
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoactive chlorin e6 is a multifunctional modulator of amyloid-β aggregation and toxicity via specific interactions with its histidine residues†
Guy
Leshem‡
a,
Michal
Richman‡
a,
Elvira
Lisniansky
a,
Merav
Antman-Passig
a,
Maram
Habashi
a,
Astrid
Gräslund
b,
Sebastian K. T. S.
Wärmländer
*b and
Shai
Rahimipour
 *a
*a
aDepartment of Chemistry, Bar-Ilan University, Ramat-Gan 5290002, Israel. E-mail: rahimis@biu.ac.il
bDepartment of Biochemistry and Biophysics, Arrhenius Laboratories, Stockholm University, S-106 91 Stockholm, Sweden. E-mail: sebastian.warmlander@dbb.su.se
First published on 3rd October 2018
Abstract
The self-assembly of Aβ to β-sheet-rich neurotoxic oligomers is a main pathological event leading to Alzheimer's disease (AD). Selective targeting of Aβ oligomers without affecting other functional proteins is therefore an attractive approach to prevent the disease and its progression. In this study, we report that photodynamic treatment of Aβ in the presence of catalytic amounts of chlorin e6 can selectively damage Aβ and inhibit its aggregation and toxicity. Chlorin e6 also reversed the amyloid aggregation process in the dark by binding its soluble and low molecular weight oligomers, as shown by thioflavin T (ThT) fluorescence and photoinduced cross-linking of unmodified protein (PICUP) methods. Using HSQC NMR spectroscopy, ThT assays, amino acid analysis, SDS/PAGE, and EPR spectroscopy, we show that catalytic amounts of photoexcited chlorin e6 selectively damage the Aβ histidine residues H6, H13, and H14, and induce Aβ cross-linking by generating singlet oxygen. In contrast, photoexcited chlorin e6 was unable to cross-link ubiquitin and α-synuclein, demonstrating its high selectivity for Aβ. By binding to the Aβ histidine residues, catalytic amounts of chlorin e6 can also inhibit the Cu2+-induced aggregation and toxicity in darkness, while at stoichiometric amounts it acts as a chelator to reduce the amount of free Cu2+. This study demonstrates the great potential of chlorin e6 as a multifunctional agent for treatment of AD, and shows that the three N-terminal Aβ histidine residues are a suitable target for Aβ-specific drugs.
Introduction
Misfolding of proteins and their subsequent aggregation to cross β-sheet conformation are the hallmark of more than thirty different devastating diseases, including many neurodegenerative and peripheral diseases, such as Alzheimer's, Parkinson's, and Huntington's diseases and type II diabetes. Among these diseases, Alzheimer's disease (AD) is the most prevalent neurodegenerative disease with approximately 35 million people diagnosed worldwide. The neuropathology of AD is characterized by two types of lesions – senile plaques and neurofibrillary tangles (NFTs). The NFTs are composed of aberrantly phosphorylated tau, while the plaques consist mainly of aggregates of β-amyloid (Aβ) – a 39–43-amino acid protein generated from enzymatic degradation of the amyloid precursor protein (APP).1 The self-assembly and aggregation of Aβ to form soluble oligomers and fibrils are strongly associated with the onset of the disease, where the soluble oligomers are believed to be the most toxic species to the neurons.2 Although the exact mechanism by which Aβ oligomers cause neurotoxicity is still under scientific debate, specific targeting of toxic Aβ oligomers has shown promising therapeutic results.3 During the past two decades, numerous approaches have been envisioned and adopted to reduce the amount of Aβ in AD patients and to inhibit Aβ aggregation and accumulation.2b,4 These efforts include the development of β- and γ-secretase inhibitors that target the production of Aβ from APP, anti-Aβ immunotherapy, chelation therapy, and β-sheet blockers that inhibit the oligomerization of soluble Aβ. However, none of these approaches has been approved yet for clinical application, mainly due to nonspecific effects of the agents on other important biological targets.Since many misfolded proteins have important biological activities in their correctly folded conformations, selective targeting of the specific protein at its pathological site might reduce systemic toxicity and increase patient safety. Photodynamic therapy (PDT) has been used for decades in cancer therapy and different skin diseases to improve drug selectivity.5 In PDT a combination of light, oxygen, and a photosensitizer is used to generate reactive oxygen species (ROS), mainly singlet oxygen (1O2). ROS and especially HO• radicals are very powerful oxidizing agents that can oxidize and degrade biopolymers such as DNA and proteins. The free radicals have a very short half-life, and are therefore most effective along a path of <15 Å.6 Oxidation of amino acid residues in proteins leads to generation of cross-linked aggregates, cleavage of the polypeptide chain, and modifications of the proteins and peptide fragments.7 Thus, a system generating ROS at well-defined loci should be capable of producing targeted local damage at the subcellular and/or molecular level. Selective damage at the subcellular level could be easily achieved via a focused light source of suitable wavelength and a corresponding photosensitizer. Superior selectivity could further be achieved through conjugation of the photosensitizer to vectors that bind with high affinity and selectivity to an over-expressed protein or biomolecule on the cell surface.8
We and others have recently demonstrated that photo-irradiation of Aβ in the presence of a photosensitizer dramatically attenuates its aggregation and toxicity in vitro as well as in a Drosophila model.9 To further increase the specificity and selectivity, the photosensitizers were cleverly conjugated to an Aβ binding peptide such as KLVFF.9c,f,g Nevertheless, conjugation of a photosensitizer to a targeting peptide may alter its pharmacokinetics and bioavailability, such as the blood–brain barrier permeability, highlighting the need for more selective photosensitizers. Moreover, little attention has been given to the interactions between the photosensitizers and Aβ at a molecular level and the mechanism by which they attenuate Aβ aggregation under light and dark conditions.
In this study, we demonstrate that the photosensitizer chlorin e6 (Ce6) binds Aβ40 with high affinity and selectivity, and show that the ability of Ce6 to inhibit Aβ's aggregation and toxicity significantly increases upon illumination with light. We also studied the mechanism by which Ce6 selectively inhibits Aβ aggregation and toxicity, and explored molecular-level modes of interaction between Ce6 and Aβ. Ce6 and its derivatives are well known for their biochemical and biophysical properties and photo-induced antitumor activity.10 Unlike porphyrins with 22 π electrons and 4 pyrroles, chlorins are characterized by 20 π electrons at their cores that are composed of three pyrroles and one pyrroline bridged by four sp2 hybridized carbon atoms without the presence of a metal (Scheme S1†). It has minimal dark toxicity, high stability and solubility in injectable solutions, and exhibits a high quantum yield of singlet molecular oxygen production.11 An injectable formulation of a Ce6–polyvinylpyrrolidone complex (Photolon®), was reported to easily cross the intact blood–brain barrier in rats and accumulate in tissues of the central nervous system,12 while mono-L-aspartyl Ce6 was approved in Japan in 2003 as a PDT agent for treatment of lung cancer.13
