
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDissolving uptake-hindering surface defects in metal–organic frameworks†
Kai
Müller
 a,
Nina
Vankova
bc,
Ludger
Schöttner
a,
Thomas
Heine
bc and
Lars
Heinke
*a
a,
Nina
Vankova
bc,
Ludger
Schöttner
a,
Thomas
Heine
bc and
Lars
Heinke
*a
aInstitute of Functional Interfaces (IFG), Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany. E-mail: Lars.Heinke@KIT.edu
bTheoretische Chemie, Technische Universität Dresden, Bergstraße 66c, König-Bau, 01062 Dresden, Germany. E-mail: Thomas.Heine@tu-dresden.de
cWilhelm-Ostwald-Institut für Physikalische und Theoretische Chemie, Universität Leipzig, Linnéstraße 2, 04103 Leipzig, Germany
First published on 10th October 2018
Abstract
Metal–organic frameworks (MOFs) have unique properties which make them perfectly suited for various adsorption and separation applications; however, their uses and efficiencies are often hindered by their limited stability. When most MOFs are exposed to water or humid air, the MOF structure, in particular at the surface, is destroyed, creating surface defects. These surface defects are surface barriers which tremendously hinder the uptake and release of guest molecules and, thus, massively decrease the performance in any application of MOFs. Here, the destruction by exposure to water vapor is investigated by using well-defined MOF films of type HKUST-1 as a model system for uptake experiments with different-sized probe molecules as well as for spectroscopic investigations, complemented by density functional theory calculations of the defect structure. In addition to the characterization of the surface defects, it is found that the pristine MOF structure can be regenerated. We show that the surface defects can be dissolved by exposure to the synthesis solvent, here ethanol, enabling fast uptake and release of guest molecules. These findings show that the storage of MOF materials in a synthesis solvent results in healing of surface defects and enables ideal performance of MOF materials.
Introduction
Metal–organic frameworks (MOFs) are nanoporous, crystalline hybrid materials which are composed of metal nodes connected by organic linker molecules.1 MOFs are very light materials with high porosities and with very large specific surface areas.2 This material class has a large variety and more than 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 different MOF structures have been published in the last 20 years.3 Due to their unique properties, their chemical functionality and flexible architecture, MOFs are intensively investigated with respect to various applications. These applications range from the storage of small molecules like methane and hydrogen4 and catalysis5 to sensor applications.6 For all these MOF applications, the interaction with guest molecules in the pores is crucial. Therefore, the uptake and release of guest molecules are vital and reduced uptake rates tremendously limit the performance during applications.
000 different MOF structures have been published in the last 20 years.3 Due to their unique properties, their chemical functionality and flexible architecture, MOFs are intensively investigated with respect to various applications. These applications range from the storage of small molecules like methane and hydrogen4 and catalysis5 to sensor applications.6 For all these MOF applications, the interaction with guest molecules in the pores is crucial. Therefore, the uptake and release of guest molecules are vital and reduced uptake rates tremendously limit the performance during applications.
Many MOF structures are destroyed under technical and even laboratory conditions. Thus, the major drawback of many MOFs is their limited stability, especially with respect to water.7 In addition, it was found that most MOF materials possess surface barriers which limit the uptake of guest molecules.8 These surface barriers presumably are the reason why for identical guest–host systems, i.e. the same guest molecules in apparently identical MOF structures, often diffusion coefficients are measured which differ by many orders of magnitude from each other. For instance, for hydrogen in MOFs of type MOF-5, diffusion coefficients ranging over four orders of magnitude, i.e. between 10−5 m2 s−1 and 10−9 m2 s−1, were determined.9 This large difference presumably is caused by a thin layer at the surface with a reduced permeability, hindering the uptake and release of guest molecules, although structural investigations, e.g. by X-ray diffraction (XRD), show no indication for changes and defects. For one of the most popular and most intensively investigated MOFs, HKUST-1, it was shown that the exposure to humid air or water vapor results in a destruction of the surface, hindering the uptake of guest molecules and, thus, limiting the performance of the MOF.10 On the other hand, it was found that the structure of the HKUST-1 powder can be composed and decomposed in ethanol and water, respectively.11 This exploits the fact that bindings in MOFs are reversible, enabling extended crystalline frameworks. For a different MOF structure, i.e. MOFs of type Co-MOF-74, it could be shown in uptake experiments with single crystals that exposure to the vapor of the synthesis solvent (methanol) results in an increase of the uptake rate by the MOF crystals which initially possess surface barriers.12
Here, the phenomenon of surface barriers and the healing of these performance-limiting defects in MOFs (Fig. 1) are thoroughly investigated. For precise quantification and a high degree of reproducibility, we use a well-defined model system, namely thin MOF films prepared in a step-by-step fashion, referred to as surface-mounted MOFs, SURMOFs.13 Moreover, this approach enables a clean synthesis, where the sample is never exposed to air, excluding any uncontrolled influence from the environment. The sample surface is destroyed by controlled exposure to water vapor. We demonstrate that the destroyed MOF surface can be dissolved resulting in uptake rates of the MOF structure as fast as those of the pristine MOF. Based on this finding, we suggest storing MOF materials in a synthesis solvent, here ethanol. Therewith, in addition to protecting MOF materials from the exposure to humid air and other harmful conditions, destroyed MOF materials can be repaired.
