
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLysine carbonylation is a previously unrecognized contributor to peroxidase activation of cytochrome c by chloramine-T†
Victor
Yin
 ,
Safee H.
Mian
and
Lars
Konermann
*
,
Safee H.
Mian
and
Lars
Konermann
*
Department of Chemistry and Department of Biochemistry, The University of Western Ontario, London, Ontario N6A 5B7, Canada. E-mail: konerman@uwo.ca
First published on 7th January 2019
Abstract
The peroxidase activity of cytochrome c (cyt c) plays a key role during apoptosis. Peroxidase catalysis requires a vacant Fe coordination site, i.e., cyt c must undergo an activation process involving structural changes that rupture the native Met80–Fe contact. A common strategy for dissociating this bond is the conversion of Met80 to sulfoxide (MetO). It is widely believed that this MetO formation in itself is sufficient for cyt c activation. This notion originates from studies on chloramine-T-treated cyt c (CT-cyt c) which represents a standard model for the peroxidase activated state. CT-cyt c is considered to be a “clean” species that has undergone selective MetO formation, without any other modifications. Using optical, chromatographic, and mass spectrometry techniques, the current work demonstrates that CT-induced activation of cyt c is more complicated than previously thought. MetO formation alone results in only marginal peroxidase activity, because dissociation of the Met80–Fe bond triggers alternative ligation scenarios where Lys residues interfere with access to the heme. We found that CT causes not only MetO formation, but also carbonylation of several Lys residues. Carbonylation is associated with −1 Da mass shifts that have gone undetected in the CT-cyt c literature. Proteoforms possessing both MetO and Lys carbonylation exhibit almost fourfold higher peroxidase activity than those with MetO alone. Carbonylation abrogates the capability of Lys to coordinate the heme, thereby freeing up the distal site as required for an active peroxidase. Previous studies on CT-cyt c may have inadvertently examined carbonylated proteoforms, potentially misattributing effects of carbonylation to solely MetO formation.
Introduction
Cytochrome c (cyt c) is a 12 kDa mitochondrial heme protein whose primary role is to serve as an electron shuttle in the respiratory chain.1 Cyt c also possesses a second biologically relevant function as a peroxidase.2–8 Peroxidases catalyze the oxidation (H abstraction) of substrates in the presence of H2O2. Catalytic turnover involves heme cycling between the Fe(III) and Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O states.9 The peroxidase activity of cyt c is critical for triggering apoptosis (programed cell death) via oxidation of the mitochondrial membrane lipid cardiolipin.7,10–12 Apoptosis is an essential process, e.g., for suppressing tumor growth.13
O states.9 The peroxidase activity of cyt c is critical for triggering apoptosis (programed cell death) via oxidation of the mitochondrial membrane lipid cardiolipin.7,10–12 Apoptosis is an essential process, e.g., for suppressing tumor growth.13
Peroxidases require a pentacoordinated heme with a vacant distal coordination site where H2O2 can bind, and where the Fe(IV)O oxygen can be accommodated.14 The heme in native cyt c is hexacoordinated, as the distal site is occupied by the Met80 sulfur (Fig. 1).15 Dissociation of this Met80–Fe contact is a mandatory prerequisite for converting cyt c into a peroxidase.14,16,17 There are different ways to achieve displacement of the distal Met80, such as unfolding,14,16 chemical,5,17–20 or mutational modifications,4,7,18,21–26 as well as cardiolipin binding.3,27–29
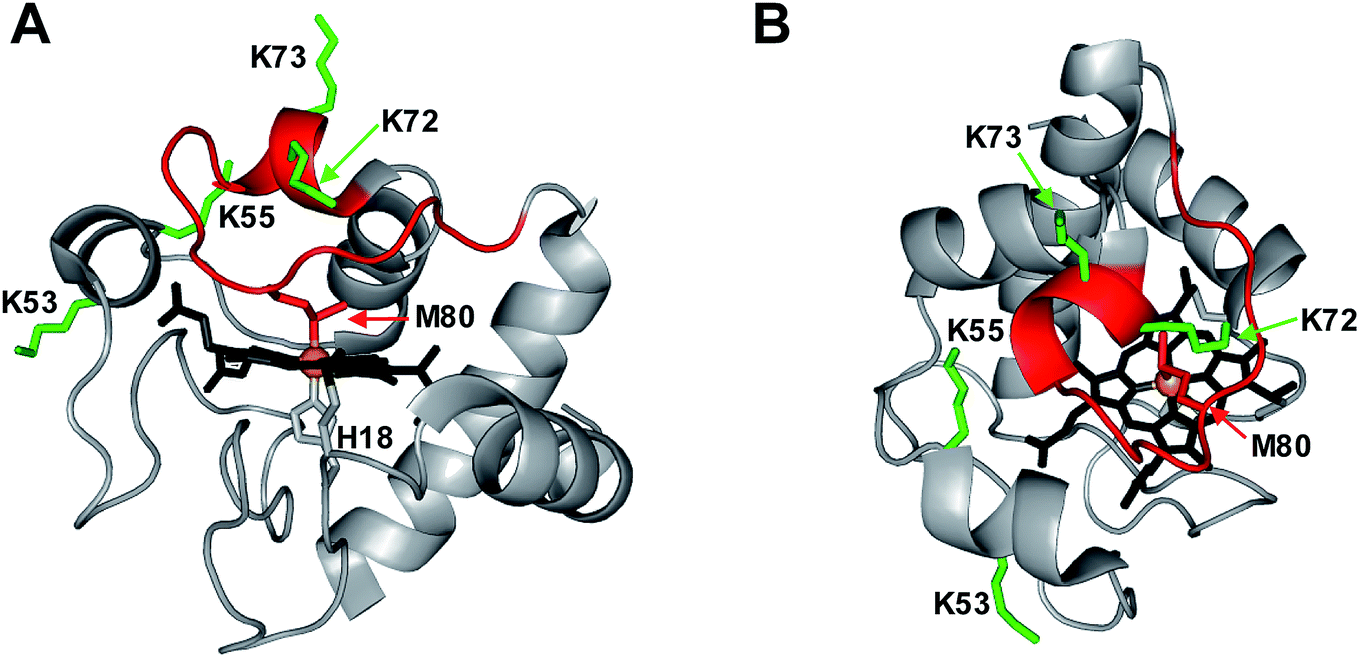 | ||
| Fig. 1 Crystal structure of equine cyt c (1HRC) with key residues highlighted. (A) Side view. (B) Top view. In the native state, the heme co-factor is ligated by Met80 and His18. Lys72/73 and Lys53/55 represent possible alternative distal ligands that may interact with Fe instead of Met80 under certain conditions. The flexible 71–85 Ω loop is shown in red. | ||
Exposure to basic pH induces the formation of “alkaline” conformations where Lys72, 73 or 79 serve as distal ligands instead of Met80.20,30–34 Chemical or mutational modifications can shift this alkaline transition into the neutral range.5,19,20,35–40
We recently demonstrated41 that H2O2 converts native cyt c into an active peroxidase via a reaction sequence that starts with Tyr67 oxidation, followed by dissociation of the Met80–Fe contact. Subsequently, Lys72 or 73 occupy the distal coordination site. Carbonylation then abrogates the capability of Lys72/73 to serve as Fe ligands, thereby generating the free distal site that is required for an active peroxidase.41 Lys carbonylation corresponds to the oxidation of the ε-amino group to an aldehyde (LysCHO, aminoadipic semialdehyde, ΔM = −1 Da).42,43
The most straightforward method to rupture the Met80–Fe bond is via Met oxidation to methionine sulfoxide (MetO). It is widely believed that this oxidative modification is sufficient for generating the open distal site that is required for peroxidase activation of cyt c in vitro and in vivo.17,18,37,38,44–47 In other words, this Met80-centric view envisions a simple causal relationship between MetO formation and peroxidase activity.17,18,37,38,45–47
A key pillar of the Met80-centric view comes from work on chloramine-T (CT) oxidized cyt c (CT-cyt c). CT is a mild oxidizing agent48 that transforms into other reactive species in aqueous solution.49 Among these, OCl− appears to be mainly responsible for cyt c oxidation, evident from the fact that cyt c exposure to CT and to OCl− causes similar effects.17 CT-cyt c is a potent peroxidase.17,37 Several studies have shown that CT-cyt c carries a sulfoxide at both Met80 and Met65, while claiming that no other chemical modifications are present.17,48,50 These claims go back to amino acid analyses conducted in the 1970s, which concluded that CT specifically oxidizes Met, while leaving all other residues unmodified (except for –SH groups, which are absent in cyt c).51 Those early studies17,48,50,51 cemented the widely held belief that Met80 oxidation, without any other covalent changes, generates an active peroxidase.17,18,37,45–47 As a result, CT-cyt c has become a widely used model for peroxidase-activated cyt c.17,37,38,47
Considering the far-reaching implications of work in this area (including the role of cyt c in apoptosis and tumorigenesis),13 it is imperative to fully understand the properties of CT-cyt c. Is it true that Met80 oxidation is sufficient for peroxidase activation? Is it true that MetO formation is the only CT-induced oxidative modification? Several lines of evidence hint at the involvement of additional factors. (1) Disruption of the Met80–Fe bond tends to produce “alkaline” ligation scenarios, where even at near-neutral pH the distal site becomes occupied by side chains such as Lys72/73.19,35,36,38 These alternative ligation scenarios are promoted by the flexible nature of the 71–85 Ω loop (Fig. 1).52,53 Hence, it is not obvious why Met80 oxidation in itself would produce a vacant distal site that is required for peroxidase activity. (2) The purported strict selectivity of CT for Met is somewhat suspicious, considering that other oxidants can induce various modifications at different types of residues.54–58 (3) Typical CT-cyt c literature protocols utilize strong cation exchange (SCX) chromatography, which separates proteins according to their positive surface charge. Surprisingly, CT-cyt c undergoes SCX fractionation into several distinct bands.37,38,47,48 MetO formation does not directly affect charge. The fact that CT-cyt c produces several SCX fractions thus suggests that the protein might contain additional, hitherto unidentified modifications. The possible existence of such modifications and their implications for peroxidase activation have been ignored thus far in the literature.
In this work we critically examined the structure and function of CT-cyt c. Various mass spectrometry (MS) techniques were applied to characterize CT-induced covalent modifications in unprecedented detail. We were able to pinpoint previously unidentified features of the peroxidase activation process. Met80 oxidation alone was found to result in low peroxidase activity, because the distal site remained impeded by alkaline-like Lys ligation. Only after opening of the distal site by CT-induced LysCHO formation did the protein show higher levels of activity. Previous studies on CT-cyt c that assumed a “clean” MetO protein have—in all likelihood—inadvertently characterized samples that contained both MetO and LysCHO sites. Our findings argue against the prevailing notion that Met80 oxidation alone is sufficient for peroxidase activation. Instead, Lys carbonylation is a key peroxidase co-activator.
Methods
Materials
Horse heart ferri-cyt c, H2O2, guaiacol (o-methoxyphenol), ammonium acetate, and chloramine-T (N-chloro-4-toluol-sulfonamide), were from Sigma Aldrich (St. Louis, MO). Solvents were from Fisher Scientific (Nepean, ON). Potassium phosphate was supplied by Caledon Laboratories (Georgetown, ON), and MS-grade modified trypsin was from Promega (Madison, WI). All experiments were conducted at 22 ± 2 °C.Preparation of CT-cyt c
Treatment of cyt c with CT was performed as described,37 with minor alterations. Briefly, a 5 mM aqueous solution of CT was added to 1 mM cyt c (both in 50 mM Tris buffer, pH 8.4) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio and reacted at room temperature for 3 hours. The reaction was then halted by dialysis against 10 mM potassium phosphate (pH 7.4) using 10 kDa MWCO filters (EMD Millipore, Etobicoke, ON). The CT-cyt c produced in this way was either used directly, or subjected to SCX chromatography using a 5 mL HiTrap sulfopropyl (SP) cartridge (GE Healthcare) on an ÄKTApurifier FPLC. The HiTrap SP column was conditioned as per manufacturer instructions, and proteins were loaded at 0.25 mL min−1. The column was then washed with H2O until the absorbance (280 nm) reached the pre-loading baseline. SCX elution was performed using a linear gradient of 500 mM ammonium acetate (pH 9) over 5 column volumes while collecting the eluent in 1 mL fractions.
:1 volume ratio and reacted at room temperature for 3 hours. The reaction was then halted by dialysis against 10 mM potassium phosphate (pH 7.4) using 10 kDa MWCO filters (EMD Millipore, Etobicoke, ON). The CT-cyt c produced in this way was either used directly, or subjected to SCX chromatography using a 5 mL HiTrap sulfopropyl (SP) cartridge (GE Healthcare) on an ÄKTApurifier FPLC. The HiTrap SP column was conditioned as per manufacturer instructions, and proteins were loaded at 0.25 mL min−1. The column was then washed with H2O until the absorbance (280 nm) reached the pre-loading baseline. SCX elution was performed using a linear gradient of 500 mM ammonium acetate (pH 9) over 5 column volumes while collecting the eluent in 1 mL fractions.
Optical spectroscopy
UV-Vis spectra were acquired on a Cary-100 spectrophotometer (Varian, Mississauga, ON). Soret measurement were performed on 10 μM protein solutions in 50 mM potassium phosphate buffer (pH 7.4). Absorbance measurements at 695 nm were performed at 40 μM protein. Peroxidase activity was measured as described59 by tracking the oxidation of 9 mM guaiacol in the presence of 100 μM H2O2. Additional experiments were conducted using 500 μM H2O2. This H2O2 concentration range is in line with previous work,7,32 and it is comparable to physiological H2O2 levels at the onset of apoptosis.60 In these assays the formation of tetraguaiacol was probed by UV-Vis at 470 nm.59 All activity assays contained 1 μM protein as determined by absorption measurements at the Soret maximum. Kinetic measurements at each H2O2 concentration were repeated in triplicate. Circular dichroism (CD) spectra were recorded on a Jasco J-810 spectropolarimeter (JASCO, Easton, MD), on 10 μM protein solutions in phosphate buffer (pH 7.4).Mass spectrometry
MS experiments were performed on a Synapt G2 ESI quadrupole-time-of-flight instrument (Waters, Milford, MA) operated in resolution mode. The capillary, sample cone, and extraction cone voltages were set at +2.8 kV, 25 V, and 4 V, respectively. MS/MS experiments were performed using collision-induced dissociation (CID) with argon as collision gas. Protein samples for bottom-up MS experiments were prepared by overnight digestion using trypsin (50:1 protein/trypsin) at 37 °C, and subsequently analyzed by liquid chromatography-mass spectrometry (LC/MS) using a nanoACQUITY UPLC (Waters) with a C18 column. Samples for intact protein MS were prepared as 10 μM solutions in a 50:50 solution of H2O:ACN with 0.1% formic acid, and directly infused at 5 μL min−1 using a syringe pump. For ion mobility-assisted top-down MS/MS experiments (top-down CID-IM-MS), 16+ protein ions were quadrupole-selected prior to CID in the trap collision cell. The collision energy used was 23 V for all samples. Ion mobility spectrometry (IMS) separation of CID products was performed in N2 buffer gas at a wave velocity of 350 m s−1 and wave height of 13 V. Simulated isotope distributions were generated using ProteinProspector (UCSF). All simulated spectra correspond to the ferric (Fe(III)) state of cyt c.
Results and discussion
Chloramine-T-induced Met80 oxidation
The CT-cyt c used in this study was prepared using established procedures.37,38,47 An initial characterization was conducted by subjecting unfractionated samples to a standard tryptic digestion/LC-MS/MS workflow. As expected, these assays revealed MetO formation (+16 Da) at Met80 and Met65, in line with earlier reports (Fig. S1†).17,37,46,47 No other +16 Da modifications were detected. These observations appear to support the widely held belief that CT selectively oxidizes Met.17,48,50,51 However, from the data discussed below it will be seen that standard mapping experiments (such as those of Fig. S1†) provide an incomplete view of the CT-induced oxidation events.SCX fractionation and optical characterization
SCX elution profiles of CT-cyt c exhibited three major peaks (Fig. 2A), consistent with previous observations.37,38,47,48 Close examination of the highest intensity region revealed the presence of two partially overlapping signals (around 23 mL, equivalent to “peak B” in ref. 37). In total, therefore, SCX revealed the existence of four chromatographically distinguishable CT-cyt c forms. We will refer to these fractions as I, II, III, and IV in the order of decreasing retention. Fraction I had the largest retention volume, i.e., it interacted most strongly with the stationary phase, implying the most highly cationic character. The SCX behavior of fraction I was indistinguishable from that of unmodified cyt c. Conversely, fraction IV showed the weakest interaction with the stationary phase, i.e., proteins in this fraction carried the least amount of positive charge. From the A280 values in the SCX chromatograms, the relative abundance of fractions IV, III, II, and I can be estimated to be roughly 10%, 35%, 35%, and 20%, respectively. For subsequent experiments we isolated thin slices out of the chromatograms for targeted experiments on each fraction (Fig. 2A).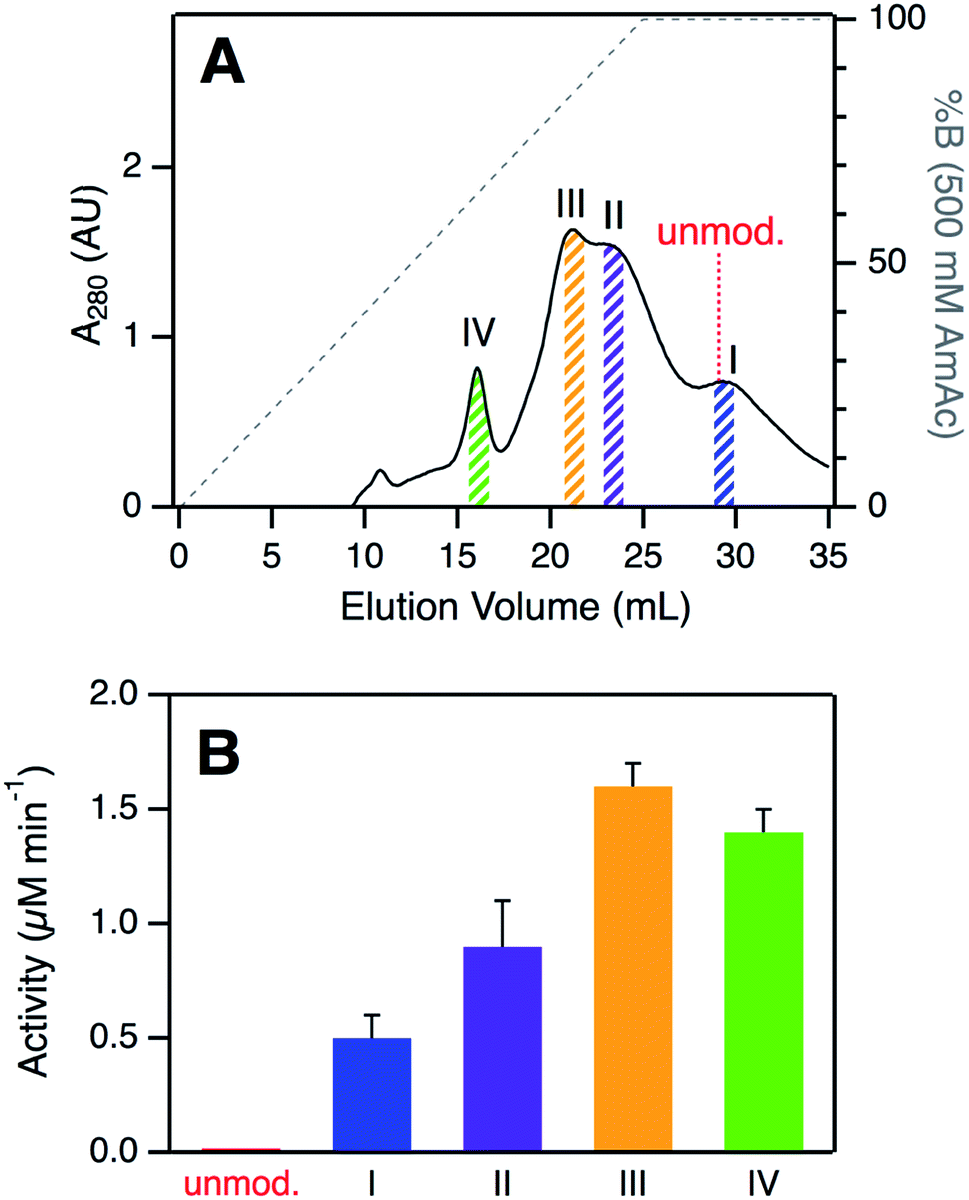 | ||
| Fig. 2 (A) Analysis of CT-cyt c by SCX chromatography. Four major species (I–IV) were discernible. Colored bars indicate collected fractions used for subsequent experiments. The elution volume of unmodified cyt c is indicated for comparison. (B) Peroxidase activity of CT-cyt c fractions measured in 100 μM H2O2. See Fig. S3A and ESI Table S1† for more information. Error bars represent standard deviations of three independent experiments. | ||
Optical spectra of the four CT-cyt c fractions were very similar to one another, but markedly different from those of unmodified cyt c (Fig. S2†). CT-cyt c showed a ∼30% CD intensity loss at 222 nm, revealing some decrease of α-helicity upon oxidation.61 There was no concomitant increase in the random coil region (∼200 nm), suggesting that CT-induced modifications did not cause unfolding, but transitions to alternative folded structures.61 A blue shift of the UV-vis Soret band from 409 nm to ∼405 nm indicated alterations in the heme environment. In addition, all CT-cyt c fractions exhibited loss of the 695 nm absorption band, signaling disruption of the Met80–Fe bond.62
In summary, the spectroscopic data of Fig. S2† indicate the formation of non-native, structured conformers in the CT-treated samples. The CD spectra were similar to those of Lys-ligated alkaline cyt c.19,35,36 The blue-shifted Soret band is reminiscent of alkaline cyt c as well.34,38 Resonance Raman (RR) data in the literature showed fraction I to be Lys-ligated, whereas the remaining fractions were reported to have an open (OH−/H2O coordinated) distal site.37 All these observations support the view that CT-cyt c shares structural features with previously characterized alkaline cyt c conformers.19,34,36 Recent NMR and EPR work by Zhong et al. arrived at an analogous conclusion.38
Peroxidase activity of CT-cyt c fractions
Despite their similar CD and UV-Vis spectroscopic properties (Fig. S2†), the four CT-cyt c fractions exhibited very different levels of peroxidase activity. Kinetic measurements were performed in the presence of 100 μM H2O2 (Fig. 2B and S3A†).59 Fraction I showed a relatively low activity; that of fraction II was twofold higher, and for fraction III an additional ∼twofold increase was observed. Fractions III and IV had similar activities. Under the low H2O2 concentration conditions of Fig. 2B, turnover was barely detectable for unmodified cyt c. This activity trend can be summarized as follows: unmodified cyt c < fraction I < fraction II < fraction III ≈ fraction IV. The same trend was observed when the experiments were repeated at a higher H2O2 concentration of 500 μM, where even untreated cyt c showed non-zero activity (Fig. S3B†).A number of earlier studies implied that CT treatment of cyt c generates a homogeneous product that is oxidized only at Met80 and Met65.17,48,50 The data presented here demonstrate that this view is not correct. Instead, CT-cyt c is a mixture of at least four species that can be separated by SCX (Fig. 2A) and that exhibit different peroxidase activities (Fig. 2B). While SCX fractionation of CT-cyt c has been demonstrated previously,37,38,47,48 the chemical origin of this phenomenon and the associated activity differences remain unexplained. SCX separates proteins according to their cationic surface charge. CT-induced Met → MetO conversion does not directly alter the ionic nature of the protein (Fig. S4†). Conformational factors could, in principle, contribute to the SCX fractionation.48 However, the nearly indistinguishable CD and UV-Vis spectra point to similar overall structures for fractions I–IV (Fig. S2†), making conformational factors an unlikely culprit for the observed SCX behavior.
SCX fractions represent specific proteoforms
For uncovering the origin of the SCX properties and activity differences of fractions I–IV we applied a range of MS methods. As an initial step, intact protein mass spectra were acquired (Fig. 3). At first glance, the mass distributions of the four fractions appear to be almost identical, reminiscent of the optical data in Fig. S2.† Each spectrum in Fig. 3 is dominated by two signals that are spaced +16 Da apart, as expected for MetO formation at Met80 and/or Met65. In addition, each fraction showed a low intensity signal close to the mass of unmodified cyt c. In Fig. 3 these signals were annotated according to the number (0, 1, 2) of MetO formation events. This interpretation reflects the existing literature, which assumes that CT specifically targets Met80 and Met65 while leaving other residues unmodified.17,37,38,47,48,50,51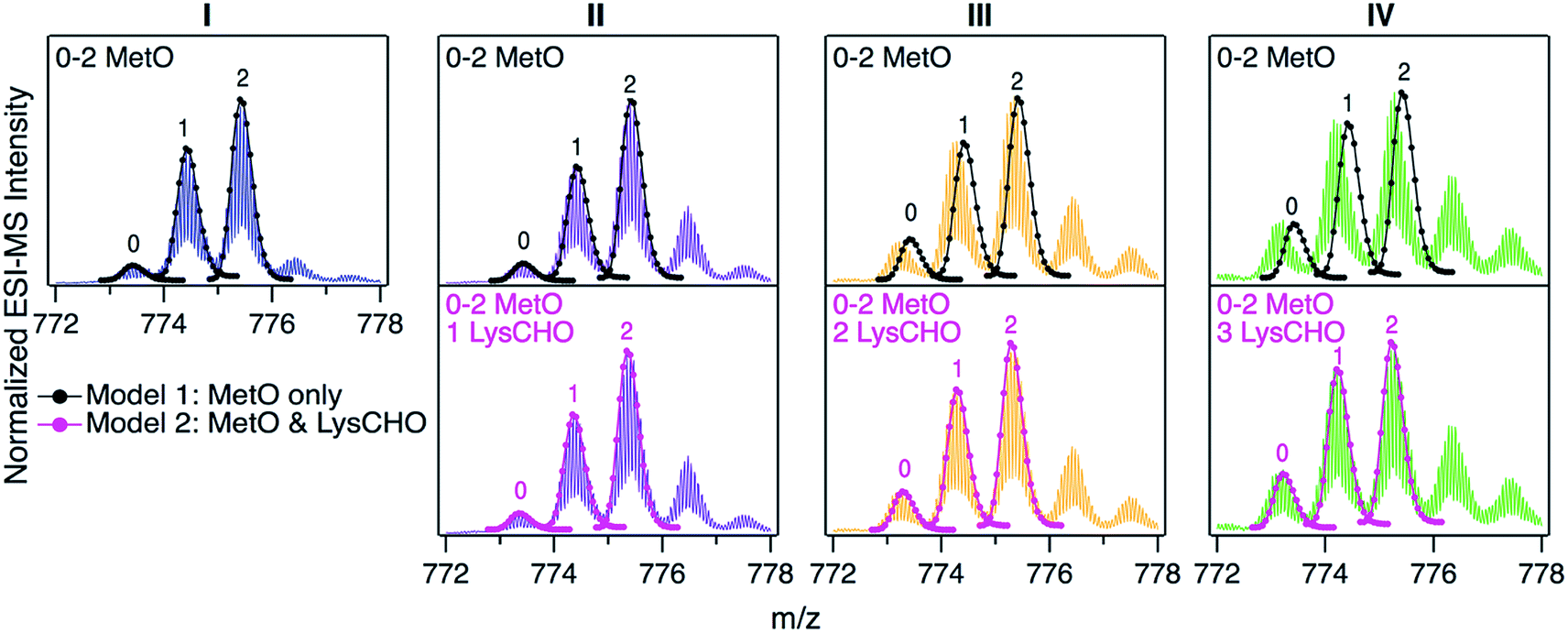 | ||
| Fig. 3 Intact protein mass spectra of CT-cyt c fractions I–IV, focusing on the 16+ charge state, which yielded the highest signal intensity. Labels 0, 1, or 2 above peaks refer to the number of MetO sites (+16 Da). Top: Black lines/dots represent theoretical isotope distributions for MetO formation. Bottom: Magenta lines/dots represent theoretical isotope distributions for MetO formation plus one, two, or three LysCHO formation events (Lys carbonylation, −1 Da). Top and bottom panels in each column show the same experimental data with different isotope models. | ||
To test the viability of this simple MetO-centric interpretation we calculated theoretical isotope distributions for unmodified cyt c, a +16 Da variant (one MetO), and a +32 Da variant (two MetO). These theoretical distributions were superimposed on the experimental spectra (Fig. 3, top row). Fraction I showed excellent agreement with the theoretical data. In contrast, fractions II, III, and IV exhibited gradually increasing mass defects, i.e., the measured peaks had a lower mass than expected. The magnitude of this discrepancy correlated with the SCX elution behavior: Fraction IV showed the largest discrepancy, while fraction I exhibited no discrepancy. The mismatch between experimental spectra and the theoretical “clean” +16 Da adducts implies that MetO formation is not the only type of covalent modification in fractions II–IV. Control experiments confirmed that mass discrepancies were already observable for unfractionated CT-cyt c, i.e., the observed effects were not an SCX-related artifact (Fig. S5†).
The missing piece of the puzzle that explains the properties of CT-cyt c is Lys carbonylation (Fig. S4†). This reaction is known to be facilitated by metal centers (i.e., heme in cyt c).43 It involves a radical intermediate63 that gets converted to ε-hydroxylysine. Elimination of ammonia then produces LysCHO.64 Each LysCHO formation event is accompanied by a −1 Da shift.41,64 Compilations of known oxidative protein modifications reveals LysCHO formation to be the only process associated with a −1 Da shift.42,43 The direct MS-based detection of LysCHO sites will be discussed below. Returning to Fig. 3, revised modeling of the mass spectra to account for a combination of MetO (+16 Da) and LysCHO formation (−1 Da) resulted in excellent agreement between experimental and simulated spectra for all fractions (Fig. 3, bottom row). The isotope models suggest that fractions I, II, III, and IV carry 0, 1, 2, and 3 LysCHO groups, respectively. These modifications are in addition to MetO formation at Met80 and Met65.17,37,38,47,48,50,51
The different numbers of LysCHO groups provide an explanation for the SCX behavior of the four fractions. The SCX strategy used here separates proteins on the basis of their overall cationic charge; each LysCHO site reduces this charge by one unit.41,42,64 In fraction I, the lack of Lys modifications causes the most positive surface charge, similar to that of unmodified cyt c (pI 9.6).65 Hence, fraction I (and unmodified cyt c) interact most tightly with the SCX column, resulting in the largest elution volume (Fig. 2A). On the other extreme, fraction IV with its three LysCHO has lost three positive charges, causing those proteins to interact most weakly with the column. The behavior of fractions II and III falls in-between these two extremes.
In summary, LysCHO formation accounts for the previously unexplained observation37,38,47,48 that CT-cyt c can be fractionated by SCX. LysCHO has been shown to play a role in the peroxidase activation of cyt c by H2O2.41 However, LysCHO formation in cyt c has not been reported for oxidizing agents other than H2O2. The number of LysCHO sites is the only detectable difference in the covalent structures of fractions I–IV, implying that LysCHO formation must be responsible for their different activities. Combining the information of Fig. 1B and 3, the peroxidase activity can be ranked according to the number of LysCHO: fraction I (0 LysCHO) < fraction II (1 LysCHO) < fraction III (2 LysCHO) ≈ fraction IV (3 LysCHO).
Top-down CID-IM-MS for proteoform-selective analysis
Mapping of LysCHO sites poses significant analytical challenges.66 Typical bottom-up MS assays struggle with this task due to LysCHO-induced interference with tryptic cleavage and losses of ionization/fragmentation efficiency.42,67,68 Thus, it is not surprising that previous MS investigations failed to detect LysCHO in CT-cyt c.17,18,37,38,45–48,50,51 To address these challenges we applied top-down MS/MS, i.e., gas phase fragmentation of undigested proteins.69 Initial attempts were unsuccessful due to the complexity of the fragment ion spectra. This problem was solved by incorporating ion mobility (IM) spectrometry to provide an additional separation dimension, thereby implementing a “top-down CID-IM-MS” workflow (Fig. S6†). Similar experiments have previously been applied to simple model systems,70–72 but this work marks the first time that such an approach was used for tracking unknown biologically relevant modifications.For validation, top-down CID-IM-MS was initially applied to peak 1 of fraction I (which is devoid of LysCHO) to examine the initial MetO formation event (Fig. 4). All C-terminal fragments starting at y25 showed a +16 Da shift relative to unmodified cyt c. The y25 to y24 transition allowed the oxidation site to be identified as Met80 with single-residue resolution. The presence of other oxidation sites in peak 1 (including Met65) could be ruled out. Measurements on peak 2 (+32 Da) verified Met80 and Met65 as the only two oxidation sites in fraction I (Fig. S8†). Taken together, these data imply that CT-induced MetO formation is a sequential process, with Met80 as the initial oxidation target whereas Met65 is affected at a later stage.
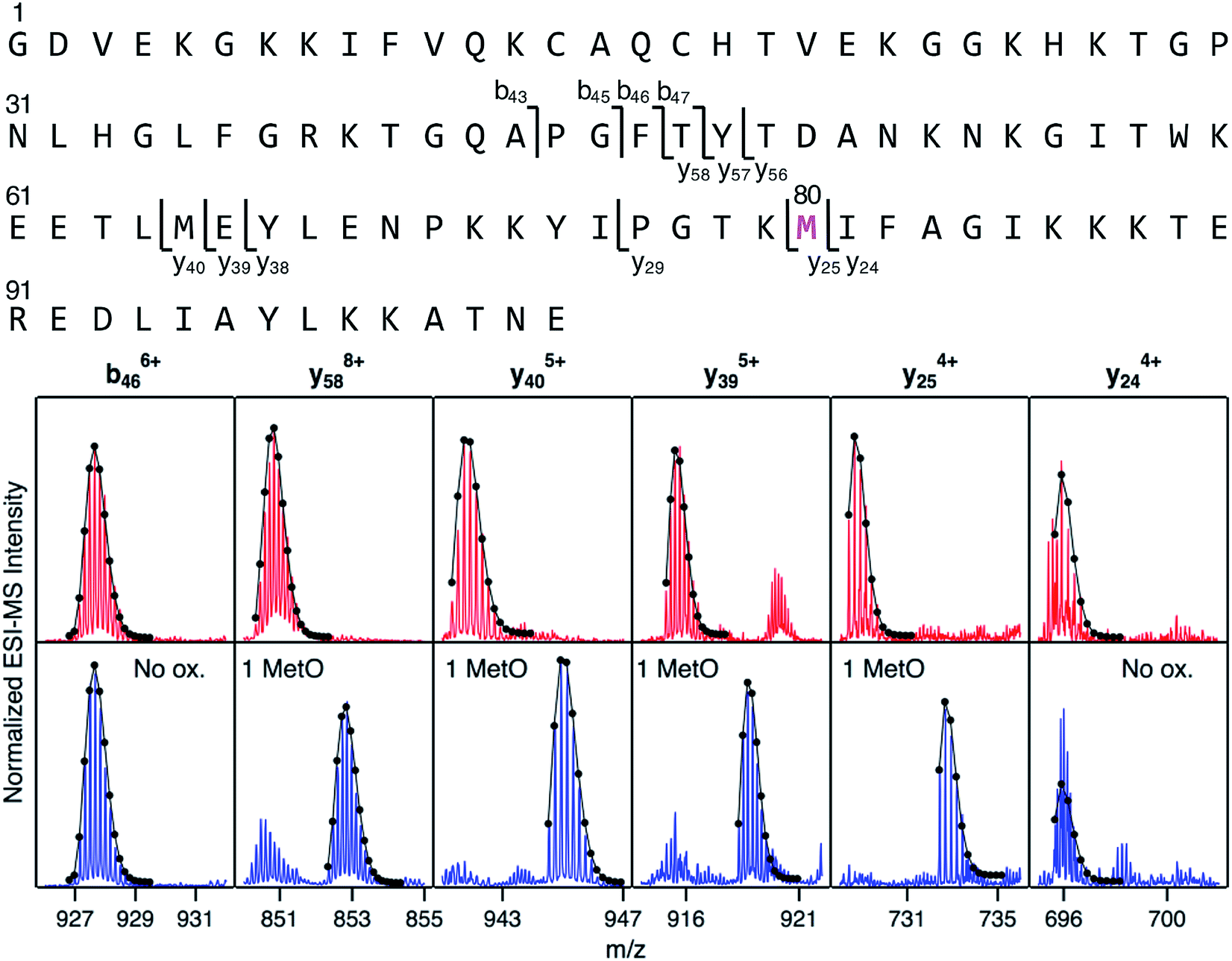 | ||
| Fig. 4 Top-down CID-IM-MS data that pinpoint MetO formation in fraction I. The sequence is shown along the top, with key fragments indicated. Bottom: selected fragment ions from an unmodified control (red), and from peak 1 of fraction I (blue, see Fig. 3). Lines/dots represent theoretical isotope distributions calculated for unmodified fragments (“no ox.”), or for MetO containing fragments. Met80 is the only covalent modification in peak 1 of fraction I. | ||
Mapping of LysCHO sites
We next sought to identify the locations of LysCHO groups in fractions II, III, and IV by top-down CID-IM-MS. According to Fig. 3, these fractions possess 1, 2, and 3 carbonylation sites, respectively. LysCHO sites can be identified from −1 Da mass shifts of fragment ions. The experiments were facilitated by the fact that, fortuitously, peak 1 in all fractions corresponded exclusively to MetO formation at Met80 (Fig. S9†). Thus, LysCHO sites in the various fractions could be mapped via comparisons with fraction I fragments. By using this internal reference we could unequivocally rule out calibration errors as an explanation for the observed −1 Da shifts. Our assignments were further verified by simulated isotope distributions that were overlaid onto the experimental spectra.
Fig. 5 reveals that for all fractions, N-terminal fragments up to b46, as well as C-terminal fragments up to y29 were indistinguishable from those of fraction I. This finding confines all LysCHO sites to residues 47–75 which comprise five out of the total 19 Lys residues. In fraction II, y-ions from y38 to y58 showed a −1 Da shift. The y38 to y29 transition narrowed down the location of this modification to 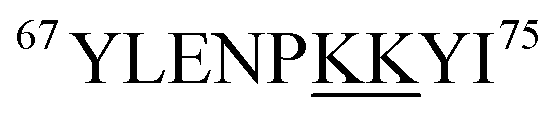 . Hence, the single LysCHO site in fraction II is located on Lys72 or Lys73 (Fig. 5, second row).
. Hence, the single LysCHO site in fraction II is located on Lys72 or Lys73 (Fig. 5, second row).