Results and discussion
Anti-aggregation activity of Ce6 under dark and light conditions
We used a fluorescent titration experiment and the thioflavin T (ThT) assay to examine the association of Ce6 with Aβ and its ability to inhibit Aβ amyloid aggregation. The binding constant between Ce6 and Aβ and number of the binding sites were determined according to Bose et al.14 Addition of increasing concentrations of Ce6 (1–100 μM) to a constant concentration of Aβ (50 μM) dose-dependently quenched the Aβ fluorescence at 316 nm (excitation at 280 nm, Fig. S1a†). Fig. S1b† shows the linear quenching of Aβ fluorescence by increasing concentration of Ce6 with a Y-axis intercept of 0.9977, indicating single class of interaction between Aβ and Ce6.14 The fluorescence studies also provided a binding constant of 5.5 μM between Ce6 and Aβ and the number of binding sites of 0.995 (Fig. S1c†). The antiamyloidogenic activity of Ce6 was determined by exposing monomeric Aβ to increasing concentrations of Ce6 for 1 h in the dark, followed by exposure to visible light. Samples kept in the dark were used as controls. The extent of Aβ aggregation was then determined after 72 h incubation using ThT fluorescence in which a large increase in fluorescence is generated upon binding of ThT to cross β-sheet structures.15 While incubation of Aβ with Ce6 in the dark dose-dependently decreased the ThT fluorescence intensity of the samples (EC50 = 2 μM), incubation following visible light exposure significantly increased the antiamyloidogenic potency of Ce6 (EC50 = 0.4 μM; Fig. 1a).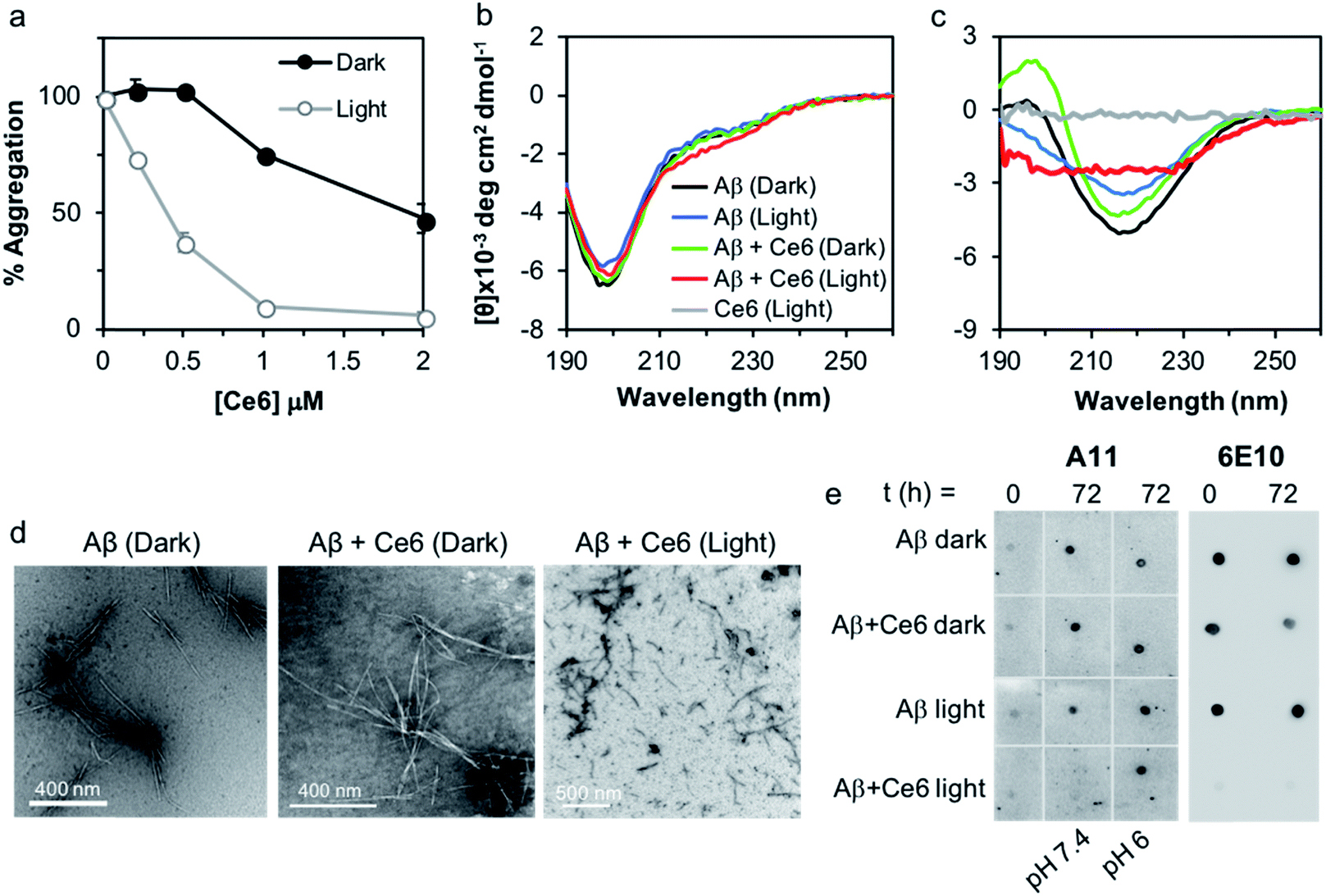 | ||
| Fig. 1 The inhibitory activity of Ce6 on Aβ aggregation. (a) Increasing amounts of Ce6 (0.2–2 μM) were incubated in PBS (50 mM, pH 7.4) with monomeric Aβ40 (20 μM) in darkness or photoirradiated for 1 h. Samples were incubated with constant shaking for further 72 h, and the extent of Aβ aggregation was then determined by the ThT assay. Results are mean ± SD of three experiments (n = 3 each). (b and c) Effect of photoexcited Ce6 on the secondary structure of Aβ. Time dependent far-UV CD spectra of freshly prepared Aβ40 monomer (10 μM) incubated in the absence or presence of photoexcited Ce6 (10 μM) in phosphate buffer (50 mM, pH 7.4) and analyzed after (b) 0 and (c) 72 h of aging. (d) TEM images of Aβ (20 μM) incubated in the absence or presence of Ce6 (2 μM) in darkness or after 1 h photoirradiation following 72 h incubation in darkness. Negatively stained samples are shown. (e) Effect of Ce6 and photoexcited Ce6 on antibody binding to Aβ at different pH values. Aβ40 (20 μM) was aged for 0 or 72 h in the absence or presence of Ce6 or photoexcited Ce6 (20 μM) at different pH values, spotted onto nitrocellulose membranes, and probed with either the A11 or 6E10 antibody. | ||
We also used circular dichroism (CD) spectroscopy to study the effect of Ce6 on the structural transformation of Aβ from random coil to β-sheet, both in the dark and after photoirradiation (Fig. 1b and c). Aging of monomeric Aβ for 72 h changed its conformation from random coil, characterized by a negative peak at 198 nm (Fig. 1b), to β-sheet conformation with a positive band around 198 nm and a negative band at 222 nm (Fig. 1c). Presence of an equimolar concentration of Ce6 in the mixture under dark conditions did not significantly affect this transition. In contrast, incubation of the Aβ/Ce6 mixture under photoirradiation conditions generated a distinct CD signal, which was very different from that of a β-sheet. Ce6 did not produce any CD signal by its own, and photoirradiation of Aβ alone had no significant effect on its structural transformation (Fig. 1c). These results indicate that photoexcited Ce6 prevents Aβ aggregation by inhibiting its structural transition to a β-sheet conformation.
The strong antiamyloidogenic activity of Ce6 under photoirradiation was confirmed by TEM, using the Aβ samples from the ThT assay. We found that aging of Aβ (20 μM) alone and in the presence of Ce6 (2 μM) generated well-defined fibrils under dark conditions. In contrast, aging of the Aβ and Ce6 mixtures under light generated aggregates that were morphologically different and significantly shorter (Fig. 1d).
Photoexcited Ce6 decreases the amount of Aβ toxic oligomers and disassembles aggregated Aβ
Next, we tested whether photoexcited Ce6 can modulate the self-assembly of Aβ to generate toxic oligomers, using a dot blot assay and the oligomer-specific polyclonal antibody A11. This conformational antibody was raised against oligomeric Aβ species and was found to reduce their toxicity.16 Aging of Aβ (20 μM) in darkness for 72 h, with or without Ce6 (20 μM), generated A11-reactive aggregates. This shows that stoichiometric amount of Ce6 has a small effect on Aβ oligomerization under dark conditions. In stark contrast, photoirradiation of the Aβ–Ce6 mixture dramatically reduced its reactivity to A11 (Fig. 1e). Interestingly, under light conditions Ce6 completely inhibits binding of the 6E10 antibody to both Aβ monomers (t = 0 h) and Aβ oligomers (t = 72 h), with some effect also on the Aβ oligomers under dark conditions (Fig. 1e). As the 6E10 antibody binds to the N-terminal Aβ1–17 segment,17 these results indicate that Ce6 also binds to this segment.Having demonstrated that Ce6 strongly binds to Aβ (Fig. 1a and S1†), we examined whether it can disassemble aggregated or fibrillar Aβ under dark as well as light conditions. Aβ40 was aged for three days to generate aggregates and fibrils with strong ThT fluorescence signal. The aggregated Aβ was then incubated with Ce6 for 1 h, either in darkness or under light irradiation. The amount of remaining aggregated Aβ was then determined by ThT assay after 72 h of incubation. Under dark conditions, Ce6 dose-dependently disaggregated the Aβ aggregates. However, disaggregation of Aβ aggregates by Ce6 under light conditions was observed only for higher Ce6 concentration (50 μM; Fig. 2a). Surprisingly, at low concentration, photoexcited Ce6 caused a significantly stronger ThT fluorescent signal than that obtained in the absence of Ce6 or at the higher Ce6 concentration, possibly suggesting that photoirradiation of Aβ aggregates in the presence of small amounts of Ce6 (10%) generates greater amounts of ThT-active Aβ species (Fig. 2a). However, although ThT fluorescence intensity is often used to quantify the amount of amyloid material present in a solution, it is nonlinear and depends on other factors including solvent and the presence of additional small molecules – in particular those that interact with Aβ.18
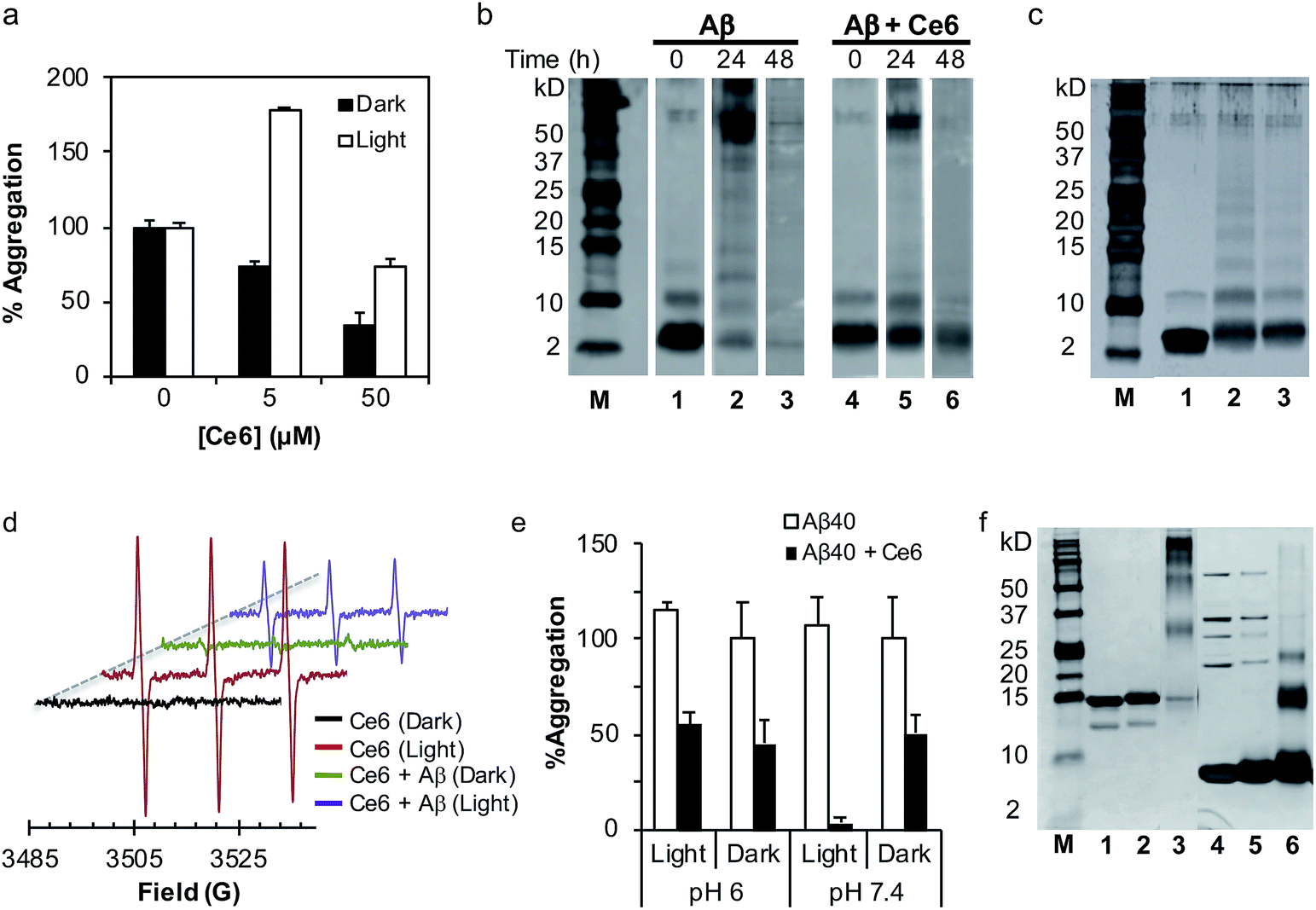 | ||
