
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Host–guest selectivity in a series of isoreticular metal–organic frameworks: observation of acetylene-to-alkyne and carbon dioxide-to-amide interactions†
Jack D.
Humby‡
 a,
Oguarabau
Benson‡
b,
Gemma L.
Smith
a,
Stephen P.
Argent
c,
Ivan
da Silva
d,
Yongqiang
Cheng
e,
Svemir
Rudić
d,
Pascal
Manuel
d,
Mark D.
Frogley
f,
Gianfelice
Cinque
f,
Lucy K.
Saunders
f,
Iñigo J.
Vitórica-Yrezábal
a,
George F. S.
Whitehead
a,
Timothy L.
Easun
g,
William
Lewis
b,
Alexander J.
Blake
b,
Anibal J.
Ramirez-Cuesta
d,
Sihai
Yang
*a and
Martin
Schröder
*a
a,
Oguarabau
Benson‡
b,
Gemma L.
Smith
a,
Stephen P.
Argent
c,
Ivan
da Silva
d,
Yongqiang
Cheng
e,
Svemir
Rudić
d,
Pascal
Manuel
d,
Mark D.
Frogley
f,
Gianfelice
Cinque
f,
Lucy K.
Saunders
f,
Iñigo J.
Vitórica-Yrezábal
a,
George F. S.
Whitehead
a,
Timothy L.
Easun
g,
William
Lewis
b,
Alexander J.
Blake
b,
Anibal J.
Ramirez-Cuesta
d,
Sihai
Yang
*a and
Martin
Schröder
*a
aSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: Sihai.Yang@manchester.ac.uk; M.Schroder@manchester.ac.uk
bSchool of Chemistry, University of Nottingham, Nottingham, NG7 2RD, UK
cDepartment of Chemistry, University of Warwick, CV4 7AL, UK
dISIS Facility, STFC Rutherford Appleton Laboratory, Oxfordshire OX11 0QX, UK
eOak Ridge National Laboratory, Oak Ridge, TN 37831, USA
fDiamond Light Source, Harwell Science and Innovation Campus, Oxfordshire, OX11 0DE, UK
gSchool of Chemistry, Cardiff University, Cardiff CF10 3XQ, UK
First published on 12th October 2018
Abstract
In order to develop new porous materials for applications in gas separations such as natural gas upgrading, landfill gas processing and acetylene purification it is vital to gain understanding of host–substrate interactions at a molecular level. Herein we report a series of six isoreticular metal–organic frameworks (MOFs) for selective gas adsorption. These materials do not incorporate open metal sites and thus provide an excellent platform to investigate the effect of the incorporation of ligand functionality via amide and alkyne groups on substrate binding. By reducing the length of the linker in our previously reported MFM-136, we report much improved CO2/CH4 (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) and CO2/N2 (15:85) selectivity values of 20.2 and 65.4, respectively (1 bar and 273 K), in the new amide-decorated MOF, MFM-126. The CO2 separation performance of MFM-126 has been confirmed by dynamic breakthrough experiments. In situ inelastic neutron scattering and synchrotron FT-IR microspectroscopy were employed to elucidate dynamic interactions of adsorbed CO2 molecules within MFM-126. Upon changing the functionality to an alkyne group in MFM-127, the CO2 uptake decreases but the C2H2 uptake increases by 68%, leading to excellent C2H2/CO2 and C2H2/CH4 selectivities of 3.7 and 21.2, respectively. Neutron powder diffraction enabled the direct observation of the preferred binding domains in MFM-126 and MFM-127, and, to the best of our knowledge, we report the first example of acetylene binding to an alkyne moiety in a porous material, with over 50% of the acetylene observed within MFM-127 displaying interactions (<4 Å) with the alkyne functionality of the framework.
:50) and CO2/N2 (15:85) selectivity values of 20.2 and 65.4, respectively (1 bar and 273 K), in the new amide-decorated MOF, MFM-126. The CO2 separation performance of MFM-126 has been confirmed by dynamic breakthrough experiments. In situ inelastic neutron scattering and synchrotron FT-IR microspectroscopy were employed to elucidate dynamic interactions of adsorbed CO2 molecules within MFM-126. Upon changing the functionality to an alkyne group in MFM-127, the CO2 uptake decreases but the C2H2 uptake increases by 68%, leading to excellent C2H2/CO2 and C2H2/CH4 selectivities of 3.7 and 21.2, respectively. Neutron powder diffraction enabled the direct observation of the preferred binding domains in MFM-126 and MFM-127, and, to the best of our knowledge, we report the first example of acetylene binding to an alkyne moiety in a porous material, with over 50% of the acetylene observed within MFM-127 displaying interactions (<4 Å) with the alkyne functionality of the framework.
Introduction
Porous metal–organic frameworks (MOFs) have attracted considerable attention due to their promise in a wide range of applications, notably in gas adsorption, selectivity and catalysis.1–6 Modulation of organic linkers and metal nodes allows fine-tuning of properties to tailor MOFs for the application of interest. In the field of gas adsorption, MOFs have shown advantages for selective uptake of specific guests, such as CO2 (ref. 7–9) and C2H2.10–12 Capture and separation of CO2 from flue gas streams via the utilization of selective and regenerable sorbents, such as MOFs, provides a promising alternative pathway to conventional amine-scrubbers.13 Moreover, MOF adsorbents also have applications in natural gas (containing ∼15% CO2) upgrading, and the selective separation of landfill gas (40–60% CO2).14,15 Other industrially significant separations include the purification of C2H2, an important feedstock in the petrochemical industry. C2H2 is produced by the partial combustion of CH4 and hence requires separation from CH4 and CO2 to obtain high-purity C2H2.16 However, the identical kinetic diameters of 3.30 Å and similar boiling points of −84 °C and −79 °C, for C2H2 and CO2, respectively, make C2H2/CO2 separation under ambient conditions a highly challenging task.17–19Common strategies for enhancing host–guest interactions in MOFs include incorporation of open metal sites,20 polar functional groups (e.g., –NH2, –CONH–, –OH, –F)17,21–24 and narrowing pore channels by use of small ligands.25 For example, polar nitrogen-containing groups remain a favored approach to enhancing CO2 adsorption, as shown in a crystallographic study that visualized CO2 molecules directly interacting with the amine group in a Zn–MOF.26 However, we have previously reported neutron diffraction and scattering data revealing that the high CO2 uptake in amide-functionalized MFM-136 is not solely due to guest–host interactions at the amide group, but is a combination of geometry, pore size and functionality that lead to improved gas sorption properties.27 Whilst many MOFs have been reported for their gas sorption properties, it is often difficult to fully account for differences in performance owing to many variables such as surface area, porosity and pore geometry, functionality and presence of open metal sites. Thus, to aid design-based approaches for improved materials, thorough investigations of isoreticular series of MOFs such as the IRMOF,28 UiO-66 (ref. 29) and MOF-74 (ref. 30) series are important.
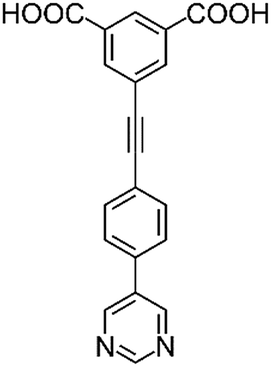
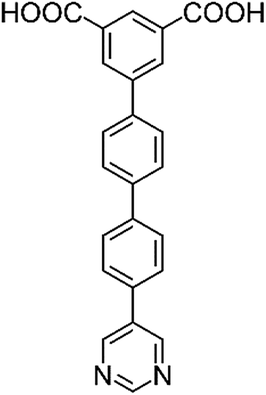
Herein we present a comprehensive investigation into the roles of functional groups, pore geometry and porosity in enhancing selective gas binding through a series of six isostructural MOFs (MFM-126–128 and MFM-136–138; Table 1). MFM-137 and MFM-138 were designed by adapting previously reported MFM-136,27 replacing the amide group with an ethynyl bond and phenyl ring, respectively. Further modification of MFM-136–138 was achieved by removal of the central phenyl unit of the linker to produce ‘shortened’ derivatives, MFM-126–128. This systematic approach allows us to isolate either the effect of varying pore size or functionality to rationalize the properties of these materials. Our approach has been to focus on the role of ligand sites for substrate binding and, significantly, we report herein the first example of binding of acetylene to the alkyne groups in a porous material at crystallographic resolution.
| MFM-126 | MFM-127 | MFM-128 | MFM-136a | MFM-137 | MFM-138 | |
|---|---|---|---|---|---|---|
| a Previously reported.27 | ||||||
| Structures of linkers |
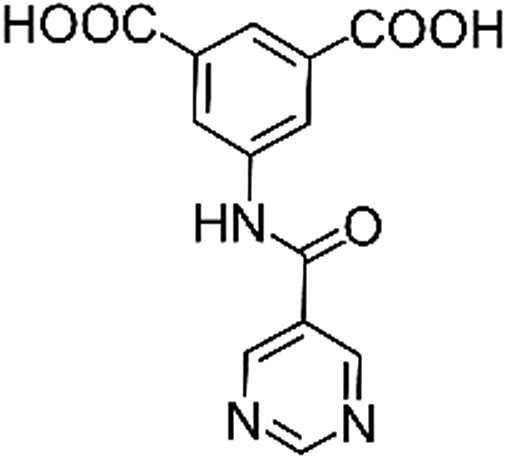
|
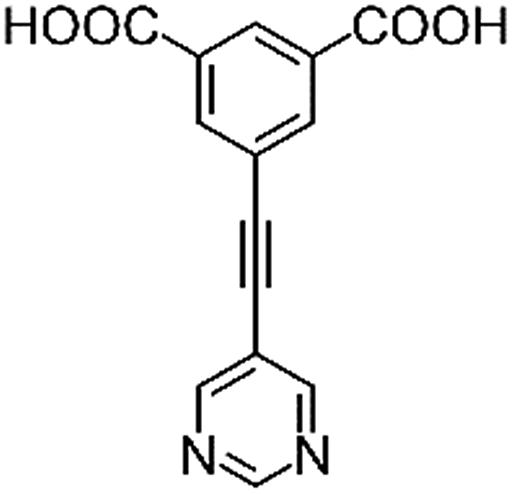
|
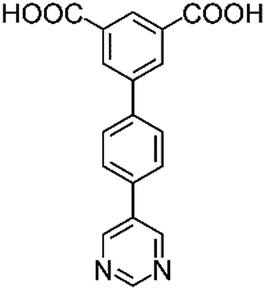
|
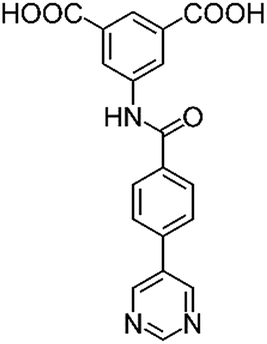
|
|
|
| H2L1 | H2L2 | H2L3 | H2L4 | H2L5 | H2L6 | |
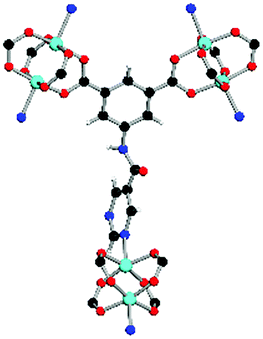
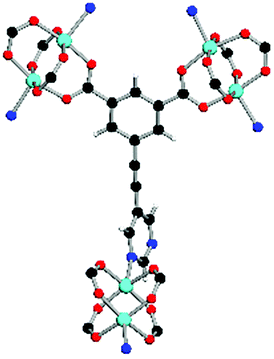
| Coordination environment |
|
|
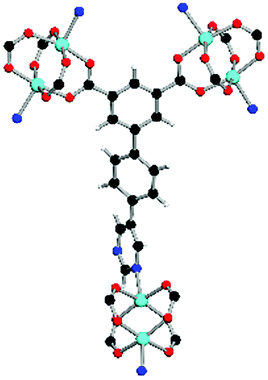
|
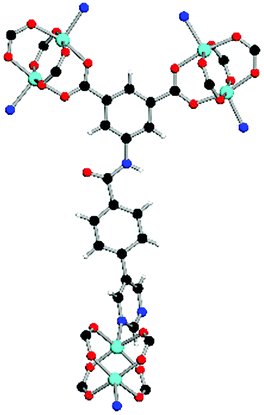
|
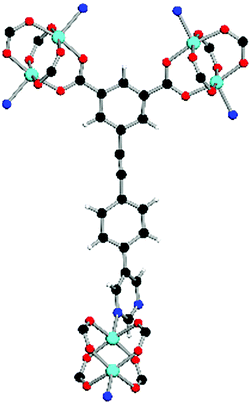
|
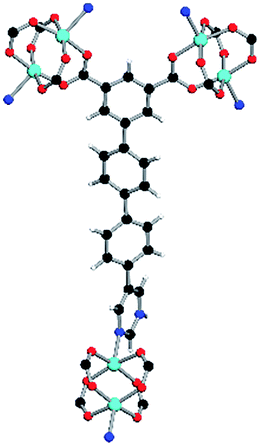
|
| Cage sizes |
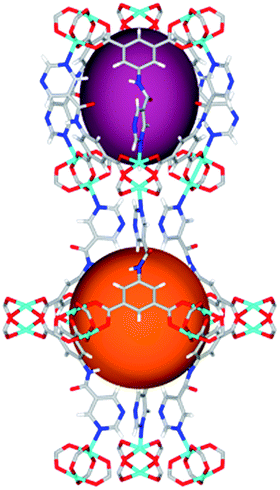
|
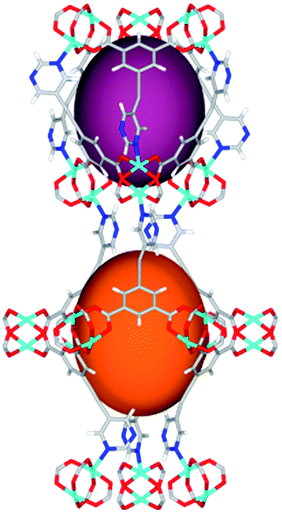
|
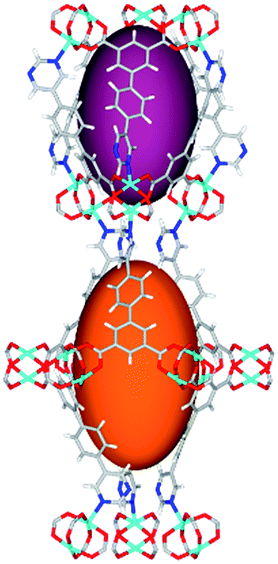
|
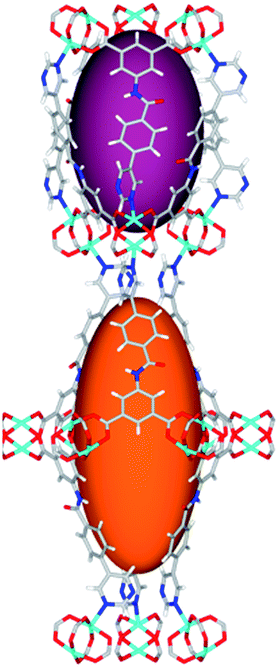
|
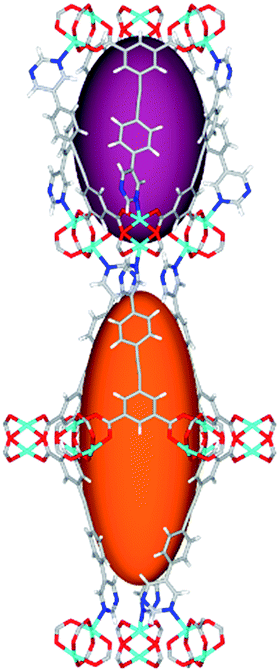
|
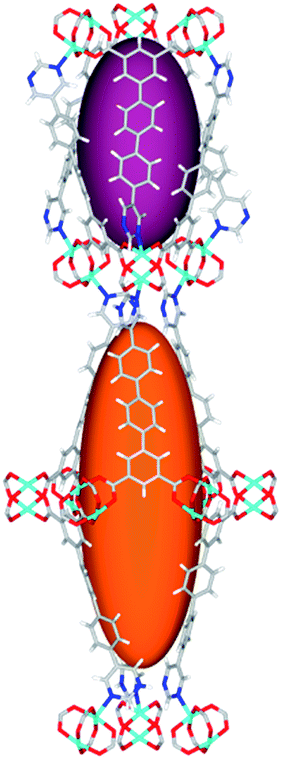
|
| Long cage (A) length/Å | 15.4 | 16.4 | 20.2 | 24.9 | 26.1 | 29.9 |
| Short cage (B) length/Å | 12.3 | 12.7 | 14.1 | 16.2 | 16.4 | 18.1 |
| BET surface area/m2 g−1 | 1004 | 1557 | 1491 | 1634 | 1749 | 1590 |
| Pore volume (N2 isotherm)/cm3 g−1 | 0.47 | 0.52 | 0.57 | 0.65 | 0.61 | 0.60 |
| Pore volume (single crystal)/cm3 g−1 | 0.52 | 0.57 | 0.57 | 0.64 | 0.67 | 0.62 |
Experimental
Neutron powder diffraction (NPD) of gas-loaded MFM-126 and MFM-127
NPD data were collected at WISH beamline at ISIS Muon and Neutron facility. Acetone-exchanged MFM-126 and MFM-127 were loaded into 11 mm diameter vanadium cans, heated at 393 K and degassed at 1 × 10−7 mbar for 2 days to activate the sample. Data for the bare framework were collected after placing the sample can into a liquid helium cryostat and cooling to 7 K. CO2, C2D2 and CD4 gases were dosed into the system after warming the system to 250 and 150 K. The gases were dosed volumetrically from a calibrated volume and, to ensure gases were fully adsorbed into the sample without condensation elsewhere, the system was cooled to 7 K slowly over 3 h. Structures of gas-loaded MFM-126 and MFM-127 were solved by sequential difference Fourier analyses followed by Rietveld refinements using TOPAS software (see ESI Section 4†).Inelastic neutron scattering of CO2-loaded MFM-126
INS data were collected on TOSCA beamline at ISIS Muon and Neutron facility. MFM-126 was loaded into an 11 mm diameter vanadium can and outgassed at 10−7 mbar at 393 K for 2 days. After placing the sample into a He-cooled cryostat, INS data of the bare framework were collected at 7 K. A loading of 1.0 CO2/Cu was dosed volumetrically from a calibrated volume at room temperature and gradually cooled to 7 K to allow the guest species to fully adsorb into MFM-126. INS data of 1.0 CO2/Cu of MFM-126 were collected at 7 K. Experimentally obtained INS data were compared with modelled data obtained via density functional theory (DFT) calculations and simulated using OClimax software (see ESI for further details†).Synchrotron FTIR microspectroscopy of CO2-loaded MFM-126
Single crystals of MFM-126 were loaded onto a ZnSe slide and placed into a Linkam FTIR600 variable temperature gas-tight cell fitted with ZnSe windows. The MOF sample was activated in situ under a flow of N2 whilst heating the Linkam stage to 413 K for 6 h. Partial pressures of zeolite-dried gases N2 and CO2 were controlled by varying the volumetric flow of the two gases via separate mass flow controllers. FTIR spectra were collected at the B22 MIRIAM beamline at Diamond Light Source using a polarized and highly bright synchrotron IR source connected to a Bruker Hyperion 3000 IR microscope with a 15× objective and MCT detector (liq. N2 cooled). Spectra (256 scans) were measured at room temperature with a 20 × 20 μm beam, in the spectral range of 4000–650 cm−1 (4 cm−1 resolution).Gas breakthrough
Breakthrough experiments were performed using a Hiden Isochema Automated Breakthrough Analyzer with integrated mass spectrometer. MFM-126 (950 mg) was packed into a stainless steel column and the sample was activated by heating to 393 K under a flow of He (100 mL min−1) overnight. Breakthrough experiments were conducted using gas mixtures of CO2/N2 (15:85) and equimolar CO2/CH4 which were flowed over MFM-126 at a total flow rate of 10 mL min−1 at 298 K and 1.0 bar (see ESI† for further detail).
Results and discussion
Structures
Solvothermal reactions of isophthalate–pyrimidyl linkers H2L1–H2L6 (Table 1) with Cu(NO3)2 affords isostructural (3,6)-connected MOFs of eea topology. All these frameworks are constructed from Cu(II) cations bridged by four carboxylate groups from four independent linkers and capped by two pyrimidyl nitrogen donors to form elongated octahedral [Cu2(RCOO)4(NR)2] nodes (Table 1, Fig. 1). The isophthalate units bridge adjacent {Cu2} paddlewheels to form a Kagomé lattice net (Fig. 1d), and these are connected by coordination of a pyrimidyl nitrogen center to an axial position of the {Cu2} paddlewheels. The capping of the {Cu2} paddlewheels at both axial positions results in the absence of any open Cu(II) sites. Lu et al. recently reported HHU-2,31 a (3,4,6)-connected Cu(II)-based MOF using H2L1, but its structure has both pyrimidine N atoms (instead of just one as in MFM-126) bound to Cu(II) ions and therefore does not have free N centers pointing into the pore as observed in MFM-126 and all MOFs in this current series. MFM-137 and MFM-138 are formed by replacement of the amide group of MFM-136 with alkyne and phenyl moieties, respectively. The removal of a central phenyl ring from MFM-136–138 affords the corresponding shortened derivatives MFM-126–128, respectively.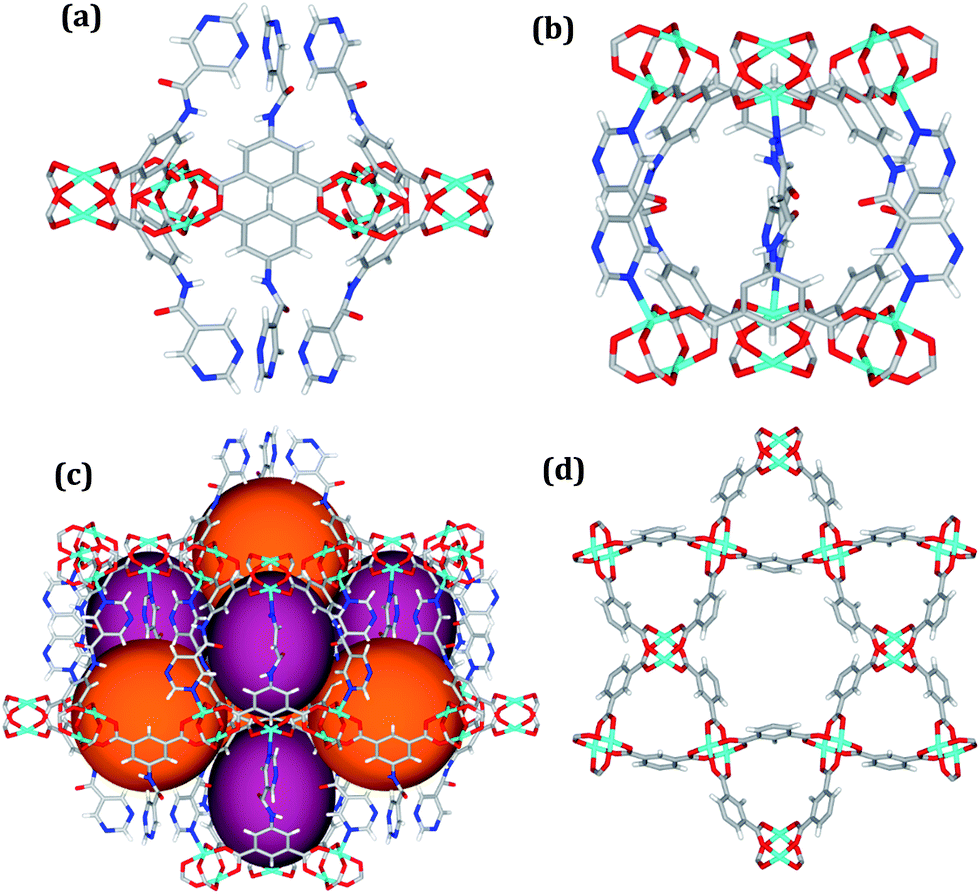 | ||
| Fig. 1 Views of crystal structure of MFM-126. (a) Cage A; (b) cage B. (c) View of the alternate packing of cages A (void space coloured orange) and B (void space coloured plum). (d) View along the c-axis of the Kagomé lattice in MFM-126. Colours: C, grey; H, white; O, red; N, blue; Cu, teal. | ||
The MOFs in this series all incorporate two types of cages, the larger of which, cage A (Fig. 1a) is comprised of six ligands and six [Cu2(RCOO)4(NR)2] paddlewheel units forming a hexagonal bipyramid. The six {Cu2} units form the six equatorial vertices of this cage and the hexagonal window of the Kagomé lattice (Fig. 1d). Six pyrimidyl units form the apical vertices, whereby ligands form six of the twelve faces of the hexagonal bipyramid. The smaller cage B (Fig. 1b) is constructed from six ligands and six {Cu2} paddlewheels forming a ditrigonal scalenohedral cage, whereby two sets of three {Cu2} paddlewheels bridged by three linker isophthalate units form triangular windows of neighboring Kagomé lattices. The overall structure of these MOFs is comprised of discrete cages A and B, which are packed in an alternating manner (Fig. 1c), giving highly porous and robust 3D frameworks. Views of the structure along the principal crystallographic axes of MFM-126 are shown in Fig. S3.† The phase purity of all material samples was confirmed by powder X-ray diffraction (PXRD) data (Fig. S4†).
Modulation of porosity and CO2 adsorption properties
All MOFs were solvent-exchanged with acetone or ethanol before heating at 393 K under dynamic vacuum to produce desolvated materials. The N2 (77 K) isotherms (Fig. S6†) reveal that the desolvated MOFs have BET surface areas of 1000–1750 m2 g−1 and pore volumes of 0.47–0.65 cm3 g−1 (Table 1). As expected, MFM-136–138 have higher BET surface areas compared to their isostructural shorter derivatives MFM-126–128. The higher BET surface area and pore volume of MFM-127 (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C linker) compared with MFM-126 (C(O)–NH linker) can be attributed to the presence of the space-efficient alkyne group in the linker, which has been shown previously to increase the hypothetical maximum surface area of MOFs.32 The measured pore volumes compare favorably with those calculated directly from single crystal structures, confirming complete desolvation and phase purity of these materials.
C linker) compared with MFM-126 (C(O)–NH linker) can be attributed to the presence of the space-efficient alkyne group in the linker, which has been shown previously to increase the hypothetical maximum surface area of MOFs.32 The measured pore volumes compare favorably with those calculated directly from single crystal structures, confirming complete desolvation and phase purity of these materials.
CO2 adsorption isotherms were measured to 20 bar at 273 and 298 K for all MOFs (Fig. S5†). Throughout the series, it was found that the CO2 uptake at 20 bar increased with increasing pore volume, indicating that the pore functionality had little effect on high pressure gas adsorption, where porosity is the dominant factor. For example, a 10% increase in CO2 uptake at 20 bar is observed in MFM-127 compared with MFM-126 (11.3 and 10.2 mmol g−1 at 298 K, respectively), corresponding well with the 11% pore volume increase from MFM-126 to MFM-127. However, at 1.0 bar and 298 K, comparing phenyl-functionalized MFM-138 and MFM-128 (2.89 and 3.19 mmol g−1, respectively), it was found that the CO2 uptake is greater in MFM-128, indicating a stronger interaction with the MFM-128 framework at ambient pressure. This suggests that pore geometry has an important role in low pressure gas uptake, with MFM-126–128 all having greater uptake at 1.0 bar CO2 than the comparative extended derivatives, MFM-136–138 (Fig. 2a). The influence of functionality on CO2 uptake can be assessed by comparing MFM-136 and MFM-137 which show similar porosity (Table 1). MFM-136 and MFM-137 show CO2 uptakes of 7.30 and 5.76 mmol g−1 (273 K), respectively, suggesting that amide-functionalized MFM-136 possesses stronger affinity for CO2 compared to the alkyne groups in MFM-137.
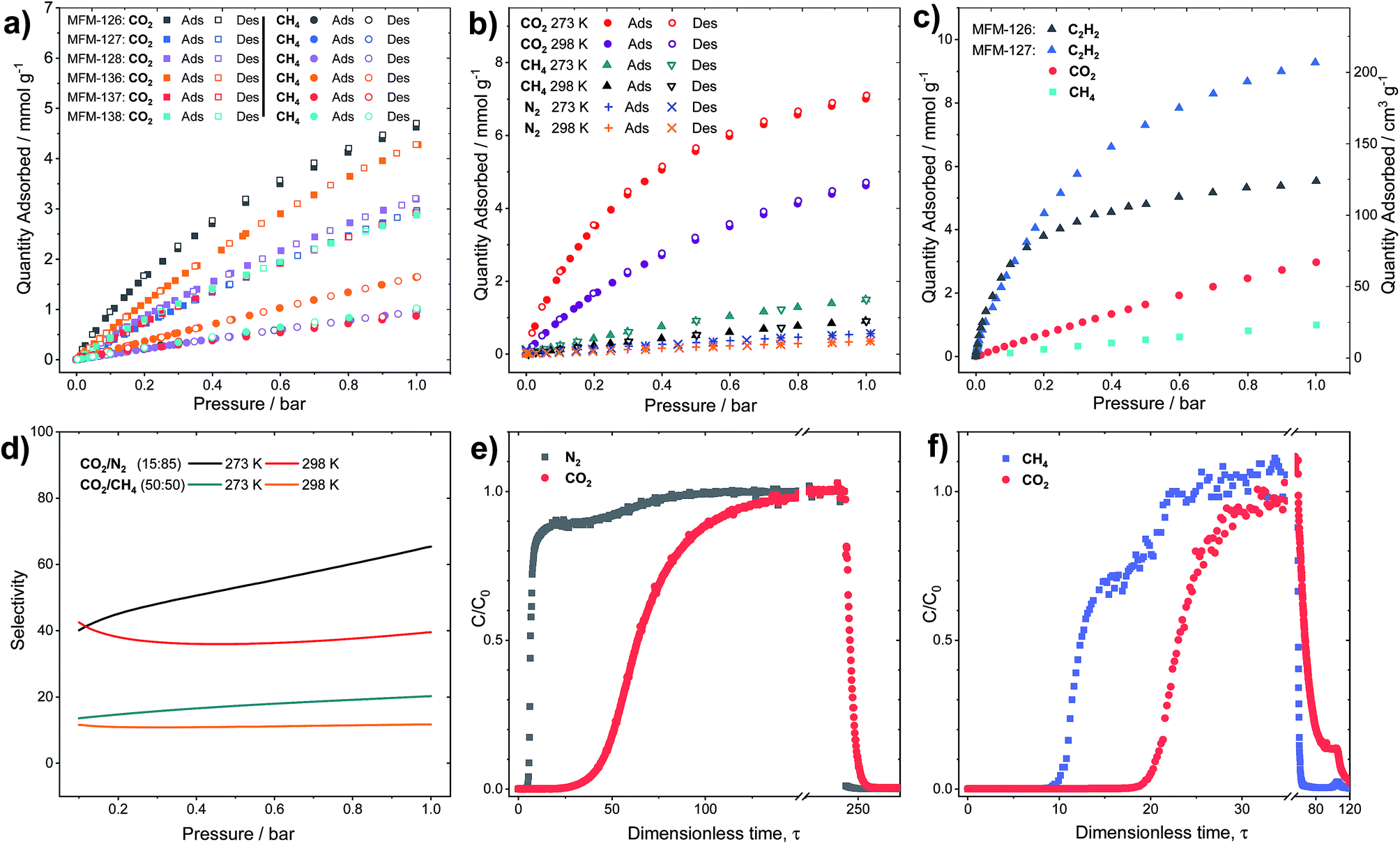 | ||
| Fig. 2 (a) Sorption isotherms for CO2 and CH4 in MFM-126–128 and MFM-136–138 at 298 K. (b) Sorption isotherms for CO2, CH4 and N2 in MFM-126 at 273 and 298 K. (c) Sorption isotherms for C2H2, CO2 and CH4 in MFM-127 and for C2H2 in MFM-126 at 273 K. (d) IAST selectivity for CO2/N2 (15:85) and equimolar CO2/CH4 in MFM-126 at 273 and 298 K. (e and f) Experimental breakthrough curves for the adsorption of CO2/N2 (15:85) and equimolar CO2/CH4 mixtures flowing through a 0.9 mL fixed-bed of MFM-126 at 298 K with a total gas flow of 10 mL min−1 at atmospheric pressure. After breakthrough of both species, a He purge was applied leading to desorption of all components. | ||
Crucially, however, MFM-126, with the smallest pore volume, has the greatest CO2 uptake at low pressure across the series, reaching 4.63 mmol g−1 at 1 bar and 298 K, presumably due to the greater overlap of attractive interactions between gas molecules and the host framework. This is exemplified further at 0.15 bar, the partial pressure of CO2 in flue gas streams, where MFM-126 has a 52% higher uptake of CO2 compared with the next best-performing MOF, MFM-136 (2.94 and 1.94 mmol g−1, respectively) in this series.
Selective sorption in MFM-126 and MFM-127
All the MOFs in this series show highly selective uptakes of CO2 with respect to CH4 and N2 (Fig. 2, Table 2). The selective uptake of CO2 over the other substrates studied is also evidenced by the analysis of heats of adsorption (Table 2 and Fig. S19†). MFM-126 exhibits the highest CO2 uptake at low pressures (<1 bar) as well as relatively low CH4 uptake in the same pressure region (Fig. 2b). The selectivity values were estimated from the single-component isotherms using ideal adsorbed solution theory (IAST) (Table 2).33 Considering the relative gas concentrations found in natural gas and flue gas streams, MFM-126 exhibits the highest selectivity values across the series for binary mixtures of both CO2/CH4 (equimolar) and CO2/N2 (15:85 composition) with selectivity values of 20.2 and 65.4, respectively at 1 bar and 273 K (Fig. 2d). MFM-126 also exhibits the highest adsorption enthalpy for CO2 of 30.7 kJ mol−1, which is significantly higher than 17.3 kJ mol−1 for CH4 (Table 2). Conversely, MFM-136 has the lowest CO2/CH4 selectivity value of 4.1 and exhibits similar values for the isosteric heat (Qst) for CO2 and CH4 adsorption, 20.1 and 18.9 kJ mol−1, respectively. When compared with other leading MOFs, MFM-126 has respectable CO2/CH4 and CO2/N2 selectivity values (Tables S18 and S19†).
| MOF | CO2 uptake/mmol g−1 (1 bar) | CH4 uptake/mmol g−1 (1 bar) |
S
CO2/CH4 (50:50, 1 bar) |
S
CO2/N2 (15:85, 1 bar) |
Q st/kJ mol−1 (virial method 1) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 273 K | 298 K | 273 K | 298 K | 273 K | 298 K | 273 K | 298 K | CO2 | CH4 | |
| MFM-126 | 7.00 | 4.63 | 1.50 | 0.897 | 20.2 | 11.7 | 65.4 | 39.6 | 30.7 | 17.3 |
| MFM-127 | 5.72 | 2.97 | 1.71 | 0.991 | 5.08 | 3.33 | 10.6 | 7.65 | 25.8 | 13.0 |
| MFM-128 | 5.76 | 3.20 | 1.77 | 0.953 | 5.46 | 4.53 | 35.8 | 18.9 | 20.4 | 22.7 |
| MFM-136 | 7.29 | 4.28 | 2.93 | 1.64 | 4.07 | 3.35 | 37.0 | 23.2 | 20.1 | 18.9 |
| MFM-137 | 5.76 | 2.92 | 1.41 | 0.870 | 6.09 | 4.08 | 27.6 | 15.7 | 19.2 | 17.1 |
| MFM-138 | 6.08 | 2.89 | 1.75 | 1.02 | 5.42 | 3.87 | 17.1 | 15.5 | 30.0 | 18.8 |
To evaluate the dynamic separation performance of MFM-126, breakthrough experiments were performed with CO2/N2 (15:85) and equimolar CO2/CH4 mixtures (Fig. 2e and f). The gas mixtures were flowed over a fixed-bed packed with MFM-126 with a total flow of 10 mL min−1 at 298 K (1 bar). These breakthrough experiments confirmed the separation potential for both gas mixtures as predicted by IAST selectivity calculations (Fig. 2d).
Although the amide-functionalised MFM-126 exhibits higher CO2 uptake compared with MFM-127, the functional groups were also investigated for their effect on acetylene adsorption. Interestingly, alkyne-functionalised MFM-127 exhibits a 68% higher acetylene uptake than MFM-126 at 273 K and 1 bar (9.28 and 5.54 mmol g−1, respectively, Fig. 2c). IAST analysis for MFM-127 reveals selectivity values for equimolar mixtures of C2H2/CO2 and C2H2/CH4 to be 3.7 and 21.2, respectively (Fig. S20†). This result is comparable to that observed for [Cu2(EBTC)] (H4EBTC = 1,1′-ethynebenzene-3,3′,5,5′-tetracarboxylic acid),34 which shows enhanced acetylene uptake compared with the non-alkyne bridged analogue MOF-505/MFM-100.35,36 Whilst the presence of CC⋯CC interactions between acetylene gas and an alkyne organic linker may be postulated, this has not yet been confirmed or observed experimentally.
In situ neutron powder diffraction (NPD) of gas-loaded MFM-126 and MFM-127
The absence of open metal sites in this series of MOFs affords an excellent platform to study the role of functional group on gas binding. In situ neutron powder diffraction (NPD) has been applied to determine the preferred binding sites of CO2 and CD4 in MFM-126 and MFM-127 at loadings of 1.2 CO2/Cu and 1.0 CD4/Cu, and C2D2 in MFM-127 at a loading of 1.0 C2D2/Cu. NPD data for the desolvated samples confirm the complete removal of guest solvents and an absence of structural distortion in the parent solvated material. Relatively low loadings of 1.0–1.2 adsorbate/Cu were used to assess the strongest binding sites within the material without involving notable adsorbate–adsorbate interactions. These loadings represent typically 25–40% of the saturated capacities of each adsorbate. Fourier difference map analysis of the NPD patterns afforded the location of guest CO2, CD4 and C2D2 molecules which, after further development by Rietveld refinement, allowed unambiguous determination of gas positions, orientations and crystallographic occupancies within each sample.MFM-126 displays four binding sites for CO2, 1–4 (in decreasing order of occupancy; Fig. 3). CO2-1 is situated in small cage B and exhibits co-operative binding between crystallographically equivalent CO2 molecules [CCO2⋯OCO2 = 3.30(3) Å] where the linear bodies of the CO2 molecules lie parallel with an interaction to an adjacent amide [OCO2⋯Namide = 3.86(5) Å, <C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯N = 111°]. In addition, there are short contacts between a pyrimidyl ring [C–Hpyrimidine⋯OCO2 = 2.32(5) Å, <C–H⋯O = 143°] and an isophthalate ring [OCO2⋯centroidisophthal. = 3.15(4) Å, <CO⋯centroid = 111°]. CO2-2 is located in the triangular window of cage B, with a close contact to a C–H group of the isophthalate unit [OCO2⋯H–Cisophthal. = 1.71(10) Å, <O⋯H–C = 152°] and a side-on interaction with two adjacent pyrimidine rings [OCO2⋯H–Cpyrimidine = 2.17(5) Å, <O⋯H–C = 128°]. CO2-3 is situated in a pocket between cages A and B with two short contacts to amido N–H units [OCO2⋯H–Namide = 3.77(6) Å, <O⋯H–N = 124° and OCO2⋯H–Namide = 3.98(8) Å, <O⋯H–N = 138°]. Further to this, there are two other close contacts with a pyrimidyl ring [OCO2⋯Cpyrimidine = 2.44(5) Å, CO⋯C = 115°] and a side-on interaction with an isophthalate ring [CCO2⋯H–Cisophthal. = 3.34(12) Å, <C⋯H–C = 172°]. CO2-4 is positioned at the periphery of larger cage A with a hydrogen bond to an amido N–H [OCO2⋯H–N = 4.14(9) Å, <O⋯H–N = 149°] as well as lying in a crevice between two isophthalate units [OCO2⋯Cisophthal. = 3.14(11) Å, CO⋯C = 81° and OCO2⋯Cisophthal. = 3.25(10) Å, CO⋯C = 96°].
O⋯N = 111°]. In addition, there are short contacts between a pyrimidyl ring [C–Hpyrimidine⋯OCO2 = 2.32(5) Å, <C–H⋯O = 143°] and an isophthalate ring [OCO2⋯centroidisophthal. = 3.15(4) Å, <CO⋯centroid = 111°]. CO2-2 is located in the triangular window of cage B, with a close contact to a C–H group of the isophthalate unit [OCO2⋯H–Cisophthal. = 1.71(10) Å, <O⋯H–C = 152°] and a side-on interaction with two adjacent pyrimidine rings [OCO2⋯H–Cpyrimidine = 2.17(5) Å, <O⋯H–C = 128°]. CO2-3 is situated in a pocket between cages A and B with two short contacts to amido N–H units [OCO2⋯H–Namide = 3.77(6) Å, <O⋯H–N = 124° and OCO2⋯H–Namide = 3.98(8) Å, <O⋯H–N = 138°]. Further to this, there are two other close contacts with a pyrimidyl ring [OCO2⋯Cpyrimidine = 2.44(5) Å, CO⋯C = 115°] and a side-on interaction with an isophthalate ring [CCO2⋯H–Cisophthal. = 3.34(12) Å, <C⋯H–C = 172°]. CO2-4 is positioned at the periphery of larger cage A with a hydrogen bond to an amido N–H [OCO2⋯H–N = 4.14(9) Å, <O⋯H–N = 149°] as well as lying in a crevice between two isophthalate units [OCO2⋯Cisophthal. = 3.14(11) Å, CO⋯C = 81° and OCO2⋯Cisophthal. = 3.25(10) Å, CO⋯C = 96°].
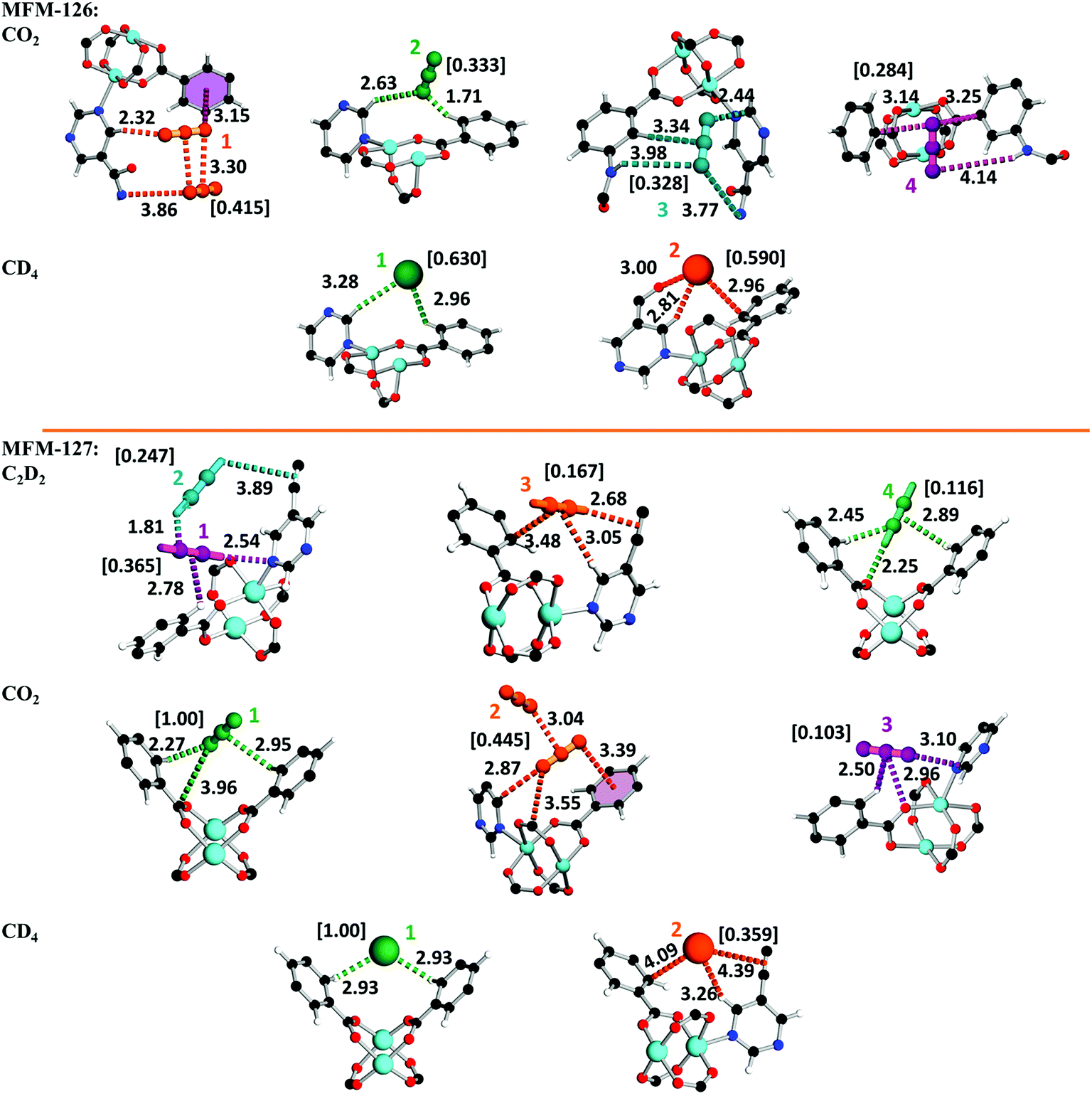 | ||
| Fig. 3 Binding sites of guests in MFM-126 and MFM-127 derived from Rietveld refinement of NPD data with quoted distances in Å. Colors: C, black; H, white; O, red; N, blue; Cu, teal. Refined occupancies of guest molecules are shown in square brackets. | ||
Analysis of CD4-loaded MFM-126 reveals seven sites 1–7, the first two of which have significantly higher crystallographic occupancies of 0.630 and 0.590 than the rest (Fig. 3). CD4-1, resides in cage B in an equivalent site to that of CO2-2, also with close interactions to isophthalate and pyrimidyl rings [D4C⋯H–Cisophthal. = 2.96(11) Å, <C⋯H–C = 122° and D4C⋯H–Cpyrimidine = 3.28(4) Å, <C⋯H–C = 159°]. CD4-2 is found in an identical site to that of CO2-1 located in the center of cage B with two key interactions to an isophthalate ring and an amido carbonyl of the framework [D4C⋯H–Cisophthal. = 2.81(4) Å, <C⋯H–C = 146° and D4C⋯OCamide = 3.00(5) Å, <C⋯OC = 140°].
Refinement of NPD data of C2D2-loaded MFM-127 unveiled five sites, 1–5 (Fig. 3). Interestingly, over 50% of C2D2 were found having short contacts (<4 Å) to the framework alkyne units. C2D2-1 is situated in a narrow window between cages A and B, with closest interactions to pyrimidyl and isophthalate rings [DC2–D⋯Npyrimidine = 2.54(5) Å, <C–D⋯Npyrimidine = 145° and η2-D2C2⋯H–Cisophthal. = 2.78(2) Å, <C–H⋯CCcentroid = 108°]. Cooperative binding is visualized between C2D2-1 and C2D2-2 [1-D2CC⋯D–C2D-2 = 1.81(3) Å, <C1⋯D–C2 = 127°]. C2D2-2 also binds to the framework alkyne [DC2–D⋯η2-CCframework = 3.89(5) Å, <C–D⋯CCcentroid = 123°] suggesting the framework alkyne has an integral role in the uptake of acetylene in MFM-127. C2D2-3 is located in the center of cage B with H-bonds to a framework alkyne moiety [DC2–D⋯η2-CCframework = 2.68(5) Å, <C–D⋯CCcentroid = 120°] as well as close contacts to pyrimidyl and isophthalate rings [η2-D2CC⋯H–Cpyrimidine = 3.05(8) Å, <C–H⋯CCcentroid = 130° and η2-CCD2⋯Cisophthal. = 3.48(3) Å]. C2D2-4 occupies the hydrophobic triangular window of cage B with strong binding to an oxygen of the {Cu2} paddlewheel [DC2–D⋯Opaddlewheel = 2.25(5) Å, <C–D⋯O = 150°] as well as to adjacent isophthalate rings [D2C2⋯H–Cisophthal. = 2.45(2) Å, <C–H⋯C = 120° and η2-CCD2⋯H–Cisophthal. = 2.89(1) Å, <C–H⋯CCcentroid = 124°].
Three CO2 binding domains, 1–3, were found in MFM-127 (Fig. 3). CO2-1 is situated in an equivalent site to C2D2-4 with a close contact to a carboxylate Cδ+ [OCO2⋯CCOO = 3.96(6) Å, <C–O⋯C = 100°] as well as side-on interactions with isophthalate rings [OCO2⋯H–Cisophthal. = 2.27(1) Å, <C–H⋯O = 135° and CCO2⋯H–Cisophthal. = 2.95(7) Å, <C–H⋯C = 114°]. CO2-2 occupies the corresponding site to CO2-1 in MFM-126. As for MFM-126, cooperative binding is exhibited between crystallographically equivalent CO2-2 molecules in cage B of MFM-127 [CCO2⋯OCO2 = 3.04(2) Å, <CO⋯C = 128°]. Crucially, these results reveal the key role of the amide groups on selective adsorption of CO2 in MFM-126 as well as confirming that alkyne moieties do not act as effective adsorption sites for CO2 in MFM-127.
NPD data for CD4-loaded MFM-127 exposed only two noteworthy sites of interaction. Both sites correspond directly to CD4 sites 1 and 2 observed in MFM-126, respectively. In MFM-127, CD4-1 resides in the hydrophobic triangular windows of cage B [D4C⋯H–Cisophthal. = 2.93(1) Å, <C–H⋯C = 120°], further showing that this pocket provides an optimal environment for CD4. CD4-2 is found in the center of cage B and interacts weakly to the framework alkyne [D4C⋯η2-CCframework = 4.39(1) Å] revealing that the alkyne moiety has little effect on CD4 adsorption.
Thus, significantly, the NPD study reveals that it is a combination of cooperative binding as well as the amide functionality that leads to enhanced interaction of CO2 with MFM-126. On the other hand, NPD data reveal that alkyne-functionalized MFM-127 exhibits much weaker interaction with CO2, but that the alkyne moieties play critical roles in acetylene binding, with over half of the adsorbed acetylene molecules exhibiting interactions (<4 Å) to alkyne moieties in the pore of MFM-127. This represents the first example of direct visualization of acetylene binding to an alkyne moiety in porous materials.
Inelastic neutron scattering (INS) studies of CO2-loaded MFM-126
To gain further understanding into CO2 binding in MFM-126, inelastic neutron scattering (INS) was measured as a function of CO2-loading (Fig. S18†). The INS spectrum for the bare MOF exhibits numerous vibrational modes which have been assigned by comparison with a DFT-calculated INS spectrum (Fig. S17†). Interestingly, unlike previously reported with isostructural MFM-136,27 MFM-126 exhibits significant interactions with guest CO2. Upon CO2 loading, there is a shift in the peak at 82.8 meV (assigned to the in-plane bending of the amide group) to 84.1 meV signifying an interaction between adsorbed CO2 and the amide group, such as those visualized in the structure model of CO2-loaded MFM-126. This result contrasts with that of MFM-136 (ref. 27) and confirms the importance of cooperativity between functional groups (in this case amide) and the pore geometries on guest binding.In situ synchrotron FT-IR microspectroscopy of MFM-126
To investigate further the nature of host–guest interactions, in situ synchrotron FT-IR microspectroscopy was conducted on a single crystal of activated MFM-126. FTIR spectra were collected as a function of CO2-loading by increasing the partial pressure of CO2 in N2 from 0 to 1.0 bar (ppCO2; Fig. 4). The ν1 + ν3 (3695 cm−1) and 2ν2 + ν3 (3590 cm−1) combination bands of CO2 (Fig. 4a) were used to monitor CO2 sorption as the fundamental antisymmetric stretch at ∼2348 cm−1 saturates at ppCO2 above 0.2 bar. These combination bands are red-shifted from their free values (3714 and 3612 cm−1, respectively) at all partial pressures of CO2 loading and increase in intensity as a function of ppCO2, confirming that CO2 interacts strongly with the framework (Fig. 4a). Activated MFM-126 has a ν(N–H) stretch at 3306 cm−1 and upon increasing ppCO2 from 0 to 1 bar, this band shifts by 36 cm−1 to 3270 cm−1. In addition, the observed shift of the amide ν(CO) stretching band by 10 cm−1 (from 1684 cm−1 in the absence of CO2 to 1674 cm−1 at 1.0 bar ppCO2; Fig. 4c) further indicates CO2 adsorption directed by the amide group in MFM-126. Overall, these observations are highly consistent with the NPD and INS results.
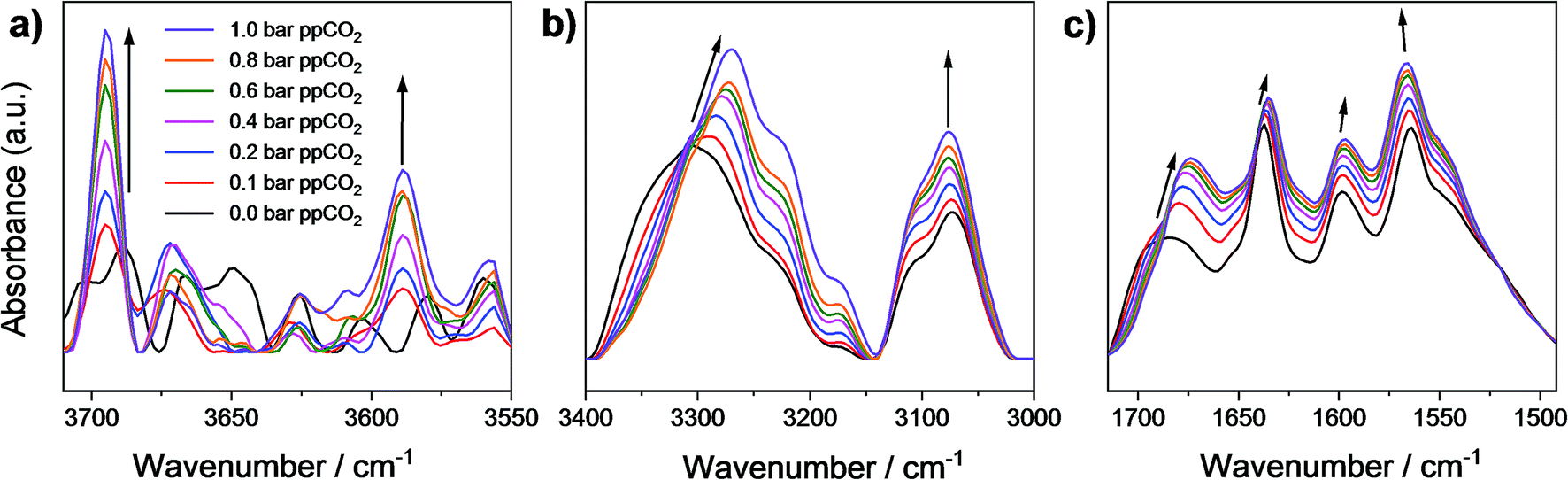 | ||
| Fig. 4 Micro-FTIR spectra of a single crystal of MFM-126 as a function of CO2-loading incrementally from 0 to 1 bar ppCO2 focusing on (a) the CO2 combination bands at 3695 and 3590 cm−1; (b) ν(N–H) stretching band shifting from 3306 cm−1 at 0 bar ppCO2 (in N2) of CO2 (in N2) to 3270 cm−1 at 1 bar ppCO2; (c) the ν(N–H) bending mode shifting from 1684 cm−1 at 0 bar ppCO2 (in N2) to 1674 cm−1 at 1 bar ppCO2. All spectra were recorded referenced to the blank cell as a function of CO2 loading to remove contributions of free gaseous CO2. | ||
Conclusions
A series of isoreticular (3,6)-connected pyrimidyl isophthalate Cu(II)-based MOFs, of rare eea topology have been synthesized and characterized. Although MFM-136 exhibits the highest CO2 uptake at 20 bar, it was found that upon shortening the linker unit to form MFM-126, selectivity of CO2 uptake at 1 bar increases dramatically. The relative ease of synthesis of MFM-126 coupled with its high selectivity for binary mixtures of CO2/CH4 and CO2/N2, position this MOF as a viable candidate for CO2 separations. Furthermore, the CO2 separation performance of MFM-126 has been confirmed by dynamic breakthrough experiments. NPD data of CO2-loaded MFM-126 has revealed that cooperative binding of CO2 (at position CO2-1) and binding to amide groups that decorate cage B both have strong effects on the observed selective CO2 binding. The in situ spectroscopic studies using INS and FTIR also establish that adsorbed CO2 interacts strongly with the amide groups of MFM-126, with significant shifts of amide (CO and N–H) vibrational bands on CO2 loading. Replacing the amide-group in MFM-126 with an alkyne-group to give MFM-127 leads to a decrease in both CO2 and CH4 uptake capacities relative to MFM-126, but affords a 68% greater C2H2 capacity than MFM-126. NPD experiments reveal for the first time that acetylene interacts directly with alkyne moieties of MFM-127 in the pore. Notably, over 50% of the acetylene observed within MFM-127 displays strong interactions (<4 Å) with the alkyne functionality of the framework. The understanding gained in this study provides further insights into the development of materials showing improved gas binding via specific interaction to ligand sites within the MOF.
Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
We thank EPSRC (EP/I011870, EP/K038869, EP/P001386), ERC (AdG 742041), the Royal Society (6866) and the Universities of Manchester and Nottingham for funding. O. B. thanks Niger Delta University and TETFund, Nigeria for a PhD scholarship. We are especially grateful to STFC/ISIS Neutron Facility for access to the Beamlines TOSCA and WISH and to Diamond Light Source for access to the Beamlines I19 and B22. The computing resources were made available through the VirtuES and ICEMAN projects, funded by Laboratory Directed Research and Development program at ORNL.References
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- L.-B. Sun, X.-Q. Liu and H.-C. Zhou, Chem. Soc. Rev., 2015, 44, 5092–5147 RSC.
- T. L. Easun, F. Moreau, Y. Yan, S. Yang and M. Schröder, Chem. Soc. Rev., 2017, 46, 239–274 RSC.
- X. Han, H. G. W. Godfrey, L. Briggs, A. J. Davies, Y. Cheng, L. L. Daemen, A. M. Sheveleva, F. Tuna, E. J. L. McInnes, J. Sun, C. Drathen, M. W. George, A. J. Ramirez-Cuesta, K. M. Thomas, S. Yang and M. Schröder, Nat. Mater., 2018, 17, 691–696 CrossRef CAS PubMed.
- Y. Yan, D. I. Kolokolov, I. da Silva, A. G. Stepanov, A. J. Blake, A. Dailly, P. Manuel, C. C. Tang, S. Yang and M. Schröder, J. Am. Chem. Soc., 2017, 139, 13349–13360 CrossRef CAS PubMed.
- A. H. Chughtai, N. Ahmad, H. A. Younus, A. Laypkov and F. Verpoort, Chem. Soc. Rev., 2015, 44, 6804–6849 RSC.
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2010, 132, 38–39 CrossRef CAS PubMed.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- S. Chand, A. Pal and M. C. Das, Chem.–Eur. J., 2018, 24, 5982–5986 CrossRef CAS PubMed.
- J. W. Zhang, M. C. Hu, S. N. Li, Y. C. Jiang, P. Qu and Q. G. Zhai, Chem. Commun., 2018, 54, 2012–2015 RSC.
- F. Chen, Y. Wang, D. Bai, M. He, X. Gao and Y. He, J. Mater. Chem. A, 2018, 6, 3471–3478 RSC.
- D.-M. Chen, C.-X. Sun, N.-N. Zhang, H.-H. Si, C.-S. Liu and M. Du, Inorg. Chem., 2018, 57, 2883–2889 CrossRef CAS PubMed.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- A. Pal, S. Chand, S. M. Elahi and M. C. Das, Dalton Trans., 2017, 46, 15280–15286 RSC.
- H. H. Storch, Ind. Eng. Chem., 1934, 26, 56–60 CrossRef CAS.
- R.-B. Lin, L. Li, H. Wu, H. Arman, B. Li, R.-G. Lin, W. Zhou and B. Chen, J. Am. Chem. Soc., 2017, 139, 8022–8028 CrossRef CAS PubMed.
- W. Yang, A. J. Davies, X. Lin, M. Suyetin, R. Matsuda, A. J. Blake, C. Wilson, W. Lewis, J. E. Parker, C. C. Tang, M. W. George, P. Hubberstey, S. Kitagawa, H. Sakamoto, E. Bichoutskaia, N. R. Champness, S. Yang and M. Schröder, Chem. Sci., 2012, 3, 2993–2999 RSC.
- H.-M. Wen, H. Wang, B. Li, Y. Cui, H. Wang, G. Qian and B. Chen, Inorg. Chem., 2016, 55, 7214–7218 CrossRef CAS PubMed.
- D. Britt, H. Furukawa, B. Wang, T. G. Glover and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20637–20640 CrossRef CAS PubMed.
- Q. Yan, Y. Lin, C. Kong and L. Chen, Chem. Commun., 2013, 49, 6873–6875 RSC.
- B. Zheng, Z. Yang, J. Bai, Y. Li and S. Li, Chem. Commun., 2012, 48, 7025–7027 RSC.
- S. Yang, J. Sun, A. J. Ramirez-Cuesta, S. K. Callear, W. I. F. David, D. P. Anderson, R. Newby, A. J. Blake, J. E. Parker, C. C. Tang and M. Schröder, Nat. Chem., 2012, 4, 887–894 CrossRef CAS PubMed.
- S. Yang, A. J. Ramirez-Cuesta, R. Newby, V. Garcia-Sakai, P. Manuel, S. K. Callear, S. I. Campbell, C. C. Tang and M. Schröder, Nat. Chem., 2015, 7, 121–129 CrossRef CAS PubMed.
- L. Du, Z. Lu, K. Zheng, J. Wang, X. Zheng, Y. Pan, X. You and J. Bai, J. Am. Chem. Soc., 2013, 135, 562–565 CrossRef CAS PubMed.
- R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed.
- O. Benson, I. Da Silva, S. P. Argent, R. Cabot, M. Savage, H. G. W. Godfrey, Y. Yan, S. F. Parker, P. Manuel, M. J. Lennox, T. Mitra, T. L. Easun, W. Lewis, A. J. Blake, E. Besley, S. Yang and M. Schröder, J. Am. Chem. Soc., 2016, 138, 14828–14831 CrossRef CAS PubMed.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed.
- S. J. Garibay and S. M. Cohen, Chem. Commun., 2010, 46, 7700–7702 RSC.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gandara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
- Z. Lu, J. Zhang, J. Duan, L. Du and C. Hang, J. Mater. Chem. A, 2017, 5, 17287–17292 RSC.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydin and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS PubMed.
- N. F. Cessford, N. A. Seaton and T. Düren, Ind. Eng. Chem. Res., 2012, 51, 4911–4921 CrossRef CAS.
- Y. Hu, S. Xiang, W. Zhang, Z. Zhang, L. Wang, J. Bai and B. Chen, Chem. Commun., 2009, 45, 7551–7553 RSC.
- B. Chen, N. W. Ockwig, A. R. Millward, D. S. Contreras and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4745–4749 CrossRef CAS PubMed.
- X. Lin, J. Jia, X. B. Zhao, K. M. Thomas, A. J. Blake, N. R. Champness, P. Hubberstey and M. Schröder, Angew. Chem., Int. Ed., 2006, 45, 7358–7364 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Correspondence and requests for materials should be addressed to S. Y. and M. S. CCDC 1857732–1857743. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03622e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |