
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnderstanding the desulphurization process in an ionic porous aromatic framework†
Yuyang
Tian‡
 a,
Jian
Song‡
a,
Youliang
Zhu
b,
Huanyu
Zhao
c,
Faheem
Muhammad
a,
Tingting
Ma
a,
Mo
Chen
a and
Guangshan
Zhu
*a
a,
Jian
Song‡
a,
Youliang
Zhu
b,
Huanyu
Zhao
c,
Faheem
Muhammad
a,
Tingting
Ma
a,
Mo
Chen
a and
Guangshan
Zhu
*a
aKey Laboratory of Polyoxometalate Science of the Ministry of Education, Faculty of Chemistry, Northeast Normal University, Changchun 130024, P. R. China. E-mail: zhugs100@nenu.edu.cn
bState Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China
cLaboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Changchun 130023, P. R. China
First published on 23rd October 2018
Abstract
An ionic porous aromatic framework, iPAF-1, was successfully synthesized from a designed monomer with imidazolium functional groups. The iPAF-1 exhibits the highest dibenzothiophene uptake among all reported adsorptive desulphurization adsorbents. The so-called precursor designed synthetic route provides the stoichiometric and homogeneous introduction of desired functional groups into the framework. Molecular dynamics simulation was performed to understand the structure and the desulphurization process within the amorphous iPAF-1. The insight into the key role of the moderate bonding interaction between the adsorbate and the functional groups of iPAF-1 for improved uptake is highlighted in this work.
Introduction
Porous materials are extensively applied for removal of contaminants in water and air as primary adsorbents.1–4 Classic porous materials including activated carbons, zeolites, and metal organic frameworks (MOFs) have attracted wide attention as valuable adsorbents for various molecules that are of environmental concern, such as carbon dioxide, ammonia, volatile organic compounds and heavy metals. Sulphur emission during fossil fuel combustion has led to environmental pollution, fog and haze around the world. Thus, it is necessary to control the growing environmental issues, particularly the automobile exhaust emission pollutants in the air.5 Due to their harmful effects, many countries mandate the reduction of sulphur compounds in fuel oils below 10 ppm and anticipate sulphur-free gasoline and diesel in the future.6 Therefore, desulphurization from fuel oils has become a vital research topic. Numerous processes have been developed to remove sulphur compounds from fuel oils.7,8 Among them, adsorptive desulphurization is promising, due to its mild operating conditions, low energy consumption and effective removal of aromatic sulphur pollutants.9Activated carbons have been extensively reported for adsorptive desulphurization of fuel oils. Furthermore, zeolites and MOFs as functional porous adsorbent materials have been explored for use in desulphurization, with reported adsorption capacities of about 8–50 mg g−1 in the form of S uptake.10 However, they suffer from either relatively low sulphur compound uptake or low chemical stability. Therefore, the design of new and effective adsorbents is highly demanded. Porous organic frameworks (POFs)11 with sub-class materials, including porous aromatic frameworks (PAFs),12–16 conjugated microporous polymers (CMPs),17,18 covalent organic frameworks (COFs),19–21 covalent triazine frameworks22 and hyper-crosslinked polymers,23 possess large specific surface areas and nanosized pores. Therefore, they are intensively studied as new adsorbents for various molecules.24,25 A representative material is the first porous aromatic framework named PAF-1, which was synthesized via cross-coupling of tetrakis(4-bromophenyl)methane through the Yamamoto-type Ullmann reaction.12 Due to the rigidity of the tetrahedral building blocks and the high stability of the C–C bonds, PAF-1 possesses a specific BET surface area of 5600 m2 g−1 and exhibits high uptake capacities for hydrogen, carbon dioxide and benzene.
Besides their practical application as effective adsorbents, the fundamental investigations of adsorption in PAFs are particularly important for further rational design of new functional adsorbents. Compared to activated carbons and zeolites, certain chemical functionalities can be introduced into POFs via functional group modification or structural engineering.26,27 For example, imidazolium salts are exclusively used for desulphurization of aromatic sulphur compounds from transport oils because of the affinity between electron-rich thiophene-based compounds and electrophilic imidazolium compounds.28 On the other hand, PAFs are entirely composed of organic building units, and this simplifies the studies on the adsorption and diffusion behaviors of guest molecules in the channels. Currently, the complicated amorphous structure of PAFs hampers the understanding of these behaviors, and employing suitable computational simulations is especially an interesting research topic.
In this work, in an effort to understand sulphur uptake from gasoline with PAF materials, we present a viable synthetic approach for an ionic porous aromatic framework material with imidazolium functional groups, referred to as iPAF-1, using a precursor-designed synthetic method. Specifically, the precursor-designed approach stoichiometrically introduces functional groups and ensures evenly distributed adsorptive active sites in the products, thus a computational simulation based on molecular dynamics has made application to the functionalized PAF structures possible for the first time. The simulation results agree well with the experimental results of the porous structures and the adsorptive capacity of iPAF-1 for dibenzothiophene (DBT). In particular, iPAF-1 exhibits the highest adsorption desulphurization capacity of 769.23 mg g−1 for dibenzothiophene (DBT) compared with all other reported porous materials to date.10,29–44 Meanwhile, with the aid of computational simulation, the effect of binding energy on the DBT uptake during desulphurization was investigated to gain insight into the structural engineering of porous aromatic frameworks and emerging applications for environmentally friendly processes.