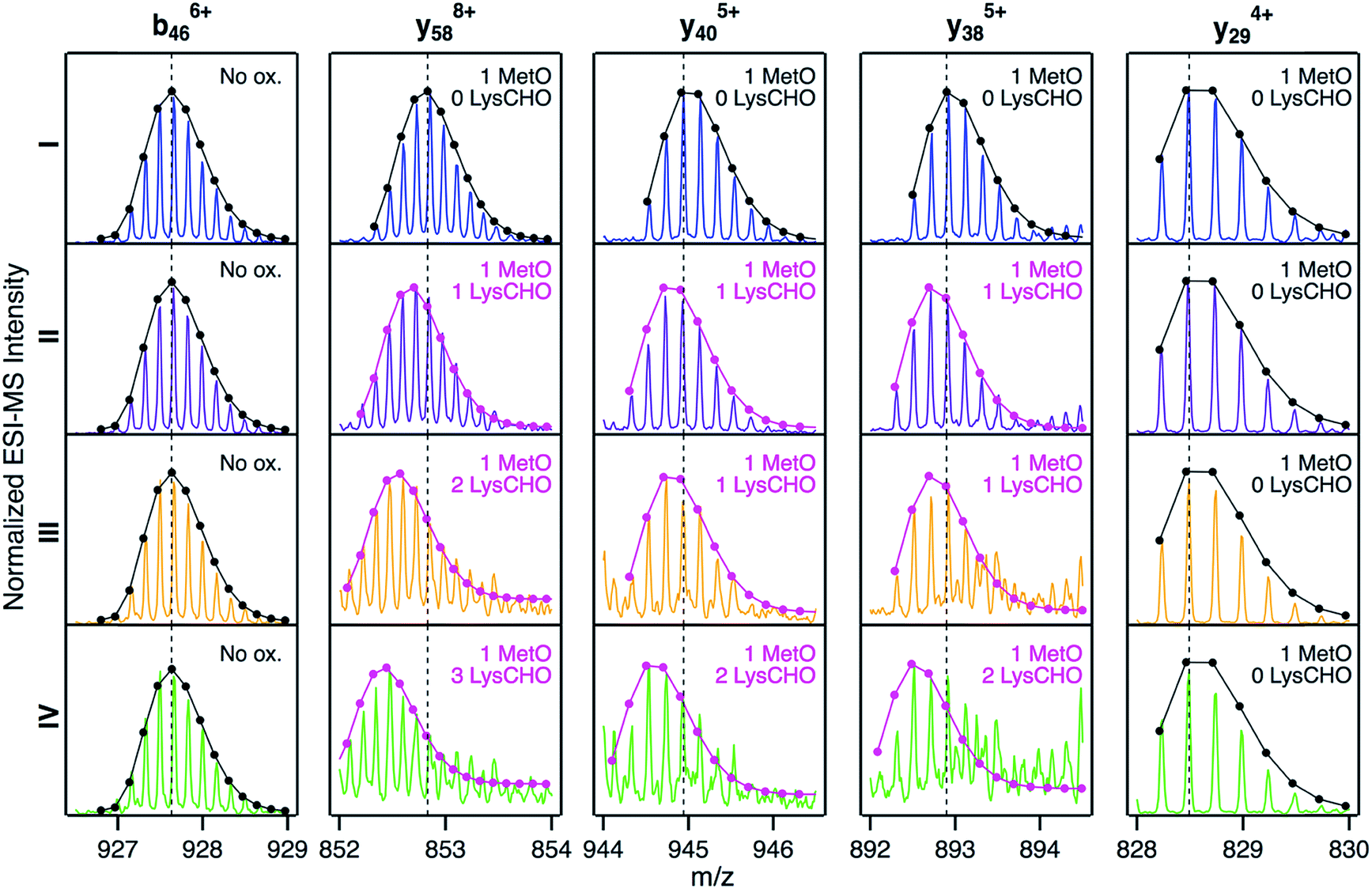 | ||
| Fig. 5 Selected top-down CID-IM-MS ions generated by fragmentation of peak 1 from fractions I (blue), II (violet), III (orange), and IV (green). Each fragment ion is overlaid with its simulated isotopic envelope (lines/dots). Vertical dashed lines were included as visual aid to highlight LysCHO-related −1 Da mass shifts. Complete CID-IM-MS data are summarized in Fig. S7.† | ||
Fraction III also showed a y38 to y29 −1 Da transition, implying one LysCHO modification on Lys72/73. An additional transition took place between y40 and y58, localizing the second LysCHO site to the region 49TDAN![[K with combining low line]](https://www.rsc.org/images/entities/char_004b_0332.gif) NGITWEETLM65. Thus, the second modification site in fraction III is located on Lys53, Lys55, or Lys60 (Fig. 5, third row).
NGITWEETLM65. Thus, the second modification site in fraction III is located on Lys53, Lys55, or Lys60 (Fig. 5, third row).
Pinpointing the three LysCHO sites in Fraction IV was complicated by diminished fragmentation efficiencies that translated into low S/N ratios. This phenomenon likely reflects the loss of protonation sites after LysCHO formation, keeping in mind that the proximity to H+ sites favors b/y ion formation.73–75 y58 exhibited a mass shift of −3 Da (Fig. 5, bottom row), confirming the presence of three LysCHO sites, as inferred from Fig. 3. From the fragment ion mass shifts in Fig. 5 (bottom row), the predominant modification pattern of fraction IV appears to be a single LysCHO in the Lys53/55/60 range, plus two LysCHO at Lys72/73. However, we cannot rule out that the final LysCHO formation event also affects Lys53/55/60 to some extent. The similar peroxidase activities of fractions III and IV suggest that this third LysCHO is of minor functional importance. Experiments analogous to those of Fig. 5 were conducted on peak 2 of each fraction, yielding LysCHO patterns similar to those of peak 1 (Fig. S10 and S11†).
Fig. 5 implies that CT-mediated LysCHO formation is a well-ordered process. Starting from LysCHO-free protein (fraction I), carbonylation initially affects Lys72/73 (fraction II). The next carbonylation event takes place in the Lys53/55/60 region, generating fraction III. The final LysCHO site may affect Lys72/73 or Lys53/55/60. As noted above, MetO formation is also a sequential process, initially affecting Met80 and subsequently Met65.
LC-MS/MS peptide mapping revisited
The detection of LysCHO sites by top-down CID-IM-MS may seem at odds with previous studies on CT-cyt c that only found MetO formation.17,37,38,47 In intact protein MS data the small (−1 Da) mass shifts associated with LysCHO formation are easily overlooked, unless the spectra are carefully scrutinized via isotope modeling (Fig. 3). Similarly, LysCHO detection by traditional tryptic digestion LC–MS/MS (Fig. S1†) is difficult due to reduced fragmentation resulting from charge site loss, and suppression of tryptic cleavage.42,73–75 These issues are exacerbated for the situation encountered here, where up to three LysCHO sites are confined to a fairly narrow sequence range (Lys53/55/60/72/73).Armed with the information from top-down CID-IM-MS, we revisited the tryptic digestion LC–MS/MS workflow with careful optimization of experimental parameters. Gratifyingly, we were able to confirm carbonylation of Lys72 in the missed-cleavage peptide T13–14 (residues 61–73, Fig. 6). Consistent with the data reported above, this LysCHO peptide was found in fractions II–IV, but not in fraction I. No other LysCHO sites were detected by LC-MS/MS. Thus, the top-down CID-IM-MS workflow is superior for this type of application, as it allowed the detection of all three carbonylation sites (Fig. 5).
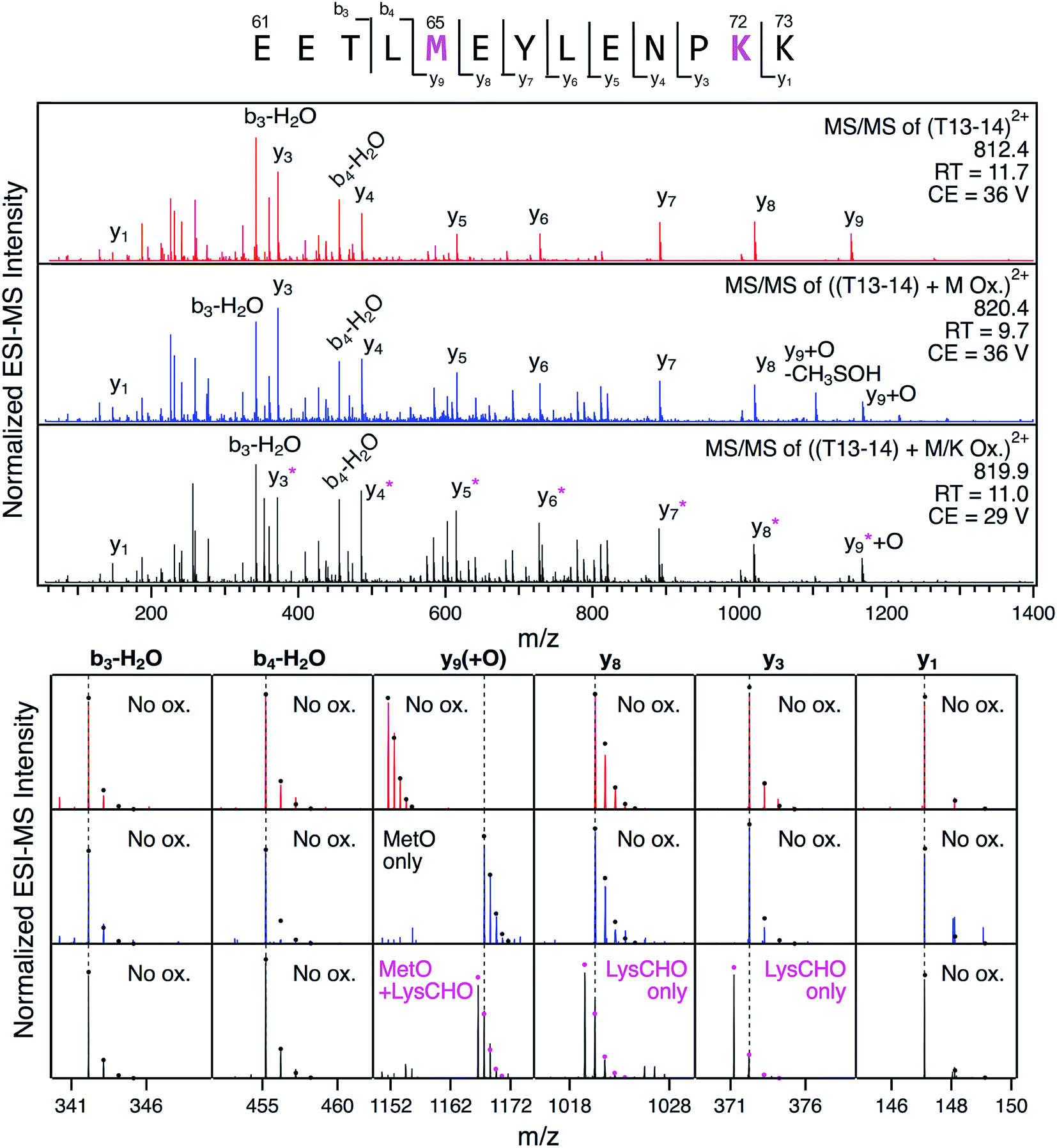 | ||
Fig. 6 LC-MS/MS analysis of tryptic peptide T13–14 (61EETL![[M with combining low line]](https://www.rsc.org/images/entities/char_004d_0332.gif) EYLENPK73) in unfractionated CT-cyt c. Three LC separated variants were interrogated: unmodified (red), with Met65 oxidation (blue), and with both Met65 oxidation and Lys72 carbonylation (black). Top: fragment ion spectra. Bottom: Magnified views of representative fragment ions, highlighting +16 Da (MetO) and −1 Da (LysCHO) mass shifts. Ions denoted with an asterisk contain the LysCHO site. Simulated isotopic envelopes are shown as circles. Much lower collision energies (CE) were required for adequate fragmentation of the carbonylated peptide. EYLENPK73) in unfractionated CT-cyt c. Three LC separated variants were interrogated: unmodified (red), with Met65 oxidation (blue), and with both Met65 oxidation and Lys72 carbonylation (black). Top: fragment ion spectra. Bottom: Magnified views of representative fragment ions, highlighting +16 Da (MetO) and −1 Da (LysCHO) mass shifts. Ions denoted with an asterisk contain the LysCHO site. Simulated isotopic envelopes are shown as circles. Much lower collision energies (CE) were required for adequate fragmentation of the carbonylated peptide. | ||
Mechanism of CT-induced peroxidase activation
The data of this work provide a clear view of the sequential CT-induced events that convert cyt c into an active peroxidase. This mechanism, summarized in Fig. 7, is governed by the undisputed fact that peroxidase activity requires the distal coordination site to be accessible.14,16,17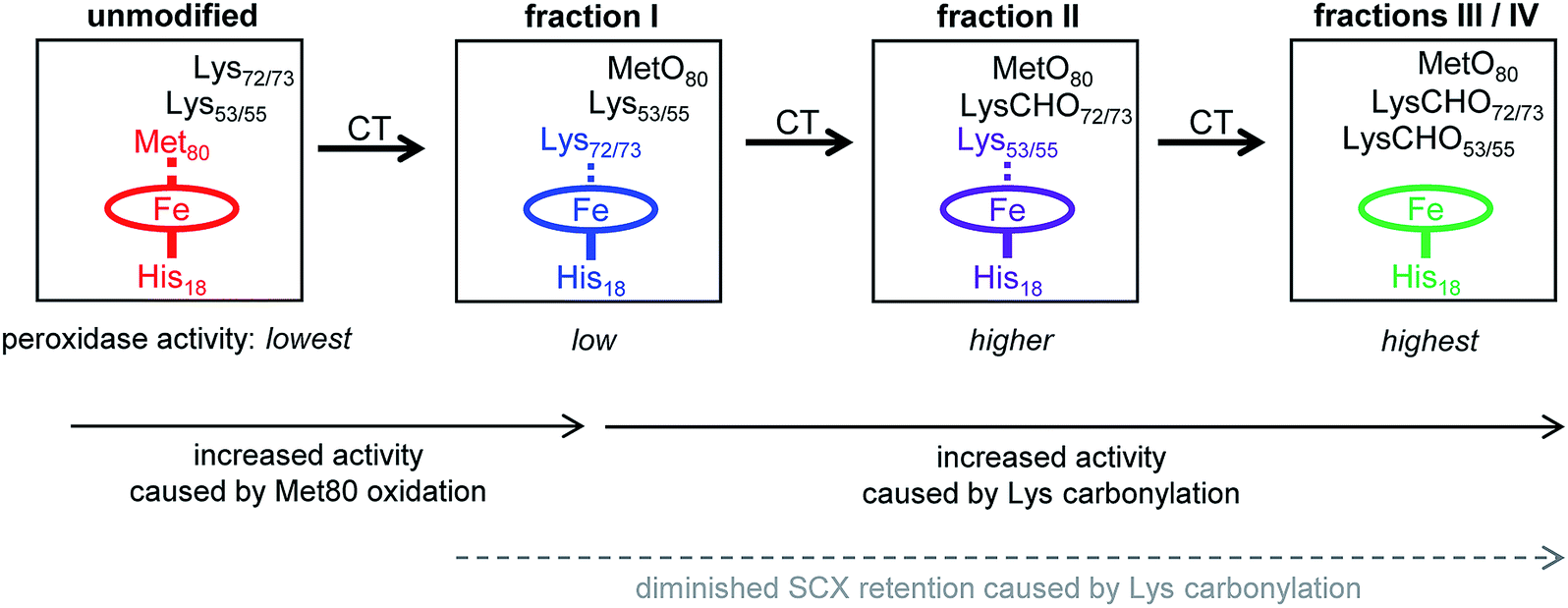 | ||
| Fig. 7 Model of CT-induced peroxidase activation of cyt c. SCX fractions I–IV exhibit different activity due to distal ligation changes. In unmodified cyt c the distal ligation site is blocked by Met80. In fraction I MetO formation has displaced Met80 from the heme iron, and distal ligation is provided by Lys72/73. In fraction II carbonylation of Lys72/73 causes a transition to distal ligation by Lys53/55. In fractions III and IV the heme has an open coordination site because carbonylation has abrogated the capability of Lys72/73 and Lys53/55 to act as distal ligands. Peroxidase activity increases from left to right, concomitant with increased accessibility of the distal ligation site. Dashed lines indicate Fe coordination bonds that can transiently open as a result of protein conformational fluctuations. | ||
Unmodified (native) cyt c has the lowest peroxidase activity of all the forms studied here because its distal ligation site is blocked by Met80. Very slow substrate turnover under these conditions may take place as the result of conformational fluctuations52,53 that occasionally disrupt the Met80–Fe bond.40,41,76
CT-induced oxidation of Met80 generates fraction I, an “alkaline” conformer where Lys72 or Lys73 has moved into the ligation site previously occupied by Met80. This ligation switch is supported by RR data37 as well as NMR and EPR experiments.38 While the enhanced peroxidase activity of alkaline Lys–Fe conformers is well documented,38 the origin of this effect is somewhat unclear because transient opening of the Lys–Fe contact takes place more slowly than for Met–Fe.34,77 Likely, the enhanced activity of fraction I is not caused by hexacoordinated Lys–Fe species per se, but by pentacoordinated structures that are part of the fraction I ensemble.14,38 RR spectra are consistent with this interpretation, suggesting the presence of ∼15% pentacoordinated protein in the predominantly Lys–Fe coordinated fraction I (“peak A” in Fig. S13 of ref. 37). Other factors may contribute to the peroxidase activity of Lys-ligated cyt c as well,41 one of these is the increased heme pocket volume which facilitates substrate access.19,77
As the next step in Fig. 7, carbonylation abrogates the iron ligating capability of Lys72/73, thereby generating fraction II. Fraction II still does not represent a genuine pentacoordinated conformer, evident from the fact that subsequent Lys53/55 carbonylation further enhances peroxidase activity (our MS data suggested the possible participation of Lys60, but we exclude this residue for reasons outlined below). Our data are thus consistent with a scenario where Lys53/55 participate in iron ligation in fraction II. However, these Lys53/55–Fe contacts must be quite labile, evident from the fact that fraction II has a higher activity than both fraction I and unmodified cyt c (Fig. 2B).
Carbonylation of Lys53/55 subsequently produces fraction III which possesses a permanently open distal site and shows the highest peroxidase activity. RR data confirm that fraction III is pentacoordinated, as expected for a functional peroxidase.37 A final carbonylation event produces fraction IV which retains both an open distal site and peroxidase activity.
The essential point in the activation cascade of Fig. 7 is that CT-induced Met80 oxidation only results in a moderate level of peroxidase activity, because the distal site remains congested by Lys ligation. Subsequent CT-induced LysCHO formation frees up the distal site, thereby producing a genuine pentacoordinated heme. Previous work attributed the peroxidase activity of CT-cyt c exclusively to MetO formation,17,18,37,38,44–47 failing to recognize the occurrence and mechanistic significance of Lys carbonylation. Our data reveal that MetO formation only represents an initial step en route toward peroxidase-activated CT-cyt c. Lys carbonylation represents a co-activator that boosts the activity by a factor of ∼4 (fractions III/IV) relative to the MetO-only form (fraction I, see Fig. 2).
The model of Fig. 7 provides a simple explanation for the correlation between the activity of the CT-cyt c fractions and their SCX behavior: both factors are directly tied to the number of LysCHO sites. When accounting for the SCX fractionation behavior, one has to remember that while the protein interacts with the anionic stationary phase, it may adopt structures and ligation scenarios that differ from the bulk solution conformations of Fig. 7.78
The ability of cyt c to adopt various distal ligation scenarios is consistent with the foldon model that dissects the protein into regions of different stability (Fig. S12†).52 The gray and red foldons (40–57 and 71–85 Ω loops, respectively) show the highest flexibility.52 Not surprisingly, the alternative Fe ligands identified above are located in these two flexible regions, i.e., Lys72/73 (red) and Lys53/55 (gray). Opening of gray79 and red53 has previously been linked to alkaline conformers. The flexibility of gray and red also governs the CT-induced transitions outlined in Fig. 7. In native cyt c the red foldon is anchored by the Met80–Fe linkage.52,53,79 Our data suggest that MetO formation sterically perturbs this (already labile)52 region, thereby triggering rearrangements that allow Lys72/73 to move into the distal site.53 The foldon hierarchy dictates that red can only open up when gray is already unfolded.52,53,79 Hence, after carbonylation of Lys72/73, the plasticity of the unfolded gray foldon will allow Lys53/55 to move into the vacated distal site. Our MS data suggest that Lys60 could also participate in Fe ligation, but its placement in the relatively rigid yellow foldon renders such a scenario less likely (Fig. S12†).
Our data do not provide evidence for carbonylation of Lys79. In principle, this residue represents another possible ligand that might block access to the iron after Met80 oxidation.38 However, evidence for such Lys79–Fe contacts comes primarily from work on K72A yeast iso-1 cyt c which has coordination propensities that are different from those of the horse wild type studied here.38 Our findings suggest that Lys79–Fe contacts do not play a major role in Met80-oxidized horse cyt c, such that peroxidase activation is attainable without Lys79 carbonylation.
Conclusions
Our discovery of multiple, previously unreported carbonylation events in CT-cyt c calls into question the prevailing Met80-centric view of peroxidase activation, according to which oxidation of Met80 alone is sufficient for turning cyt c into a pentacoordinated peroxidase.17,18,37,38,44,45,47,48 The conclusions from a number of those reports have relied on the alleged fidelity of CT for specific Met oxidation. Several studies pooled the highest intensity region in the SCX chromatogram, assuming that samples obtained in this way represent the expected “clean” MetO species.37,38,47,48 Our data strongly suggest that such samples correspond to fractions II/III, with oxidation at Met80, Met65, and with LysCHO sites in the Lys72/73 and Lys53/55 range. Carbonylation of these samples enhances their peroxidase activity almost fourfold relative to fraction I, which represents the true MetO-only species (Fig. 2 and 7). It appears that past studies inadvertently studied carbonylated CT-cyt c proteoforms, misattributing some of the carbonylation-induced effects to MetO formation.Previous studies on CT-cyt c yielded conflicting results, suggesting that the distal site is open37 or occupied by Lys.38 The present work helps resolve these discrepancies. CT-cyt c comprises multiple species with different coordination patterns (Fig. 7). Thus, any results will depend on the precise experimental conditions used for isolating protein from the CT-cyt c reaction mixture.
Only four out of the 19 Lys residues in cyt c were identified as possible carbonylation sites. In native cyt c these four residues are relatively close to the distal face of the heme, which represents the site of peroxidase catalysis (Fig. 1). Considering that all 19 Lys residues are solvent-exposed (Fig. S13†), the spatial selectivity of LysCHO formation rules out diffusion-mediated oxidation events, where modifications are governed by solvent accessibility.55 It instead appears that LysCHO formation is heme-dependent.43,58 This line of thought bolsters our conclusions, as carbonylation of Lys53/55/72/73 confirms the capability of these residues to come in direct contact with the heme by serving as alternative Fe ligands (Fig. 7).
There are certain parallels, but also differences between the CT-induced peroxidase activation of cyt c studied here, and the H2O2-induced activation that we explored earlier.41 The initial transformation triggered by H2O2 is oxidation of Tyr67, while this residue remains unmodified during CT exposure. Peroxidase activation by both CT and H2O2 involves sequential modifications and transiently populated alkaline conformers, ultimately producing a vacant distal site. One key difference is that CT-induced activation relies on MetO formation for disruption of the Met80–Fe bond (Fig. 7). In contrast, H2O2 causes this bond to break as a result of Tyr67 oxidation, without Met80 oxidation. The reasons underlying the different initial reactivities of the two oxidants remain to be fully explored. Likely, H2O2 oxidation of Tyr67 proceeds with catalysis by the iron center,43 whereas CT can oxidize Met without heme involvement.51 A common feature of CT and H2O2-induced activation is LysCHO formation which prevents the distal site from being blocked by Lys side chains. It is likely that LysCHO formation plays a similar role for the conversion of cyt c into an apoptotic peroxidase under other conditions, e.g., after cardiolipin binding3,27–29in vitro and in vivo. However, this hypothesis remains to be verified experimentally.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Funding was provided by the Natural Sciences and Engineering Research Council of Canada (RGPIN-2018-04243).References
- D. Alvarez-Paggi, L. Hannibal, M. A. Castro, S. Oviedo-Rouco, V. Demicheli, V. Tórtora, F. Tomasina, R. Radi and D. H. Murgida, Chem. Rev., 2017, 117, 13382–13460 CrossRef CAS PubMed.
- R. Radi, L. M. Thomson, H. Rubbo and E. Prodanov, Arch. Biochem. Biophys., 1991, 288, 112–117 CrossRef CAS PubMed.
- V. E. Kagan, H. A. Bayır, N. A. Belikova, O. Kapralov, Y. Y. Tyurina, V. A. Tyurin, J. Jiang, D. A. Stoyanovsky, P. Wipf, P. M. Kochanek, J. S. Greenberger, B. Pitt, A. A. Shvedova and G. G. Borisenko, Free Radical Biol. Med., 2009, 46, 1439–1453 CrossRef CAS PubMed.
- L. C. Godoy, C. Muñoz-Pinedo, L. Castro, S. Cardaci, C. M. Schonhoff, M. King, V. Tórtora, M. Marín, Q. Miao, J. F. Jiang, A. A. Kapralov, R. Jemmerson, G. G. Silkstone, J. N. Patel, J. E. Evans, M. l. T. Wilson, D. R. Green, V. E. Kagan, R. Radi and J. B. Mannick, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2653–2658 CrossRef CAS PubMed.
- L. Hannibal, F. Tomasina, D. A. Capdevila, V. Demicheli, V. Tórtora, D. Alvarez-Paggi, R. Jemmerson, D. H. Murgida and R. Radi, Biochemistry, 2016, 55, 407–428 CrossRef CAS PubMed.
- M. D. Liptak, R. D. Fagerlund, E. C. Ledgerwood, S. M. Wilbanks and K. L. Bren, J. Am. Chem. Soc., 2011, 133, 1153–1155 CrossRef CAS PubMed.
- L. J. McClelland, T.-C. Mou, M. E. Jeakins-Cooley, S. R. Sprang and B. E. Bowler, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6648–6653 CrossRef CAS PubMed.
- C. M. Jenkins, K. Yang, G. Liu, S. H. Moon, B. G. Dilthey and R. W. Gross, J. Biol. Chem., 2018, 293, 8693–8709 CrossRef CAS PubMed.
- N. C. Veitch, Phytochemicals, 2004, 65, 249–259 CrossRef CAS.
- J. P. Kitt, D. A. Bryce, S. D. Minteer and J. M. Harris, J. Am. Chem. Soc., 2017, 139, 3851–3860 CrossRef CAS PubMed.
- G. Oemer, K. Lackner, K. Muigg, G. Krumschnabel, K. Watschinger, S. Sailer, H. Lindner, E. Gnaiger, S. B. Wortmann, E. R. Werner, J. Zschocke and M. A. Keller, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 4158–4163 CrossRef CAS PubMed.
- N. A. Belikova, Y. A. Vladimirov, A. N. Osipov, A. A. Kapralov, V. A. Tyurin, M. V. Potapovich, L. V. Basova, J. Peterson, I. V. Kurnikov and V. E. Kagan, Biochemistry, 2006, 45, 4998–5009 CrossRef CAS PubMed.
- R. S. Y. Wong, J. Exp. Clin. Cancer Res., 2011, 30 Search PubMed.
- R. E. M. Diederix, M. Ubbink and G. W. Canters, Biochemistry, 2002, 41, 13067–13077 CrossRef CAS PubMed.
- G. W. Bushnell, G. V. Louie and G. D. Brayer, J. Mol. Biol., 1990, 214, 585–595 CrossRef CAS PubMed.
- K. L. Bren and E. L. Raven, Science, 2017, 356, 1236 CrossRef CAS PubMed.
- Y.-R. Chen, L. J. Deterding, B. E. Sturgeon, K. B. Tomer and R. P. Mason, J. Biol. Chem., 2002, 277, 29781–29791 CrossRef CAS PubMed.
- Z. Wang, Y. Ando, A. D. Nugraheni, C. Ren, S. Nagao and S. Hirota, Mol. BioSyst., 2014, 10, 3130–3137 RSC.
- J. F. Amacher, F. F. Zhong, G. P. Lisi, M. Q. Zhu, S. L. Alden, K. R. Hoke, D. R. Madden and E. V. Pletneva, J. Am. Chem. Soc., 2015, 137, 8435–8449 CrossRef CAS PubMed.
- L. A. Abriata, A. Cassina, V. Tortora, M. Marin, J. M. Souza, L. Castro, A. J. Vila and R. Radi, J. Biol. Chem., 2009, 284, 17–26 CrossRef CAS PubMed.
- T. M. Josephs, I. M. Morison, C. L. Day, S. M. Wilbanks and E. C. Ledgerwood, Biochem. J., 2014, 458, 259–265 CrossRef CAS PubMed.
- A. I. Karsisiotis, O. M. Deacon, M. T. Wilson, C. Macdonald, T. M. A. Blumenschein, G. R. Moore and J. A. R. Worrall, Sci. Rep., 2016, 6, 30447 CrossRef CAS PubMed.
- B. Moreno-Beltran, A. Guerra-Castellano, A. Diaz-Quintana, R. Del Conte, S. M. Garcia-Maurino, S. Diaz-Moreno, K. Gonzalez-Arzola, C. Santos-Ocana, A. Velazquez-Campoy, M. A. De la Rosa, P. Turano and I. Diaz-Moreno, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E3041–E3050 CrossRef CAS PubMed.
- L. Tognaccini, C. Ciaccio, V. D'Oria, M. Cervelli, B. D. Howes, M. Coletta, P. Mariottini, G. Smulevich and L. Fiorucci, J. Inorg. Biochem., 2016, 155, 56–66 CrossRef CAS PubMed.
- C. Ciaccio, L. Tognaccini, T. Battista, M. Cervelli, B. D. Howes, R. Santucci, M. Coletta, P. Mariottini, G. Smulevich and L. Fiorucci, J. Inorg. Biochem., 2017, 169, 86–96 CrossRef CAS PubMed.
- Z.-H. Wang, Y.-W. Lin, F. I. Rosell, F.-Y. Ni, H.-J. Lu, P.-Y. Yang, X.-S. Tan, X.-Y. Li, Z.-X. Huang and A. G. Mauk, ChemBioChem, 2007, 8, 607–609 CrossRef CAS PubMed.
- M. Abe, R. Niibayashi, S. Koubori, I. Moriyama and H. Miyoshi, Biochemistry, 2011, 50, 8383–8391 CrossRef CAS PubMed.
- L. Serpas, B. Milorey, L. A. Pandiscia, A. W. Addison and R. Schweitzer-Stenner, J. Phys. Chem. B, 2016, 120, 12219–12231 CrossRef CAS PubMed.
- J. M. Bradley, G. Silkstone, M. T. Wilson, M. R. Cheesman and J. N. Butt, J. Am. Chem. Soc., 2011, 133, 19676–19679 CrossRef CAS PubMed.
- M. Assfalg, I. Bertini, A. Dolfi, P. Turano, A. G. Mauk, F. I. Rosell and H. B. Gray, J. Am. Chem. Soc., 2003, 125, 2913–2922 CrossRef CAS PubMed.
- S. M. Nold, H. Lei, T.-C. Mou and B. E. Bowler, Biochemistry, 2017, 56, 3358–3368 CrossRef CAS PubMed.
- J. M. García-Heredia, I. Díaz-Moreno, P. M. Nieto, M. Orzáez, S. Kocanis, M. Teixeira, E. Pérez-Payá, A. Díaz-Quintana and M. A. De la Rosa, Biochim. Biophys. Acta, 2010, 1797, 981–993 CrossRef PubMed.
- S. Döpner, P. Hildebrandt, F. I. Rosell and A. G. Mauk, J. Am. Chem. Soc., 1998, 120, 11246–11255 CrossRef.
- F. I. Rosell, J. C. Ferrer and A. G. Mauk, J. Am. Chem. Soc., 1998, 120, 11234–11245 CrossRef CAS.
- J. Gu, D.-W. Shin and E. V. Pletneva, Biochemistry, 2017, 56, 2950–2966 CrossRef CAS PubMed.
- J. M. Garcia-Heredia, A. Diaz-Quintana, M. Salzano, M. Orzaez, E. Perez-Paya, M. Teixeira, M. A. De la Rosa and I. Diaz-Moreno, J. Biol. Inorg Chem., 2011, 16, 1155–1168 CrossRef CAS PubMed.
- D. A. Capdevila, W. A. Marmisolle, F. Tomasina, V. Demicheli, M. Portela, R. Radi and D. H. Murgida, Chem. Sci., 2015, 6, 705–713 RSC.
- F. Zhong and E. V. Pletneva, Inorg. Chem., 2018, 57, 5754–5766 CrossRef CAS PubMed.
- D. A. Capdevila, D. Alvarez-Paggi, M. A. Castro, V. Tortora, V. Demicheli, D. A. Estrin, R. Radi and D. H. Murgida, Chem. Commun., 2014, 50, 2592–2594 RSC.
- O. M. Deacon, D. A. Svistunenko, G. R. Moore, M. T. Wilson and J. A. R. Worrall, Biochemistry, 2018, 57, 4276–4288 CrossRef CAS PubMed.
- V. Yin, G. S. Shaw and L. Konermann, J. Am. Chem. Soc., 2017, 139, 15701–15709 CrossRef CAS PubMed.
- A. Bachi, I. Dalle-Donne and A. Scaloni, Chem. Rev., 2013, 113, 596–698 CrossRef CAS PubMed.
- I. M. Møller, A. Rogowska-Wrzesinska and R. S. P. Rao, J. Proteomics, 2011, 74, 2228–2242 CrossRef PubMed.
- G. H. Lushington, A. B. Cowley, S. Silchenko, G. S. Lukat-Rodgers, K. R. Rodgers and D. R. Benson, Inorg. Chem., 2003, 42, 7550–7559 CrossRef CAS PubMed.
- A. V. Birk, W. M. Chao, S. Y. Liu, Y. Soong and H. H. Szeto, Biochim. Biophys. Acta, 2015, 1847, 1075–1084 CrossRef CAS PubMed.
- A. D. Nugraheni, C. G. Ren, Y. Matsumoto, S. Nagao, M. Yamanaka and S. Hirota, J. Inorg. Biochem., 2018, 182, 200–207 CrossRef CAS PubMed.
- R. D. Parakra, G. N. L. Kleffmann, T. Jameson and E. C. Ledgerwood, Dalton Trans., 2018, 47, 9128–9135 RSC.
- J. Pande, K. Kinnally, K. K. Thallum, B. C. Verma, Y. P. Myer, L. Rechsteiner and H. R. Bosshard, J. Protein Chem., 1987, 6, 295–319 CrossRef CAS.
- S. P. Mushran, M. C. Agrawal and B. Prasad, J. Chem. Soc. B, 1971, 1712–1714 RSC.
- B. Michel, A. E. I. Proudfoot, C. J. A. Wallace and H. R. Bosshard, Biochemistry, 1989, 28, 456–462 CrossRef CAS PubMed.
- Y. Shechter, Y. Burstein and A. Patchornik, Biochemistry, 1975, 14, 4497–4503 CrossRef CAS PubMed.
- W. B. Hu, Z. Y. Kan, L. Mayne and S. W. Englander, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3809–3814 CrossRef CAS PubMed.
- H. Maity, J. N. Rumbley and S. W. Englander, Proteins, 2006, 63, 349–355 CrossRef CAS PubMed.
- L. M. Jones, J. B. Sperry, J. A. Carroll and M. L. Gross, Anal. Chem., 2011, 83, 7657–7661 CrossRef CAS PubMed.
- G. Xu and M. R. Chance, Chem. Rev., 2007, 107, 3514–3543 CrossRef CAS PubMed.
- M. Kathiresan and A. M. English, Chem. Sci., 2017, 8, 1152–1162 RSC.
- J. S. Sharp, J. M. Becker and R. L. Hettich, Anal. Chem., 2004, 76, 672–683 CrossRef CAS PubMed.
- J. D. Bridgewater, J. Lim and R. W. Vachet, Anal. Chem., 2006, 78, 2432–2438 CrossRef CAS PubMed.
- D. A. Baldwin, H. M. Marques and J. M. Pratt, J. Inorg. Biochem., 1987, 30, 203–217 CrossRef CAS PubMed.
- M. Giorgio, M. Trinei, E. Migliaccio and P. G. Pelicci, Nat. Rev. Mol. Cell Biol., 2007, 8, 722A–728A CrossRef PubMed.
- S. W. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, 2005, 1751, 119–139 CrossRef CAS PubMed.
- L. S. Kaminsky, V. J. Miller and A. J. Davison, Biochemistry, 1973, 12, 2215–2221 CrossRef CAS PubMed.
- Y. R. Chen, C. L. Chen, X. P. Liu, H. T. Li, J. L. Zweier and R. P. Mason, Free Radical Biol. Med., 2004, 37, 1591–1603 CrossRef CAS PubMed.
- J. R. Requena, C.-C. Chao, R. L. Levine and E. R. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 69–74 CrossRef CAS.
- M. Graf, R. G. Garcia and H. Watzig, Electrophoresis, 2005, 26, 2409–2417 CrossRef CAS PubMed.
- W. G. Chung, C. L. Miranda and C. S. Maier, Electrophoresis, 2008, 29, 1317–1324 CrossRef CAS PubMed.
- A. Temple, T.-Y. Yen and S. Gronert, J. Am. Soc. Mass Spectrom., 2006, 17, 1172–1180 CrossRef CAS PubMed.
- K. Artemenko, J. Mi and J. Bergquist, Free Radical Res., 2015, 49, 477–493 CrossRef CAS PubMed.
- N. Siuti and N. L. Kelleher, Nat. Methods, 2007, 4, 817–821 CrossRef CAS PubMed.
- N. F. Zinnel, P.-J. Pai and D. H. Russell, Anal. Chem., 2012, 84, 3390–3397 CrossRef CAS PubMed.
- D. A. Polasky, F. Lermyte, M. Nshanian, F. Sobott, P. C. Andrews, J. A. Loo and B. T. Ruotolo, Anal. Chem., 2018, 90, 2756–2764 CrossRef CAS PubMed.
- F. Halgand, J. Habchi, L. Cravello, M. Martinho, B. Guigliarelli and S. Longhi, Anal. Chem., 2011, 83, 7306–7315 CrossRef CAS PubMed.
- A. R. Dongré, J. L. Jones, Á. Somogyi and V. H. Wysocki, J. Am. Chem. Soc., 1996, 118, 8365–8374 CrossRef.
- B. Paizs and S. Suhai, Mass Spectrom. Rev., 2005, 24, 508–548 CrossRef CAS PubMed.
- S. M. Greer, D. D. Holden, R. Fellers, N. L. Kelleher and J. S. Brodbelt, J. Am. Soc. Mass Spectrom., 2017, 28, 1587–1599 CrossRef CAS PubMed.
- N. Tomášková, R. Varhač, V. Lysáková, A. Musatov and E. Sedlák, Biochim. Biophys. Acta, 2018, 1866, 1073–1083 CrossRef PubMed.
- Y. Deng, F. Zhong, S. L. Alden, K. R. Hoke and E. V. Pletneva, Biochemistry, 2018, 57, 5827–5840 CrossRef CAS PubMed.
- J. Guo and G. Carta, J. Chromatogr. A, 2015, 1388, 184–194 CrossRef CAS PubMed.
- M. M. G. Krishna, Y. Lin, J. N. Rumbley and S. W. Englander, J. Mol. Biol., 2003, 331, 29–36 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc03624a |
| This journal is © The Royal Society of Chemistry 2019 |