
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWetting the lock and key enthalpically favours polyelectrolyte binding†
Emeric
Jeamet
a,
Jean
Septavaux
 a,
Alexandre
Héloin
a,
Marion
Donnier-Maréchal
a,
Melissa
Dumartin
a,
Benjamin
Ourri
a,
Pradeep
Mandal
b,
Ivan
Huc
b,
Emmanuelle
Bignon
cd,
Elise
Dumont
*c,
Christophe
Morell
d,
Jean-Patrick
Francoia
e,
Florent
Perret
a,
Laurent
Vial
*a and
Julien
Leclaire
*a
a,
Alexandre
Héloin
a,
Marion
Donnier-Maréchal
a,
Melissa
Dumartin
a,
Benjamin
Ourri
a,
Pradeep
Mandal
b,
Ivan
Huc
b,
Emmanuelle
Bignon
cd,
Elise
Dumont
*c,
Christophe
Morell
d,
Jean-Patrick
Francoia
e,
Florent
Perret
a,
Laurent
Vial
*a and
Julien
Leclaire
*a
aInstitut de Chimie et Biochimie Moléculaires et Supramoléculaires, UMR 5246 CNRS, Université Claude Bernard Lyon1, CPE Lyon, 43 Boulevard du 11 Novembre 1918, 69622 Villeurbanne Cedex, France. E-mail: laurent.vial@univ-lyon1.fr; julien.leclaire@univ-lyon1.fr
bInstitut de Chimie et Biologie des Membranes et des Nano-objets, UMR 5248 CNRS, Université de Bordeaux, IPB, 2 rue Escarpit, 33600 Pessac, France
cLaboratoire de Chimie, UMR 5182 CNRS, Ecole Normale Supérieure de Lyon, Université Claude Bernard Lyon 1, 46 Allée d’Italie, 69364 Lyon Cedex 07, France. E-mail: elise.dumont@ens-lyon.fr
dInstitut des Sciences Analytiques, UMR 5280 CNRS, Université Claude Bernard Lyon 1, Ecole Nationale Supérieure de Lyon, 5, rue de la Doua, 69100 Villeurbanne, France
eInstitut des Biomolécules Max Mousseron, UMR 5247 CNRS, Université de Montpellier, ENSCM, Place Eugène Bataillon, 34296 Montpellier Cedex 5, France
First published on 8th October 2018
Abstract
By using a combination of readily accessible experimental and computational experiments in water, we explored the factors governing the association between polyanionic dyn[4]arene and a series of α,ω-alkyldiammonium ions of increasing chain length. We found that the lock-and-key concept based on the best match between the apolar and polar regions of the molecular partners failed to explain the observed selectivities. Instead, the dissection of the energetic and structural contributions demonstrated that the binding events were actually guided by two crucial solvent-related phenomena as the chain length of the guest increases: the expected decrease of the enthalpic cost of guest desolvation and the unexpected increase of the favourable enthalpy of complex solvation. By bringing to light the decisive enthalpic impact of complex solvation during the binding of polyelectrolytes by inclusion, this study may provide a missing piece to a puzzle that one day could display the global picture of molecular recognition in water.
Introduction
Medicinal chemistry predominantly relies on the design and screening of synthetic guests to efficiently bind to the solvent-shielded cavities of biomacromolecular hosts, mostly proteins.1 While numerous lifesaving drugs have successfully been elaborated following this strategy over the past century, the reports of highly efficient artificial hosts targeting biomolecules are more seldom.2 Indeed, the design of synthetic hosts working in highly dissociative aqueous media requires not only the anticipation of the nature and strength of the non-covalent interactions with the target guest molecules, but also grasping the crucial role of the multiple water molecules surrounding the binding partners during the process.3 In the past few years, cucurbiturils have joined the restricted circle of cavitands, such as cyclodextrins and functionalized cyclophanes, successfully operating in water.2a These objects are made of two uncharged hydrophilic rims decorating a concave hydrophobic pocket. Efficient guest binding was proved to obey the lock-and-key principle, with a perfect match between the apolar and polar regions of the partners upon substantial water removal. In fact, the efficiency of these associations appeared to rely on two desolvation-based driving forces: an enthalpic gain due to the expulsion of high-energy water molecules from the cavity (so-called non-classical hydrophobic effect) and the entropically favoured release of the solvation layers from the rims into bulk water (so-called classical hydrophobic effect).4 In addition, Lee et al. recently exposed an additional role of water in the binding of α,ω-alkyldiammonium ions to cucurbiturils, where the structural mismatch between hosts and guests is compensated by supplemental hydrogen bonds between the guests and water molecules rather than by the distortion of the alkyl chains to maximize ion–dipole interactions.5The factors controlling the inclusion binding events between anionic or cationic cyclophanes with guest molecules of opposite polarity in water have similarly been extensively examined in the literature.6 In these studies, water was reported to agonistically contribute to the binding mostly through classical hydrophobic effects, i.e. via the entropically favourable release of water from the binding partners. Apart from the non-classical hydrophobic effect, desolvation of the partners is known to potentially dramatically hamper the binding enthalpy between charged species.7 For instance, the strength of the association between linear polyelectrolytes was reported to linearly increase with the number of salt bridges formed upon binding, but the corresponding enthalpic increment was shown to be two orders of magnitude lower in water than in the gas phase (i.e. 1.2 kcal mol−1 instead of 120 kcal mol−1).8 This penalty is commonly perceived as the inevitable enthalpic cost to remove water from the hydrophilic zones of the reactants, allowing their solvent-free pairing into a dry and stable complex. This scenario involves an enthalpy–entropy trade-off which results in an entropically driven association. Water and binding partners are therefore pictured to have an exclusive relationship: full desolvation of the hydrophobic zones maximizes the binding, while the full desolvation of the polar zones is the inevitable price to pay for maximizing their association. Careful examination of the X-ray structures of anionic p-sulfonatocalix[4]arene bound to lysine residues in proteins by McGovern et al.9 revealed that some water molecules are involved in the direct coordination environment of the host/guest ensemble. Beyond the classical hydrophobic effect which favourably contributes to the association, these specific solvent molecules also seem to participate in the binding as a true partner by playing the role of “bridging water”, a term coined for water mediating the assembly between biomolecules.10 Rationalizing and predicting the role of water in such a context require moving from a host/guest-centric to a solvent-centric point of view of the binding phenomena, or at least consideration of the solvent as a true binding partner.11,12
Herein, we collected experimental and computational data on the formation of inclusion complexes between dyn[4]arene 14 – displaying two polyanionic rims surrounded by a concave hydrophobic pocket as a charged analogue of cucurbiturils – and a series of α,ω-alkyldiammonium ions 2–8 of various chain lengths in water (Fig. 1). By dissecting the information generated from readily accessible tools, we elucidated for the first time the enthalpic contribution of water molecules to both the efficiency and the selectivity of the binding events, providing an unexpected and new solvent-centric binding scenario for the formation of tightly bound inclusion complexes between charged partners in water.