Fig. 2 The mechanism of action of Ce6 in darkness and after irradiation. (a) Ce6 disassembles aggregated Aβ under dark and light conditions. Aβ (50 μM) was aged for 3 days to generate Aβ aggregates with high ThT fluorescence. Ce6 (5 or 50 μM) was then added and the mixtures were kept in the dark or photoirradiated for 1 h. The amount of Aβ fibrils was then determined after 3 days by the ThT assay. The data represent mean ± SD values of an experiment out of four carried in duplicate. (b) Effect of Ce6 on oligomer distribution of Aβ. Treatment of Aβ with Ce6 significantly reduces its self-assembly under dark conditions. Aβ (50 μM) was aged in the dark for 0, 24, or 48 h in the absence (lanes 1–3) or presence (lanes 4–6) of Ce6 (50 μM) and analyzed by the Tris–tricine SDS–PAGE PICUP method. (c) Tris–tricine SDS–PAGE separation of Aβ (50 μM) before (lane 1) and after PICUP cross-linking using Ru2+ complex (lane 2). Lane 3 represents Tris–tricine SDS–PAGE separation of Aβ (50 μM) incubated with Ce6 (50 μM) and photoirradiated for 1 min in the absence of Ru2+. (d) Effect of Aβ40 on photogeneration of singlet oxygen from Ce6. EPR spectra obtained from photoirradiation of Ce6 (40 μM) and TEMP (40 μM) in the absence or presence of Aβ (40 μM) in PBS/D2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9). EPR conditions: microwave power, 20 mW; modulation amplitude, 1.0 G; receiver gain, 4 × 105; time constant, 0.64 s; scan range, 100 G; center of field, 3300 G; temperature, 25 °C. (e) Effect of pH on antiamyloidogenic activity of Ce6 in darkness and after photoirradiation, using the ThT assay. Details are identical to those described in Fig. 1a legend. (f) SDS–PAGE separation of α-synuclein (6 μM, lane 1) treated either with photoexcited Ce6 (33 μM, lane 2) or Ru2+ catalyst (33 μM, lane 3). Lanes 4–6 demonstrate respectively SDS–PAGE separation of ubiquitin (30 μM, lane 4) treated either with photoexcited Ce6 (33 μM, lane 5) or Ru2+ catalyst (33 μM, lane 6). :9). EPR conditions: microwave power, 20 mW; modulation amplitude, 1.0 G; receiver gain, 4 × 105; time constant, 0.64 s; scan range, 100 G; center of field, 3300 G; temperature, 25 °C. (e) Effect of pH on antiamyloidogenic activity of Ce6 in darkness and after photoirradiation, using the ThT assay. Details are identical to those described in Fig. 1a legend. (f) SDS–PAGE separation of α-synuclein (6 μM, lane 1) treated either with photoexcited Ce6 (33 μM, lane 2) or Ru2+ catalyst (33 μM, lane 3). Lanes 4–6 demonstrate respectively SDS–PAGE separation of ubiquitin (30 μM, lane 4) treated either with photoexcited Ce6 (33 μM, lane 5) or Ru2+ catalyst (33 μM, lane 6). | ||
Effect of Ce6 on Aβ oligomer distribution under dark and light conditions
In order to shed light on the mechanism by which Ce6 affects Aβ aggregation under dark and light conditions, its effect on Aβ oligomer distribution was determined using the photoinduced cross-linking of unmodified proteins (PICUP) method.7b,19 Under dark conditions and in the absence of Ce6, freshly prepared Aβ maintains a dynamic equilibrium between several oligomeric species, with monomers as the most abundant species (Fig. 2b, lane 1). Following the aging process, a time-dependent decrease in the amounts of Aβ monomers and small oligomers (2–5-mers) was evident (Fig. 2b, lanes 1–3). The presence of Ce6 in the mixture stabilized the 1–3-mers (Fig. 2b, lanes 4–6), suggesting that binding of Ce6 to Aβ monomers and early soluble oligomers is responsible for the reduced aggregation and possible disaggregation of Aβ fibrils, most probably by shifting the equilibrium toward the small, soluble oligomers (Fig. 2b).20The effect of Ce6 on the oligomer distribution was also tested under light conditions. Monomeric Aβ was incubated without or with Ce6, photoirradiated for 1 min, and analyzed with SDS–PAGE under the same conditions used in the PICUP analysis. Interestingly, photoirradiation of Aβ in the presence of Ce6 but in the absence of Ru2+, which was used as a cross-linker in the PICUP experiments, also generated SDS-stable cross-linked oligomers very similar to those generated in the presence of Ru2+ (Fig. 2c). Since the cross-linking mechanism of the Ru2+ complex involves photo-generation of radical intermediates,7b these results suggest that photo-induced cross-linking might be the mechanism by which Ce6 inhibits aggregation of Aβ into toxic oligomers. It has previously been shown that metalloporphyrins mediate cross-linking of proteins upon photoirradiation, and this effect is in part responsible for their mode of action in photodynamic therapy (PDT).21
Molecular mechanism of Aβ cross-linking by photoexcited Ce6
The cross-linking of Aβ by Ce6 prompted us to study its mechanism of action. To show that it is mediated through type II photo-oxidation involving singlet oxygen,14 mixtures of Aβ and Ce6 were photoirradiated in the presence of singlet oxygen quenchers, such as His and NaN3, and analyzed by SDS–PAGE (Fig. S2a,† compare lane 3 to lanes 4 and 5). Addition of His and NaN3 to the solution of Aβ and Ce6 prior to photoirradiation reduced the cross-linking efficacy of Ce6 (Fig. S2a†). On the other hand, neither H2O2 nor HO• (generated from H2O2 and Fe2+ or from photocleavage of H2O2) caused the cross-linking of Aβ (Fig. S2b†). Moreover, in order to show that type II-mediated generation of 1O2 is the main oxidative mechanism involved in the Aβ cross-linking process, the mixture of Aβ and Ce6 was photoirradiated in the absence of oxygen. Fig. S2c† clearly shows that under anaerobic conditions Ce6 fails to cross-link Aβ, strongly suggesting that type II-mediated oxidation of Aβ is most likely responsible for its cross-linking. Photogeneration of 1O2 by Ce6 and its consumption by Aβ to generate protein-cross links were also confirmed by EPR, using 2,2,6,6-tetramethylpiperidine (TEMP) as a specific spin trap of 1O2 (Fig. 2d).22 While photoirradiation of Ce6 in the presence of TEMP generated a corresponding three-line spectrum, the presence of Aβ significantly decreased the intensity of the signal, indicating that Aβ competes with TEMP to react with the 1O2 that cross-links Aβ.To further study the mechanism by which Ce6 cross-links Aβ at a molecular level, a solution of Aβ and Ce6 was photoirradiated in the presence of excess Tyr and analyzed by PICUP and PAGE. Previous studies have demonstrated that Ru2+-based cross-linking of Aβ occurs mainly through Tyr–Tyr interaction via photogeneration of Tyr radicals.7b,23 Indeed, as expected, a high excess of Tyr in the reaction mixture significantly reduced the cross-linking of Aβ induced by the Ru2+-complex (Fig. S2a,† lane 8), although it had only a small effect on the Ce6-induced Aβ cross-linking (Fig. S2a†, lane 6). Together with the significant inhibitory effect of His (Fig. S2a,† lane 4), these results strongly suggest that cross-linking of Aβ in the presence of Ce6 and light is mediated mainly through His residues. This possibility was further investigated via amino acid analysis, which showed that although photoirradiation of monomeric Aβ alone had no effect on its amino acid composition, in the presence of Ce6 it selectively affected the His residues (Table S1†).
To confirm that the inhibitory effect of photoexcited Ce6 on Aβ aggregation is mediated through interaction with, and modification of the Aβ His residues, the ThT and dot blot assays were repeated at a pH of 6. We hypothesized that at a pH below the pKa of the His residues, Ce6 will bind Aβ weakly, and will thus affect Aβ aggregation less while photoirradiated. Thus, Aβ was incubated with Ce6 at pH 6, photoirradiated with visible light, and analyzed by ThT and dot blot assays, using the A11 antibody. Fig. 2e shows that while photoirradiation of the Ce6–Aβ mixture at pH 7.4 significantly enhanced the anti-amyloidogenic activity, as compared to the dark conditions, it had no effect on Aβ aggregation when the incubation was carried out at pH 6. Similarly, the dot blot assay also demonstrated that photoexcited Ce6 is less effective in reducing the amount of toxic Aβ oligomers at pH 6 (Fig. 1e, lane 3). These results indicate that interactions between Ce6 and unprotonated histidines of Aβ and their subsequent modifications/cross-linking are at least partially responsible for the anti-amyloidogenic activity of Ce6.
Targeting the toxic oligomeric state of amyloidogenic proteins is vital for successful treatment,3 as many of these proteins, such as amylin and transthyretin exhibit important biological functions when folded correctly. We used SDS–PAGE to show that in contrast to the Ru-mediated PICUP that photo-crosslinks many proteins, Ce6 selectively cross-links Aβ via residue-specific binding. Ubiquitin and the Parkinson's disease-associated protein α-synuclein (α-syn) were incubated with Ru2+ and Ce6, photoirradiated, and analyzed by SDS–PAGE. While photoirradiation of α-syn and ubiquitin with Ru2+ induced cross-linking as expected (Fig. 2f, lanes 3 and 6), photoexcited Ce6 failed to cross-link these proteins under similar conditions (Fig. 2f, lanes 2 and 5). This suggests that the cross-linking effect of Ce6 on Aβ involves specific binding between Ce6 and Aβ.
NMR studies
The molecular interactions between Ce6 and Aβ were further elucidated using NMR spectroscopy. The 1H, 15N-HSQC NMR spectra of uniformly 15N-labeled Aβ clearly show that under dark conditions, addition of Ce6 has no significant effect on the monomeric Aβ peptide (Fig. 3a and b, blue peaks). However, clear changes in the NMR spectrum were observed after photoirradiation of the mixture for 1 h (Fig. 3b, red peaks). Significant losses of NMR signal intensity were observed for crosspeaks corresponding to the N-terminal Aβ residues, e.g. F4, R5, D7, S8, G9, Y10, E11 and V12, indicating the site where Ce6 binds. This is also consistent with the lack of interaction between the 6E10 antibody and the photoirradiated Aβ–Ce6 mixture (Fig. 1e). Similar measurements carried out at pH 5.0 showed no effects on the NMR crosspeaks, neither before nor after 1 h of light exposure (Fig. 3c and d), indicating that Ce6 does not interact with the Aβ N-terminal segment when the His residues are protonated. Moreover, similar 1H, 15N-HSQC NMR measurements carried out with [H6A, H13A and H14A]-mutated Aβ40 and Ce6 suggested no interaction between Ce6 and the mutated Aβ analog under both dark and light conditions (Fig. S3†).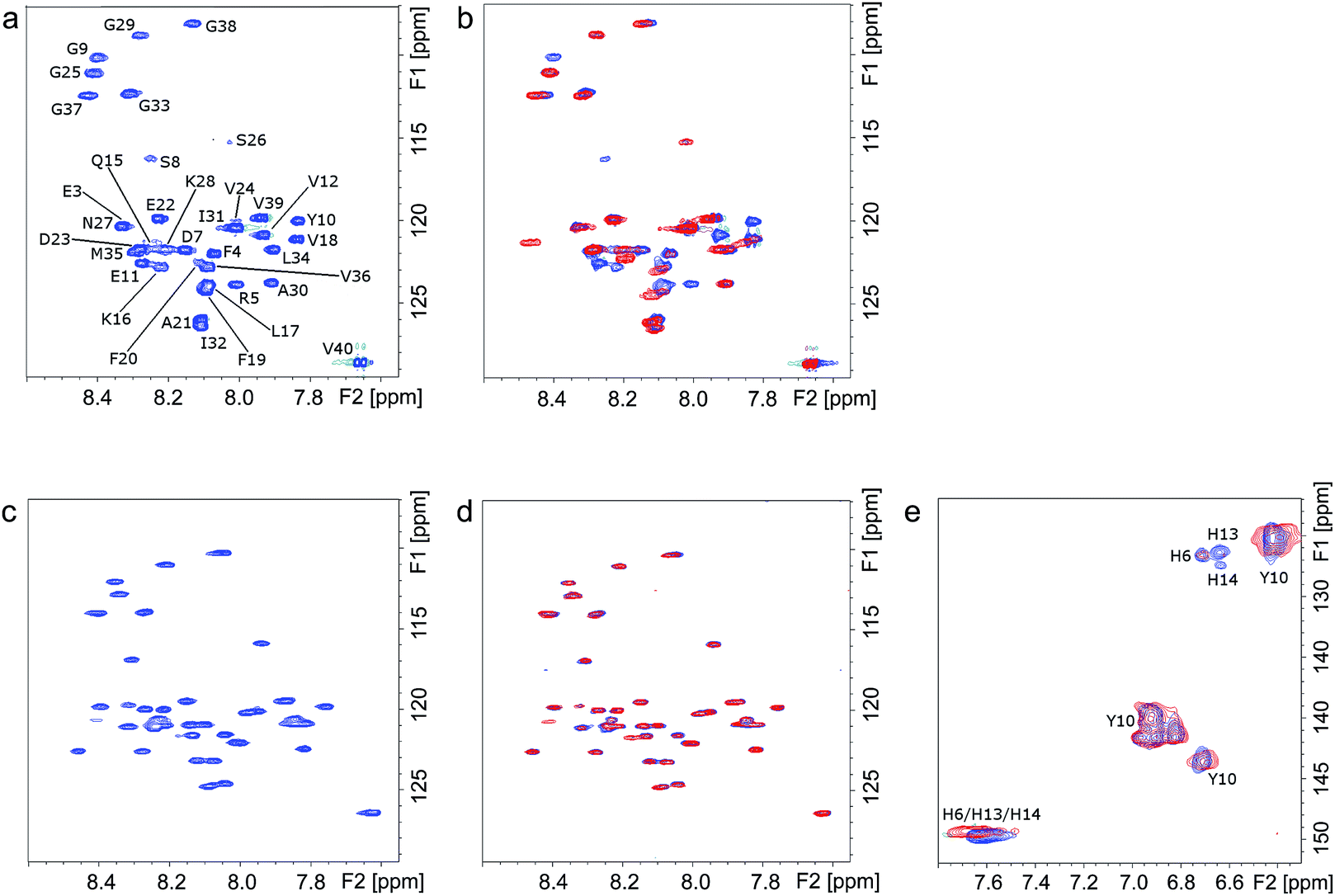 | ||
| Fig. 3 NMR investigations of the molecular interactions between the monomeric Aβ40 and Ce6 in darkness and following 1 h photoirradiation. (a) 1H, 15N-HSQC NMR spectrum of Aβ40 (75 μM) in PB (20 mM, pH 7.3). (b) 1H, 15N-HSQC NMR spectra of Aβ40 (75 μM) after addition of Ce6 (1 mg) under dark conditions (blue peaks), and following 1 h of photoirradiation (red peaks). (c) 1H, 15N-HSQC NMR spectrum of Aβ40 (75 μM) in PB (20 mM, pH 5.0). (d) 1H, 15N-HSQC NMR spectra of Aβ40 (75 μM) in PB after addition of Ce6 (1 mg) under dark conditions (blue peaks), and after 1 h exposure to light (red peaks). (e) 1H, 13C-HSQC NMR spectra of Aβ40 (75 μM) and Ce6 (1 mg) in PB (20 mM, pH 7.3) before (blue peaks) and after 1 h exposure to light (red peaks). | ||