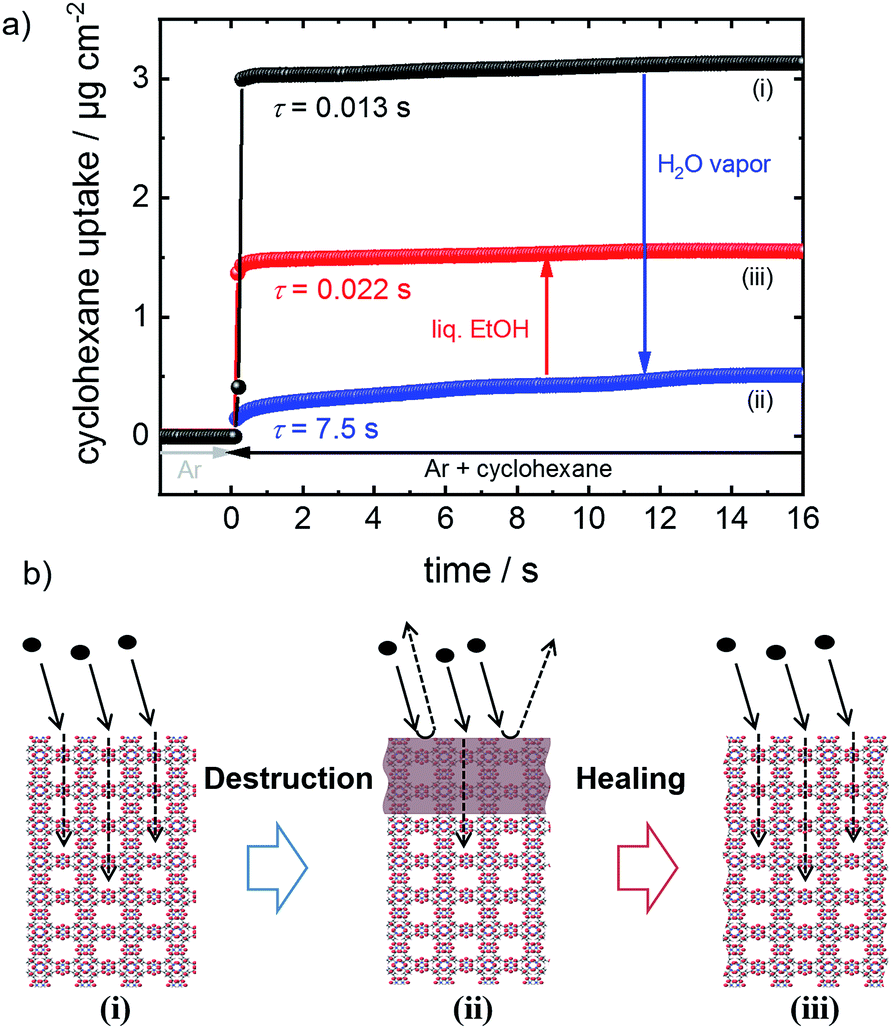 | ||
| Fig. 1 (a) Uptake of cyclohexane by the HKUST-1 SURMOF in the pristine form (black, i), after exposure to water vapor (blue, ii) and after healing in ethanol at 65 °C for 5 days (red, iii). The uptake time constants τ, determined from an exponential fit, are indicated. The uptake by the MOF film is significantly slowed down, by exposing the sample to water vapor. The initial fast uptake is recovered after dissolving the surface barriers of the MOF in liquid ethanol. (b) Model of HKUST-1. Left (i) uptake by the pristine HKUST-1 MOF without defects. Center (ii) surface barriers of the MOF hinder the molecules entering the pores. Right (iii) healing/dissolution of surface defects results in unhindered, fast uptake of the guest molecules. | ||
Experimental
The MOF films were prepared on modified substrate surfaces in a step-by-step fashion by alternatively exposing the samples to an ethanolic solution of metal nodes, i.e. 1 mM copper(II) acetate, and to an ethanolic solution of organic linker molecules, i.e. 0.2 mM trimesic acid (also referred to as BTC: benzene-1,3,5-tricarboxylic acid).13 In between, the samples are rinsed with pure ethanol. The SURMOF syntheses are performed in closed cells of a quartz crystal microbalance (QCM) of type Q-Sense E4 by subsequently pumping the metal node and linker solutions as well as pure ethanol through the QCM cells. This enables an in situ measurement of the SURMOF mass during the synthesis and determination of the SURMOF thickness, see ESI (SI1).† The SURMOF substrates are gold-coated QCM sensors functionalized by an 11-mercapto-1-undecanol (MUD) self-assembled monolayer (SAM).The QCM is also used to measure the uptake of guest molecules, where the initially pure argon gas flow is switched to an argon flow enriched with the vapor of guest molecules.10,14 The experiments were performed at a temperature of 50 °C with a gas flow rate of 100 cm3 min−1. Before each uptake experiment, the SURMOF was activated, i.e. the guest and solvent molecules were removed, in the pure argon flow at 50 °C overnight. The exposure of the SURMOF to water vapor, i.e. the destruction of the MOF structure, was performed in the QCM cell by enriching the argon flow with water vapor of a partial pressure of approximately 28 mbar for 1 h. The healing of the surface defects was carried out in the QCM cell by pumping liquid ethanol through the QCM cell with a flow rate of 20 μl min−1 at a temperature of 65 °C. All experiments were carried out without opening the QCM cell, preventing the exposure of the SURMOF to laboratory air or any environmental conditions. The uptake experiments (except for the data shown in Fig. 3) were performed with 4 samples in parallel and were repeated at least 3 times, resulting in a large database enabling solid and reliable statistics. The data shown in Fig. 3 were obtained with only one sample each. The average values with the standard deviations as error bars are shown in the figures.