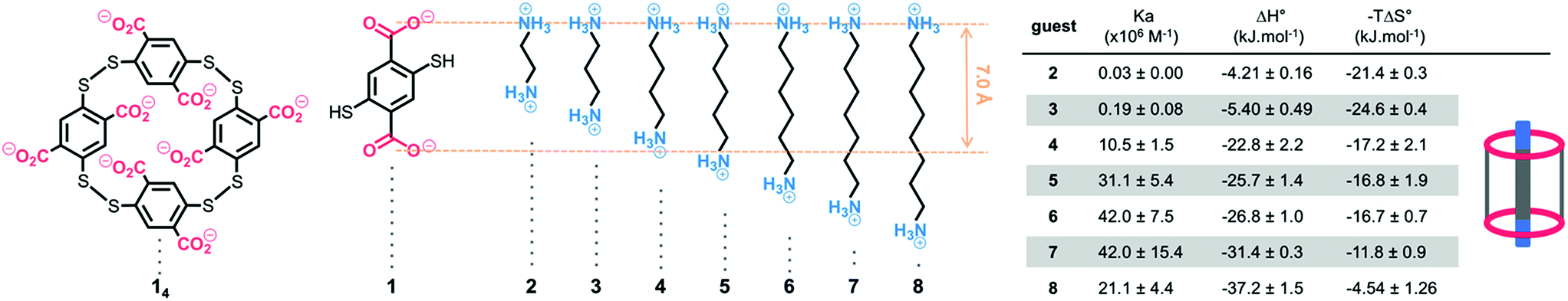 | ||
| Fig. 1 Dyn[4]arene 14, bisthiophenol 1, α,ω-alkyldiammonium ions 2–8, and their respective thermodynamic binding parameters measured by ITC in 200 mM TRIS buffer at a physiological pH of 7.4. | ||
Results and discussion
Experimental data
Dyn[4]arene 14, which could be isolated on a gram-scale and without chromatographic purification from a dynamic combinatorial library made from bisthiophenol 1,13 was reported by some of us to display promising biological properties such as the ability to modulate polyamine-mediated DNA helicity and aggregation states14 and also to detect and discriminate lysine derivatives through induced circular dichroism.15 Both properties were directly related to the sub-micromolar affinity of 14 for biogenic polyamines in water at neutral pH, where these strong binding constants were imputed to the formation of pseudorotaxane-type complexes that optimize the formation of salt bridges between the molecular partners. New experiments allowed us to further characterize dyn[4]arene 14 and its corresponding complexes with polyaminated guests. First, potentiometric titrations revealed that the macrocycle is fully deprotonated at a physiological pH of 7.4 as previously postulated (degree of protonation Θ < 0.5%, see the ESI†). The proof of the formation of inclusion complexes between fully deprotonated 14 and aliphatic guests bearing hydrophilic end groups was provided by the X-ray diffraction analyses of a rac-Lys-NH2⊂14 complex (see the ESI†). It confirms that the strong ring-current shielding effect (i.e. a Δ∂ of −2.5 ppm on average) observed by NMR for the central protons of guest molecules upon binding is symptomatic of this inclusion phenomenon.16From a naïve lock-and-key perspective based on only the best distance match occurring between partner-borne charges, we expected that – among α,ω-alkyldiammonium ions 2–8 – guest 4 or 5 should display a higher affinity for macrocyclic 14 (Fig. 1). Isothermal calorimetry titrations (ITC) actually revealed that the association constants increased from 1,2-diaminobutane 2 until reaching an optimum value for 1,6-diaminohexane 6. Further elongation of the carbon chain surprisingly did not result in significant variation of the binding constant. Examination of the individual thermodynamic parameters indicated that the binding enthalpy (i.e. ΔH°) continuously increased with the chain length of the guest. In turn, the binding entropy (i.e. −TΔS°) followed an opposite trend, suggesting a traditional enthalpy–entropy compensation effect at first sight.17
Experimental vs. computed data
In order to obtain some structural and energetic information that could explain these experimental trends, we performed molecular dynamics (MD) simulations on dyn[4]arene 14 and its various complexes with guests 2–8. MD trajectories of 100 ns within a truncated octahedral box of ∼8000 water molecules (10 Å buffer) were collected with the Amber 12 software package,18 using the GAFF19 and parm99 (ref. 20) force–field parameters. Our dynamics were post-processed for the last converged 10 ns of the trajectories in order to extract free energies of binding using the MM/GBSA method (see the ESI† for full details).21,22 Such calculations are known to provide inaccurate absolute energies of complex formation, since the MM/GBSA approach includes two major thermodynamic approximations. Indeed, it does not provide information about the evolution of the entropy from water molecules during the binding processes and also does not take into account any potential conformational induced fit upon complexation. Therefore, the only valid comparison between such computed data from MD trajectories with the experimental data from ITC titrations must be drawn on the relative thermodynamic parameters (i.e. ΔΔH° and −TΔΔS°) between guests 2–8 and guest 5 as a reference of intermediate chain length. In this respect, an excellent agreement between computed and experimental differential energies could be observed (Fig. 2), suggesting that both the entropy from water molecules and the energetic penalties associated with conformational induced fit upon complexation – although they certainly contributed to the binding events – remained constant along the series of guests (vide infra). Therefore, readily accessible MD trajectories on dyn[4]arene 14 and its various complexes with guests 2–8 could be exploited for an in-depth structural analysis of the binding events.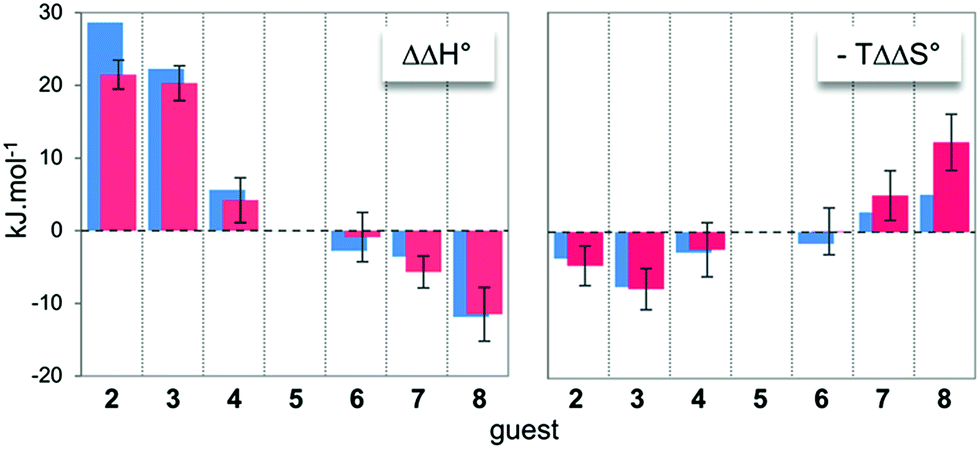 | ||
| Fig. 2 Comparison between the experimental (red) and computed (blue) relative free energies of binding between host 14 and guests 2–8. Guest 5 was selected as a reference. | ||
Assessing the lock-and-key model
First, the time-averaged solvent-accessible surface areas (SASAs) of the complexes, as well as the time-averaged ammonium–carboxylate distances between the partners, were extracted along the trajectories (Fig. 3, left). Such parameters were chosen for their ability to further apprehend the structural complementarity between the host and the guests. Accordingly, the enthalpy of association between charged partners was predicted to be optimal when both the SASA and the mean distance between the cationic and anionic centres are at their lowest. As first intuitively anticipated, guest 4 displayed an optimal structural complementarity for host 14, marking the frontier between a “filling” and a “protruding” regime, while its experimental binding enthalpy was surprisingly higher by 13 kJ mol−1 in comparison to the longest guest 8 (Fig. 1).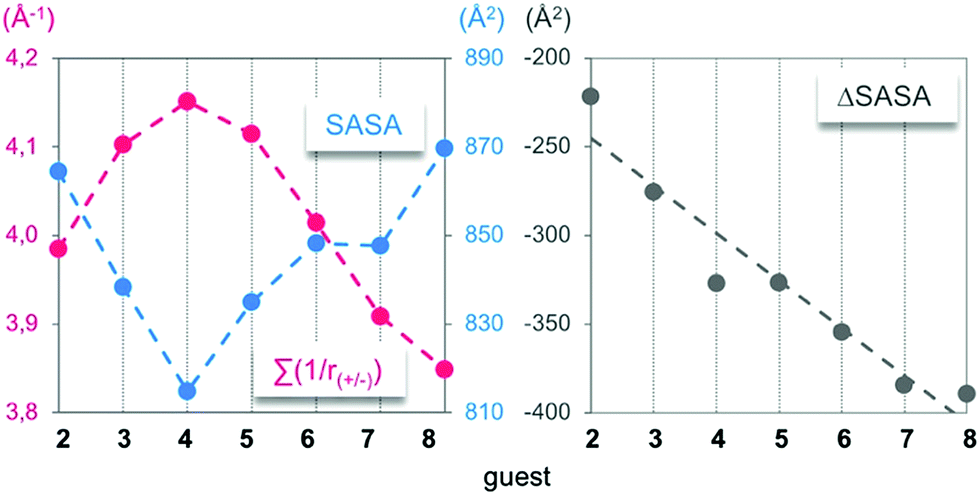 | ||
| Fig. 3 Left: Time-averaged solvent-accessible surface area (SASA) and sum of the time-averaged mean distances between the cationic and anionic centres (inverse function) of the complexes between 14 and 2–8. Right: ΔSASA = SASA of the complex − SASA of the host − SASA of the guest. | ||
Further structural analysis of the X-ray and computed structures established that the perfect match between the rectangular cross sections of both the aliphatic linear guests and the host's cavity in the complexed state strongly limited the extent of guest-dependent induced fit of the partners during the binding. Energetically speaking, the most contributing phenomenon appears to be the guest-independent transition of 14 from an empty diamond to a filled parallelepipedic shape accompanied by the expulsion of high-energy water.23 On the host, although bending, tilting or twisting of terephthalic units could be observed in snapshots to improve the contact with short chain guests, it clearly does not lead to significant enthalpic contribution to the binding in favour of these species. On the guest side, the cross section match rules out any spring-like compression of the guest within the cavity. Instead, an excessive chain length was structurally compensated by end-chain gauche conformations (e.g. with torsion angles of 178° and 74° in 4⊂14 and 8⊂14, respectively) that maintained the protruding polar groups of the guest in contact with the axial part of the rims of the host. Hence one may hypothesize that the enthalpic preference for long chain guests may partly be due to a stronger syn vs. anti salt bridge.24
The lock and key picture does not only imply an optimized match between the binding sites of opposite polarities, but it also encompasses the classical hydrophobic effect. In this respect, long-chain diamines bending toward the axial regions of the host's rim may be expected to display a stronger hydrophobic effect than the short homologues upon binding. Since the decrease in the SASA of the complex during the host “filling” regime (i.e. for guests 2–4) was twice as important as its increase during the guest “protruding” regime (i.e. for guests 5–8), it demonstrated, in terms of the surface, an incremental growth of the key within the lock. As witnessed by the ΔSASA which is the standard metric for the classical hydrophobic effect (Fig. 3, right),25 it can consequently be considered as constant during the protruding regime.
In summary, even when incorporating some potential induced fit, the lock-and-key concept based on the best match between the apolar and polar regions of the molecular partners failed to explain the observed association constants, and the thermodynamics of inclusion complexation should follow an unknown scenario, which may involve water as a third and overwhelming binding partner.
Thermodynamic deconvolution of the binding events
In order to appreciate the role of water, the binding process (Fig. 4, A) was divided into the following thermodynamic cycle: the desolvation of the isolated partners (Fig. 4, B), their subsequent association in the gas phase (Fig. 4, C), and the resolvation of the resulting complex (Fig. 4, D). An enthalpic (i.e. ΔH°) and an entropic (i.e. −TΔS°) term which may be favourable or unfavourable to the global process were assigned to each step (Fig. 4, A), according to eqn (1) and (2).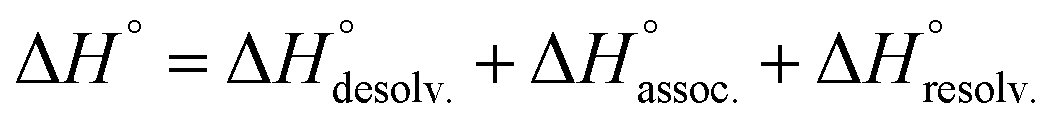 | (1) |
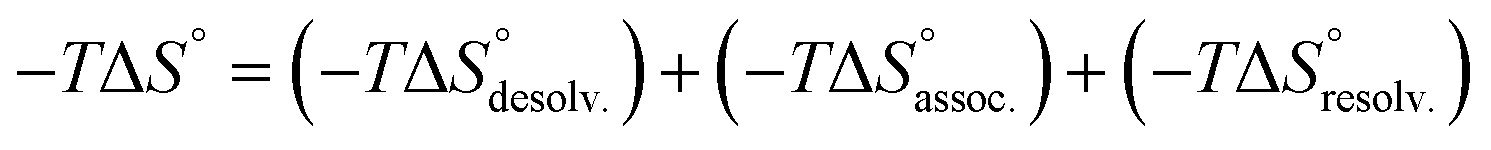 | (2) |
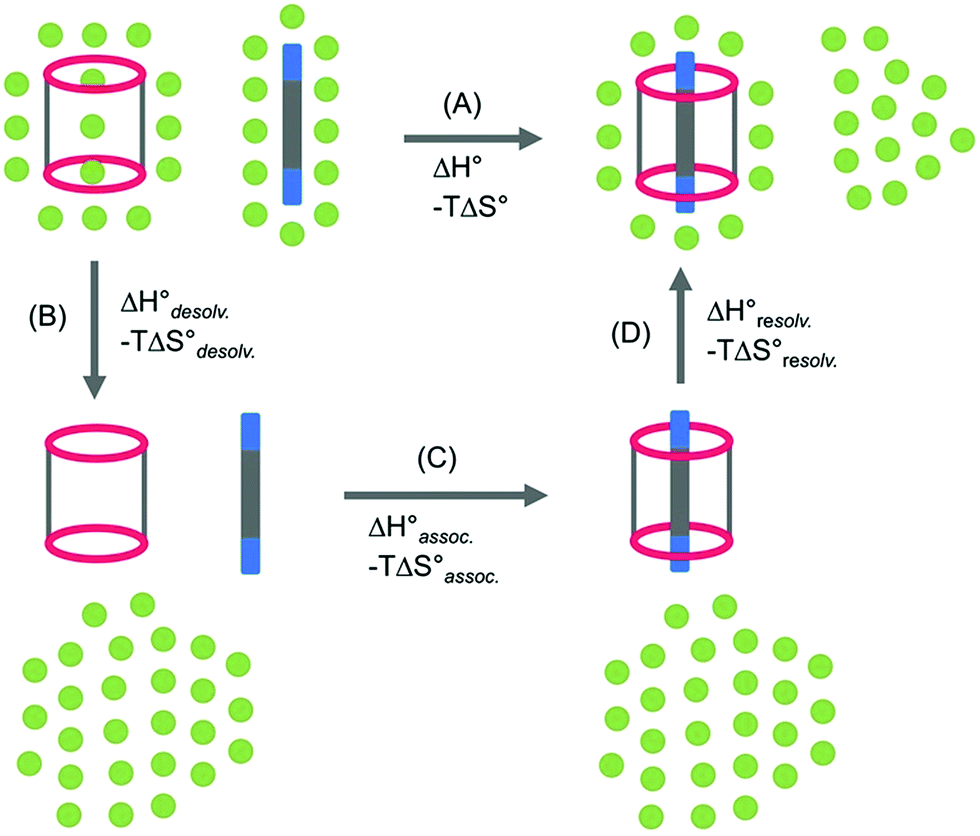 | ||
| Fig. 4 Schematic representation of the binding process between host 14 and guest 2–8 (A), divided along a thermodynamic cycle into the desolvation of the partners (B), their association in the gas phase (C), and the solvation of the complexes (D). Each step was associated with the corresponding enthalpic (i.e. ΔH°) and entropic (i.e. ΔS°) contributions. | ||
The increasing entropic penalty with the guest chain length principally resulted from the increasing loss of degrees of freedom of the guest upon binding (e.g. translational freedom or internal rotation freedom) and not from the host. As mentioned above, the MM/GBSA approach did not provide any access to the entropic parameters of the desolvation/resolvation steps. Nevertheless, the strong correspondence observed between the experimental relative entropy of complex formation in solution (i.e. −TΔΔS°) and the computed relative entropy of association in the gas phase (i.e. −TΔΔS°) seems to indicate that  , which encompasses the classical hydrophobic effect and the release of water molecules from the polar areas remained constant along the series of guests (Fig. 2).
, which encompasses the classical hydrophobic effect and the release of water molecules from the polar areas remained constant along the series of guests (Fig. 2).
Regarding the binding enthalpy in the gas phase (i.e.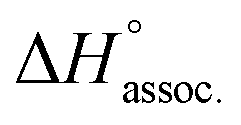 ), it was mainly of electrostatic nature (Fig. 5, light grey), and decreased in amplitude with the chain length of the guest. This trend could be explained by the increase of the inductive effect exerted by the hydrocarbon spacing chain on the ammonium end groups, which progressively attenuated the carboxylate–ammonium interactions. As a compensation effect, the van der Waals interactions between the central moieties of the partners increased with the length of the guest, presumably through the multiplication of CH–π interactions between the aliphatic axle and the aromatic wheel (Fig. 5, dark grey). Nevertheless, this latter contribution remained marginal, even for the complex formed with the most lipophilic guest 8 (i.e. < 2%). Overall,
), it was mainly of electrostatic nature (Fig. 5, light grey), and decreased in amplitude with the chain length of the guest. This trend could be explained by the increase of the inductive effect exerted by the hydrocarbon spacing chain on the ammonium end groups, which progressively attenuated the carboxylate–ammonium interactions. As a compensation effect, the van der Waals interactions between the central moieties of the partners increased with the length of the guest, presumably through the multiplication of CH–π interactions between the aliphatic axle and the aromatic wheel (Fig. 5, dark grey). Nevertheless, this latter contribution remained marginal, even for the complex formed with the most lipophilic guest 8 (i.e. < 2%). Overall, 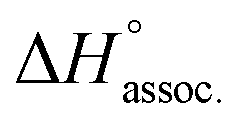 decreased with the chain length of the guest, while the experimental enthalpy of complex formation exactly followed the opposite trend. Although being counter-intuitive, solvent–solute interactions (i.e.
decreased with the chain length of the guest, while the experimental enthalpy of complex formation exactly followed the opposite trend. Although being counter-intuitive, solvent–solute interactions (i.e.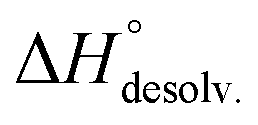 and
and 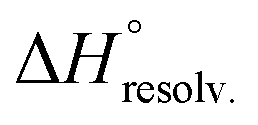 ) should therefore be the discriminant contribution to the global enthalpy ΔH° of complexation between polyelectrolytes 14 and 2–8.
) should therefore be the discriminant contribution to the global enthalpy ΔH° of complexation between polyelectrolytes 14 and 2–8.
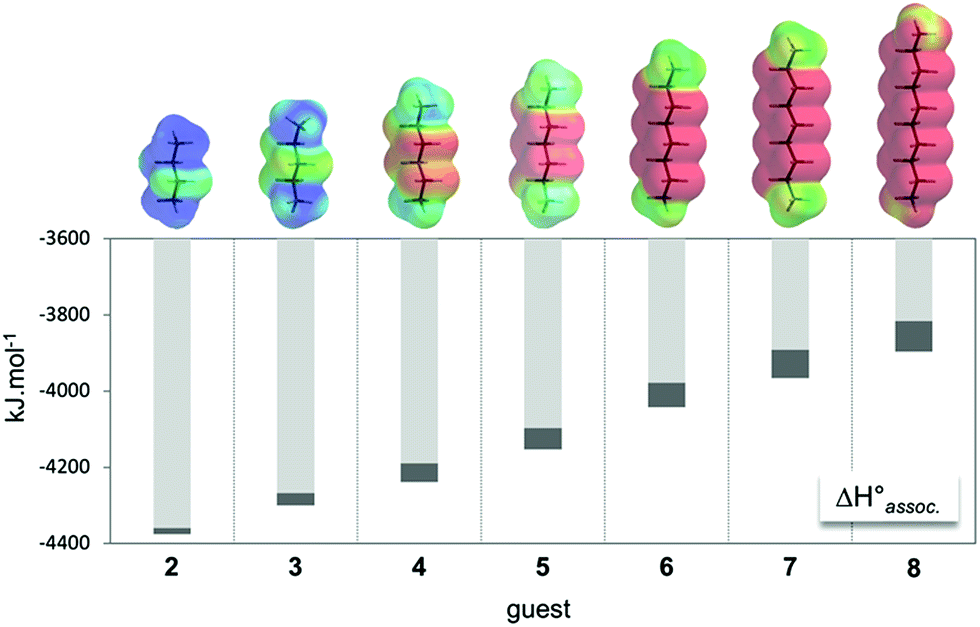 | ||
Fig. 5 Evolution of the computed enthalpy of association in the gas phase between host 14 and guest 2–8 from MD trajectories. The coulombic and van der Waals contributions to 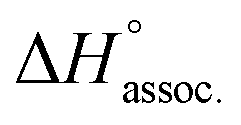 are highlighted in light and dark grey, respectively. Each guest is displayed with its corresponding electrostatic potential surface calculated using Spartan'14 software. Color scale from 720 (red) to 980 (blue) kJ mol−1; isovalue = 0.002 au. are highlighted in light and dark grey, respectively. Each guest is displayed with its corresponding electrostatic potential surface calculated using Spartan'14 software. Color scale from 720 (red) to 980 (blue) kJ mol−1; isovalue = 0.002 au. | ||
Following Hess's law, the relative enthalpy of solvation between the bound and unbound states is the sum of the enthalpy of desolvation of the free polyelectrolytes 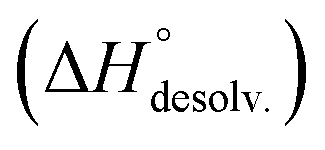 and the enthalpy of solvation of the resulting complex
and the enthalpy of solvation of the resulting complex 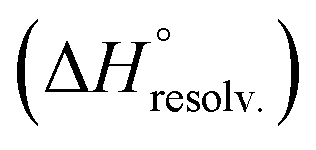 . As deduced from the comparison between the trends in the gas-phase and in solution, the experimental enthalpy of binding followed the relative enthalpy of solvation between bound and unbound states. These computed values decreased with the size of the guest and were markedly dominated by polar contributions (i.e. >99.8%), corresponding to the desolvation/solvation of the polar areas of the different partners (Fig. 6, blue). Regarding the individual species, the enthalpic cost of desolvation of the receptor is independent of the nature of the guest, and the enthalpic penalty of desolvation of the guest decreased linearly with its chain length, correlating with its hydrophobicity (i.e. hydrocarbon chain elongation and concomitant reduction of the effective charge on the ammonium groups) (Fig. 6, red). Interestingly, the desolvation of the guest alone could not account for the steep decrease in the relative enthalpy of solvation between bound and unbound states induced by the chain elongation. Mathematically, the difference corresponds to the increasingly favourable enthalpy of solvation of the complex with the guest's chain length, despite its progressive protrusion from the cavity and the consequent exposure of its hydrophobic surface to the bulk solution.
. As deduced from the comparison between the trends in the gas-phase and in solution, the experimental enthalpy of binding followed the relative enthalpy of solvation between bound and unbound states. These computed values decreased with the size of the guest and were markedly dominated by polar contributions (i.e. >99.8%), corresponding to the desolvation/solvation of the polar areas of the different partners (Fig. 6, blue). Regarding the individual species, the enthalpic cost of desolvation of the receptor is independent of the nature of the guest, and the enthalpic penalty of desolvation of the guest decreased linearly with its chain length, correlating with its hydrophobicity (i.e. hydrocarbon chain elongation and concomitant reduction of the effective charge on the ammonium groups) (Fig. 6, red). Interestingly, the desolvation of the guest alone could not account for the steep decrease in the relative enthalpy of solvation between bound and unbound states induced by the chain elongation. Mathematically, the difference corresponds to the increasingly favourable enthalpy of solvation of the complex with the guest's chain length, despite its progressive protrusion from the cavity and the consequent exposure of its hydrophobic surface to the bulk solution.
 | ||
| Fig. 6 Evolution of the computed enthalpy of desolvation of guests 2–8, and of the corresponding relative enthalpy of solvation between bound and unbound states with host 14. | ||
Solvation picture of the complexes
The guest-dependent evolution of the solvation for the complexes' polar areas could be visualized. To put it simply, a mesh incorporating the positions of each oxygen atom from the solvent molecules was extracted for each complex from their respective MD trajectory. Then, a continuum of values corresponding to the water molecule density was interpolated from this mesh. From a plane intersecting the carboxylate rims or the facing ammonium heads, we summed these values on a 3 Å-thick vertical slice of the water box. The isodensity contours of the slice were finally plotted using the average bulk density as a reference (Fig. 7, see the ESI† for the full set). A distance of 3 Å is generally considered as the maximum length for an intermolecular hydrogen bond in water. Therefore, the slices encompass the first solvation layer around the partners.26 It turned out that the density of water molecules in these solvation layers became higher around the polar heads as the chain length of the guest increased, with densities reaching ca. twice that of bulk water for 8⊂14. In addition, the percentage of overlapping for the computed slices from the carboxylate rim and its facing ammonium head was 55% for 2⊂14, 93% for 4⊂14, and 59% for 8⊂14. Since the enthalpy of ion–water and water–water interactions respectively display an inverse distance dependence,27 the increased density of water molecules around the ionic heads of the complexes should result in both stronger and more numerous solute–solvent and solvent–solvent interactions (i.e. related to a more favourable enthalpy of solvation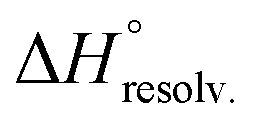 ). To summarize, a lock-and-key model represented by complex 4⊂14 did not lead to the ideal resolvation scenario for the inclusion binding of charged partners, which seemed to require exclusive solvation layers, and consequently maximized the solvent exposure of the polar heads for both partners. Finally, this increased density of water molecules engaged in the first solvation layers of the complex should come with an increased entropic cost, which should be counterbalanced by the entropic benefit in the desolvation of the guest as its chain length increased (i.e. corresponding to a classical hydrophobic effect), potentially explaining the compensation between
). To summarize, a lock-and-key model represented by complex 4⊂14 did not lead to the ideal resolvation scenario for the inclusion binding of charged partners, which seemed to require exclusive solvation layers, and consequently maximized the solvent exposure of the polar heads for both partners. Finally, this increased density of water molecules engaged in the first solvation layers of the complex should come with an increased entropic cost, which should be counterbalanced by the entropic benefit in the desolvation of the guest as its chain length increased (i.e. corresponding to a classical hydrophobic effect), potentially explaining the compensation between 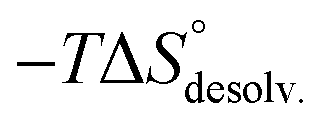 and
and 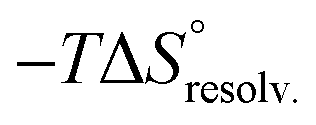 observed along the series of guests.
observed along the series of guests.
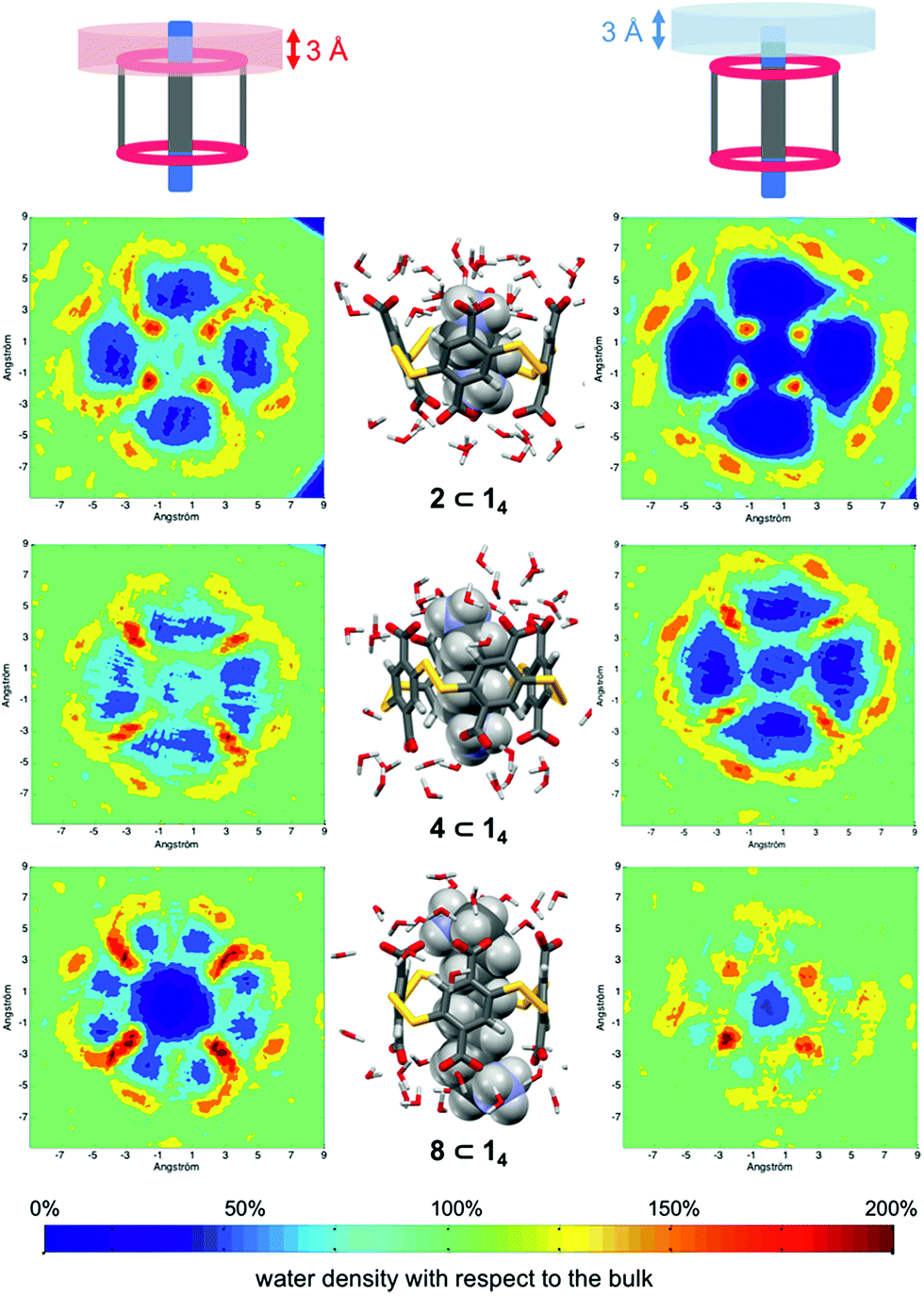 | ||
| Fig. 7 Time-averaged density of water molecules around the complexes formed between the dyn[4]arene 14 and α,ω-alkyldiammonium ions 2, 4 and 8 from the MD trajectories (see the ESI† for the full set). Left and right maps correspond to the first solvation layers of the carboxylate rims and the ammonium heads, respectively. Corresponding snapshots from the MD trajectories are displayed in the middle. Bottom: increased water density scale from dark blue (0%) to dark red (200%), with respect to the bulk (green) as the average value (100%). | ||
Conclusions
We demonstrated that a solvent-centric analysis may be conducted with basic tools, allowing us to decipher the energetic and structural contributions that govern the formation of inclusion complexes between polyanionic dyn[4]arene and α,ω-alkyldiammonium ions in water at physiological pH. Despite a pairing through multiple salt bridges, which is often considered as one of the strongest non-covalent bonds in the supramolecular toolbox, these associations do not follow a classical lock-and-key model. While the binding enthalpy is more favourable for the smallest guests in the gas phase, the trend is completely reversed in solution, with a marked preference for long diamines whose end groups protrude from the cavity. Dissection of the various enthalpic contributions indicates that this counter-intuitive selectivity was dictated by two crucial solvent–related phenomena as the chain length of the guest increases: the expected decrease of the enthalpic cost of guest desolvation and the unexpected increase of the favourable enthalpy of complex solvation. Remarkably, this latter phenomenon could be qualitatively appreciated by comparing the densities of water layers around the binding sites of the different complexes. Such a picture, which gathers not two but three essential molecular components, points out the need for a fresh perspective when it comes to the inclusion binding of charged molecular partners in water.Conflicts of interest
There are no conflicts to declare.References
- (a) Protein-Ligand Interactions, in Methods and Principles in Medicinal Chemistry Series, ed. S. Gohlke, Wiley-VCH, Weinheim, 2012, vol. 53 Search PubMed; (b) C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061–5084 CrossRef CAS.
- For recent reviews about, respectively, cucurbit[n]urils, pillar[n]arenes, cyclodextrins, calix[n]arenes, and deep-cavity cavitands, see: (a) S. J. Barrow, S. Kasera, M. J. Rowland, J. Del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS; (b) L. L. Tan and Y. W. Yang, J. Inclusion Phenom. Macrocyclic Chem., 2015, 81, 13–33 CrossRef CAS; (c) G. Crini, Chem. Rev., 2014, 114, 10940–10975 CrossRef CAS; (d) F. Perret and A. W. Coleman, Chem. Commun., 2011, 47, 7303 RSC; (e) B. C. Gibb, Beilstein J. Org. Chem., 2016, B, 684–701 CrossRef.
- K. Kanagaraj, M. Alagesan, Y. Inoue and C. Yang, Solvation Effects in Supramolecular Chemistry, in Comprehensive Supramolecular Chemistry II, ed. J. Atwood, G. W. Gokel and L. Barbour, 2017, vol. 1, pp. 11–60 Search PubMed.
- (a) F. Biedermann, W. M. Nau and H.-J. Schneider, Angew. Chem., Int. Ed., 2014, 53, 11158–11171 CrossRef CAS; (b) F. Biedermann, V. D. Uzunova, O. A. Scherman, W. M. Nau and A. De Simone, J. Am. Chem. Soc., 2012, 134, 15318–15323 CrossRef CAS.
- S. J. C. Lee, J. W. Lee, H. H. Lee, J. Seo, D. H. Noh, Y. H. Ko, K. Kim and H. I. Kim, J. Phys. Chem. B, 2013, 117, 8855–8864 CrossRef CAS.
- For selected recent references, see: (a) Q. Duan, W. Zhao and K. Lu, Tetrahedron Lett., 2017, 58, 4403–4406 CrossRef CAS; (b) X. Y. Hu, S. Peng, D. S. Guo, F. Ding and Y. Liu, Supramol. Chem., 2015, 27, 336–345 CrossRef CAS; (c) K. Wang, S.-Y. Xing, X.-G. Wang and H.-X. Dou, Org. Biomol. Chem., 2015, 13, 5432–5443 RSC; (d) B. Gõmez, V. Francisco, F. Fernández-Nieto, L. Garcia-Rio, M. Martín-Pastor, M. R. Paleo and F. J. Sardina, Chem.–Eur. J., 2014, 20, 12123–12132 CrossRef; (e) C. Bonaccorso, A. Ciadamidaro, V. Zito, C. Sgarlata, D. Sciotto and G. Arena, Thermochim. Acta, 2012, 530, 107–115 CrossRef CAS; (f) K. Fucke, K. M. Anderson, M. H. Filby, M. Henry, J. Wright, S. A. Mason, M. J. Gutmann, L. J. Barbour, C. Oliver, A. W. Coleman, J. L. Atwood, J. A. K. Howard and J. W. Steed, Chem.–Eur. J., 2011, 17, 10259–10271 CrossRef CAS.
- As a result, the binding enthalpy associated with the formation of a salt bridge in water is lower than that for cation–π interactions, see: S. M. Ngola, P. C. Kearney, S. Mecozzi, K. Russell and D. A. Dougherty, J. Am. Chem. Soc., 1999, 121, 1192–1201 CrossRef CAS.
- (a) F. Biedermann and H. J. Schneider, Chem. Rev., 2016, 116, 5216–5300 CrossRef CAS; (b) J. P. Gallivan and D. A. Dougherty, J. Am. Chem. Soc., 2000, 122, 870–874 CrossRef CAS.
- (a) R. E. McGovern, B. D. Snarr, J. A. Lyons, J. McFarlane, A. L. Whiting, I. Paci, F. Hof and P. B. Crowley, Chem. Sci., 2015, 6, 442–449 RSC; (b) R. E. McGovern, H. Fernandes, A. R. Khan, N. P. Power and P. B. Crowley, Nat. Chem., 2012, 4, 527–533 CrossRef CAS.
- S. H. Chong and S. Ham, Sci. Rep., 2017, 7, 1–10 CrossRef CAS.
- P. W. Snyder, M. R. Lockett, D. T. Moustakas and G. M. Whitesides, Eur. Phys. J.: Spec. Top., 2014, 223, 853–891 Search PubMed.
- Such a paradigm shift has already been adopted for the design of drugs targeting water-exposed protein surfaces, where it is admitted as a rule of thumb that ordered water molecules undergoing four bonding interactions to the protein cannot be replaced by ligands, see: E. Persch, O. Dumele and F. Diederich, Angew. Chem., Int. Ed., 2015, 54, 3290–3327 CrossRef CAS.
- P. T. Skowron, M. Dumartin, E. Jeamet, F. Perret, C. Gourlaouen, A. Baudouin, B. Fenet, J. V. Naubron, F. Fotiadu, L. Vial and J. Leclaire, J. Org. Chem., 2016, 81, 654–661 CrossRef CAS.
- L. Vial, R. F. Ludlow, J. Leclaire, R. Pérez-Fernández and S. Otto, J. Am. Chem. Soc., 2006, 128, 10253–10257 CrossRef CAS.
- L. Vial, M. Dumartin, M. Donnier-Maréchal, F. Perret, J.-P. Francoia and J. Leclaire, Chem. Commun., 2016, 52, 14219–14221 RSC.
- The analogous dyn[3]arene 13, which was unable to accommodate guest molecules within its small cavity, displayed much lower binding affinities with polyamines in comparison with 14, see: M. Donnier-Maréchal, J. Septavaux, E. Jeamet, A. Héloin, F. Perret, E. Dumont, J.-C. Rossi, F. Ziarelli, J. Leclaire and L. Vial, Org. Lett., 2018, 20, 2420–2423 CrossRef.
- D. H. Leung, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 2798–2805 CrossRef CAS PubMed.
- D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco, 2012 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS.
- J. Wang, P. Cieplak and P. A. Kollman, J. Comput. Chem., 2000, 21, 1049–1074 CrossRef CAS.
- (a) S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed; (b) J. Wang, T. Hou and X. Xu, Curr. Comput.-Aided Drug Des., 2006, 2, 287–306 CrossRef CAS.
- We also used the MM/PBSA approach that yielded similar results (data not shown), see ref. 19.
- The computed data showed that a single molecule of water was present within the cavity of the empty host, where its release during the binding events could result in a non-classical hydrophobic effect that, if applicable, remained constant along the series of guests. See: H. J. Schneider, Int. J. Mol. Sci., 2015, 16, 6694–6717 CrossRef CAS.
- The syn lone pair of a carboxylate was reported to be several orders of magnitude more basic than the anti, see: Y. Li and K. N. Houk, J. Am. Chem. Soc., 1989, 111, 4505–4507 CrossRef CAS.
- J. M. Fox, K. Kang, M. Sastry, W. Sherman, B. Sankaran, P. H. Zwart and G. M. Whitesides, Angew. Chem., Int. Ed., 2017, 56, 3833–3837 CrossRef CAS.
- W. Kabsch and C. Sander, Biopolymers, 1983, 22, 2577–2637 CrossRef CAS.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental and computational details. CCDC 1554746. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc02966k |
| This journal is © The Royal Society of Chemistry 2019 |