
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid synthesis of gold nanoparticles with carbon monoxide in a microfluidic segmented flow system†
He
Huang
 a,
Hendrik
du Toit
a,
Sultan
Ben-Jaber
b,
Gaowei
Wu
a,
Luca
Panariello
a,
Nguyen Thi Kim
Thanh
cd,
Ivan P.
Parkin
b and
Asterios
Gavriilidis
*a
a,
Hendrik
du Toit
a,
Sultan
Ben-Jaber
b,
Gaowei
Wu
a,
Luca
Panariello
a,
Nguyen Thi Kim
Thanh
cd,
Ivan P.
Parkin
b and
Asterios
Gavriilidis
*a
aDepartment of Chemical Engineering, University College London, Torrington Place, London WC1E 7JE, UK. E-mail: a.gavriilidis@ucl.ac.uk
bDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
cBiophysics Group, Department of Physics and Astronomy, University College London, Gower Street, London WC1E 6BT, UK
dUCL Healthcare Biomagnetic and Nanomaterials Laboratories, 21 Albemarle Street, London W1S 4BS, UK
First published on 25th March 2019
Abstract
A microfluidic reactor was developed to enhance the speed (reaction time <4 min) and control of the synthesis of gold nanoparticles (3–25 nm) with or without capping agents using carbon monoxide as gaseous reductant, which is easy to remove from the reaction mixture by venting. Gas–liquid segmented flow was formed inside a 1 mm inner diameter coiled flow inverter with aqueous gold precursor and the reducing gas to produce gold nanoparticles with polydispersity as low as 5%. The uncapped gold nanoparticles make the product attractive for surface-enhanced Raman scattering, and showed an average enhancement factor of 1.94 × 105. Various capping agents (tri-sodium citrate, polysorbate 80, oleylamine and poly(ethyleneglycol) 2-mercaptoethyl ether acetic acid) and different operational parameters were also tested, demonstrating that the proposed synthesis is flexible and can continuously produce a variety of gold nanoparticles, with potential for other applications.
Introduction
Due to their unique optical, electronic, catalytic and biocompatibility properties, gold nanoparticles have a wide range of applications in many areas such as catalysis,1 analytical chemistry,2 medical diagnostics and therapies.3 Gold ions (Au3+ or Au+) in solution are typically reduced by either a strong reducing agent (e.g., NaBH4) to form gold nanoparticles under 5 nm in diameter,4 or mild reducing agent (e.g., tri-sodium citrate)5 to form larger gold nanoparticles with sizes ranging between 10–40 nm. Once formed, they generally require stabilisation to prevent aggregation. This is achieved using capping agents which bind temporally or permanently to the gold nanoparticle surfaces and provide steric or electrostatic repulsion.6 In general, the formation of gold nanoparticles is divided into two stages: nucleation and growth.7 A key element to obtain monodisperse particles is the separation between nucleation and growth processes by the transition between gold species with different reactivity to terminate nucleation and encourage growth on the existing gold surface.8 A detailed study into gold nanoparticle synthesis by reduction with gaseous carbon monoxide (CO) in a batch system has been presented by Young et al.9 The CO was introduced via a gas diffuser with 60 μm pore size into the aqueous HAuCl4 solution at different flow rates. Compared with typical gold nanoparticle synthesis routes, which rely on liquid reducing agents, reduction by CO has various advantages, such as no impurity or side product from reducing agent is left after the synthesis, room temperature synthesis and shorter reaction times (3–5 min) compared to the typical Turkevich method (around 30 min).5 However, mixing between CO and aqueous HAuCl4 solution in a batch system, where small bubbles of gaseous reducing agent are formed and dispersed in the solution, could lead to coalescence of the CO bubbles. The changing bubble size resulting from coalescence leads to decreasing surface-to-volume ratio, which can influence the reduction rate at different locations in the batch reactor leading to nanoparticle polydispersity. The use of microfluidic reactors can alleviate this problem. Taifur-Rahman et al. utilized CO in a membrane-based droplet microfluidic system to produce 10 nm gold nanoparticles, as well as grow Au shells on silica nanoparticles with pre-attached gold nanoparticle seeds.10 The residence time was controlled by the gas–liquid contact time and was much larger than the calculated mixing time, enabling controlled dosing of CO into each droplet.In this study, we present a simple method of producing gold nanoparticles in a coiled flow inverter (CFI) reactor at room temperature via CO reduction. Such a multiphase microfluidic reactor with a large surface-to-volume ratio is expected to enhance the mass transfer between gas and liquid.11 Additionally, the combination of segmented flow and CFI should improve mixing and narrow the residence time distribution,12 resulting in more monodisperse gold nanoparticles.
Experimental
All the chemicals were purchased from Sigma-Aldrich and used without any further treatment or purification. A schematic of the segmented flow system utilised in this work is shown in Fig. S1.† A CFI made of fluorinated ethylene propylene (FEP) tubing, 1 mm internal diameter (I.D.), 2 mm outside diameter (O.D.), 3.6 m length (VICI Jour) was utilized. There were 100 coils in total formed on a PTFE support plate (9.5 cm × 7.5 cm) and the coil diameter was 1 cm. A 90° bend between successive coiled sections, each one containing 5 coils, produced a compact design, with the added advantage of improving mixing and residence time distribution.13 An aqueous HAuCl4 solution was mixed with CO gas (99.5%, BOC gases) through a Tefzel T-junction (0.5 mm I.D., P632, IDEX), which then formed segmented flow inside the CFI at atmospheric pressure. The liquid flow rate was fixed at 0.5 ml min−1 using a syringe pump (Legato 270P, KD Scientific; 25 ml glass syringe, Mod 1025 Syr 25Ml Tll Terminus, Biochrom Ltd.) and different liquid to gas volumetric flow rate ratios (2 to 20) were attained by tuning the gas flow rate in the range of 0.025 to 0.25 ml min−1 with a mass flow controller (EL Flow, Bronkhorst). The residence time was adjusted to 3–5 min by altering only the gas flow rate. The flow inside the reactor was regarded as plug flow, since the liquid slugs where mixing occurred by recirculation, were isolated from each other due to the wall hydrophobicity. The residence time was measured based on the time for one slug to pass through the whole CFI. The solution pH was measured by a pH meter (SevenCompact™ pH/Ion S220).UV-Vis absorption spectra of gold nanoparticles were recorded immediately after collection from the reactor using an Ocean Optics UV-Vis spectrometer (USB 2000+ spectrometer and DT-Mini-2-GS light source). The resulting gold nanoparticle size was measured by differential centrifugal sedimentation (DCS) (CPS 24000 Disc Centrifuge, CPS Instruments); the total number of particles counted was in the order of 1011. Transmission electron microscopy (TEM) (Jeol 2010) at 200 kV was used to determine morphology, particle size and polydispersity (defined as standard deviation over the average particle size). Zeta potential measurement was performed with a data acquisition time of 10 s with a dynamic light scattering apparatus (DLS) (DelsaMax-Pro, Beckman Coulter) using the supplied flow cell system in batch mode at 22 °C. For the synthesis at different initial pH, the precursor was mixed with K2CO3 one day in advance to ensure the gold species achieved equilibrium. ATR-FTIR spectroscopy of liquid sample was performed using a Perkin-Elmer 1605 FT-IR spectrometer with a resolution of 0.5 cm−1 and wavenumber range 0–5000 cm−1. Raman spectra were obtained with a Renishaw Raman inVia microscope with a 633 nm He–Ne excitation laser (1.9 eV, 0.7 mW, pore size ∼4.4 μm2). A Rhodamine 6G (Rh6G) solution with concentration 4 × 10−6 M was mixed with gold nanoparticle solutions at a volume ratio of 2/3 and then stirred for 20 min to allow enough time for the Rh6G molecules to bind onto the gold nanoparticle surface. The mixed solutions were dropped onto glass substrates and dried at ambient conditions to create surface-enhanced Raman scattering (SERS) hotspots. The commercial citrate-capped gold nanoparticles used for comparison were from BBI Solutions (UK).
Results and discussion
Synthesis of gold nanoparticles without capping agents
| 2[AuCl4−x(OH)x]− + 3CO + 3H2O → 2Au0 + 3CO2 + 6H+ + 2xOH− + 2(4−x)Cl− | (1) |
The conditions of 0.54 mM initial concentration of gold precursor and the highest liquid to gas volumetric flow rate ratio (L/G) of 20, gave the lowest CO/Au3+ inlet molar flow rate ratio of 3.8. Considering a stoichiometry of CO/Au3+ for the reduction reaction of 3/2, it is evident that the CO available is in significant excess for all experiments. Thus, the gaseous CO acted as a reservoir to continuously dose the liquid slugs throughout the whole reduction process. Fig. 1 shows the product characterization results from a 0.27 mM HAuCl4 solution at different liquid to gas flow rate ratios, at room temperature (maintained between 20–22 °C). Nonspherical particles were observed which is consistent with previous literature,9,15 rather than spherical shape from citrate reduction. The lowest polydispersity (based on Differential Centrifugal Sedimentation (DCS) analysis) observed was <5%, which is smaller than the CO-produced gold nanoparticles in batch by Young et al.9 with similar initial concentration of gold precursor (0.3 mM, polydispersity of 11%), likely due to the good mixing provided by the recirculation in the liquid slugs11 and helical flow inversion.12 Experiments with a slightly larger (internal diameter 1.5 mm) glass tube with 4 coils and no flow inversion (see section 2, ESI†), using the same experimental conditions (residence time, reactant concentration and liquid to gas volumetric flow rate ratio) led to similar nanoparticle size (7.8 nm in coiled glass tube vs. 7.7 nm in FEP CFI). However, the polydispersity increased from 4.1% in the CFI to 14.2% in the glass tube, likely due to the inferior mixing in the slugs and the existence of liquid film.
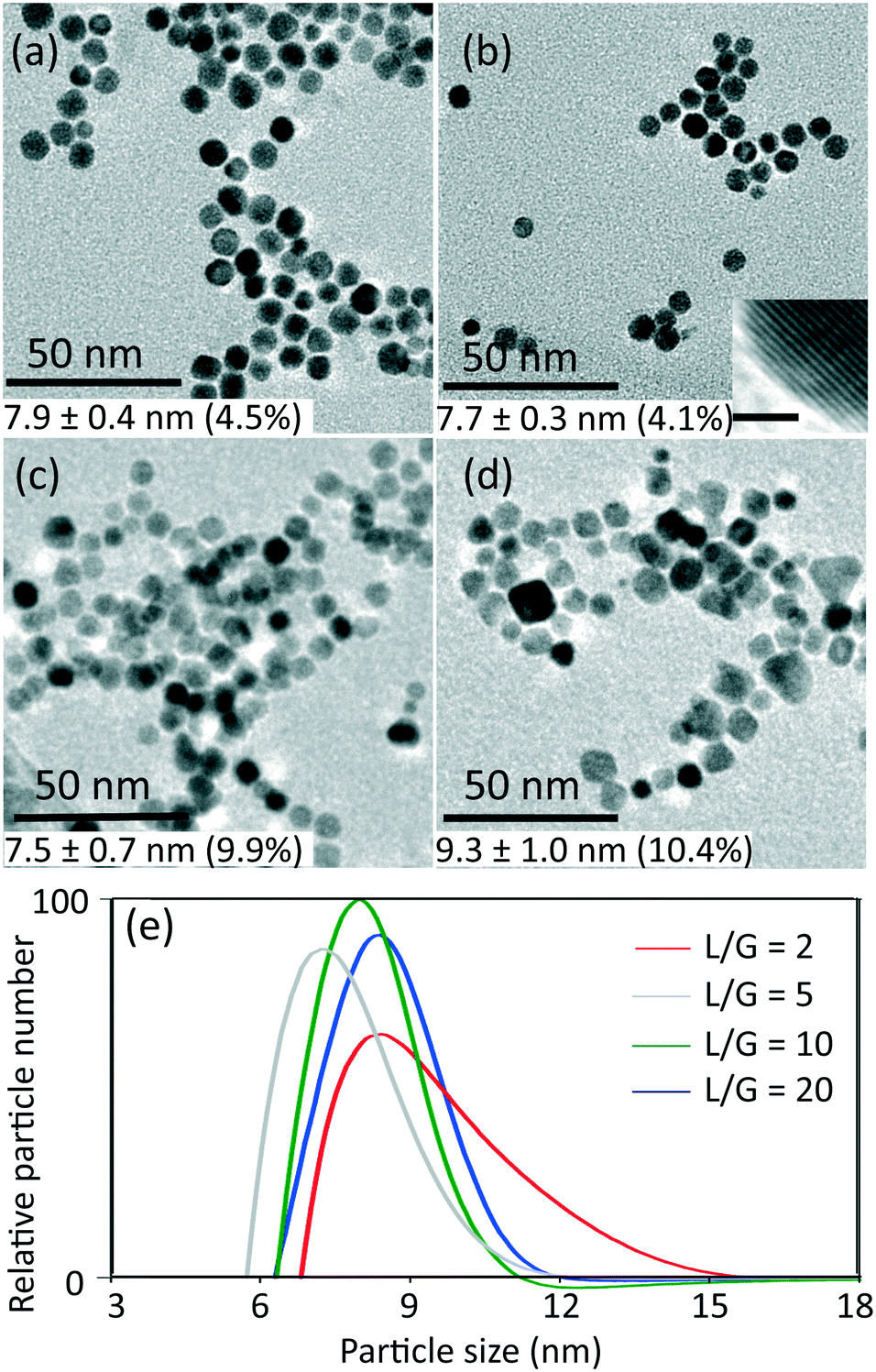 | ||
| Fig. 1 TEM images of gold nanoparticles synthesized at different liquid to gas volumetric flow rate ratios (L/G): a) 20 (residence time 5 min), b) 10 (residence time 4.4 min), c) 5 (residence time 4.2 min) and d) 2 (residence time 3.4 min) with inlet concentration of HAuCl4 0.27 mM, at room temperature. Insert in b) shows the lattice plane spacing of a gold nanoparticle, which indicates Au [111] surface (scale bar 1 nm). e) Particle size distribution obtained from DCS (normalised by area in each curve) for samples obtained at the L/G indicated. The values of average particle size and polydispersity in the TEM images (a–d) are obtained from DCS (e). | ||
For L/G ratio in the range of 20 to 2, the flow patterns remained as segmented flow, but with different liquid slug lengths (see Fig. S3, ESI†). Increasing the gas flow rate with constant liquid flow rate led to more frequent and shorter liquid slugs with larger interfacial area-to-volume ratio, which enhanced the mass transfer between gas and liquid phase. The volumetric mass transfer coefficient increased (see section 3, ESI†) as the L/G decreased from 20 to 2, leading to faster dosing of CO into the liquid phase.
The effect of initial concentration of HAuCl4 on particle size and size distribution was studied and the results are shown in Fig. 2. Since DCS uses higher sample volume (total counted particle number is much larger than those observed in TEM), most of the particle size and size distribution measurements were based on DCS for higher accuracy. However, since DCS is limited to gold nanoparticles above 3 nm,16 the particle size at 0.030 mM concentration of gold precursor was based on TEM (see Fig. S4, ESI†).
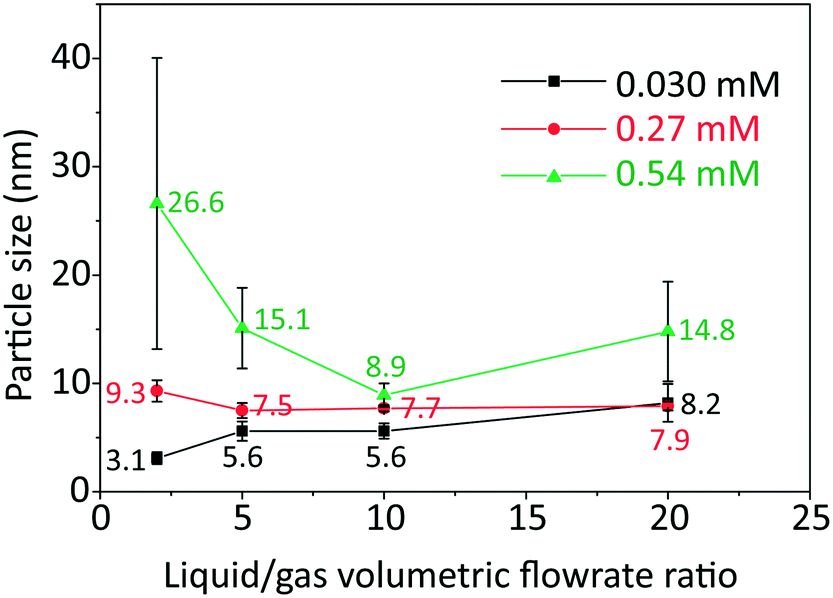 | ||
| Fig. 2 Particle size (labelled near the points) and standard deviation of size distribution (denoted by error bar) obtained from TEM and DCS (Fig. S4 and S5, ESI†) as a function of liquid to gas volumetric flow rate ratio, with inlet concentrations of HAuCl4 indicated, at room temperature and residence time 3–5 min. | ||
As the L/G ratio decreases, the mass transfer becomes faster, as discussed above. Thus, a higher overall CO concentration in the slugs is expected for low L/G ratios. As the increase in concentration of CO acidifies the solution,14 the pH decreases for lower values of the L/G ratio. In light of the above, we can interpret the trends of particle size in Fig. 2, based on the effect of pH on the gold species present in solution.
For HAuCl4 concentrations of 0.54 mM and 0.27 mM, the observed trend of the nanoparticle size and polydispersity vs. the L/G ratio (low L/G ratios lead to low pH) is analogous to that reported by Kettemann et al.17 who observed a minimum in particle size and polydispersity when varying the pH of the Turkevich synthesis. At low pH the reactivity of HAuCl4 is high,8 hence the separation of nucleation and growth steps is unlikely. This results in polydisperse and large particles. As the L/G ratio increases, the pH increases and this enables a decoupling of nucleation and growth steps, leading to smaller size and more monodisperse nanoparticles. If one increases the pH beyond a certain point, the redistribution of the precursor between AuCl4− and AuCl4−xOHx− changes in favor of the more hydroxylated forms.8 The latter are less reactive and lead to larger nanoparticles (since nucleation has slowed down) and higher polydispersity (due to overlapping of nucleation and growth). So, the minimum in particle size and polydispersity observed in Fig. 2 for 0.54 mM and 0.27 mM precursor concentration is attributed to optimal pH conditions. The experiment at 0.54 mM HAuCl4 and L/G = 10 showing a minimum particle size (8.9 ± 1.1 nm, 12.4%) was repeated to confirm that a minimum truly existed. The repeated experiment produced a size and polydispersity of 8.6 ± 1.4 nm, 16.5%.
One should also note that the position of the minimum in the nanoparticle size vs. L/G data in Fig. 2, shifts to the left for decreasing precursor concentration. This agrees with the proposed interplay between the nucleation/growth mechanism and mass transfer: the initial pH of the solution is higher for lower initial concentration of HAuCl4, hence the pH that minimizes the particle dispersity by separating nucleation and growth is reached at a lower value of L/G ratio. For a HAuCl4 concentration of 0.03 mM the shift of the minimum to the left causes the corresponding L/G ratio to coincide with (or be lower than) the lowest value of L/G employed; at L/G = 2 the smallest nanoparticle size was obtained (3.1 ± 0.5 nm).
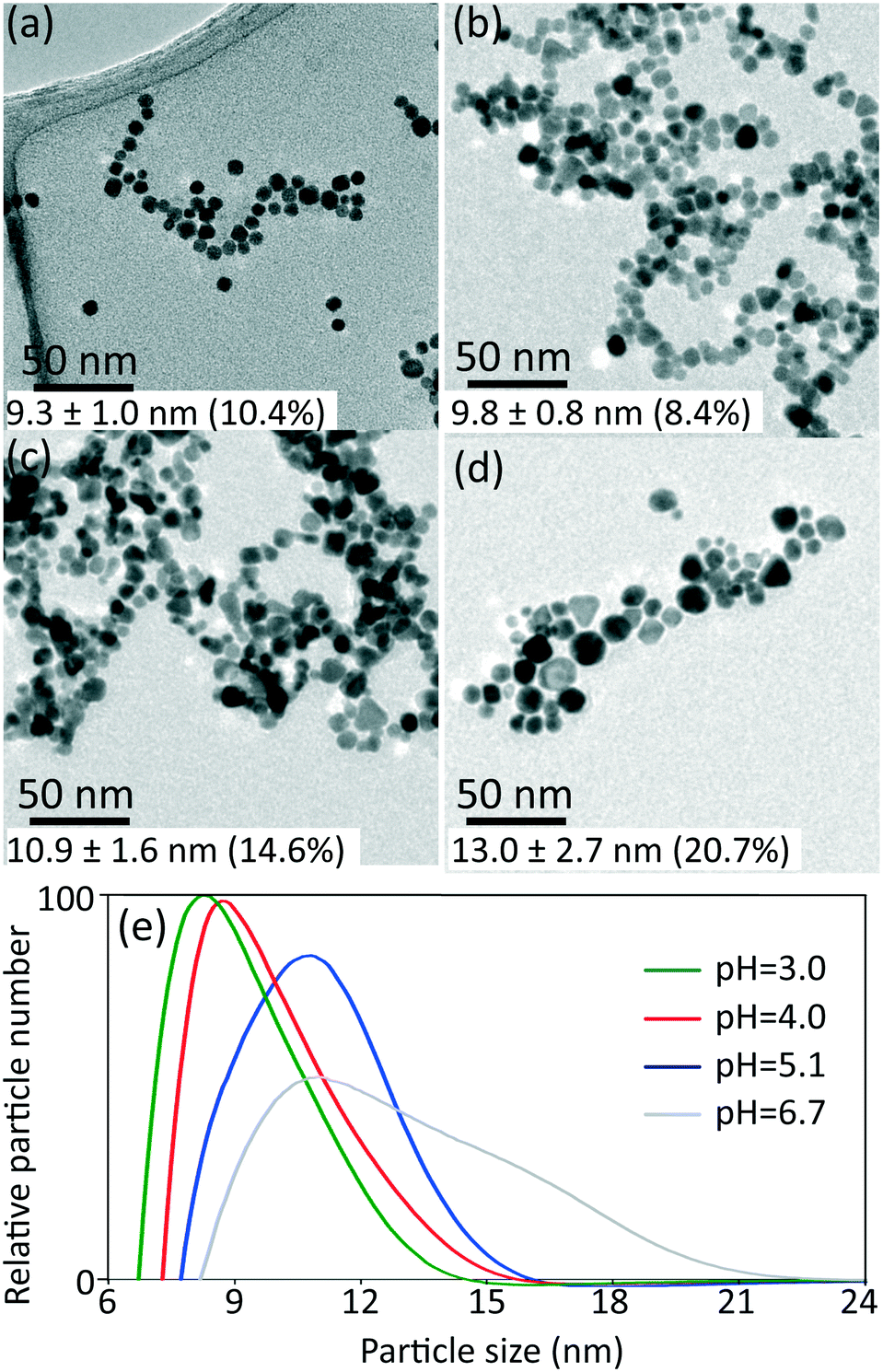 | ||
| Fig. 3 TEM images of gold nanoparticles synthesized at different initial pH: a) 3.0, b) 4.0, c) 5.1 and d) 6.7, with inlet concentration of HAuCl4 0.27 mM, liquid to gas volumetric flow rate ratio 2 and room temperature. e) Particle size distribution from DCS (normalised by area in each curve) for samples obtained at initial pH indicated. The values of average particle size and polydispersity indicated in the TEM images (a–d) are obtained from DCS (e). | ||
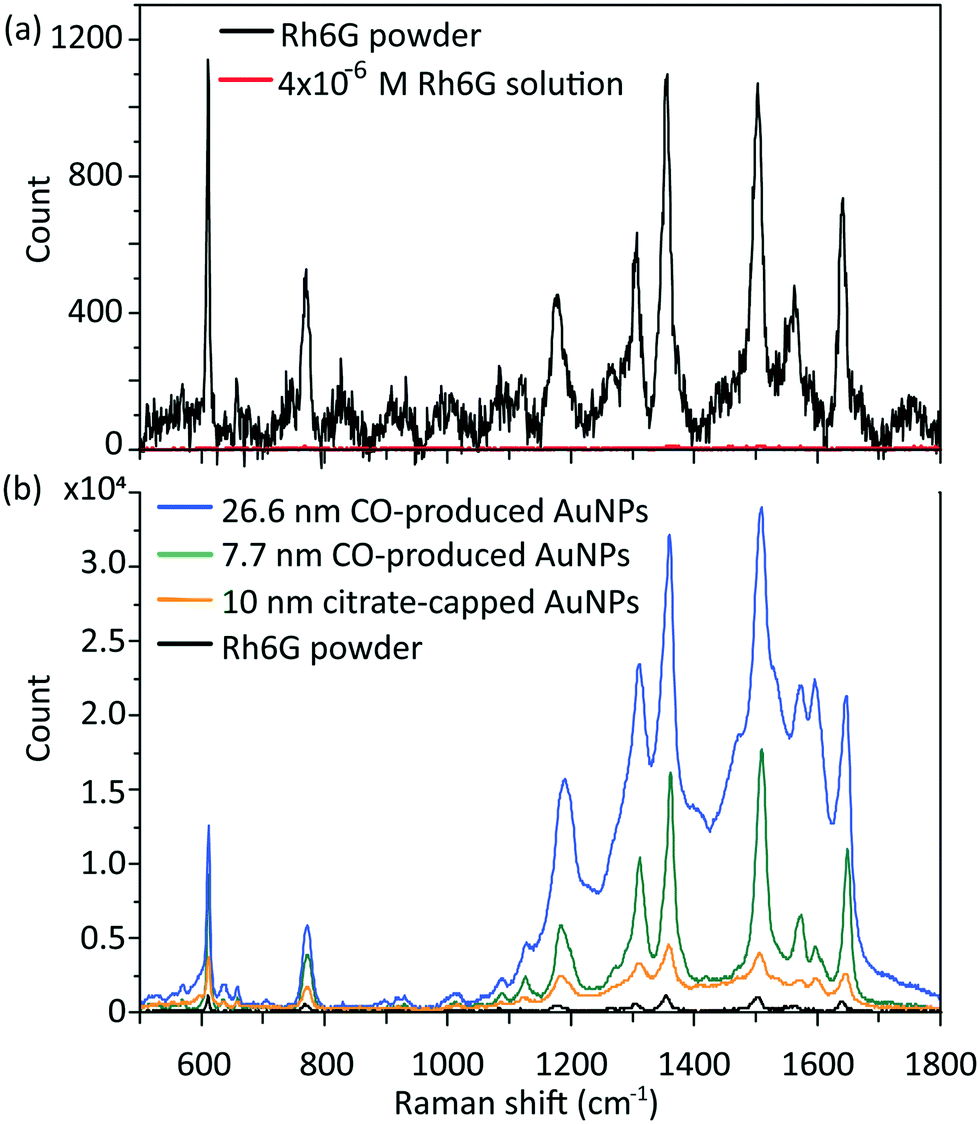 | ||
| Fig. 4 (a) Typical Raman signal of Rh6G powder (in black) and 4 × 10−6 M Rh6G solution (in red, near background); (b) SERS spectra of 4 × 10−6 M Rh6G solution enhanced by 26.6 nm CO-produced gold nanoparticles (in blue), 7.7 nm CO-produced nanoparticles (in green) and 10 nm citrate-capped gold nanoparticles (in orange), compared to Rh6G powder (in black) without enhancement. | ||
Synthesis of gold nanoparticles with various capping agents
The synthesized gold nanoparticles without any capping agents (shown in Fig. 1c) were monitored by UV-Vis and proven to be stable for 1 week at room temperature and for at least one month when stored at 4 °C. The reason for the stability of the uncapped nanoparticles is not clear. No CO was found on the surface of gold nanoparticles (as measured by IR spectroscopy, see Fig. S6†).Longer shelf lives could be obtained by adding different capping agents, either before or immediately after the synthesis. Fig. 5 shows gold nanoparticles with different sizes obtained using different capping agents, namely trisodium citrate, polysorbate 80 (TWEEN 80), oleylamine and poly(ethyleneglycol) 2-mercaptoethyl ether acetic acid (thiol-PEG-COOH). All experiments were performed at room temperature apart from the oleylamine-capped gold nanoparticles synthesis (for synthesis details see section 6, ESI†). The size of the citrate-capped gold nanoparticles (11.6 ± 1.6 nm, 14.0%) shown in Fig. 5a was higher compared to the no-capping-agent system (7.7 ± 0.3 nm, 4.1% in Fig. 1b) for the same operation conditions. This is because adding the alkaline citrate increases the pH of the solution, which leads to a shift of equilibrium to the gold species with less reactivity8 and decrease of the nucleation rate.9 With fewer nuclei forming and higher amount of gold precursor available for growth, larger particle size was obtained. TWEEN 80 was used to change the surface tension of the solvent as a non-ionic surfactant, leading to smaller bubble size (insert in Fig. 5b) compared to that when no capping agents were used (Fig. S3†). The enhanced mass transfer between gas and liquid due to the increased surface-to-volume ratio and the good protecting ability of TWEEN 80 produced gold seeds with size of 2.7 ± 0.4 nm (14.6%) at residence time 4.2 min and TWEEN 80/gold precursor ratio of 0.5 (Fig. S7†). As TWEEN 80 has been shown to be a weaker reducing agent (reaction time of 6 h at room temperature) compared to CO for gold nanoparticles synthesis,23 seed particles subsequently grew to 4.0 ± 0.4 nm (9.9%) inside the container reduced by excess TWEEN 80 within 8 days. Stability of the gold nanoparticles after growth (4.0 nm) was more than 1 month at room temperature.
 | ||
| Fig. 5 TEM images of gold nanoparticles capped by a) trisodium citrate premixed with HAuCl4 (insert shows the flow pattern in the CFI), b) TWEEN 80 (insert shows the flow pattern in the CFI), c) oleylamine in octane and d) thiol-PEG-COOH. For experimental details see ESI.† The values of average particle size and polydispersity were obtained by TEM. | ||
Using the same set-up, hydrophobic gold nanoparticles could also be synthesized by using oleylamine as a capping agent at 100 °C. The different synthesis procedure, including higher reaction temperature, led to spherical shape gold nanoparticles rather than gold nanowires formed via CO at 60 °C in previous batch synthesis.24 A TEM image of the synthesized gold nanoparticles with size of 8.6 ± 0.6 nm (7.4%) is shown in Fig. 5c. A comparative experiment using N2 instead of CO was conducted under the same experimental conditions (Fig. S8, ESI†). Nanowires formed without CO as observed for oleylamine reduction in previous literature.25 Thiol-PEG-COOH gold nanoparticles were also synthesized, because they are widely used in drug and gene delivery26,27 as relatively non-toxic carriers. They could be obtained with our system by post-synthetic modification methods. To synthesize PEG-capped gold nanoparticles, poly(sodium 4-styrenesulfonate) (PSS) was used during the CO reduction stage to stabilize the gold nanoparticles temporarily. Subsequently, thiol-PEG-COOH was added into the gold nanoparticle solution obtained and stirred overnight. Because of the stronger binding strength of thiols, the thiol-PEG-COOH was adsorbed onto the gold nanoparticles without size alteration (compared to PSS-capped gold nanoparticles with size of 7.8 ± 1.2 nm, 15.4% in Fig. S9b, ESI†), and no further aggregation was observed (Fig. 5d).
Conclusions
We demonstrated a room temperature synthesis of gold nanoparticles with carbon monoxide in a microfluidic segmented flow system. The reducing strength of CO is between that of NaBH4 and citrate, which aids the rapid synthesis of sub-10 nm gold nanoparticles. The use of microfluidic segmented flow in a coiled flow inverter under optimised conditions can lead to monodisperse particles (<5%). Our procedure can produce gold nanoparticles, whose size and hydrophilicity can be tuned by capping agents like citrate, TWEEN 80, oleylamine and thiol-PEG-COOH. Since this approach can produce highly controlled and impurity-free gold nanoparticles, it provides advantages for surface-enhanced Raman scattering and potentially many other applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the EPSRC for financial support (EP/M015157/1) through the Manufacturing Advanced Functional Materials (MAFuMa) scheme. HH acknowledges the financial support from UCL-CSC scholarship. We thank Dr. Tom Macdonald for his support with the IR measurement.References
- M.-C. Daniel and D. Astruc, Gold nanoparticles: assembly, supramolecular chemistry, quantum-size-related properties, and applications toward biology, catalysis, and nanotechnology, Chem. Rev., 2004, 104(1), 293–346 CrossRef CAS PubMed.
- N. T. K. Thanh and Z. Rosenzweig, Development of an aggregation-based immunoassay for anti-protein A using gold nanoparticles, Anal. Chem., 2002, 74(7), 1624–1628 CrossRef CAS PubMed.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Gold nanoparticles: Interesting optical properties and recent applications in cancer diagnostics and therapy, Nanomedicine, 2007, 2(5), 681–693 CrossRef CAS PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, Synthesis of thiol-derivatised gold nanoparticles in a two-phase liquid–liquid system, J. Chem. Soc., Chem. Commun., 1994, 0(7), 801–802 RSC.
- J. Turkevich, P. C. Stevenson and J. Hillier, A study of the nucleation and growth processes in the synthesis of colloidal gold, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- A. Leifert, Y. Pan-Bartnek, U. Simon and W. Jahnen-Dechent, Molecularly stabilised ultrasmall gold nanoparticles: Synthesis, characterization and bioactivity, Nanoscale, 2013, 5(14), 6224–6242 RSC.
- J. Wagner, T. R. Tshikhudo and J. M. Koehler, Microfluidic generation of metal nanoparticles by borohydride reduction, Chem. Eng. J., 2008, 135, S104–S109 CrossRef CAS.
- M. Wuithschick, A. Birnbaum, S. Witte, M. Sztucki, U. Vainio, N. Pinna, K. Rademann, F. Emmerling, R. Kraehnert and J. Polte, Turkevich in new robes: Key questions answered for the most common gold nanoparticle synthesis, ACS Nano, 2015, 9(7), 7052–7071 CrossRef CAS PubMed.
- J. K. Young, N. A. Lewinski, R. J. Langsner, L. C. Kennedy, A. Satyanarayan, V. Nammalvar, A. Y. Lin and R. A. Drezek, Size-controlled synthesis of monodispersed gold nanoparticles via carbon monoxide gas reduction, Nanoscale Res. Lett., 2011, 6(1), 428 CrossRef PubMed.
- M. Taifur-Rahman, P. G. Krishnamurthy, P. Parthiban, A. Jain, C. P. Park, D.-P. Kim and S. A. Khan, Dynamically tunable nanoparticle engineering enabled by short contact-time microfluidic synthesis with a reactive gas, RSC Adv., 2013, 3(9), 2897–2900 RSC.
- A. Günther, M. Jhunjhunwala, M. Thalmann, M. A. Schmidt and K. F. Jensen, Micromixing of miscible liquids in segmented gas-liquid flow, Langmuir, 2005, 21(4), 1547–1555 CrossRef PubMed.
- M. Mridha and K. Nigam, Coiled flow inverter as an inline mixer, Chem. Eng. Sci., 2008, 63(6), 1724–1732 CrossRef CAS.
- S. K. Kurt, F. Warnebold, K. D. Nigam and N. Kockmann, Gas-liquid reaction and mass transfer in microstructured coiled flow inverter, Chem. Eng. Sci., 2017, 169, 164–178 CrossRef CAS.
- L. A. Pretzer, Q. X. Nguyen and M. S. Wong, Controlled growth of sub-10 nm gold nanoparticles using carbon monoxide reductant, J. Phys. Chem. C, 2010, 114(49), 21226–21233 CrossRef CAS.
- Y. Kang, X. Ye and C. B. Murray, Size- and shape-selective synthesis of metal nanocrystals and nanowires using CO as a reducing agent, Angew. Chem., Int. Ed., 2010, 49(35), 6156–6159 CrossRef CAS PubMed.
- H. du Toit, T. Macdonald, H. Huang, I. P. Parkin and A. Gavriilidis, Continuous flow synthesis of citrate capped gold nanoparticles using UV induced nucleation, RSC Adv., 2017, 7(16), 9632–9638 RSC.
- F. Kettemann, A. Birnbaum, S. Witte, M. Wuithschick, N. Pinna, R. Kraehnert, K. Rademann and J. Polte, The missing piece of the mechanism of the Turkevich method: The critical role of citrate protonation, Chem. Mater., 2016, 28(11), 4072–4081 CrossRef CAS.
- X. Ji, X. Song, J. Li, Y. Bai, W. Yang and X. Peng, Size control of gold nanocrystals in citrate reduction: The third role of citrate, J. Am. Chem. Soc., 2007, 129(45), 13939–13948 CrossRef CAS PubMed.
- A. Campion and P. Kambhampati, Surface-enhanced Raman scattering, Chem. Soc. Rev., 1998, 27(4), 241–250 RSC.
- X.-Y. Zhang, A. Hu, T. Zhang, W. Lei, X.-J. Xue, Y. Zhou and W. W. Duley, Self-assembly of large-scale and ultrathin silver nanoplate films with tunable plasmon resonance properties, ACS Nano, 2011, 5(11), 9082–9092 CrossRef CAS PubMed.
- E. Le Ru and P. Etchegoin, Principles of Surface-Enhanced Raman Spectroscopy: and related plasmonic effects, Elsevier, Oxford, 2008 Search PubMed.
- N. R. Jana and T. Pal, Anisotropic metal nanoparticles for use as surface-enhanced Raman substrates, Adv. Mater., 2007, 19(13), 1761–1765 CrossRef CAS.
- T. Premkumar, D. Kim, K. Lee and K. E. Geckeler, Polysorbate 80 as a tool: Synthesis of gold nanoparticles, Macromol. Rapid Commun., 2007, 28(7), 888–893 CrossRef CAS.
- Y. Kang, X. Ye and C. B. Murray, Size-and shape-selective synthesis of metal nanocrystals and nanowires using CO as a reducing agent, Angew. Chem., 2010, 122(35), 6292–6295 CrossRef.
- H. Feng, Y. Yang, Y. You, G. Li, J. Guo, T. Yu, Z. Shen, T. Wu and B. Xing, Simple and rapid synthesis of ultrathin gold nanowires, their self-assembly and application in surface-enhanced Raman scattering, Chem. Commun., 2009,(15), 1984–1986 RSC.
- J. M. Bergen, H. A. Von Recum, T. T. Goodman, A. P. Massey and S. H. Pun, Gold nanoparticles as a versatile platform for optimizing physicochemical parameters for targeted drug delivery, Macromol. Biosci., 2006, 6(7), 506–516 CrossRef CAS PubMed.
- V. Dixit, J. Van den Bossche, D. M. Sherman, D. H. Thompson and R. P. Andres, Synthesis and grafting of thioctic acid− PEG− folate conjugates onto Au nanoparticles for selective targeting of folate receptor-positive tumor cells, Bioconjugate Chem., 2006, 17(3), 603–609 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8re00351c |
| This journal is © The Royal Society of Chemistry 2019 |