Two dimensional 1H, 13C-HSQC NMR spectra of uniformly 13C-labeled Aβ, which contrary to 1H, 15N-HSQC NMR spectra register His residues, clearly showed that at the concentrations used, Aβ's His residues are affected by Ce6 only after light exposure (Fig. 3e). The H13 and H14 residues are more affected than H6, suggesting a major interaction around these two central His residues. Moreover, in agreement with the amino acid analysis (Table S1†), these NMR results indicate that Y10 in Aβ is not affected by photoexcited Ce6 (Fig. 3e), further suggesting that Aβ cross-linking occurs via the His residues. Together, the NMR and amino acid analysis data suggest that Ce6 selectively binds to and damages the N-terminal His residues, while other amino acids such as F4, R5, D7, S8, G9, Y10 and E11 are affected by their proximity to the His residues. It has been suggested that H13 (or H14), R5 and Y10 interact with porphyrin-based heme and are responsible for heme–Aβ peroxidase activity.24 In another study, F19 was found to be vital in Aβ–heme binding through π–π interaction with the porphyrin ring, which disrupts Aβ aggregation.25 Notably, in most of these studies, the interaction of Aβ with heme involves coordination of the related amino acid side chains with the central metal (Fe3+). Since Ce6 contains no metal, the specific interactions between Ce6 and Aβ must depend on mechanisms other than metal coordination.26
Cytoprotecting effect of Ce6 against Aβ-induced toxicity
Because Ce6 inhibits Aβ aggregation and reduces the amounts of toxic oligomers, we next examined whether it can also reduce Aβ cell toxicity, in darkness or upon photoexcitation. First, Aβ40 was incubated in the absence or presence of Ce6, either in darkness or photoirradiated for 1 h. Next, the mixtures were further aged for 48 h, to allow Aβ aggregation, and then incubated for 48 h with rat pheochromocytoma PC12 cells. Finally, cell viability was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Fig. 4). Incubation of the cells with 2 days aged Aβ (without Ce6) killed about 45% of the cells. Photoirradiation of Aβ alone, prior to incubation with cells, had no significant effect on cell survival. In contrast, incubation of the cells with the photoirradiated Aβ–Ce6 mixture (10 μM each) significantly increased cell survival (p < 0.05), while aged Aβ–Ce6 mixture kept in darkness had no effect on cell survival. In control experiments, Ce6 alone (10 μM) had no effect on cell survival, neither in darkness nor following 1 h photoirradiation. The highest protecting effect of Ce6 on Aβ's toxicity (∼30%) was achieved when Aβ (10 μM) was incubated with 10-fold excess of Ce6 and photoirradiated for 1 h. However, at this high Ce6 concentration (100 μM), some toxicity was observed even when the cells were incubated in darkness, suggesting that complexation of Aβ with Ce6 reduces the toxicity of both components. | ||
| Fig. 4 Effect of Ce6 and photoexcited Ce6 on the toxicity of Aβ40 to rat pheochromocytoma PC12 cells. Freshly prepared Aβ40 (100 μM) was aged for 48 h with Ce6 under either dark or light conditions (100 and 1000 μM) in PBS. The samples were then diluted 10 times in the wells containing 10000 cells. Cell viability was then determined after 48 h of incubation by MTT. *p < 0.005 compared to the same treatment in darkness and with Aβ only. All samples contained 0.25% DMSO, which was found to have no effect on cell survival (100% cell survival). | ||
Effect of Ce6 on Aβ–Cu2+ interaction and toxicity
Metal ions and metal homeostasis play a vital role in AD etiology and progression.27 In particular, Cu and Fe ions bind Aβ, modulate its aggregation, and generate ROS that are toxic to neurons.28 AD plaques contain a variety of metals including redox-active Cu and Fe ions, and the increased levels of these metals in AD brain tissue correlate with AD pathology.29 Monomeric Aβ peptides bind metal ions with high affinity at their N-terminal, involving the three histidine residues H6, H13, and H14, which act as specific binding ligands, e.g. for Cu2+, Mn2+, Pb4+ and Zn2+ ions.30 Moreover, H13 and H14 of Aβ have been associated with Aβ-mediated ion channel/pore formation and cytotoxicity, and blocking these channels with small peptides or molecules prevented Ca2+ influx and neurotoxicity.31 Having demonstrated that Ce6 can selectively bind Aβ's H13 and H14 residues and damage them under photoirradiation, we tested whether it can also modulate Cu2+ binding to Aβ and reduce Cu2+-induced aggregation and toxicity. A solution of monomeric Aβ (5 μM) in HEPES buffer was incubated in darkness with Cu2+ (1.5 μM) in the absence or presence of Ce6 (0.125 μM), and the extent of aggregation was followed by ThT fluorescence. Ce6 is known for its capability to chelate metals such as Cu2+, and this property has been used to generate probes for PET and MRI diagnostics.32 Therefore, catalytic amounts of Ce6 were used to rule out the possibility that the anti-aggregation activity of Ce6 derives only from its chelation activity. In the absence of Cu2+ and under the conditions used (20 mM HEPES, pH 7.4), Aβ40 aggregated with a lag phase of about 24 hours (Fig. 5a). Co-incubation of Aβ (5 μM) with catalytic amount of Ce6 (0.125 μM) in the absence of Cu2+ dramatically delayed the lag phase to about 42 h, suggesting that Ce6 binds most likely with Aβ monomers, early forms of oligomers, or Aβ seeds and converts them to incompetent structures that are not ThT reactive. In the presence of Cu2+ (0.3 eq.), Aβ aggregates with an accelerated rate with a lag phase of about 17 hours, indicating that Cu2+ facilitated formation of ThT-active β-sheet structure as expected (Fig. 5a). The presence of catalytic amounts of Ce6 (0.025 eq.) in the Aβ–Cu2+ mixtures extended the lag phase to about 22 hours and reduced its aggregation, suggesting that in the early Aβ aggregation process, Ce6 binds the Aβ His residues with higher affinity than Cu2+. However, catalytic damage and possible cross-linking of Aβ's His residues by Ce6, which would affect Cu2+ binding, could not be excluded due to possible light exposure.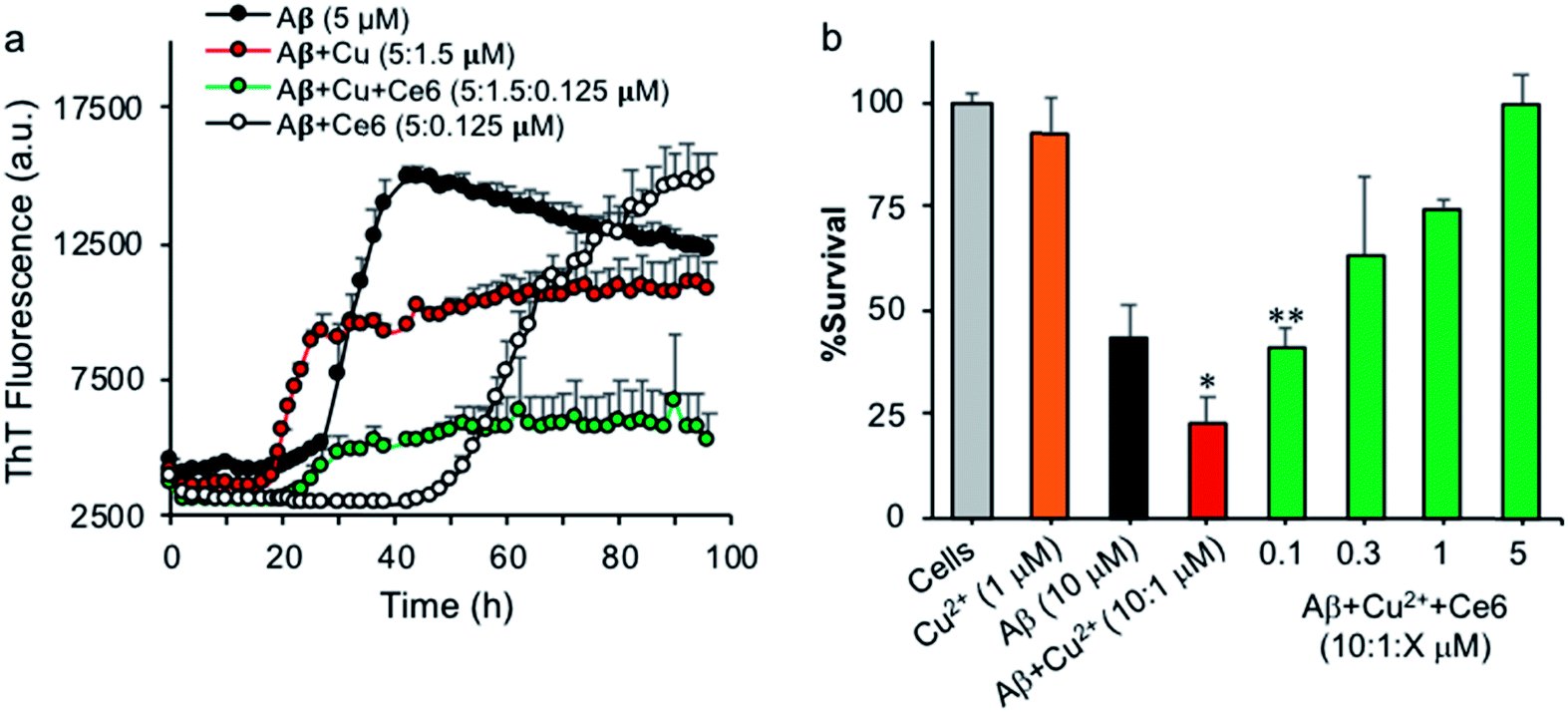 | ||
| Fig. 5 Effect of Ce6 on Cu2+-induced Aβ aggregation and toxicity. (a) Monomeric Aβ (5 μM) was incubated in the dark with Cu2+ (1.5 μM) in the absence or presence of a catalytic amount of Ce6 (0.125 μM) in HEPES buffer (50 mM, 160 mM NaCl, pH 7.4), and the extent of amyloid formation was monitored over time using the ThT fluorescence. (b) Ce6 counters the effect of Cu2+ on Aβ-induced PC12 toxicity. Aβ (10 μM) was aged for 48 h with Cu2+ in absence or presence of increasing amounts of Ce6 and exposed to PC12 cells for a further 4 days. Cell viability was then determined by the MTT assay. All samples contained 0.25% of DMSO, which was found to be harmless (100% cell survival). The results are presented as mean ± SD of two experiments (n = 3–5 each). Significance (*,**p < 0.05) were calculated relative to Aβ40 (10 μM) or Aβ + Cu2+ (10:1 μM) toxicity, respectively. | ||
Next, the protective effect of Ce6 against Cu2+-induced Aβ toxicity was evaluated using PC12 cells. Aβ (10 μM) was aged for 48 hours with Cu2+ (1 μM), in the absence or presence of increasing concentrations of Ce6 (0.1–5 μM) in HEPES buffer (50 mM, pH 7.4) and then incubated with PC12 cells for a further 4 days. Cell survival was then determined by the MTT assay. Under the conditions tested, the presence of Cu2+ significantly increased the toxicity of Aβ from 55 to 77%, which is in agreement with previous reports.28c,33 The presence of Ce6 in the mixture dose-dependently decreased the Cu2+-induced toxicity even at substoichiometric concentrations (0.1 μM, Fig. 5b). In control experiments, Cu2+ alone did not induce any significant toxicity to the cells. These results confirm the observations from the ThT assay and suggest that Ce6 can revert the effects of Cu2+.
To further demonstrate that Ce6 affects the binding affinity of Cu2+ to the Aβ His residues, 1H, 15N-HSQC NMR experiments were carried out in the presence of Cu2+. Addition of Cu2+ to monomeric Aβ caused the N-terminal crosspeaks to disappear completely, as expected (Fig. 6b),30b while addition of excess Ce6 to the mixture under dark conditions allowed these crosspeaks to partially re-appear (Fig. 6c). These results indicate that a high excess of Ce6 is able to remove Cu2+ ions from Aβ via competitive binding.
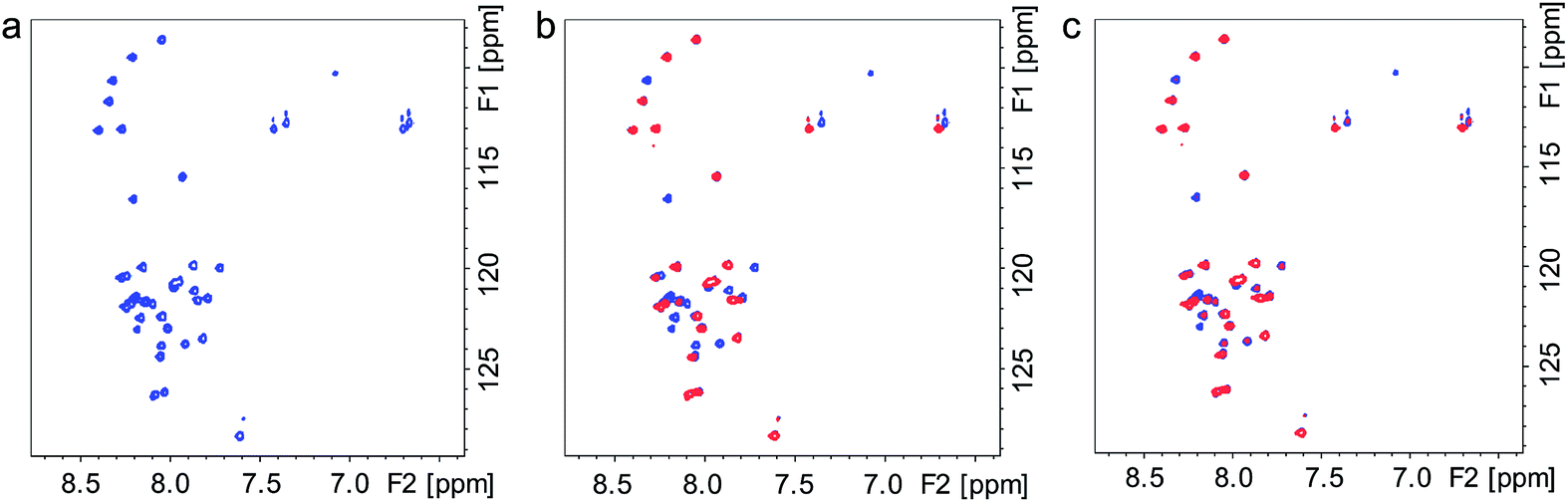 | ||
| Fig. 6 Effect of Cu2+ and Ce6 on the Aβ NMR spectrum. (a) 1H, 15N-HSQC NMR spectrum of Aβ40 (80 μM) in HEPES buffer (40 mM, pH 7.3). (b) NMR spectra of Aβ40 (80 μM) in HEPES buffer, before (blue) and after (red) addition of Cu2+ (40 μM). Most N-terminal Aβ crosspeaks have completely disappeared. (c) NMR spectrum of Aβ (80 μM) and Cu2+ (40 μM) in HEPES buffer, before (blue) and after (red) addition of Ce6 (800 μM). Many N-terminal Aβ crosspeaks have partially returned, indicating that Ce6 can compete with Cu2+ ions for binding to the Aβ N-terminal segment. | ||
Altogether, our results indicate that photoexcited Ce6 catalytically damages the His residues in the early stage of Aβ self-assembly, i.e. before its further aggregation into fibrils. This is in agreement with the reduced immunoreactivity of the Aβ–Ce6 complex to the A11 antibody, the generation of Aβ cross-links, and the amino acid analysis results (Fig. 1 and 2). The specific damage to the Aβ His residues in the early aggregation phase by catalytic amounts of Ce6 reduces the capability of Aβ to bind Cu2+, and thus diminishes the Cu-induced Aβ aggregation. High Ce6 concentrations can furthermore reduce the effects of Cu2+ on Aβ aggregation via competitive binding (Fig. 6).
The specific targeting of Ce6 to Aβ, rather than to α-synuclein for instance, is noteworthy. Most amyloid proteins aggregate into similar cross-β structures, which is why they are often able to cross-seed each other's aggregation.34 Thus, many natural and designed amyloid-inhibiting molecules, such as β-sheet breakers, affect a wide range of amyloid proteins.2b,20,35 The specificity of Ce6 for Aβ is arguably related to the interaction with the three N-terminal Aβ histidines, which separate Aβ from other amyloid proteins and confer important functionality: in addition to coordinating metal ion binding,30 they are also required for Aβ oligomer formation.36 As the N-terminal segment is known to be dangling outside when Aβ interacts with cell membranes,28b we expect that Ce6 will be able to target Aβ not only in solution, but also in membrane environments. Thus, one important finding of this study is that the three Aβ histidines are a suitable target when developing Aβ-specific anti-amyloidogenic drugs.
Conclusion
In the present study, we have shown that Ce6 potently inhibits the aggregation and toxicity of Aβ under dark conditions, while irradiation with visible light significantly enhanced its antiamyloidogenic activity. Taken together, our CD, NMR, amino acid analysis, and immunochemical studies using the oligomer-specific A11 antibody indicate that photoexcited Ce6 dramatically delays the formation of Aβ β-sheet structures that are reactive to the A11 antibody, by binding to and damaging the Aβ N-terminal segment. SDS/PAGE analysis revealed that Ce6 selectively cross-links Aβ monomers without affecting other proteins such as α-synuclein or ubiquitin. The Aβ cross-linking most likely originates from singlet oxygen generation by light-exposed Ce6, as shown by the EPR studies. The ThT and NMR experiments carried out at neutral and acidic pH show that the N-terminal histidines, especially H13 and H14, are the Aβ residues mostly affected by the photoexcited Ce6. We speculate that by binding and damaging the His residues that are responsible for Cu2+ binding and Cu-induced channel/nanopore formation,31b,37 catalytic amounts of photoexcited Ce6 can inhibit Cu-induced Aβ aggregation and toxicity. However, at high concentrations, Ce6 can either compete with Cu2+ to bind Aβ or act as a chelator to reduce the local concentration of Cu, as shown by the NMR studies. By selective binding to Aβ, Ce6 acts as a multifunctional agent that can both damage Aβ and remove toxic Cu. While further studies are required in AD animal models to assess the potential clinical applications of Ce6, the specific targeting of Ce6 to the Aβ His residues, and not the cross-β sheet structure present in many amyloid aggregates, shows that the three N-terminal histidines are a promising target for Aβ-specific anti-amyloidogenic drugs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Ronit Lavi (Bar-Ilan University) for assistance with the EPR experiments. This study was supported in part by the Swedish Research Council (Grant 2017-04102) to AG, Magnus Bergvall Foundation to SW and the Israel Ministry of Science – Infrastructure Grants (Grant 3-12064) to SR.Notes and references
- (a) G. G. Glenner, C. W. Wong, V. Quaranta and E. D. Eanes, Appl. Pathol., 1984, 2, 357 CAS; (b) D. J. Selkoe and J. Hardy, EMBO Mol. Med., 2016, 8, 595 CrossRef CAS PubMed.
- (a) M. P. Lambert, A. K. Barlow, B. A. Chromy, C. Edwards, R. Freed, M. Liosatos, T. E. Morgan, I. Rozovsky, B. Trommer, K. L. Viola, P. Wals, C. Zhang, C. E. Finch, G. A. Krafft and W. L. Klein, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6448 CrossRef CAS PubMed; (b) I. W. Hamley, Chem. Rev., 2012, 112, 5147 CrossRef CAS PubMed; (c) C. Haass and D. J. Selkoe, Nat. Rev. Mol. Cell Biol., 2007, 8, 101 CrossRef CAS PubMed; (d) M. Fandrich, J. Mol. Biol., 2012, 421, 427 CrossRef; (e) M. Bucciantini, E. Giannoni, F. Chiti, F. Baroni, L. Formigli, J. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson and M. Stefani, Nature, 2002, 416, 507 CrossRef CAS PubMed; (f) J. P. Cleary, D. M. Walsh, J. J. Hofmeister, G. M. Shankar, M. A. Kuskowski, D. J. Selkoe and K. H. Ashe, Nat. Neurosci., 2005, 8, 79 CrossRef CAS PubMed.
- J. Sevigny, P. Chiao, T. Bussiere, P. H. Weinreb, L. Williams, M. Maier, R. Dunstan, S. Salloway, T. Chen, Y. Ling, J. O'Gorman, F. Qian, M. Arastu, M. Li, S. Chollate, M. S. Brennan, O. Quintero-Monzon, R. H. Scannevin, H. M. Arnold, T. Engber, K. Rhodes, J. Ferrero, Y. Hang, A. Mikulskis, J. Grimm, C. Hock, R. M. Nitsch and A. Sandrock, Nature, 2016, 537, 50 CrossRef CAS.
- (a) M. S. Forman, J. Q. Trojanowski and V. M. Lee, Nat. Med., 2004, 10, 1055 CrossRef CAS; (b) E. D. Roberson and L. Mucke, Science, 2006, 314, 781 CrossRef; (c) M. J. Guerrero-Munoz, D. L. Castillo-Carranza and R. Kayed, Biochem. Pharmacol., 2014, 88, 468 CrossRef CAS; (d) T. Hamaguchi, K. Ono and M. Yamada, Cell. Mol. Life Sci., 2006, 63, 1538 CrossRef CAS; (e) S. S. Hindo, A. M. Mancino, J. J. Braymer, Y. Liu, S. Vivekanandan, A. Ramamoorthy and M. H. Lim, J. Am. Chem. Soc., 2009, 131, 16663 CrossRef CAS PubMed.
- (a) A. R. Azzouzi, E. Barret, C. M. Moore, A. Villers, C. Allen, A. Scherz, G. Muir, M. de Wildt, N. J. Barber, S. Lebdai and M. Emberton, BJU Int., 2013, 112, 766 CrossRef CAS PubMed; (b) S. Mallidi, S. Anbil, A. L. Bulin, G. Obaid, M. Ichikawa and T. Hasan, Theranostics, 2016, 6, 2458 CrossRef CAS; (c) M. R. Detty, S. L. Gibson and S. J. Wagner, J. Med. Chem., 2004, 47, 3897 CrossRef CAS.
- C. von Sonntag, The chemical basis of radiation biology, Taylor & Francis, London, 1987 Search PubMed.
- (a) E. R. Stadtman, Annu. Rev. Biochem., 1993, 62, 797 CrossRef CAS; (b) D. A. Fancy and T. Kodadek, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 6020 CrossRef CAS.
- (a) S. Rahimipour, N. Ben-Aroya, K. Ziv, A. Chen, M. Fridkin and Y. Koch, J. Med. Chem., 2003, 46, 3965 CrossRef CAS; (b) N. Solban, I. Rizvi and T. Hasan, Lasers Surg. Med., 2006, 38, 522 CrossRef.
- (a) G. Leshem, M.Sc. thesis, Bar-Ilan University, 2012; (b) Y. Ishida, T. Fujii, K. Oka, D. Takahashi and K. Toshima, Chem.–Asian J., 2011, 6, 2312 CrossRef CAS; (c) A. Hirabayashi, Y. Shindo, K. Oka, D. Takahashi and K. Toshima, Chem. Commun., 2014, 50, 9543 RSC; (d) B. I. Lee, S. Lee, Y. S. Suh, J. S. Lee, A. K. Kim, O. Y. Kwon, K. Yu and C. B. Park, Angew. Chem., Int. Ed., 2015, 54, 11472 CrossRef CAS PubMed; (e) J. S. Lee, B. I. Lee and C. B. Park, Biomaterials, 2015, 38, 43 CrossRef CAS; (f) A. Taniguchi, Y. Shimizu, K. Oisaki, Y. Sohma and M. Kanai, Nat. Chem., 2016, 8, 974 CrossRef CAS; (g) A. Taniguchi, D. Sasaki, A. Shiohara, T. Iwatsubo, T. Tomita, Y. Sohma and M. Kanai, Angew. Chem., Int. Ed., 2014, 53, 1382 CrossRef CAS.
- G. A. Kostenich, I. N. Zhuravkin and E. A. Zhavrid, J. Photochem. Photobiol., B, 1994, 22, 211 CrossRef CAS.
- H. Mojzisova, S. Bonneau, P. Maillard, K. Berg and D. Brault, Photochem. Photobiol. Sci., 2009, 8, 778 RSC.
- S. V. Shliakhtsin, T. V. Trukhachova, H. A. Isakau and Y. P. Istomin, Photodiagn. Photodyn. Ther., 2009, 6, 97 CrossRef CAS.
- H. Kato, K. Furukawa, M. Sato, T. Okunaka, Y. Kusunoki, M. Kawahara, M. Fukuoka, T. Miyazawa, T. Yana, K. Matsui, T. Shiraishi and H. Horinouchi, Lung Cancer, 2003, 42, 103 CrossRef.
- B. Bose and A. Dube, J. Photochem. Photobiol., B, 2006, 85, 49 CrossRef CAS.
- M. Biancalana and S. Koide, Biochim. Biophys. Acta, 2010, 1804, 1405 CrossRef CAS.
- R. Kayed, E. Head, J. L. Thompson, T. M. McIntire, S. C. Milton, C. W. Cotman and C. G. Glabe, Science, 2003, 300, 486 CrossRef CAS.
- K. S. Kim, G. Y. Wen, C. Bancher, C. M. J. Chen, V. J. Sapienza, H. Hong and H. M. Wisniewski, Neurosci. Res. Commun., 1990, 7, 113 CAS.
- D. J. Lindberg, A. Wenger, E. Sundin, E. Wesen, F. Westerlund and E. K. Esbjorner, Biochemistry, 2017, 56, 2170 CrossRef CAS.
- G. Bitan, M. D. Kirkitadze, A. Lomakin, S. S. Vollers, G. B. Benedek and D. B. Teplow, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 330 CrossRef CAS.
- M. Richman, S. Wilk, M. Chemerovski, S. K. Wärmländer, A. Wahlström, A. Gräslund and S. Rahimipour, J. Am. Chem. Soc., 2013, 135, 3474 CrossRef CAS.
- (a) L. A. Campbell, T. Kodadek and K. C. Brown, Bioorg. Med. Chem., 1998, 6, 1301 CrossRef CAS; (b) H. Verweij, T. M. Dubbelman and J. Van Steveninck, Biochim. Biophys. Acta, 1981, 647, 87 CrossRef CAS.
- S. Rahimipour, C. Palivan, F. Barbosa, I. Bilkis, Y. Koch, L. Weiner, M. Fridkin, Y. Mazur and G. Gescheidt, J. Am. Chem. Soc., 2003, 125, 1376 CrossRef CAS.
- D. A. Fancy, C. Denison, K. Kim, Y. Xie, T. Holdeman, F. Amini and T. Kodadek, Chem. Biol., 2000, 7, 697 CrossRef CAS.
- (a) H. Atamna and K. Boyle, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3381 CrossRef CAS; (b) C. Ghosh, M. Seal, S. Mukherjee and S. Ghosh Dey, Acc. Chem. Res., 2015, 48, 2556 CrossRef CAS; (c) D. Pramanik and S. G. Dey, J. Am. Chem. Soc., 2011, 133, 81 CrossRef CAS; (d) D. Pramanik, C. Ghosh and S. G. Dey, J. Am. Chem. Soc., 2011, 133, 15545 CrossRef CAS.
- C. Yuan and Z. Gao, Chem. Res. Toxicol., 2013, 26, 262 Search PubMed.
- (a) E. Cauet, M. Rooman, R. Wintjens, J. Lievin and C. Biot, J. Chem. Theory Comput., 2005, 1, 472 CrossRef PubMed; (b) S. M. Liao, Q. S. Du, J. Z. Meng, Z. W. Pang and R. B. Huang, Chem. Cent. J., 2013, 7, 44 CrossRef.
- (a) K. J. Barnham and A. I. Bush, Curr. Opin. Chem. Biol., 2008, 12, 222 CrossRef CAS; (b) M. A. Greenough, J. Camakaris and A. I. Bush, Neurochem. Int., 2013, 62, 540 CrossRef CAS.
- (a) D. G. Smith, R. Cappai and K. J. Barnham, Biochim. Biophys. Acta, 2007, 1768, 1976 CrossRef CAS; (b) A. Tiiman, J. Luo, C. Wallin, L. Olsson, J. Lindgren, J. Jarvet, P. Roos, S. B. Sholts, S. Rahimipour, J. P. Abrahams, A. E. Karlstrom, A. Gräslund and S. K. Wärmländer, J. Alzheimer's Dis., 2016, 54, 971 CAS; (c) C. J. Sarell, S. R. Wilkinson and J. H. Viles, J. Biol. Chem., 2010, 285, 41533 CrossRef CAS.
- (a) R. Squitti, D. Lupoi, P. Pasqualetti, G. Dal Forno, F. Vernieri, P. Chiovenda, L. Rossi, M. Cortesi, E. Cassetta and P. M. Rossini, Neurology, 2002, 59, 1153 CrossRef CAS; (b) G. J. Brewer, J. Alzheimer's Dis., 2015, 46, 593 CAS; (c) J. Dong, C. S. Atwood, V. E. Anderson, S. L. Siedlak, M. A. Smith, G. Perry and P. R. Carey, Biochemistry, 2003, 42, 2768 CrossRef CAS.
- (a) B. Alies, E. Renaglia, M. Rozga, W. Bal, P. Faller and C. Hureau, Anal. Chem., 2013, 85, 1501 CrossRef CAS; (b) S. Wärmländer, A. Tiiman, A. Abelein, J. Luo, J. Jarvet, K. L. Soderberg, J. Danielsson and A. Gräslund, ChemBioChem, 2013, 14, 1692 CrossRef; (c) C. Wallin, Y. S. Kulkarni, A. Abelein, J. Jarvet, Q. Liao, B. Strodel, L. Olsson, J. Luo, J. P. Abrahams, S. B. Sholts, P. M. Roos, S. C. Kamerlin, A. Gräslund and S. K. Wärmländer, J. Trace Elem. Med. Biol., 2016, 38, 183 CrossRef CAS; (d) C. Wallin, S. B. Sholts, N. Österlund, J. Luo, J. Jarvet, P. M. Roos, L. Ilag, A. Gräslund and S. K. Wärmländer, Sci. Rep., 2017, 7, 14423 CrossRef.
- (a) R. Lal, H. Lin and A. P. Quist, Biochim. Biophys. Acta, 2007, 1768, 1966 CrossRef CAS; (b) J. C. Diaz, J. Linnehan, H. Pollard and N. Arispe, Biol. Res., 2006, 39, 447 CAS; (c) J. C. Diaz, O. Simakova, K. A. Jacobson, N. Arispe and H. B. Pollard, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3348 CrossRef CAS; (d) N. Arispe, J. Diaz, S. R. Durell, Y. Shafrir and H. R. Guy, Biochemistry, 2010, 49, 7847 CrossRef CAS.
- (a) I. A. Nikolaeva, A. Y. Misharin, G. V. Ponomarev, V. P. Timofeev and Y. V. Tkachev, Bioorg. Med. Chem. Lett., 2010, 20, 2872 CrossRef CAS; (b) L. Cheng, A. Kamkaew, H. Sun, D. Jiang, H. F. Valdovinos, H. Gong, C. G. England, S. Goel, T. E. Barnhart and W. Cai, ACS Nano, 2016, 10, 7721 CrossRef CAS PubMed.
- Q. Zhang, X. Hu, W. Wang and Z. Yuan, Biomacromolecules, 2016, 17, 661 CrossRef CAS.
- J. Luo, S. K. Wärmländer, A. Gräslund and J. P. Abrahams, J. Biol. Chem., 2016, 291, 16485 CrossRef CAS.
- (a) M. Chemerovski-Glikman, E. Rozentur-Shkop, M. Richman, A. Grupi, A. Getler, H. Y. Cohen, H. Shaked, C. Wallin, S. K. Wärmländer, E. Haas, A. Gräslund, J. H. Chill and S. Rahimipour, Chemistry, 2016, 22, 14236 CrossRef CAS; (b) J. Bieschke, J. Russ, R. P. Friedrich, D. E. Ehrnhoefer, H. Wobst, K. Neugebauer and E. E. Wanker, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7710 CrossRef CAS; (c) F. Yang, G. P. Lim, A. N. Begum, O. J. Ubeda, M. R. Simmons, S. S. Ambegaokar, P. P. Chen, R. Kayed, C. G. Glabe, S. A. Frautschy and G. M. Cole, J. Biol. Chem., 2005, 280, 5892 CrossRef CAS.
- K. Brännström, T. Islam, L. Sandblad and A. Olofsson, FEBS Lett., 2017, 591, 1167 CrossRef.
- N. Arispe, E. Rojas and H. B. Pollard, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 567 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: General details on the materials and methods, and any associated references and supporting scheme, table and figures. See DOI: 10.1039/c8sc01992d |
| ‡ Guy Leshem and Michal Richman contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |