI-7†).
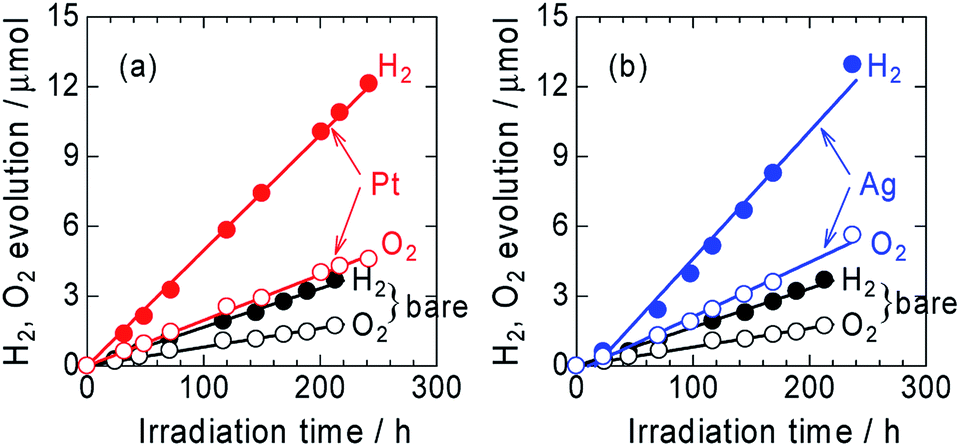 | ||
| Fig. 4 Time courses of photocatalytic evolution of H2 and O2 from water over Pt/ZnRh2O4/Ag/Bi4V2O11 (a) and Ag/ZnRh2O4/Ag/Bi4V2O11 (b) irradiated with 700 nm LED light. For comparison, the evolution of H2 and O2 over bare ZnRh2O4/Ag/Bi4V2O11 is also shown. | ||
| Light source | Light/mW cm−2 | H2 evolution rate/μmol h−1 | AQE/% |
|---|---|---|---|
| ZnRh2O4/Ag/Bi4V2O11 | 2.9 | 1.7 × 10−2 | 1.5 × 10−2 |
| Pt/ZnRh2O4/Ag/Bi4V2O11 | 3.5 | 5.0 × 10−2 | 4.1 × 10−2 |
| Ag/ZnRh2O4/Ag/Bi4V2O11 | 3.5 | 5.5 × 10−2 | 4.6 × 10−2 |
We previously reported that the treatment of ZnRh2O4/Ag/Bi4V2O11 with nitric acid (HNO3) is required to achieve overall water splitting (ESI-1†). The treatment removes excess Ag containing Ag+ which is a sacrificial agent for O2 evolution.8,9 Thus, without HNO3 treatment, excess Ag remaining on Bi4V2O11 in ZnRh2O4/Ag/Bi4V2O11 evolves O2 even from water, resulting in more O2 being released than that of the stoichiometric amount of O2 to H2 of ∼1 to 2. However, in the present study, H2 and O2 were evolved at a 2:1 ratio, providing further evidence that Ag was selectively deposited onto ZnRh2O4. Notably, Ag-deposited ZnRh2O4 is unable to evolve O2 from water because the VB top potential of ZnRh2O4 lies at ∼0.1 V (vs. SHE, pH = 0),16 which is thermodynamically unfavorable for O2 evolution.
In conclusion, Pt or Ag as a cocatalyst was selectively photo-deposited onto ZnRh2O4 in ZnRh2O4/Ag/Bi4V2O11 and resulted in the enhancement of stoichiometric H2 and O2 evolution from water. ZnRh2O4/Ag/Bi4V2O11 also has the potential to reduce CO2 under visible-light irradiation to carbon monoxide (CO), formate, methanol, methane, and other hydrocarbons using water as an electron source. Notably, when present on ZnRh2O4, Ag acts as a selective cocatalyst for the conversion of CO2 to CO. In contrast, as Pt lacks this property, different reaction products can be detected. Such studies are currently underway in our laboratory.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by JSPS KAKENHI (Grant-in-Aid for Scientific Research) (B), Grant Number 17H03126. We express gratitude to Mr G. Newton for the careful reading of the manuscript.Notes and references
- R. M. Navarro, M. A. Pena and J. L. G. Fierro, Chem. Rev., 2007, 107, 3952–3991 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- O. Khaselev and J. A. Turner, Science, 1998, 280, 425–427 CrossRef CAS PubMed; W. J. Youngblood, S.-H. A. Lee, Y. Kobayashi, E. A. Hernandez-Pagan, P. G. Hoertz, T. A. Moore, A. L. Moore, D. Gust and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 926–927 CrossRef PubMed; S. Y. Reece, J. A. Hamel, K. Sung, T. D. Jarvi, A. J. Esswein, J. J. H. Pijpers and D. G. Nocera, Science, 2011, 334, 645–648 CrossRef PubMed; M. R. Singh, K. Papadantonakis, C. Xiang and N. S. Lewis, Energy Environ. Sci., 2015, 8, 2760–2767 RSC; S. Haussener, C. Xiang, J. M. Spurgeon, S. Ardo, N. S. Lewis and A. Z. Weber, Energy Environ. Sci., 2012, 5, 9922–9935 RSC; M. R. Singh, C. Xiang and N. S. Lewis, Sustainable Energy Fuels, 2017, 1, 458–466 RSC; M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef PubMed; T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef PubMed; K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef.
- K. Maeda, K. Teramura, D. Lu, T. Takata, N. Saito, Y. Inoue and K. Domen, Nature, 2006, 440, 295 CrossRef CAS PubMed.
- L. Liao, Q. Zhang, Z. Su, Z. Zhao, Y. Wang, Y. Li, X. Lu, D. Wei, G. Feng and Q. Yu, et al., Nat. Nanotechnol., 2014, 9, 69–73 CrossRef CAS PubMed; C. Pan, T. Takata, M. Nakabayashi, T. Matsumoto, N. Shibata, Y. Ikuhara and K. Domen, Angew. Chem., Int. Ed., 2015, 54, 2955–2959 CrossRef PubMed; J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. Kang, Science, 2015, 347, 970–974 CrossRef PubMed.
- H. Tada, T. Mitsui, T. Kiyonaga, T. Akita and K. Tanaka, Nat. Mater., 2006, 5, 782–786 CrossRef CAS PubMed.
- Y. Sasaki, H. Nemoto, K. Saito and A. Kudo, J. Phys. Chem. C, 2009, 113, 17536–17542 CrossRef CAS; A. Iwase, Y. H. Ng, Y. Ishiguro, A. Kudo and R. Amal, J. Am. Chem. Soc., 2011, 133, 11054–11057 CrossRef PubMed; Z. Pan, T. Hisatomi, Q. Wang, S. Chen, M. Nakabayashi, N. Shibata, C. Pan, T. Takata, M. Katayama, T. Minegishi, A. Kudo and K. Domen, ACS Catal., 2016, 6, 7188–7196 CrossRef; Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef PubMed; S. Sun, T. Hisatomi, Q. Wang, S. Chen, G. Ma, J. Liu, S. Nandy, T. Minegishi, M. Katayama and K. Domen, ACS Catal., 2018, 8, 1690–1696 CrossRef.
- R. Kobayashi, K. Kurihara, T. Takashima, B. Ohtani and H. Irie, J. Mater. Chem. A, 2016, 4, 3061–3067 RSC.
- R. Kobayashi, T. Takashima, S. Tanigawa, S. Takeuchi, B. Ohtani and H. Irie, Phys. Chem. Chem. Phys., 2016, 18, 27693–28378 RSC.
- K. Kamijyo, T. Takashima, M. Yoda, J. Osaki and H. Irie, Chem. Commun., 2018, 54, 7999–8002 RSC.
- Q. Jia, K. Iwashima and A. Kudo, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11564–11569 CrossRef CAS PubMed.
- L. Ye, K. Deng, F. Xe, L. Tian, T. Peng and L. Zan, Phys. Chem. Chem. Phys., 2012, 14, 82–85 RSC.
- G. B. Houflund, Z. F. Hazos and G. N. Salaita, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 4482–4486 Search PubMed; K. Kuribayashi and S. Kitamura, Thin Solid Films, 2001, 400, 160–164 CrossRef CAS.
- Y. Abe, H. Yanagizawa and K. Sasaki, Jpn. J. Appl. Phys., 1998, 37, 11126–11133 Search PubMed.
- A. J. Bard, R. Parsons and J. Jordan, Standard Potentials in Aqueous Solution, 1985, New York, Marcel Dekker Search PubMed.
- Y. Takimoto, T. Kitta and H. Irie, Int. J. Hydrogen Energy, 2012, 37, 134–138 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra09421k |
| This journal is © The Royal Society of Chemistry 2019 |