Molecular vibrations in the SURMOF are investigated by infrared reflection absorption spectroscopy (IRRAS). A spectrometer of type Bruker Vertex 80 is used for ambient pressure measurements in a synthetic air atmosphere. X-ray photoelectron spectroscopy (XPS) experiments were carried out in an ultrahigh-vacuum (UHV) apparatus under a base pressure of 10−10 mbar in order to characterize the behavior of copper ions in the HKUST-1 structure. The spectroscopic measurements were performed without additional high-temperature activation of the sample in vacuum. The XPS binding energies were calibrated at the O-1s energy level of carboxylates at 532.1 eV.15 X-ray diffractograms (XRD) were recorded using a diffractometer Bruker D8 Advance with a wavelength of 0.154 nm. The samples for spectroscopic measurements were prepared ex situ in a step-by-step fashion by alternatively spraying the MOF components on gold-coated silicon substrates, functionalized by a MUD SAM.16 Immediately after synthesis, the samples were stored in pure ethanol and spectroscopic measurements were carried out subsequently. The water exposure of the samples was performed by purging a small (transport) chamber with water vapor saturated argon at room temperature.
Details of the density functional theory (DFT) calculations for a better understanding of the local (defect) structure and the electronic properties of the Cu–Cu dimers in HKUST-1 SURMOF upon treatment of the MOF films with water vapor are in the ESI (Table SI1).†
Results and discussion
Uptake investigations and dissolving of the surface barriers
The performance and the properties of the uptake by the MOF film are studied by using cyclohexane, later also n-hexane and toluene, as probe molecules. Cyclohexane was chosen because it possesses no specific interaction with the HKUST-1 framework. The loading amounts and loading rates were investigated performing QCM uptake experiments where the gas atmosphere was instantly changed from pure argon to argon enriched with the guest molecules. Typical curves of the uptake by the HKUST-1 SURMOF with a thickness of approximately 110 nm are shown in Fig. 2a (and Fig. 1a). It was found that the uptake rate and amount by the MOF film tremendously decrease upon exposure of the MOF to water vapor. The equilibrium cyclohexane uptake by the pristine MOF film is approximately 2.5 μg cm−2 and is obtained after less than 0.1 s. In contrast, the final uptake by the water-exposed MOF film is only 0.5 μg cm−2 and it takes more than 10 s to obtain the equilibrium value. By exposing the destroyed MOF film to liquid ethanol at elevated temperature for several hours and days, the uptake amount and uptake rate increase again. The liquid-ethanol exposure results in an increase of the uptake amount until roughly two thirds of the initial uptake amount by the pristine SURMOF is reached. This shows that the destruction caused by water can be repaired. It has to be stressed that prior to all uptake experiments, the samples were carefully activated, excluding any remaining water or ethanol molecules in the framework, influencing the mass transfer. Furthermore, it was found that keeping the destroyed SURMOF in argon at 65 °C for many days does not result in an increase of the uptake rate. | ||
| Fig. 2 SURMOF uptake experiments. (a) Cyclohexane uptake by the pristine HKUST-1 SURMOF (black), upon water exposure (blue) and upon healing in ethanol (red) for different durations. (b) Relative recovery, i.e. the cyclohexane uptake amount normalized to the highest degree of destruction and repair, for different ethanol immersion times. The first run is depicted in black, the second run in orange, and the third run in green. (c) Uptake rate constant k and effective diffusion coefficient Deff‡ of cyclohexane in the pristine (black), water-exposed (blue) and regenerated (red) SURMOF. | ||
Fig. 2b shows the cyclohexane uptake amount after exposing the sample to ethanol. The data for 3 consecutive runs of destruction and healing of the MOF are shown where the water exposure results in an uptake decrease to 29%, 42% and 50% of the uptake amount before water exposure. After healing in ethanol, 54%, 74% and 71% of the uptake before the water-induced-destruction was regained. The data in Fig. 2b are normalized to 0 for the uptake by the SURMOF with the maximum degree of destruction (i.e. after water vapor exposure) and to 100% for the uptake by the SURMOF upon maximum healing. The uptake amount can be described by an exponential decay function as a function of the ethanol immersion time. The time constant τ is estimated to be roughly 20 h of exposure to ethanol at 65 °C. We believe that the reason why the uptake amount after completing the dissolution of the surface barriers is smaller than the initial uptake amount is that, during the healing, the sample is exposed to an ethanol flow which is (slowly) pumped through the QCM cell. MOF components are dissolved from the destructed MOF structure and, instead of reconstructing the MOF, these MOF components are rinsed out. An estimation of the SURMOF masschanges based on the absolute values of the QCM resonance frequency was done in SI7,† where it was found that the mass decreases during the immersion in ethanol at elevated temperature. A SURMOF mass decrease of approximately 20% upon exposure in ethanol at 65 °C for 230 h was estimated. Thus, we believe that the recovery of the fast uptake is a result of dissolving the defective region which causes the surface barriers. Please note, unlike the room-temperature synthesis of HKUST-1 in solutions with high MOF-component-concentrations,17 the mass of the MOF film (slowly) decreases.
The uptake curve by the MOF film follows a mono-exponential decay function with the time constant τ. The rate constants k (= 1/τ) of the SURMOF loading, describing the mass transfer, as a function of the exposure time to liquid ethanol after the exposure to water vapor are shown in Fig. 2c. After the exposure to water vapor, resulting in the destruction of the MOF structure at the surface, the uptake rates are reduced by a factor of approximately 500, i.e. by almost three orders of magnitude. This means that the effective diffusion coefficient,‡ describing the uptake by the thin film, tremendously decreases. After only a few hours of exposure to liquid ethanol at elevated temperature, i.e. when the uptake amount is still significantly smaller than the uptake by the healed SURMOF, the uptake rate is increased to the value of the pristine SURMOF. This indicates that, after already an ethanol-immersion time which is much shorter than the time required for retaining the (final) recovered uptake amount, the water-induced surface barriers have only a minor influence on the mass transfer. Indeed, the fast healing of the surface barriers is caused by the fact that the surface barriers only have a significant impact on the mass transfer if a large majority (more than 99%) of the pore entrances are totally blocked.18 This means that already a small percentage of intact surface (>1%) is sufficient to enable large mass transfer rates. In other words, an entire defect-free surface is not required to enable fast uptake and release rates. The fast restoration of the initial uptake rate constants leads us to the speculation that the healing of the surface defects is inhomogeneous. This means that large areas of the destructed MOF surface remain defective, while a small percentage of the decomposed structure at the surface of the MOF film surface is quickly repaired.
The exponential dependence of the uptake amount on the ethanol exposure time (Fig. 2b) suggests that the retention of the defect-free MOF surface from the decomposed structure can be described as a first order reaction enabled by the exposure to liquid ethanol. This means that the increase of the uptake amount and, thus, the decrease of the destructed SURMOF structure can be considered as an activated process. Performing the dissolution of the surface barriers in ethanol at different temperatures, i.e. 45 °C, 55 °C and 65 °C, enables us to determine temperature-dependent ethanol-healing rates, see Fig. SI3.† An activation energy of 95 ± 11 kJ mol−1 is calculated from the slope of the Arrhenius plot of the rate constants, Fig. 3a. This is in agreement with the corresponding Eyring–Polanyi plot (Fig. SI3-1†). The linear temperature dependence shown in Fig. 3a enables an extrapolation of the healing-time constant. At room temperature, an ethanol-healing time constant of approximately 1 month is estimated.
 | ||
| Fig. 3 Uptake experiments at different temperatures and with different guest molecules. (a) Rate constants of uptake regeneration at temperatures of 65 °C, 55 °C and 45 °C. The linear fit in this Arrhenius plot enables the estimation of the activation energy. (b) Relative uptake of different guest molecules, i.e. cyclohexane (black), toluene (orange), and n-hexane (green), by the pristine, water-exposed and healed HKUST-1 SURMOF. | ||
The uptake experiments shown in Fig. 1–3a were performed with cyclohexane as the guest molecule. For understanding whether the surface barrier phenomenon can be generalized to other guest molecules, further QCM uptake experiments were performed with n-hexane and toluene as guest molecules in addition to cyclohexane. Although the molar masses are similar, i.e. 84, 86 and 92 g mol−1 for cyclohexane, n-hexane and toluene, respectively, the critical diameter for the diffusion in nanoporous materials is rather different. While n-hexane can enter pores with only 0.43 nm diameter, the critical pore diameters are 0.60 nm for cyclohexane and 0.585 nm for toluene.19 The uptake data of the pristine, the destroyed and the healed SURMOF samples are shown in Fig. 3b. The changes of the uptake amounts upon exposure to water and upon healing in ethanol are very similar and independent of the investigated guest molecules. Please note that if the exposure to water, i.e. the destruction of the MOF surface, would result in a reduction of the pore diameter (which also could cause the effect of surface barriers), the impact of the transport barrier would tremendously vary with the size of the guest molecules. Based on the data in Fig. 3b, we can exclude a reduction of the pore diameter as a reason for the surface barriers. Thus, we conclude that the surface barriers in HKUST-1 are caused by a total blockage of the majority of the pore entrances. This means that the destruction of the MOF structure caused by the exposure to water results in a non-porous layer at the surface. This finding is in agreement with the results from single MOF crystals of type Zn(tbip), where a total blockage of the MOF pores was found.8b,18,20
Spectroscopic and structural investigation
For reaching a better understanding of the surface barriers and the decomposed structure caused by the exposure to water, investigations of the thin MOF film by X-ray diffraction were performed. The diffractograms, Fig. 4, show only a slight change of crystallinity of the HKUST-1 SURMOF upon water exposure. The (222) reflection is decreased by 10%, which is only a very small change in contrast to the major decrease of guest molecule uptake measured in the QCM. This means that the bulk structure of the MOF film is essentially unaffected by the water exposure. Therefore, the uptake-hindering effect is not an effect of the bulk phase, so it must be caused by a small region, most likely the surface. This verifies our assumption, based on previous studies,10 that the defects are surface defects which hinder the mass transfer.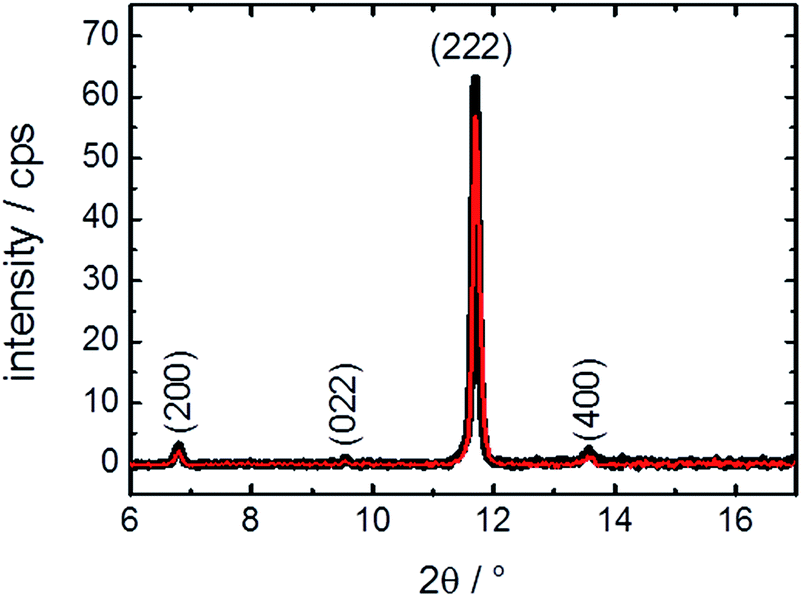 | ||
| Fig. 4 X-ray diffractograms of the pristine (black) HKUST-1 SURMOF and of the SURMOF after exposure to water vapor for 1 h. The diffractograms are measured in the out-of-plane geometry. The diffraction peaks of HKUST-1 are labelled. | ||
It might be assumed that, in addition to the (external) MOF surface which reduces the mass transfer rates, other parts of the MOF film are destroyed, also decreasing the uptake amount. However, the XRD data clearly show that the majority of the MOF structure is not affected. Thus, we propose that entire parts of the MOF film are isolated by the defects and cannot be loaded by guest molecules, decreasing the total uptake amount.
The MOF structure was spectroscopically investigated by infrared reflection absorption spectroscopy (IRRAS) and X-ray photoelectron spectroscopy (XPS). Infrared spectra of the MOF film before and after exposure to water vapor are shown in Fig. 5a. The exposure to water results in an increase of vibration bands at 1705 cm−1 and between 1500 and 1600 cm−1, which can be assigned to carbonyl groups and to the not completely (i.e. only single-or two-fold) coordinated BTC linker.7e,17b,21 This indicates that the water exposure results in a transformation of the metal carboxylate to carboxylic acid salt analogues and the bonding between the BTC linker and the Cu nodes is broken. By healing in ethanol, the vibration bands of the carbonyl groups and the not fully coordinated BTC linker decrease again, resulting in an IR spectrum, which is virtually identical to the spectra of the pristine SURMOF. This indicates that the detached carbonyl groups again are transformed to carboxylate groups, i.e. the linker–metal bond is restored and the pristine MOF structure is obtained.
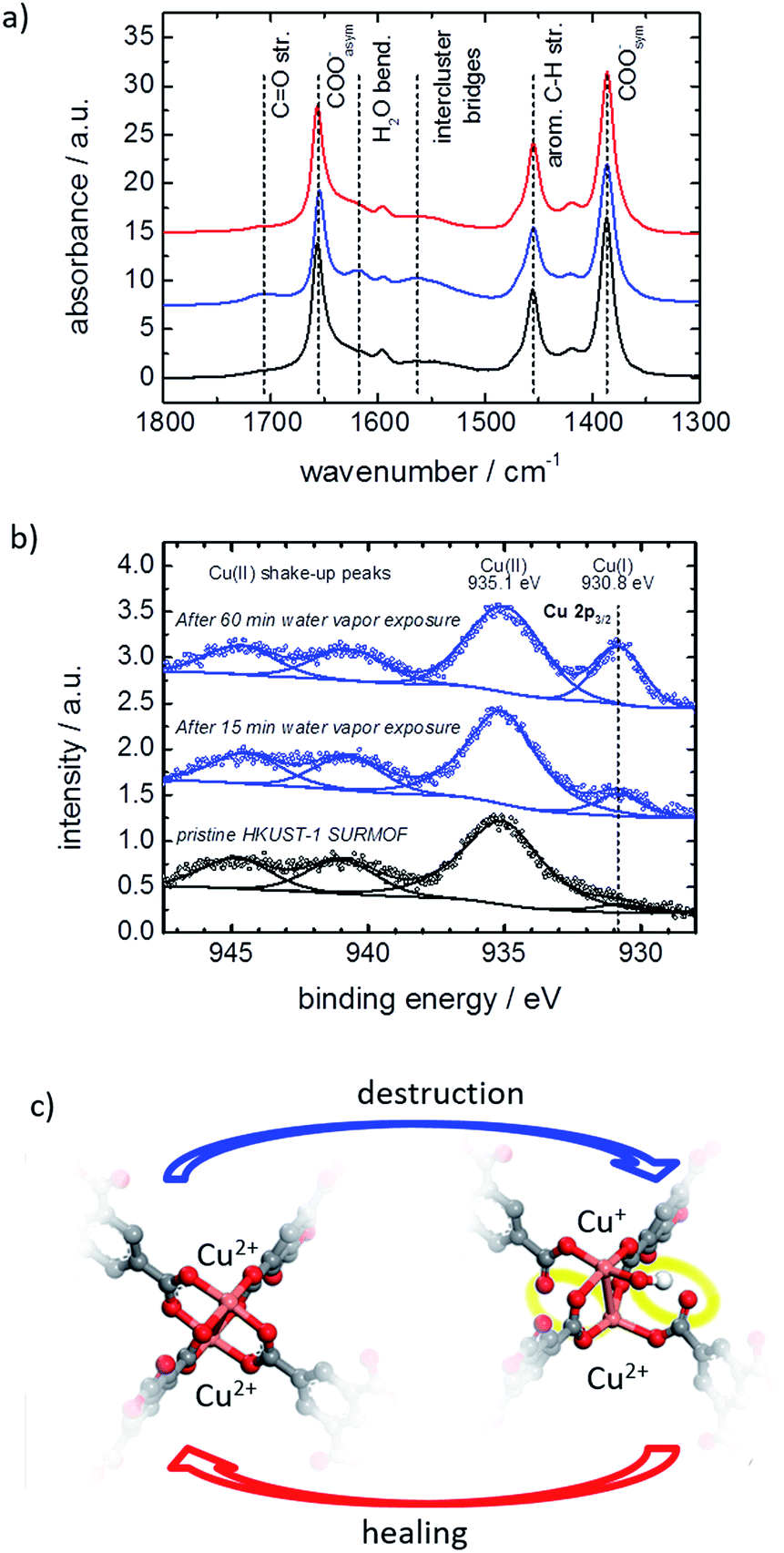 | ||
| Fig. 5 IRRAS and XPS investigations and the defect model. (a) IRRA spectra of the pristine SURMOF (black), after 1 h water exposure (blue) and after ethanol immersion at 65 °C for 3 days (red). (b) XP spectra of the pristine HKUST-1 (black) as well as of the HKUST-1 film after water vapor exposure for 15 min and 1 h (blue). The open circles depict the experimental data, and the solid lines the respective deconvoluted curves. (c) Local structure of the HKUST-1 SURMOF: upon water-vapor treatment, the defect-free fourfold symmetric Cu2+–Cu2+ dimer is transformed to a Cu2+–Cu+ defect dimer with an adsorbed OH− and only two fully coordinated BTC ligands. The broken bonds and the OH− are highlighted in yellow. The molecular model of the defect, as proposed by the DFT calculations (B3LYP-D3/ZORA scalar/TZP level), has the general formula [Cu+/Cu2+(H2BTC)4(OH)]2−. Upon healing in ethanol, the pristine Cu2+–Cu2+ metal node is restored at the surface. | ||
The water exposure also results in the generation of a vibration band at 1623 cm−1, which is assigned to water molecules trapped inside the framework.7e,22 The remaining water results from the fact that the IR measurements were performed in synthetic air right after the water exposure without previous activation. It should be noted that XPS measurements in a vacuum show no sign of water in the MOF structure, see Fig. SI4.†
The copper ions are investigated by XPS. The spectra of the pristine HKUST-1 SURMOF samples show Cu2+ states at 935.1 eV, see Fig. 5b, which is in agreement with the literature.23 After exposure to water vapor, a new peak at 930.8 eV developed. This peak may be assigned to an electronically modified Cu+ species. The Cu+:Cu2+ ratio of the pristine HKUST-1 was found to be approximately 3%. This ratio increases to 8.5% and 21% after water vapor exposure for 15 min and for 60 min, respectively.
The change of the copper oxidation state in HKUST-1 seems to be different than in the UHM-3 SURMOF, where a reduction of Cu2+ to Cu+ was observed upon thermal treatment and a Cu2+–Cu+ binding energy difference of 2.1 eV was observed.24 We conclude that the Cu-paddle-wheel structure in HKUST-1 may undergo a transformation from a four-fold bonded Cu2+–Cu2+ to a mixed configuration of an unsaturated Cu+-containing structure under loss of the characteristic antiferromagnetic metal-to-metal-coupling as described in ref. 25. This means that XPS data indicate that water acts as a reducing agent for Cu2+ and reacts under decomposition when in contact with HKUST-1. This finding based on IRRAS and XPS is in agreement with electron paramagnetic resonance (EPR) data, which also indicates that the state observed in this experiment can be attributed to the reported metastable state.26
The oxygen signal of the SURMOF after water exposure is also recorded by XPS. It shows a symmetric, Gaussian-shaped peak (Fig. SI4†), which indicates that there is no residual water. Since XPS is a very sensitive technique for the surfaces, these data precisely verify that, when the SURMOF is in a vacuum after the exposure to water, no water remains adsorbed in the pores or at the external MOF surface.
The local molecular and electronic structures of the Cu–Cu dimers in HKUST-1 SURMOF surface layers exposed to water vapor were further investigated by DFT calculations. A comprehensive set of molecular models has been considered in order to represent possible defect sites containing not only Cu2+, but also Cu+ species, as a result of the interaction of the MOF with water (see Table S1† for the full list of model structures). We have considered neutral, as well as charged types of irregular paddle-wheel structures composed of a copper dimer, four BTC linkers of which one or two are semi- or completely detached from the metal node, and one or two intact or dissociated water molecules. Modifications of the electronic states of the copper ions have been taken into account via the different spin multiplicities, M, of the molecular models. Based on the comparison of the calculated copper 2p core electronic levels in the model structures with the XPS findings for the HKUST-1 films, we conclude that the water-exposed MOF surface contains defects of the type shown in Fig. 5c (and model 10 in Table S1,† see SI5). To design such a defect site, we have assumed that upon interaction with the Cu2+–Cu2+ dimers in the HKUST-1 surface layers, water gets heterolytically dissociated. IR spectroscopy with heavy water supports the hypothesis of the dissociation of the water molecules, see SI7.† While the proton is assumed to diffuse away, the hydroxide coordinates to one of the Cu2+ ions from the dimer and causes a reduction of the other Cu2+ to a Cu+ ion. This modification of the copper electronic states is accompanied by partial destruction of the regular, four-fold paddle-wheel motif. The structure of the defect as shown in Fig. 5c was obtained after constrained geometry optimization during which all atoms except the carboxylic oxygen atoms terminating the molecular model were allowed to relax. These terminal carboxylic oxygen atoms were intentionally fixed at the positions characteristic of the fourfold, water-free, copper paddle wheels in the pristine SURMOF, in order to account, at least to some extent, for the framework rigidity. The obtained defect is characterized by a Cu2+–Cu+ distance that is substantially longer than the Cu2+–Cu2+ distance as predicted for regular, water-free, copper paddle wheels (cf. 274.2 pm vs. 251.5 pm, resp., at the B3LYP-D3/ZORA-scalar/TZP computational level). In addition, two of the four BTC ligands detach partially from the Cu2+–Cu+ node (Fig. 5c), with distances between the copper ions and the dangling carboxylic oxygens longer than 320 pm, compared to around 196 pm in the pristine paddle-wheel. Finally, the energy difference of 3.3 eV, as calculated for the copper 2p electronic states in the Cu2+–Cu+ defect dimer with an adsorbed OH−, agrees well with the experimental XPS shift of 4.3 eV, as determined for the water-exposed HKUST-1 films. We have to stress that the presented defect model (Fig. 5c) reflects all experimentally determined features, in contrast to the other investigated models in Table S1;† however, further defect structures or combination of defect structures cannot be excluded. The suggested defect structure is similar to published defect structures of HKUST-1 (ref. 25a,26,27) with the difference that, in our model, one linker molecule partially detaches from the metal node but is still part of the defect. The suggested defect model as well as other published models25a,26,27 show no features for a steric pore blockage. However, the negative charge of the defect structure seems to be an important property for the mass transfer in the pores. We believe that large counterions, like hydrated Cu2+-cation clusters and/or hydronium clusters, block the pore windows and result in the observed hindrance for the mass transfer of the guest molecules. Such an amorphous layer blocking the surface pores was previously described for MOFs of type Zn(tbip)18,28 and for MOF-74 with ethylenediamine filling the surface pores.29
Conclusions
Surface defects, which hinder the mass transfer in and the uptake by MOFs and thus hamper their performance, are thoroughly investigated by using thin MOF films of type HKUST-1. The mass transfer in the thin MOF films was investigated by QCM uptake experiments, while the structure was experimentally investigated by XRD, IRRAS and XPS supported by DFT calculations. Exposure of the MOF to water (vapor) results in surface defects, which tremendously decrease the uptake rate and amount. In this article, we show that these defects can be repaired by dissolving the surface barriers in liquid ethanol at elevated temperatures, resulting in fast uptake rates again. Furthermore, it is shown that the destruction and the healing of the MOF surface are reversible processes. The structural investigations show that these defects are caused by bond breaking between the organic linker and the metal node resulting in dangling carbonyl groups at the linker and metal centers with reduced oxidation states.Based on these findings, storage of MOF materials in the synthesis solvent, here ethanol, is recommended. This results not only in the prevention of the exposure to (humid) air and other atmospheric conditions which may destroy the MOF, but it also results in healing of the MOF structure. It can be assumed that these findings can be generalized to most water-sensitive MOF structures, in particular, to carboxylate-based MOFs, like MOF-5 and MOF-74, and Cu-paddle-wheel-based MOFs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the Funding by the German Research Foundation (SFB1176C6), the Helmholtz-Research-School “Energy-related-catalysis” and the Volkswagen Foundation. We also thank the Center for Information Services and High Performance Computing (ZIH) at TU-Dresden for generous allocation of computer time.Notes and references
-
(a) H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS
; (b) S. Kaskel, The Chemistry of Metal–Organic Frameworks: Synthesis, Characterization, and Applications, Wiley, 2016 Search PubMed
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydın and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS
- P. Z. Moghadam, A. Li, S. B. Wiggin, A. Tao, A. G. P. Maloney, P. A. Wood, S. C. Ward and D. Fairen-Jimenez, Chem. Mater., 2017, 29, 2618–2625 CrossRef CAS
- H. W. Langmi, J. W. Ren, B. North, M. Mathe and D. Bessarabov, Electrochim. Acta, 2014, 128, 368–392 CrossRef CAS
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2011, 112, 1105–1125 CrossRef
-
(a) N. C. Burtch, H. Jasuja and K. S. Walton, Chem. Rev., 2014, 114, 10575–10612 CrossRef CAS
-
(a) J. Kärger, Microporous Mesoporous Mater., 2014, 189, 126–135 CrossRef
-
(a) C. Xu, J. Yang, M. Veenstra, A. Sudik, J. J. Purewal, Y. Ming, B. J. Hardy, J. Warner, S. Maurer, U. Müeller and D. J. Siegel, Int. J. Hydrogen Energy, 2013, 38, 3268–3274 CrossRef CAS
- L. Heinke, Z. Gu and C. Wöll, Nat. Commun., 2014, 5, 4562 CrossRef CAS
- G. Majano, O. Martin, M. Hammes, S. Smeets, C. Baerlocher and J. Perez-Ramirez, Adv. Funct. Mater., 2014, 24, 3855–3865 CrossRef CAS
- C. Chmelik, A. Mundstock, P. D. C. Dietzel and J. Caro, Microporous Mesoporous Mater., 2014, 183, 117–123 CrossRef CAS
-
(a) O. Shekhah, H. Wang, S. Kowarik, F. Schreiber, M. Paulus, M. Tolan, C. Sternemann, F. Evers, D. Zacher, R. A. Fischer and C. Wöll, J. Am. Chem. Soc., 2007, 129, 15118–15119 CrossRef CAS
- W. Zhou, C. Wöll and L. Heinke, Materials, 2015, 8, 3767 CrossRef CAS
- O. Kozachuk, K. Yusenko, H. Noei, Y. M. Wang, S. Walleck, T. Glaser and R. A. Fischer, Chem. Commun., 2011, 47, 8509–8511 RSC
-
(a) H. K. Arslan, O. Shekhah, J. Wohlgemuth, M. Franzreb, R. A. Fischer and C. Wöll, Adv. Funct. Mater., 2011, 21, 4228–4231 CrossRef CAS
-
(a) D. J. Tranchemontagne, J. R. Hunt and O. M. Yaghi, Tetrahedron, 2008, 64, 8553–8557 CrossRef CAS
- L. Heinke and J. Kärger, Phys. Rev. Lett., 2011, 106, 074501 CrossRef
- M. JahandarLashaki, M. Fayaz, S. Niknaddaf and Z. Hashisho, J. Hazard. Mater., 2012, 241–242, 154–163 CrossRef CAS
- C. Chmelik, L. Heinke, P. Kortunov, J. Li, D. Olson, D. Tzoulaki, J. Weitkamp and J. Kärger, ChemPhysChem, 2009, 10, 2623–2627 CrossRef CAS
- R. J. Davey, M. Brychczynska, G. Sadiq, G. Dent and R. G. Pritchard, CrystEngComm, 2013, 15, 856–859 RSC
- N. Al-Janabi, A. Alfutimie, F. R. Siperstein and X. Fan, Front. Chem. Sci. Eng., 2016, 10, 103–107 CrossRef CAS
- M. Hanke, H. K. Arslan, S. Bauer, O. Zybaylo, C. Christophis, H. Gliemann, A. Rosenhahn and C. Wöll, Langmuir, 2012, 28, 6877–6884 CrossRef CAS
- Z. B. Wang, H. Sezen, J. X. Liu, C. W. Yang, S. E. Roggenbuck, K. Peikert, M. Froba, A. Mavrantonakis, B. Supronowicz, T. Heine, H. Gliemann and C. Wöll, Microporous Mesoporous Mater., 2015, 207, 53–60 CrossRef CAS
-
(a) P. St Petkov, G. N. Vayssilov, J. X. Liu, O. Shekhah, Y. M. Wang, C. Wöll and T. Heine, ChemPhysChem, 2012, 13, 2025–2029 CrossRef CAS
- M. Todaro, G. Buscarino, L. Sciortino, A. Alessi, F. Messina, M. Taddei, M. Ranocchiari, M. Cannas and F. M. Gelardi, J. Phys. Chem. C, 2016, 120, 12879–12889 CrossRef CAS
-
(a) M. Todaro, L. Sciortino, F. M. Gelardi and G. Buscarino, J. Phys. Chem. C, 2017, 121, 24853–24860 CrossRef CAS
- L. Heinke, D. Tzoulaki, C. Chmelik, F. Hibbe, J. M. van Baten, H. Lim, J. Li, R. Krishna and J. Kärger, Phys. Rev. Lett., 2009, 102, 065901 CrossRef
- K. Tan, S. Zuluaga, E. Fuentes, E. C. Mattson, J.-F. Veyan, H. Wang, J. Li, T. Thonhauser and Y. J. Chabal, Nat. Commun., 2016, 7, 13871 CrossRef CAS
-
J. Kärger, D. M. Ruthven and D. N. Theodorou, Diffusion in Nanoporous Materials, Wiley-VCH, 2012, p. 902 Search PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, uptake and XPS data. See DOI: 10.1039/c8sc03735c |
| ‡ The effective diffusion coefficient Deff of a homogenous film with an intracrystalline diffusion coefficient D and a thin layer on-top with a reduced (surface) permeability α can be described by 1/τ = l2/3Deff = l2/3D + l/α, where τ denotes the uptake time constant and l the thickness of the thin film (see eqn (2.35) and Table 13.1 in ref. 30). |
| This journal is © The Royal Society of Chemistry 2019 |