Experimental section
Synthesis of (3-methylphenyl)triphenylmethane (compound 1)
Trityl chloride (9.2 g, 32.9 mmol) and o-toluidine (9.4 mL, 88.8 mmol) were added to a 250 mL round-bottom flask. The mixture was stirred under reflux for 0.5 h. The resulting purple slurry reaction mixture was allowed to cool to room temperature when it solidified. The solid was ground with a spatula, and the resulting powder was combined with a mixture of 2 M HCl and methanol (25 mL/60 mL). The mixture was then heated at 80 °C for 0.5 h. After cooling to room temperature, the reaction mixture was filtered and washed with 125 mL of DI H2O to afford a light purple solid, which was briefly air-dried in an oven. After that, the crude solid was combined with 65 mL of ethanol and concentrated H2SO4 (10 mL, 96 wt%). The resulting mixture was cooled to −15 °C. Isoamyl nitrite (7.5 mL, 55.8 mmol) was added slowly, and the resulting mixture was stirred at −15 °C for 1 h. Aqueous hypophosphoric acid (15 mL, 50%) was then added to the reaction mixture at −15 °C, and the resulting mixture was brought to room temperature before being heated at 50 °C for 2 h. The resulting precipitate was collected by suction filtration and washed with DI H2O (100 mL) and ethanol (100 mL). The tan brown crude product of 1 (8.6 g, 78%) was collected without any further purification.Synthesis of (4-bromo-3-methylphenyl)tris(4-bromophenyl)methane (compound 2)
Compound 1 (3.0 g, 8.96 mmol) was added to a 150 mL 2-necked round-bottom flask equipped with a magnetic stir bar. One of the two necks was fitted with a rubber septum and the other one was fitted with a side arm adapter connected to the fume hood. Neat bromine (3.5 mL, 67.3 mmol) was added dropwise through the septum via a syringe for 5 min; subsequently, the resulting solution was stirred at room temperature for 0.5 h. Ethanol (60 mL) was then added to the reaction mixture, and the reaction was stirred for an additional 1 h. The resulting precipitate was collected by suction filtration and washed with 150 mL of ethanol. The collected crude product was then purified by column chromatography to afford compound 2 (4.9 g, 7.6 mmol, 85%) as a white solid.Synthesis of (4-bromo-3-(bromomethyl)phenyl)tris(4-bromophenyl)methane (compound 3)
Compound 2 (1.0 g, 1.53 mmol) was dissolved in anhydrous CCl4 (40 mL) in a 100 mL round-bottom flask. The mixture was degassed for 10 min. N-Bromosuccinimide (0.356 g, 1.99 mmol) and benzoyl peroxide (0.010 g, 0.04 mmol) were then added under N2, and the reaction mixture was refluxed overnight. The reaction mixture was cooled to room temperature and filtered. The filtrate was concentrated to dryness under reduced pressure to give a pale yellow oil, which was triturated with 25 mL of ethanol to give an off-white powder. The collected crude product was purified by column chromatography to afford 3 (2.6 g, 45%) as an off-white powder.Synthesis of (4-bromo-3-(3-methylimidazolemethyl)phenyl)tris(4-bromophenyl)methane (iTBPM)
Compound 3 (1.0 g, 1.53 mmol) and N-methylimidazole (0.356 g, 1.99 mmol) were dissolved with 40 mL of CH2Cl2 in a 100 mL round-bottom flask. The reaction mixture was refluxed overnight, cooled to room temperature and filtered. The white powder was washed with ethyl acetate and collected by filtration to afford iTBPM (1.10 g, 98%).Synthesis of iPAF-1
1,5-Cyclooctadiene (cod, 1.05 mL, 8.32 mmol) was added to a clear solution of bis(1,5-cyclooctadiene)nickel(0) ([Ni(cod)2], 2.25 g, 8.18 mmol) and 2,2′-bipyridyl (1.28 g, 8.18 mmol) in anhydrous DMF (120 mL). The reaction mixture was heated at 80 °C for 1 h. Dry iTBPM (1.27 g, 1.57 mmol) was added to the resulting purple solution, and the reaction mixture was stirred overnight to give a deep purple suspension. After cooling to room temperature, the residue was washed with concentrated HCl, DI H2O and THF, respectively. The resulting light yellow powder was purified by Soxhlet extraction with THF for 48 h to afford iPAF-1 (576 mg, 81%).Results and discussion
Synthesis of the materials
The idea of precursor design of PAF materials was first reported by Nguyen et al.45 as a “de novo approach”. They synthesized –CH3, –CH2OH, –CH2–NH2, –CH2N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CMe2 and phthalimide-based PAF-1 derivatives for CO2 adsorption materials. However, ionic imidazolium functional groups within PAF materials have never been reported before. Recently, the preparation of some ionic COF and PAF materials has been achieved by selecting ionic building blocks, and these materials have various applications for proton conduction, iodine adsorption and ion-exchange for water treatment.46–48 Until now, the ionic building blocks were limited to a few molecules that were either toxic or difficult to prepare, which limited their extensive applications. Herein, cationic imidazolium sites were introduced within a PAF material in order to improve the adsorption capacity of aromatic sulphur compounds. Compared to the post-modification treatment of PAFs, inhomogeneity would be avoided by precursor design. For the first time, tetrakis(4-bromophenyl)methane-based ionic porous aromatic frameworks were synthesized from designed precursors with 3-methylimidazolium ionic functional groups. To achieve this objective, we started from the synthesis of (3-methylphenyl)triphenylmethane (compound 1), (4-bromo-3-methylphenyl)tris(4-bromophenyl)methane (compound 2) and (4-bromo-3-(bromomethyl)phenyl)tris(4-bromophenyl)methane (compound 3) to build the desired monomer of (4-bromo-3-(3-methylimidazolemethyl)phenyl)tris(4-bromophenyl)methane (referred to as iTBPM), as demonstrated in the Experimental section and ESI.† Next, iPAF-1 was successfully synthesized via a Yamamoto-type Ullmann cross-coupling reaction from the iTBPM building units.
CMe2 and phthalimide-based PAF-1 derivatives for CO2 adsorption materials. However, ionic imidazolium functional groups within PAF materials have never been reported before. Recently, the preparation of some ionic COF and PAF materials has been achieved by selecting ionic building blocks, and these materials have various applications for proton conduction, iodine adsorption and ion-exchange for water treatment.46–48 Until now, the ionic building blocks were limited to a few molecules that were either toxic or difficult to prepare, which limited their extensive applications. Herein, cationic imidazolium sites were introduced within a PAF material in order to improve the adsorption capacity of aromatic sulphur compounds. Compared to the post-modification treatment of PAFs, inhomogeneity would be avoided by precursor design. For the first time, tetrakis(4-bromophenyl)methane-based ionic porous aromatic frameworks were synthesized from designed precursors with 3-methylimidazolium ionic functional groups. To achieve this objective, we started from the synthesis of (3-methylphenyl)triphenylmethane (compound 1), (4-bromo-3-methylphenyl)tris(4-bromophenyl)methane (compound 2) and (4-bromo-3-(bromomethyl)phenyl)tris(4-bromophenyl)methane (compound 3) to build the desired monomer of (4-bromo-3-(3-methylimidazolemethyl)phenyl)tris(4-bromophenyl)methane (referred to as iTBPM), as demonstrated in the Experimental section and ESI.† Next, iPAF-1 was successfully synthesized via a Yamamoto-type Ullmann cross-coupling reaction from the iTBPM building units.
Structural characterization of iTBPM and iPAF-1
The synthetic details of the monomer with designed imidazolium functional groups and iPAF-1 can be found in the Experimental section and ESI.† A schematic representation of iPAF-1 synthesis is shown in Fig. 1.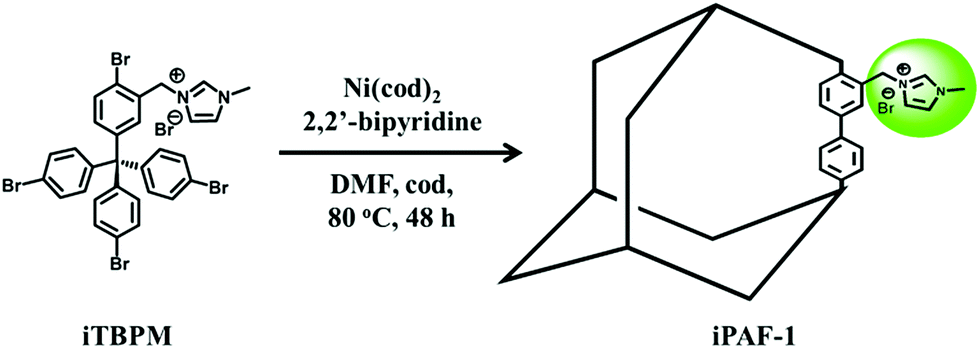 | ||
| Fig. 1 Schematic of the synthesis of iPAF-1 from the designed imidazolium-modified precursor. The iPAF-1 framework is schematically represented as an adamantane-like cage for clarity. | ||
The successful synthesis and structural characterization of the designed precursor iTBPM and iPAF-1 were confirmed using spectroscopic methods and elemental analysis techniques. The FT-IR spectra of precursor iTBPM and iPAF-1 are presented in Fig. S6.† The intense characteristic bands found at 537 and 517 cm−1 in the iTBPM spectrum do not appear in the iPAF-1 spectra, indicating that the phenyl-Br groups of the monomer iTBPM almost completely cross-coupled in the PAF-1 derivative network. Additionally, the spectra of iTBPM and iPAF-1 retain the region around 1607 cm−1 to 1160 cm−1, which are characteristic of the imidazole skeleton region: 1488 cm−1 for the CN stretching vibration, 1393 cm−1 for the deformation vibration of the C–H bond in the imidazolium ring and at 1160 cm−1 for the C–N stretching band, indicating the presence of the imidazolium group after the cross-coupling reaction. The peak for aromatic C–H stretching vibration is located at 3037 cm−1 and, the vibrations at 2980 and 2940 cm−1 are ascribed to aliphatic C–H stretching vibrations. Moreover, a broad O–H band is found around 3410.7 cm−1 due to trace amounts of absorbed water in the iPAF-1 sample. Thus, the IR data confirm the successful formation of iPAF-1 as our initial design. More detailed IR spectra of the precursor and PAF-1, with and without 3-methylimidazolium groups, are also shown in Fig. S6 and Table S1.† Elemental analyses of the iTBPM precursor and iPAF-1, with respect to C, H and N, are shown in Table S2.† The total content of these elements in iPAF-1 is 86.01%, and the remaining fraction is ascribed to the oxygen of the absorbed water, as evidenced by the IR spectra and Cl− ions as counter anions to imidazolium groups. Energy dispersive spectrometry (EDS) analysis (Fig. S7†) of iPAF-1 further confirms the presence of excess Cl− ions, rather than Br− ions, due to anion exchange when the product is washed with excess HCl solution. Moreover, the X-ray photoelectron spectroscopy (XPS) N 1s spectrum (Fig. S8†) suggests that the broad bands in the range of 399.8 to 400.6 eV were produced by the imidazolium ring. An enlarged view of Cl 1s and Br 1s XPS spectra (Fig. S8†) verifies the presence of Cl− ions, rather than Br−, in the iPAF-1 network; this result is consistent with the EDS data. Considering the above elemental analyses, the composition of iPAF-1 was confirmed to be C28.8H20.0N2Cl. Compared with the calculated molecular formula of C30H23N2Cl, the precursor design method can stoichiometrically and homogeneously introduce the desired functional groups into the final product. The thermal gravimetric (TG) profile of iPAF-1 under air (Fig. S9†) shows that the iPAF-1 network is more thermally stable than its parent monomer of iTBPM. The mass loss from 100–500 °C, which is due to the decomposition of absorbed water followed by the loss of chloride ions and imidazolium moieties, is consistent with the composition calculations. The scanning electron microscopy (SEM) images reveal the micrometer-sized aggregation of hundreds of nanometer-sized particles of iPAF-1 (Fig. S10†).
N2-sorption analysis revealed the porosity and textural properties of iPAF-1 at 77 K, as shown in Fig. 2. The obtained type I isotherm indicates a microporous network. The observed Brunauer–Emmett–Teller (BET) surface area and total micropore volume were 594 m2 g−1 and 0.280 cm3 g−1, respectively. The pore-size distribution predominantly lies in the microporous range of less than 2 nm. Three kinds of pores, centered at 0.54 nm, 0.86 nm and 1.10 nm, are calculated according to nonlocal density functional theory (NLDFT), as shown in the inset of Fig. 2. The presence of the functional group 3-methylimidazolium and heavier halide counter anions in the PAF diamond cavity reduces the BET surface area and micropore volume compared to the robust PAF-1 network.
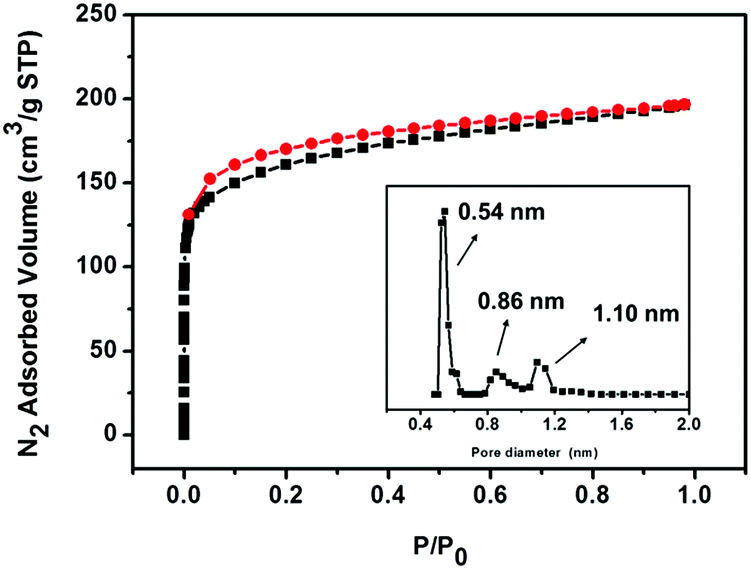 | ||
| Fig. 2 N2-sorption isotherm of iPAF-1 measured at 77 K. Square plots represent adsorption, and circle plots represent desorption. The inset shows the pore-size distribution calculated from the NLDFT method. | ||
Similar to most POF materials, iPAF-1 is amorphous, as indicated by its powder X-ray diffraction (PXRD) pattern (Fig. S11†). Although the local structure is assumed as a preferred diamond net built from tetrahedral blocks, according to the topological view, the overall structure obtained from the irreversible cross-coupling reactions possesses defects and mismatched connections. Thus, the product is an amorphous and sophisticated network. In order to elucidate its structure, we used a coarse-graining (CG) polymerization model to simulate the fabrication of iPAF-1. The CG simulations are all performed on graphics processing units (GPU) using molecular dynamics (MD) software GALAMOST.49–51 In the CG model of iTBPM, the particle A represents the quaternary carbon center linked to four benzene groups. The particle B represents the reactive brominated benzene group and has the position of the benzene group center. The particle C represents the imidazolium group and has the position of its center (see the details in Section S5.1 in the ESI†). The porous aromatic framework is formed by the cross-coupling reaction between B and B. However, the polymerization is a step growth process; therefore, not all of B can take part in the reaction for obtaining a perfect crystalline diamond structure. Instead, the tetrahedral precursors connect to a defected and complex network. Also, the entire framework can be regarded as a superposition of multiple local diamond structural networks, as shown in Fig. 3. In order to evaluate the credibility of the simulation, a new algorithm, referred to as “move-and-grow algorithm”, was designed and employed to measure the pore size distribution in the simulated structure (detailed in the ESI, Section S5.2†). The simulation result shows that three types of pores, with sizes of 0.54 nm, 0.90 nm and 1.26 nm, were detected in the simulated iPAF-1 framework (Fig. 3 inset). Specific patterns of each opening and surrounding framework are detailed in the ESI, Fig. S20.† The good agreement between the simulated pore size distribution and the experimental results from N2 adsorption indicates good credibility of the simulation model, and further simulation of the adsorption process will be conducted based on this model. The porous aromatic framework consists of three different pore sizes, which could facilitate the diffusion and absorption of sulphur compounds inside the framework.
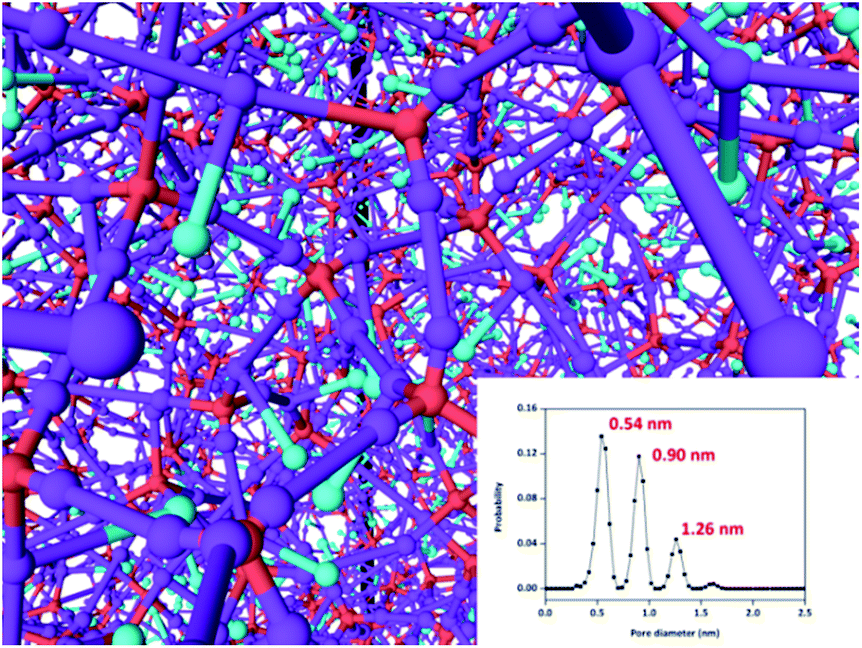 | ||
| Fig. 3 Simulated structure of iPAF-1. The inset shows the pore size distribution calculated from the move-and-grow algorithm. | ||
DBT adsorption performance of iPAF-1
Gas-chromatography (GC) was used to experimentally investigate the desulphurization performance of iPAF-1 as an adsorbent, using dibenzothiophene (DBT) as the target analyte in n-octane, a model fuel oil, at 298 K. All desulphurization experiments were performed at various solution concentrations of DBT in n-octane ranging between 0 and 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mg L−1. The results indicate that the adsorption capacity increased with increasing concentration of DBT, as shown in Fig. 4a. A linear relationship was observed between Ce/qe and Ce, with R2 > 0.9905, according to the Langmuir adsorption model (Fig. 4a inset). The maximum uptake of DBT was 769.23 mg g−1. The relationship was more closely correlated with the Langmuir adsorption model than the Freundlich model, as shown in Fig. S12.† This implies the existence of more dominant monolayer adsorption than multilayer physical adsorption, indicating strong affinity between electron-rich thiophene-based compounds and electrophilic imidazolium compounds.
000 mg L−1. The results indicate that the adsorption capacity increased with increasing concentration of DBT, as shown in Fig. 4a. A linear relationship was observed between Ce/qe and Ce, with R2 > 0.9905, according to the Langmuir adsorption model (Fig. 4a inset). The maximum uptake of DBT was 769.23 mg g−1. The relationship was more closely correlated with the Langmuir adsorption model than the Freundlich model, as shown in Fig. S12.† This implies the existence of more dominant monolayer adsorption than multilayer physical adsorption, indicating strong affinity between electron-rich thiophene-based compounds and electrophilic imidazolium compounds.
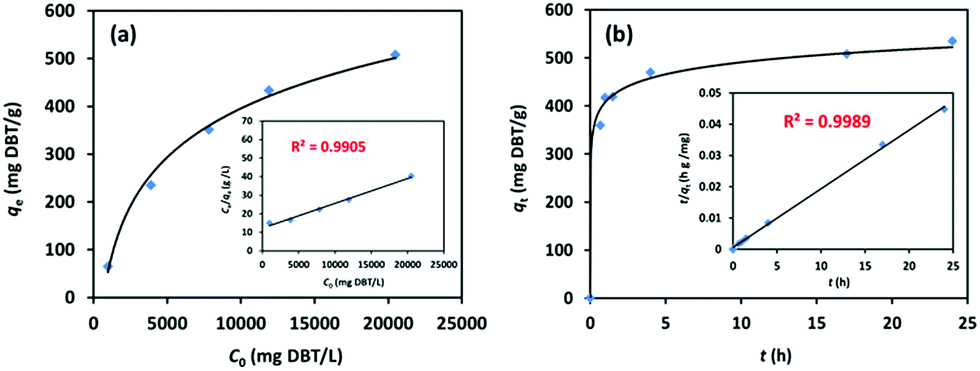 | ||
| Fig. 4 (a) DBT adsorption isotherm and Langmuir adsorption model fitting (inset) for iPAF-1 at various concentrations of DBT in n-octane. (b) Kinetic adsorption curve of DBT and the inset shows the fitting plot for the pseudo-second-order model. | ||
Furthermore, the kinetic desulphurization behaviors of iPAF-1 were examined in a solution of 20000 mg L−1 DBT in n-octane at 298 K for 24 h, as shown in Fig. 4b. The results revealed that the DBT uptake enhanced swiftly in the first hour, but when the adsorption time was extended to 24 h, the DBT adsorption almost reached equilibrium. In addition, the DBT adsorption isotherm on iPAF-1 can be described by a better fitting pseudo-second-order model, with R2 > 0.999 (Fig. 4b inset and Fig. S13†). This result further verifies the existence of finite adsorptive sites for DBT within iPAF-1, and these sites are attributed to the presence of the imidazolium group.
The calculated maximum uptake of DBT at 769.23 mg g−1 is far beyond any other adsorbent reported10,29–44 (Fig. 5a). Thus, iPAF-1 is the best adsorbent to date. Furthermore, Fig. 5b describes a brief and comprehensive comparison between well-known reported porous and non-porous adsorbents and iPAF-1, concerning the adsorption capacity value of DBT. It is understood that the uptake of an adsorbent usually scales linearly with its surface area. However, the plot of DBT uptake (qm) versus specific surface area of multiple adsorbents (SBET), shown in Fig. 5b, highlights the different behaviors of iPAF-1, which fall outside of this line. Generally speaking, compared to other reported porous adsorbents, iPAF-1 exhibits a relatively low surface area (594 m2 g−1), but it offers extraordinary DBT adsorption capacity. It is important to elucidate the key role of DBT adsorbents in light of targeted material design and engineering. With this goal, computational simulation of the desulphurization process was conducted.
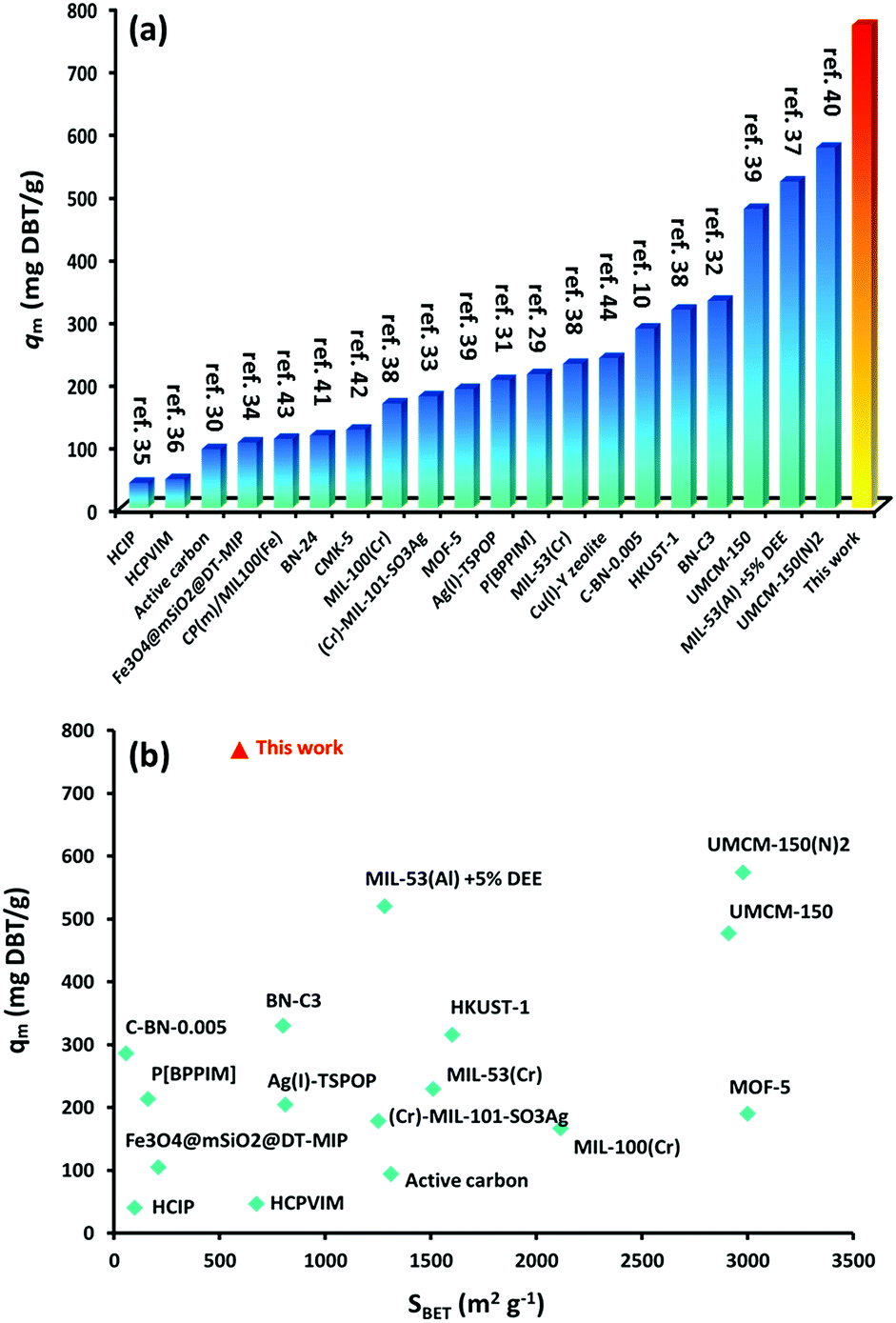 | ||
| Fig. 5 (a) Comparison of DBT desulphurization capacity of iPAF-1 with other reported materials (ref. 10 and 29–44). (b) Plot of DBT uptake (qm) versus specific surface area of multiple adsorbents (SBET). | ||
First, the binding energy (BE) was calculated using the Dmol3 module in Materials Studio 7.0 software (Accelrys Inc., San Diego). The density functional theory (DFT) method was employed using the gradient corrected (GGA) correlation function of Perdew and Wang (PW91), with the double numerical plus (DNP) polarization basis set. The convergence threshold parameters for the optimization were 2.7 × 10−4 eV (energy), 0.05 eV·Å−1 (gradient), and 0.005 Å (displacement), as reported elsewhere.29 The structures of DBT and segments of iTBPM were optimized before the BE calculations. Then, the binding structure of DBT and segments of iTBPM were constructed and optimized for BE calculation. The BE values were calculated as the energy difference between the products and the reactants in the adsorption process, as defined by BE = Etotal − Eseg − EDBT where Etotal, Eseg and EDBT represent the total energy of the monomer/adsorbates, segment and the adsorbate, respectively. The calculated BE was −36.6 kJ mol−1, where the negative value indicates an exothermic adsorption and higher absolute value means a higher adsorption strength (Fig. 6).
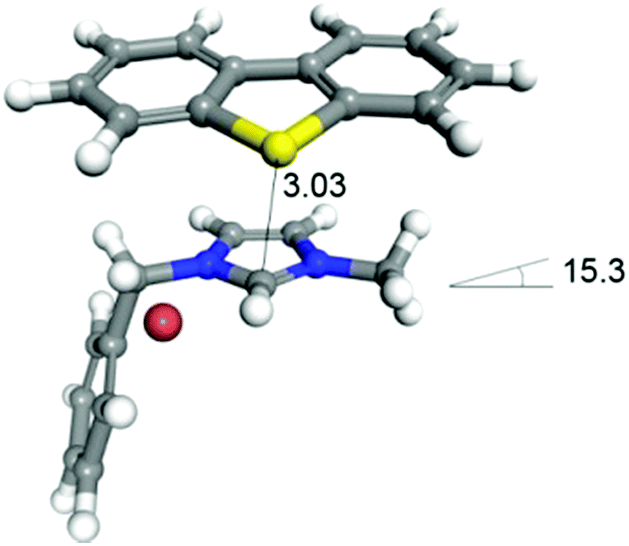 | ||
| Fig. 6 Binding configuration of DBT and the imidazolium segment. | ||
Considering the large spatial and temporal scales of the desulphurization process, we employed coarse-graining (CG) simulations to gain insight into the desulphurization phenomenon, based on the simulated model of iPAF-1. We constructed a model for desulphurization simulation, as shown in Fig. 7. The porous iPAF-1, which was obtained by previous polymerization simulation, was placed in the middle of a 15 nm × 15 nm × 30 nm box. 15000 DBT molecules were placed to the left of iPAF-1 in a 15 nm × 15 nm × 35 nm box. Wall1, Wall2 and Wall3 described by WCA interaction with ε = 1.0 kJ mol−1 and δ = 0.2 nm were employed to separate the molecules in the equilibrium simulation. The equilibrium simulation was performed for 105 time steps. After equilibrium, Wall2 and Wall3 allow the passage of DBT molecules. Then, the adsorption simulation began. The adsorption simulation was performed for approximately 2 × 109 time steps. To obtain an equilibrium state of absorption and desorption, when the DBT molecules diffused through the iPAF-1 framework and passed through Wall4, they were removed from the system.
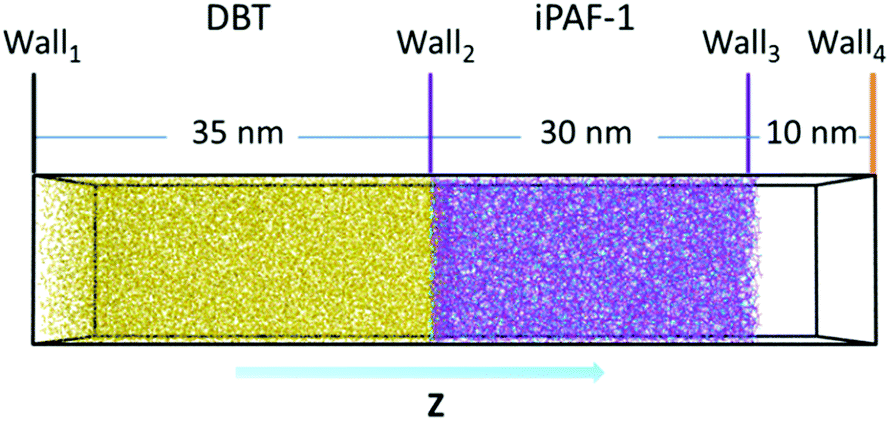 | ||
| Fig. 7 DBT desulphurization process simulation model. | ||
The adsorption doses of DBT along the Z direction within iPAF-1 for different BEs were collected. Fig. 8 shows three typical adsorption types, with different BEs. When the BE was −20 kJ mol−1, all DBT molecules entered the box of iPAF-1 but only some of them were adsorbed. When BE was −35 kJ mol−1, most of the DBT molecules entered the box and were retained inside it. When BE was −50 kJ mol−1, the diffusion of DBT molecules in iPAF-1 became quite difficult because of the stronger binding interaction between the imidazolium group and DBT. To further examine the effect of BE, a plot of DBT adsorption capability at different BE values versus the diffusion depth is shown in Fig. 8d. In general, the adsorption capability increased with increasing absolute BE value (Fig. 8). However, when the BE was −40 kJ mol−1 and −50 kJ mol−1, with stronger binding interaction, the adsorbed DBT blocked the pores and channels, preventing further diffusion. The maximum diffusion length along the Z direction was 27 nm for BE of −40 kJ mol−1 and 21 nm for BE of −50 kJ mol−1. Both lengths are shorter than the diameter of iPAF-1 particles, as observed by SEM. In this case, the inner part of the iPAF-1 bulk material will be free from DBT, which results in a lower adsorption capability overall. The maximum uptake achieved was at a BE of −35 kJ mol−1, and the simulated uptake of about 800 mg g−1 is close to the experimental result of 769.23 mg g−1, which was calculated according to the Langmuir model. This indicates the reliability of the simulation. The BE by DFT calculation is −36.6 kJ mol−1; thus, we concluded that the strength of the interaction between the imidazole group of iPAF-1 and DBT is an appropriate value for DBT adsorption, within the iPAF-1 network. The high DBT uptake of iPAF-1 may be attributed to the presence of favorable chemical interactions for DBT. Due to the diffusion control of the desulphurization process and the high sulphur uptake, the iPAF-1 is believed to be a suitable adsorbent for deep desulphurization, which is of practical significance in industry. The deep desulphurization of 100 ppm sulphur in model oil can be achieved via a sulphur breakthrough experiment. As shown in Fig. S15,† DBT molecules with concentrations below 100 ppm can be successfully removed from the model oil, indicating the potential application of iPAF-1 for deep removal of organic sulphur compounds at low concentrations.
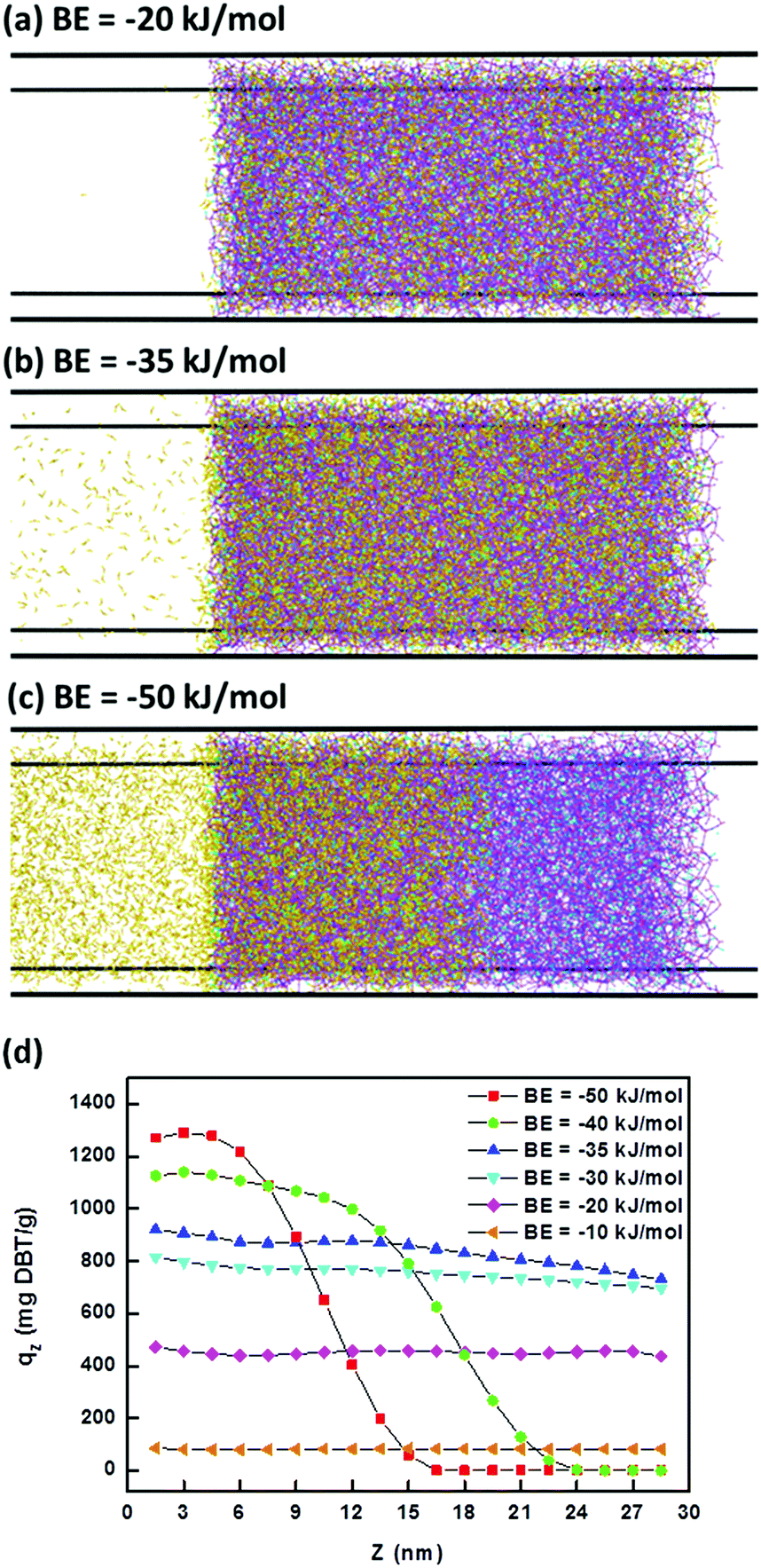 | ||
| Fig. 8 Three types of adsorption models (a–c) and the adsorption capacity for diffusion depths at different BEs (d). | ||
Conclusions
In conclusion, an imidazolium-modified building block was utilized to successfully synthesize an ionic porous aromatic framework with the goal of increasing the DBT uptake from gasoline. The synthetic route employs an engineered precursor and one-step cross coupling, so that the imidazolium functional sites are stoichiometrically and homogeneously introduced into the framework; this was confirmed by multiple characterization techniques. Compared to other porous adsorbents for adsorptive desulphurization, the ionic porous aromatic framework possesses a relatively low surface area of 594 m2 g−1 but the highest DBT uptake of 769.23 mg g−1. In order to understand the adsorptive desulphurization process and the role of the imidazolium functional group in iPAF-1, computational calculations were conducted to simulate the sophisticated structure of iPAF-1 and the desulphurization process. The simulation results indicate that the appropriate binding energy plays a key role in guest molecule diffusion and adsorption. In the case of iPAF-1, imidazolium functional groups, with a binding energy of −36.6 kJ mol−1, provided appropriate active sites for desulphurization of DBT from gasoline. This work gives insight into the design and engineering of porous aromatic frameworks for diverse applications in the future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Basic Research Program of China (973 Program, grant no. 2014CB931804) and the National Natural Science Foundation of China (NSFC Project, grant no. 91622106, 21531003 and 21601031). Y. Z. also thanks the National Natural Science Foundation of China (NSFC Project, grant no 21774129).Notes and references
- B. Aguila, Q. Sun, J. A. Perman, L. D. Earl, C. W. Abney, R. Elzein, R. Schlaf and S. Ma, Adv. Mater., 2017, 29, 1700665 CrossRef PubMed.
- B. P. Biswal, D. Becker, N. Chandrasekhar, J. S. Seenath, S. Paasch, S. Machill, F. Hennersdorf, E. Brunner, J. J. Weigand, R. Berger and X. Feng, Chem.–Eur. J., 2018, 24, 10868–10875 CrossRef PubMed.
- Q. Sun, B. Aguila, J. Perman, A. S. Ivanov, V. S. Bryantsev, L. D. Earl, C. W. Abney, L. Wojtas and S. Ma, Nat. Commun., 2018, 9, 1644 CrossRef PubMed.
- Y. Yuan, F. Sun, H. Ren, X. Jing, W. Wang, H. Ma, H. Zhao and G. Zhu, J. Mater. Chem., 2011, 21, 13498 RSC.
- X. Xu, S. Zhang, P. Li and Y. Shen, Fuel, 2014, 117, 499–508 CrossRef.
- P. Wasserscheid and A. Jess, Green Chem., 2004, 6, 316–322 RSC.
- H. Yang, B. Jiang, Y. Sun, L. Zhang, Z. Huang, Z. Sun and N. Yang, J. Hazard. Mater., 2017, 333, 63–72 CrossRef PubMed.
- M. Soleimani, A. Bassi and A. Margaritis, Biotechnol. Adv., 2007, 25, 570–596 CrossRef PubMed.
- X. L. Tang and L. Shi, Langmuir, 2011, 27, 11999–12007 CrossRef PubMed.
- J. Xiong, W. Zhu, H. Li, L. Yang, Y. Chao, P. Wu, S. Xun, W. Jiang, M. Zhang and H. Li, J. Mater. Chem. A, 2015, 3, 12738–12747 RSC.
- S. Das, P. Heasman, T. Ben and S. Qiu, Chem. Rev., 2017, 117, 1515–1563 CrossRef PubMed.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, M. Simmons Jason, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef PubMed.
- Y. Yuan, F. Sun, L. Li, P. Cui and G. Zhu, Nat. Commun., 2014, 5, 4260 CrossRef PubMed.
- J. F. Van Humbeck, T. M. McDonald, X. Jing, B. M. Wiers, G. Zhu and J. R. Long, J. Am. Chem. Soc., 2014, 136, 2432–2440 CrossRef PubMed.
- Y. Yuan, F. Sun, F. Zhang, H. Ren, M. Guo, K. Cai, X. Jing, X. Gao and G. Zhu, Adv. Mater., 2013, 25, 6619–6624 CrossRef PubMed.
- T. Ben, Y. Li, L. Zhu, D. Zhang, D. Cao, Z. Xiang, X. Yao and S. Qiu, Energy Environ. Sci., 2012, 5, 8370 RSC.
- J. X. Jiang, F. Su, A. Trewin, D. Wood Colin, L. Campbell Neil, H. Niu, C. Dickinson, Y. Ganin Alexey, J. Rosseinsky Matthew, Z. Khimyak Yaroslav and I. Cooper Andrew, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef PubMed.
- Y. Xie, T. T. Wang, X. H. Liu, K. Zou and W. Q. Deng, Nat. Commun., 2013, 4, 1960 CrossRef PubMed.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef PubMed.
- S. Y. Ding, J. Gao, Q. Wang, Y. Zhang, W. G. Song, C. Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef PubMed.
- S. Dalapati, E. Jin, M. Addicoat, T. Heine and D. Jiang, J. Am. Chem. Soc., 2016, 138, 5797–5800 CrossRef PubMed.
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef PubMed.
- H. Lau Cher, X. Mulet, K. Konstas, M. Doherty Cara, M. A. Sani, F. Separovic, R. Hill Matthew and D. Wood Colin, Angew. Chem., Int. Ed., 2016, 55, 1998–2001 CrossRef PubMed.
- B. Li, Y. Zhang, R. Krishna, K. Yao, Y. Han, Z. Wu, D. Ma, Z. Shi, T. Pham, B. Space, J. Liu, P. K. Thallapally, J. Liu, M. Chrzanowski and S. Ma, J. Am. Chem. Soc., 2014, 136, 8654–8660 CrossRef PubMed.
- Q. Sun, B. Aguila, J. Perman, L. D. Earl, C. W. Abney, Y. Cheng, H. Wei, N. Nguyen, L. Wojtas and S. Ma, J. Am. Chem. Soc., 2017, 139, 2786–2793 CrossRef PubMed.
- A. Nagai, Z. Guo, X. Feng, S. Jin, X. Chen, X. Ding and D. Jiang, Nat. Commun., 2011, 2, 536 CrossRef PubMed.
- L. H. Li, X. L. Feng, X. H. Cui, Y. X. Ma, S. Y. Ding and W. Wang, J. Am. Chem. Soc., 2017, 139, 6042–6045 CrossRef CAS PubMed.
- W. Jiang, W. Zhu, Y. Chang, H. Li, Y. Chao, J. Xiong, H. Liu and S. Yin, Energy Fuels, 2014, 28, 2754–2760 CrossRef CAS.
- L. Peng, F. Guo, C. Zhang, J. Xu, S. Xu, C. Peng, J. Hu and H. Liu, Ind. Eng. Chem. Res., 2017, 56, 4319–4326 CrossRef CAS.
- J. Xiao, C. Song, X. Ma and Z. Li, Ind. Eng. Chem. Res., 2012, 51, 3436–3443 CrossRef CAS.
- T. Jin, S. An, X. Yang, J. Hu, H. Wang, H. Liu, Z. Tian, D. e. Jiang, N. Mehio and X. Zhu, AIChE J., 2016, 62, 1740–1746 CrossRef CAS.
- J. Xiong, L. Yang, Y. Chao, J. Pang, P. Wu, M. Zhang, W. Zhu and H. Li, Green Chem., 2016, 18, 3040–3047 RSC.
- M. Huang, G. Chang, Y. Su, H. Xing, Z. Zhang, Y. Yang, Q. Ren, Z. Bao and B. Chen, Chem. Commun., 2015, 51, 12205–12207 RSC.
- J. Wang and J. Wei, J. Mater. Chem. A, 2017, 5, 4651–4659 RSC.
- J. Zhang, H. H. Xu, Y. Z. Lu, H. Meng, C. X. Li, B. H. Chen and Z. G. Lei, Energy Fuels, 2016, 30, 5035–5041 CrossRef CAS.
- J. Zhang, C. Ma, X. Zhu, Y. Lu, H. Meng, C. Li, B. Chen and Z. Lei, Ind. Eng. Chem. Res., 2016, 55, 8079–8086 CrossRef CAS.
- N. A. Khan, J. W. Yoon, J. S. Chang and S. H. Jhung, Chem. Commun., 2016, 52, 8667–8670 RSC.
- H. X. Zhang, H. L. Huang, C. X. Li, H. Meng, Y. Z. Lu, C. L. Zhong, D. H. Liu and Q. Y. Yang, Ind. Eng. Chem. Res., 2012, 51, 12449–12455 CrossRef CAS.
- K. A. Cychosz, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 6938–6939 CrossRef CAS PubMed.
- T. H. Park, K. A. Cychosz, A. G. Wong-Foy, A. Dailly and A. J. Matzger, Chem. Commun., 2011, 47, 1452–1454 RSC.
- J. Xiong, W. Zhu, H. Li, W. Ding, Y. Chao, P. Wu, S. Xun, M. Zhang and H. Li, Green Chem., 2015, 17, 1647–1656 RSC.
- N. Farzin Nejad, E. Shams, M. K. Amini and J. C. Bennett, Microporous Mesoporous Mater., 2013, 168, 239–246 CrossRef CAS.
- Z. Hasan and S. H. Jhung, ACS Appl. Mater. Interfaces, 2015, 7, 10429–10435 CrossRef CAS PubMed.
- L. Wang, B. Sun, F. H. Yang and R. T. Yang, Chem. Eng. Sci., 2012, 73, 208–217 CrossRef CAS.
- S. J. Garibay, M. H. Weston, J. E. Mondloch, Y. J. Colon, O. K. Farha, J. T. Hupp and S. T. Nguyen, CrystEngComm, 2013, 15, 1515–1519 RSC.
- H. Ma, B. Liu, B. Li, L. Zhang, Y. G. Li, H. Q. Tan, H. Y. Zang and G. Zhu, J. Am. Chem. Soc., 2016, 138, 5897–5903 CrossRef CAS PubMed.
- Y. Su, Y. Wang, X. Li, X. Li and R. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 18904–18911 CrossRef CAS PubMed.
- Z. Li, H. Li, X. Guan, J. Tang, Y. Yusran, Z. Li, M. Xue, Q. Fang, Y. Yan, V. Valtchev and S. Qiu, J. Am. Chem. Soc., 2017, 139, 17771–17774 CrossRef CAS PubMed.
- Y. L. Zhu, H. Liu, Z. W. Li, H. J. Qian, G. Milano and Z. Y. Lu, J. Comput. Chem., 2013, 34, 2197–2211 CrossRef CAS PubMed.
- Y. L. Zhu, D. Pan, Z. W. Li, H. Liu, H. J. Qian, Y. Zhao, Z. Y. Lu and Z. Y. Sun, Mol. Phys., 2018, 1–13, DOI:10.1080/00268976.2018.1434904.
- H. Liu, Y. L. Zhu, Z. Y. Lu and F. Müller-Plathe, J. Comput. Chem., 2016, 37, 2634–2646 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic details and spectra of all synthesized compounds. DBT adsorption computational details. See DOI: 10.1039/c8sc03727b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |