
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanding reversible chalcogenide binding: supramolecular receptors for the hydroselenide (HSe−) anion†
Hazel A.
Fargher‡
 ,
Nathanael
Lau‡
,
Lev N.
Zakharov
,
Michael M.
Haley
*,
Darren W.
Johnson
* and
Michael D.
Pluth
*
,
Nathanael
Lau‡
,
Lev N.
Zakharov
,
Michael M.
Haley
*,
Darren W.
Johnson
* and
Michael D.
Pluth
*
Department of Chemistry & Biochemistry, Materials Science Institute, Institute of Molecular Biology, University of Oregon, Eugene, OR 97403-1253, USA. E-mail: haley@uoregon.edu; dwj@uoregon.edu; pluth@uoregon.edu
First published on 19th November 2018
Abstract
Synthetic supramolecular receptors have been widely used to study reversible solution binding of anions; however, few systems target highly-reactive species. In particular, the hydrochalcogenide anions hydrosulfide (HS−) and hydroselenide (HSe−) have been largely overlooked despite their critical roles in biological systems. Herein we present the first example of reversible HSe− binding in two distinct synthetic supramolecular receptors, using hydrogen bonds from N–H and aromatic C–H moieties. The arylethynyl bisurea scaffold 1tBu achieved a binding affinity of 460 ± 50 M−1 for HSe− in 10% DMSO-d6/CD3CN, whereas the tripodal-based receptor 2CF3 achieved a binding affinity of 290 ± 50 M−1 in CD3CN. Association constants were also measured for HS−, Cl−, and Br−, and both receptors favored binding of smaller, more basic anions. These studies contribute to a better understanding of chalcogenide hydrogen bonding and provide insights into further development of probes for the reversible binding, and potential quantification, of HSe− and HS−.
Introduction
Synthetic supramolecular receptors have been used with great success for investigating the solution binding of biologically- and environmentally-relevant anions.1–5 By using reversible, mostly non-covalent interactions such as hydrogen bonding, electrostatic interactions, and anion–π interactions, a diverse palette of anions can be bound ranging from relatively inert anions such as halides and oxoanions6–10 to highly reactive anions.11–16 Although targeting the latter poses many challenges, reversible binding in supramolecular hosts can be used to stabilize high-energy anions through non-covalent interactions in a manner reminiscent of certain active sites in proteins.17 Despite this potential, examples of receptors targeting highly-reactive anions remain rare.11–16 In particular, the hydrochalcogenide anions hydroselenide (HSe−) and hydrosulfide (HS−) have been largely overlooked despite their considerable environmental and biological significance. These anions are weak bases that exist in equilibrium with their gaseous conjugate acids, hydrogen selenide (H2Se, pKa = 3.74) and hydrogen sulfide (H2S, pKa = 7.00).18 The anionic species dominate at physiological pH, as H2Se exists almost entirely as HSe− and HS− is favored over H2S by a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.19–21
:1 ratio.19–21
Although HSe− and HS−/H2S are highly toxic at elevated levels,19,22,23 both are essential to life at low concentrations and are produced endogenously.18–20 For example, H2S has been classified as the third gasotransmitter alongside carbon monoxide (CO) and nitric oxide (NO) and plays regulatory roles in the cardiovascular, immune, and gastrointestinal systems, among others.19,24–27 Similarly, HSe− is the common but highly-reactive intermediate generated in the metabolism of dietary selenium (Fig. 1),18,20 and it is required for the synthesis of the essential 21st amino acid selenocysteine (Se-Cys).28,29 Se-Cys is then incorporated into selenoproteins, such as thioredoxin reductases and glutathione peroxidases18,20 that play important roles in redox biochemistry.30,31 However, the high reactivity of HSe− toward both electrophiles and oxygen makes it difficult to observe directly in biological systems or to target through the design of selective synthetic receptors.20,32
 | ||
| Fig. 1 Summary of selenium metabolism in the human body.20 | ||
Understanding the reversible binding requirements for hydrochalcogenides could provide valuable insights into possible receptor motifs in biological environments. However, we are not aware of any reports showing HSe− as a viable target for molecular recognition by anion receptors. Similarly, few examples of reversible HS− binding exist,12–14 the first of which were reported by our groups using two distinct families of modular receptor scaffolds (Fig. 2). The initial report was based on a rigid arylethynyl bisurea receptor (1H)12 and the second on a flexible tripodal arylamide unit (2H),13 both of which bound HS− through N–H⋯S and aryl C–H⋯S hydrogen bonds. Building from these early insights into HS− binding, we investigated whether these receptors could also bind and stabilize the substantially more reactive HSe− anion. This was not a trivial descent down the periodic table; although sulfur and selenium share similar chemical and physical properties, HSe− is over three orders of magnitude more acidic and both a more potent nucleophile and reducing agent than HS−.18 In addition, selenium is larger and more diffuse than sulfur (Se2−: 1.84 Å; S2−: 1.70 Å),33 making non-covalent and reversible binding more difficult.34,35
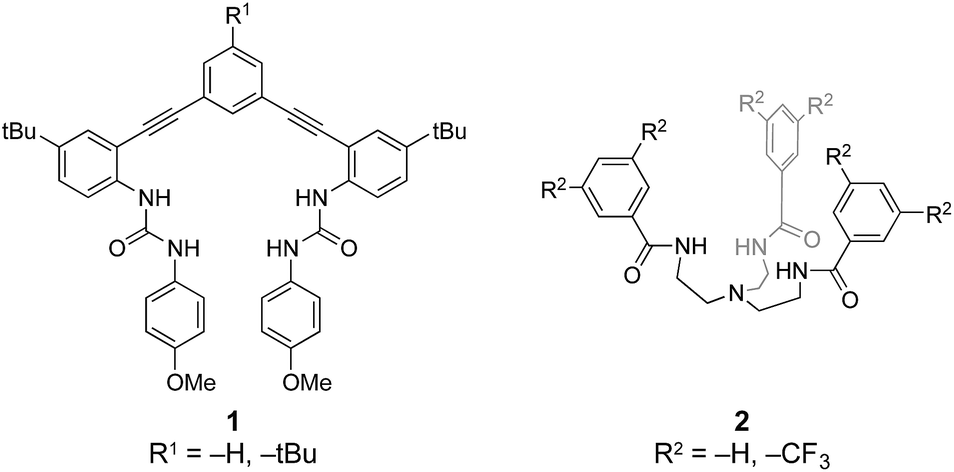 | ||
| Fig. 2 The two families of receptors used for binding HS− and HSe−.12,13,34 | ||
Herein we report the first examples of using supramolecular receptors to reversibly bind the HSe− anion, as clearly demonstrated by 1H nuclear magnetic resonance (NMR) titration studies and X-ray crystallography. The binding affinities of the receptors with other related anions (HS−, Cl−, and Br−) were also measured to determine the importance of factors such as anion size and basicity in binding. Our analysis revealed that our receptors favor smaller and more basic anions; thus, the greatest affinities observed were for HS−. Ultimately, these studies provide a starting point for designing receptors capable of selective binding to HSe−, which may provide future insights into the role of hydrochalogenide anions in biology.
Results and discussion
Synthesis of tetrabutylammonium hydroselenide (NBu4SeH)
To investigate HSe− binding to 1tBu and 2CF3, which are both insoluble in water, we prepared NBu4SeH by reducing elemental Se with NBu4BH4 in anhydrous CH3CN (Fig. 3a).36 The crude NBu4SeH oil was repeatedly washed with tetrahydrofuran (THF) to precipitate pure NBu4SeH as a white powder. Single crystals of NBu4SeH suitable for X-ray diffraction were obtained by layering a CH3CN solution of NBu4SeH with diethyl ether (Et2O) (Fig. 3b).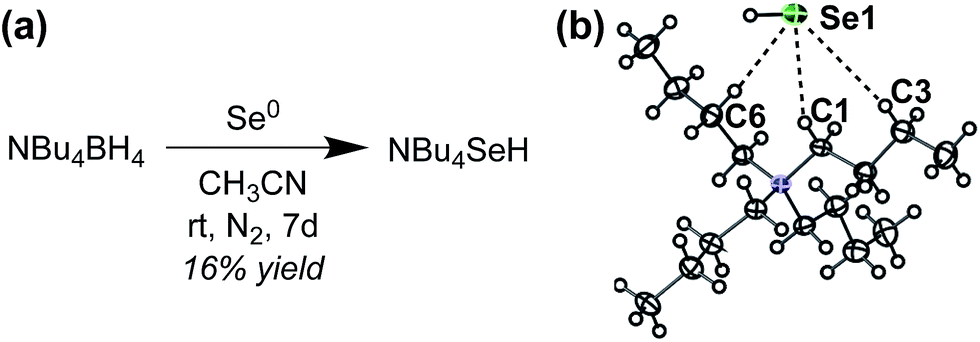 | ||
| Fig. 3 (a) Preparation of NBu4SeH. (b) Thermal ellipsoid diagram (at 50% probability) depicting the molecular structure of NBu4SeH. | ||
Much like the related structure of NBu4SH,37 short contacts (3.954–4.248 Å) between the Se atom and C1, C3, and C6 of the NBu4+ counterion are indicative of weak hydrogen bonding between the aliphatic C–H bonds of the counterion to the chalcogenide. The HSe− proton was located in the solid-state structure and found to be pointed away from the NBu4+ counterion. In addition, the 1H NMR spectrum of NBu4SeH showed the HSe− resonance at −6.61 ppm in CD3CN. The greater upfield shift of HSe− compared to that of HS− (−3.85 ppm)37 is consistent with the greater electron density around Se2− relative to S2−. We note that the salt is extremely sensitive to O2, and colorless solutions of NBu4SeH turn dark green upon exposure to the atmosphere.
Binding experiments of 1tBu and 2CF3 with HSe−
Equipped with an organic soluble source of HSe−, we next used 1H NMR spectroscopy to investigate whether 1tBu and 2CF3 could bind HSe− (Fig. 4). Solutions of each host (1.0–2.0 mM) were titrated with NBu4SeH in either anhydrous 10% DMSO-d6/CD3CN (for 1tBu) or anhydrous CD3CN (for 2CF3), due to solubility differences between the hosts. We observed a significant downfield shift in the urea N–Hb/c and aromatic C–Ha proton resonances in 1tBu and in the amide N–Ha and aromatic C–Hb proton resonances in 2CF3. Both of these results indicated that these protons are involved in binding HSe−, and matched the recognition units that were previously observed to be involved in the binding of HS− with 1H and 2H.12,13 Association constants (Ka) were determined by fitting the changes in the chemical shifts of these hydrogen bond donating moieties to a 1:1 host:guest model using Thordarson's method (Table 1, vide infra).38,39
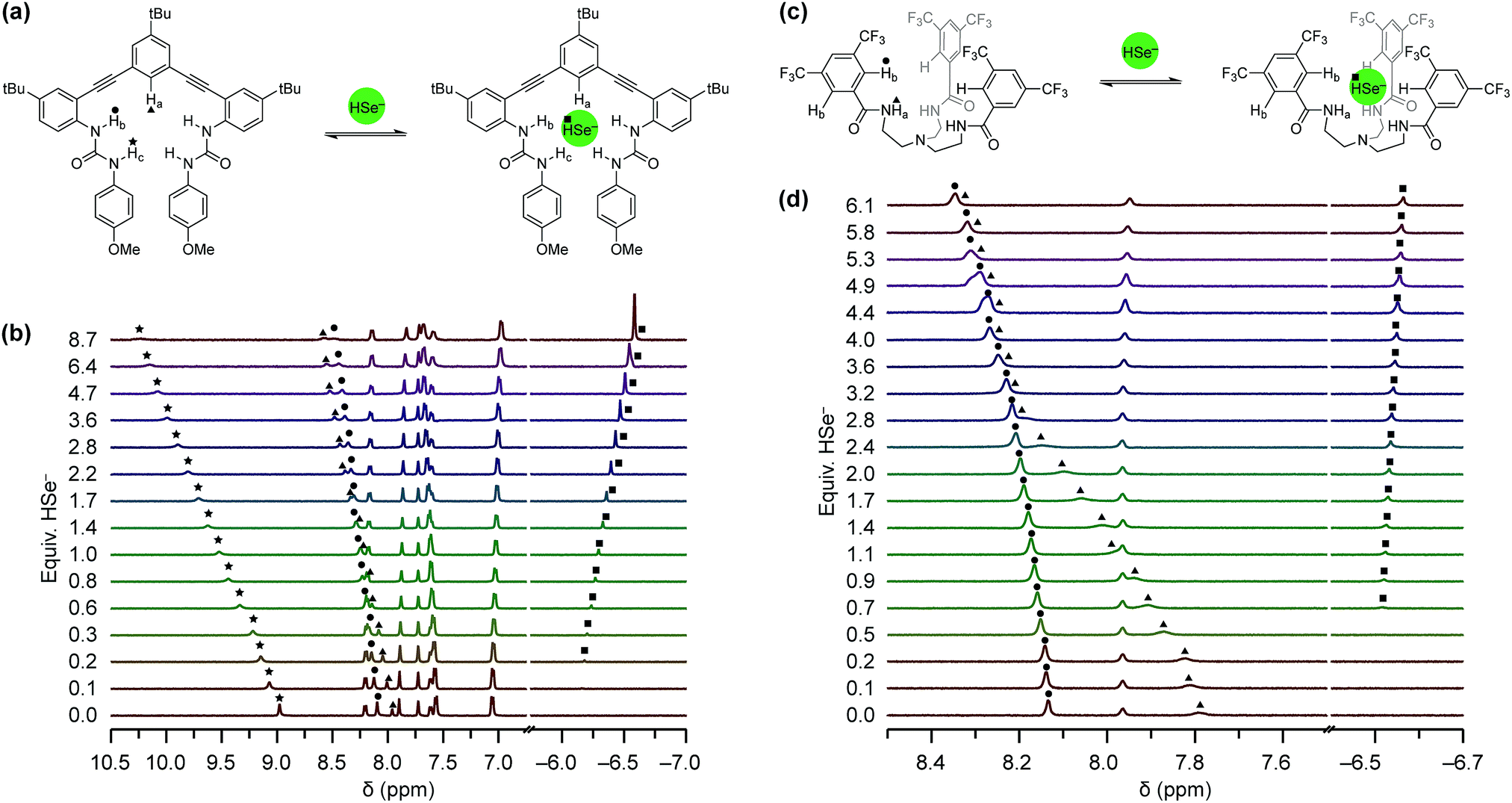 | ||
| Fig. 4 (a) Representation of the host guest equilibrium between 1tBu and HSe−. (b) 1H NMR titration of 1.6 mM 1tBu with NBu4SeH in 10% DMSO-d6 in CD3CN. (c) Representation of the host guest equilibrium between 2CF3 and HSe−. (d) 1H NMR titration of 2.0 mM 2CF3 with NBu4SeH in CD3CN. | ||
| Host | Solvent | HSe− | Br− | HS− | Cl− | ||||
|---|---|---|---|---|---|---|---|---|---|
| K a (M−1) | ΔG (kcal mol−1) | K a (M−1) | ΔG (kcal mol−1) | K a (M−1) | ΔG (kcal mol−1) | K a (M−1) | ΔG (kcal mol−1) | ||
| a The minimum error is assumed to be 10% in cases where the standard deviation is less than 10%. | |||||||||
| 1tBu | 10% DMSO-d6/CD3CN | 460 ± 50 | −3.63 ± 0.06 | 110 ± 20 | −2.79 ± 0.09 | 3600 ± 500 | −4.85 ± 0.09 | 1700 ± 200 | −4.41 ± 0.06 |
| 2CF3 | CD3CN | 290 ± 50 | −3.35 ± 0.10 | 67 ± 7 | −2.49 ± 0.06 | 840 ± 80 | −3.93 ± 0.06 | 430 ± 50 | −3.59 ± 0.07 |
To ensure that the observed binding was reversible and not due to reaction with HSe− as a nucleophile, we next looked for evidence of covalent modification of our receptors. In particular, 1tBu has several electrophilic sites, such as the urea carbonyl and alkyne moieties, that could potentially undergo nucleophilic attack by HSe−. Although no evidence of receptor modification was observed in titrations of 1H with HS−,12 treatment of 1tBu with 20 equiv. HSe− resulted in the appearance of new aromatic signals after approximately 30 min (ESI, Fig. S3†).
To determine whether 1tBu was covalently modified by HSe− over the course of the titration, 6 equiv. HSe− were added to a 2 mM solution of 1tBu in 10% DMSO-d6/CD3CN (ESI, Fig. S5†). After 1 h there was little evidence of new aromatic signals; however, after 3 h new peaks appeared in the spectra. Addition of 20 equiv. of zinc acetate (Zn(OAc)2) to the mixture removed HSe− as ZnSe. The resulting 1H NMR spectrum showed that the receptor signals return to the same shifts as unmodified 1tBu along with the presence of smaller decomposition signals, demonstrating that the binding process of HSe− is reversible within 1 h and over the timescale of the titration experiment.
To further investigate the minor decomposition products of 1tBu with HSe−, we used negative mode mass spectrometry (MS) to look for Se-containing species. We observed peaks consistent with fragments containing a molecule of HSe− added across one alkyne bond (ESI, Fig. S4†), which corroborates the observed desymmetrization of the aromatic peaks in the decomposition products in the 1H NMR spectrum of 1tBu. Furthermore, the isotope patterns and mass accuracy of these peaks unambiguously show that these species incorporate HSe−. These results underscore the challenges in binding such a highly reactive species and confirm that careful receptor choice and design (e.g., bulky t-Bu group to protect 1tBu from nucleophilic aromatic substitution) is needed to accomplish this task.
The simpler tripodal receptor proved to be more resistant to attack by HSe−, since we have not observed any evidence of modification of 2CF3 by HSe−, even though the electrophilicity of the amide carbonyl moieties should be enhanced due to the presence of the meta CF3 groups. Coupled with the resistance of 1tBu to HSe−, this result demonstrates how the presence of relatively weak, non-covalent interactions can stabilize a normally reactive species. As with 1tBu, HSe− binding was also shown to be reversible by conducting a similar Zn(OAc)2 extrusion experiment (ESI, Fig. S5†). After 2 equiv. HSe− were added to 2CF3, the addition of 12 equiv. of Zn(OAc)2 returned a 1H NMR spectrum identical to that of pure 2CF3. The ability of these two distinct receptor classes to reversibly bind HSe− demonstrates the generality of binding of this previously uninvestigated anion, despite the highly reactive and reducing nature of HSe−.
Binding experiments of 1tBu and 2CF3 with other anions
To better understand the factors influencing HSe− binding, we also measured the binding affinities of 1tBu and 2CF3 towards the related anions HS−, Cl−, and Br− (Table 1). Several notable trends emerged from these studies. For example, 1tBu maintains a higher binding affinity for HSe− than 2CF3, even in a more competitive solvent system (10% DMSO-d6 in CD3CN vs. neat CD3CN). This difference in binding affinity between the two receptors holds true for all of the other anions investigated and is consistent with our previous studies,12,13 and may reflect the increased number of N–H H-bond donors in 1tBu compared to 2CF3. Furthermore, this result underscores the importance of preorganization and directionality in hydrogen bonding in supramolecular systems, as the rigid ethynyl backbone of 1tBu offers more directed hydrogen bonds than the more flexible aliphatic backbone of 2CF3. Supporting this hypothesis, previous work on 1tBu and derivatives have shown that the central aromatic C–H hydrogen bond is unusually strong, contributing more than 1 kcal mol−1 in anion binding energy.34 In contrast, although receptor 2CF3 should donate three hydrogen bonds between three ortho aromatic C–H hydrogen atoms to a guest molecule, 1H NMR spectroscopy suggest that these interactions are relatively weak, as they are not strong enough to prevent free rotation of the aromatic rings since the ortho protons are not resolved.Interestingly, both receptors demonstrated a clear preference for binding the hydrochalcogenide anions over the halide anions in the same row. By binding affinities, 1tBu showed a two-fold preference for HS− over Cl− and a four-fold preference for HSe− over Br−, despite the nearly identical ionic radii of anions within the same periodic row (Table 1). The protonation state of each anion is unlikely to explain the preferential binding towards hydrochalcogenide anions in 1tBu because this receptor contains no hydrogen bond accepting motifs in the binding pocket. The distinguishing factor may instead be basicity, as the chalcogenides are far better bases than the halides (Table 2) and should thus form stronger hydrogen bonds with the receptors. In contrast, the ionic size of the different anions appears to be a dominant factor in determining binding affinity in 1tBu and 2CF3. In both cases, the smaller row 3 anions (HS− and Cl−) exhibit an order of magnitude stronger binding than those of the larger row 4 anions (HSe− and Br−), despite the higher basicity of HSe− over Cl−. Alternatively, because all the anions have the same charge, the row 3 anions have a higher surface charge density, which may result in greater electrostatic interactions between the anion and receptor, thus contributing to the stronger binding.
We further investigated the impact of anion size on receptor geometry in the solid-state. Single crystals of [NBu4][1tBu(SeH)] suitable for X-ray diffraction were obtained by layering an equimolar THF mixture of 1tBu and NBu4SeH under Et2O in a glovebox (Fig. 5). We compared the metrical parameters of [1tBu(SeH)]− to those of the previously reported [1H(SH)]− (ref. 12) and [1H(Cl)]− (ref. 41) to determine the effect of guest size on 1R receptors. The HSe− guest is bound by 1tBu in the pocket created by one aromatic proton and four urea protons. The C⋯Se and N⋯Se distances suggest that the strongest hydrogen bonds are formed by the distal urea protons (N2 and N4, (N⋯Se)ave = 3.385 Å), followed by the central aryl proton (C1⋯Se = 3.769 Å) then the proximal urea protons (N1 and N3, (N⋯Se)ave = 3.892 Å). These results suggested that the Se atom did not fit well inside the binding pocket of 1tBu, since the more constrained proximal urea protons had weaker interactions to the anion than the more flexible distal urea protons. Additionally, none of the C⋯H⋯Se or N⋯H⋯Se angles formed were in the preferred linear geometry (Table 3). Although similar behavior was observed for [1H(SH)]− (ref. 12) and [1H(Cl)]−,41 the larger HSe− guest distorted the binding pocket more than the smaller HS− or Cl− guests. When distances between the distal urea nitrogen atoms to the plane formed by the central aryl ring were investigated, [1tBu(SeH)]− (2.273 Å) exhibited much longer average distance than [1H(SH)]− (2.109 Å) or [1H(Cl)]− (2.029 Å). In tandem, these results suggest that the larger HSe− guest distorts the binding cavity more than related row 3 anions, perhaps explaining the poorer binding affinity for HSe− in these systems.42,43
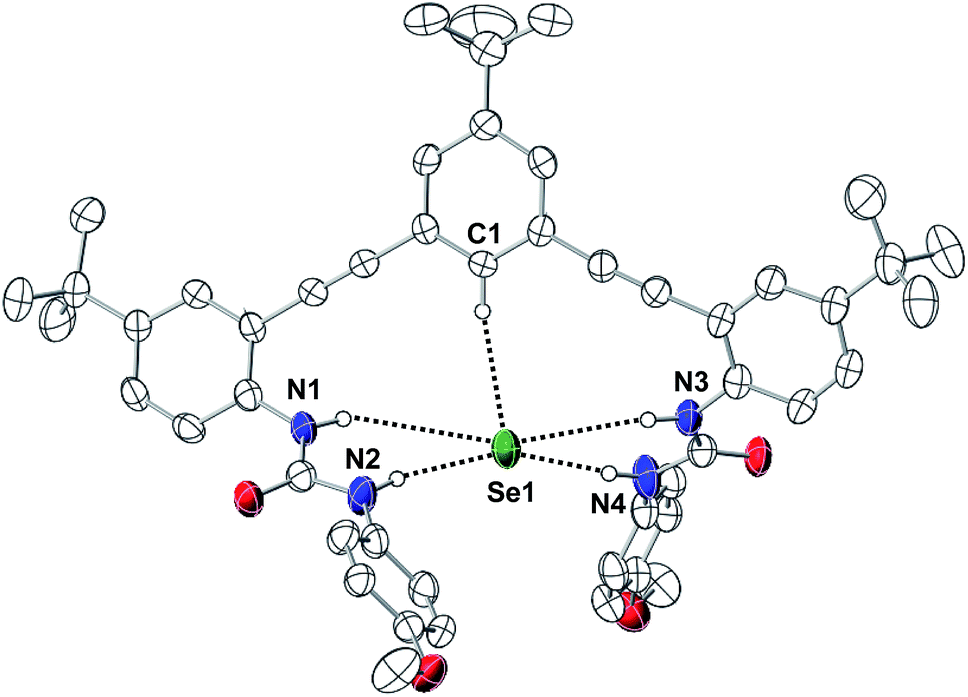 | ||
| Fig. 5 Thermal ellipsoid diagram (at 50% probability) depicting the molecular structure of [1tBu(SeH)]−. Hydrogen atoms not interacting with the bound HSe− are omitted for clarity. | ||
| Atomic distance (Å) | Bond angle (°) | |
|---|---|---|
| C1(H)⋯Se1 | 3.769 | 168.4 |
| N1(H)⋯Se1 | 4.073 | 144.2 |
| N2(H)⋯Se1 | 3.373 | 173.2 |
| N3(H)⋯Se1 | 3.710 | |
| N4(H)⋯Se1 | 3.397 | 172.7 |
Conclusions
In this study we have presented the first example of reversible HSe− binding with two separate supramolecular receptors. Both receptors interact with HSe− through N–H and aryl C–H hydrogen bonds and the ability of two structurally distinct receptors to bind HSe− demonstrates the generality of this type of reversible supramolecular interaction. Additional studies with the related anions HS−, Cl−, and Br− suggested basicity and anion size impact the binding affinities of the receptors in polar, aprotic organic solvents. Both receptors show the greatest binding affinity for the smallest and most basic anion, HS−. The dramatic decrease in binding affinity for larger anions suggests that smaller anions fit better in these systems, giving our receptors a preference for HS− over HSe−. The size of the anion appears to impact binding more significantly than basicity, as the binding affinity of the relatively basic anion HSe− is surprisingly almost four times less than that of the substantially less basic but smaller anion Cl−. The predictability of these trends suggests clear enthalpic driving forces behind binding preference, but the role of entropy cannot be discounted. The analysis of entropy versus enthalpy in our hosts will be followed up in a future report.These results, coupled with the development of the first synthesis for NBu4SeH, provide a solid platform for development of future supramolecular HSe− receptors. Reversible receptors for HSe− certainly require scaffolds resistant to nucleophilic attack and should be able to bind selenium through suitable hydrogen bond donors such as urea N–H, amide N–H, or aromatic C–H groups, likely among many others. Furthermore, receptors more selective for HSe− may require binding cavities larger than either 1tBu or 2CF3 possess. Such developments will ultimately provide better tools toward understanding the supramolecular chemistry of the biologically- and environmentally-relevant hydrochalcogenide anions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation (CHE-1454747 to M. D. P.), the Dreyfus Foundation (M. D. P), and NIH (R01-GM087398 to D. W. J./M. M. H.). This work was also supported by the Bradshaw and Holzapfel Research Professorship in Transformational Science and Mathematics to DWJ. Mass spectrometry capibilities in the CAMCOR facility are supported by the NSF (CHE-1625529).Notes and references
- A. E. Hargrove, S. Nieto, T. Zhang, J. L. Sessler and E. V. Anslyn, Chem. Rev., 2011, 111, 6603–6782 CrossRef CAS PubMed.
- P. A. Gale and C. Caltagirone, Chem. Soc. Rev., 2015, 44, 4212–4227 RSC.
- C. L. Gibb, E. E. Oertling, S. Velaga and B. C. Gibb, J. Phys. Chem. B, 2015, 119, 5624–5638 CrossRef CAS PubMed.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486–516 CrossRef CAS PubMed.
- P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907–9972 CrossRef CAS PubMed.
- A. Pramanik, D. R. Powell, B. M. Wong and M. A. Hossain, Inorg. Chem., 2012, 51, 4274–4284 CrossRef CAS PubMed.
- P. Blondeau, M. Segura, R. Perez-Fernandez and J. de Mendoza, Chem. Soc. Rev., 2007, 36, 198–210 RSC.
- P. Sabater, F. Zapata, A. Caballero, I. Fernandez, C. Ramirez de Arellano and P. Molina, J. Org. Chem., 2016, 81, 3790–3798 CrossRef CAS PubMed.
- J. Y. C. Lim and P. D. Beer, New J. Chem., 2018, 42, 10472–10475 RSC.
- L. Gonzalez, F. Zapata, A. Caballero, P. Molina, C. Ramirez de Arellano, I. Alkorta and J. Elguero, Chem.–Eur. J., 2016, 22, 7533–7544 CrossRef CAS PubMed.
- N. Lopez, D. J. Graham, R. McGuire Jr, G. E. Alliger, Y. Shao-Horn, C. C. Cummins and D. G. Nocera, Science, 2012, 335, 450–453 CrossRef CAS PubMed.
- M. D. Hartle, R. J. Hansen, B. W. Tresca, S. S. Prakel, L. N. Zakharov, M. M. Haley, M. D. Pluth and D. W. Johnson, Angew. Chem., Int. Ed., 2016, 55, 11480–11484 CrossRef CAS PubMed.
- N. Lau, L. N. Zakharov and M. D. Pluth, Chem. Commun., 2018, 54, 2337–2340 RSC.
- J. Vazquez and V. Sindelar, Chem. Commun., 2018, 54, 5859–5862 RSC.
- S. O. Kang, D. Powell, V. W. Day and K. Bowman-James, Angew. Chem., Int. Ed., 2006, 45, 1921–1925 CrossRef CAS PubMed.
- S. O. Kang, V. W. Day and K. Bowman-James, Inorg. Chem., 2010, 49, 8629–8636 CrossRef CAS PubMed.
- R. H. Holm and E. I. Solomon, Chem. Rev., 2004, 104, 347–348 CrossRef CAS PubMed.
- H. J. Reich and R. J. Hondal, ACS Chem. Biol., 2016, 11, 821–841 CrossRef CAS PubMed.
- R. Wang, Physiol. Rev., 2012, 92, 791–896 CrossRef CAS PubMed.
- C. M. Weekley and H. H. Harris, Chem. Soc. Rev., 2013, 42, 8870–8894 RSC.
- M. D. Hartle and M. D. Pluth, Chem. Soc. Rev., 2016, 45, 6108–6117 RSC.
- I. M. Arnold, R. M. Dufresne, B. C. Alleyne and P. J. Stuart, J. Occup. Med., 1985, 27, 373–376 CrossRef CAS.
- N. R. Council, Acute Exposure Guideline Levels for Selected Airborne Chemicals, The National Academies Press, Washington, DC, 2014, vol. 16, ch. Hydrogen Selenide, p. 398, DOI:10.17226/18707.
- R. Wang, FASEB J., 2002, 16, 1792–1798 CrossRef CAS PubMed.
- R. C. Zanardo, V. Brancaleone, E. Distrutti, S. Fiorucci, G. Cirino and J. L. Wallace, FASEB J., 2006, 20, 2118–2120 CrossRef CAS PubMed.
- E. Distrutti, L. Sediari, A. Mencarelli, B. Renga, S. Orlandi, G. Russo, G. Caliendo, V. Santagada, G. Cirino, J. L. Wallace and S. Fiorucci, J. Pharmacol. Exp. Ther., 2006, 319, 447–458 CrossRef CAS PubMed.
- Z. Zhang, H. Huang, P. Liu, C. Tang and J. Wang, Can. J. Physiol. Pharmacol., 2007, 85, 1248–1253 CrossRef CAS PubMed.
- Z. Veres, L. Tsai, T. D. Scholz, M. Politino, R. S. Balaban and T. C. Stadtman, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 2975–2979 CrossRef CAS.
- R. S. Glass, W. P. Singh, W. Jung, Z. Veres, T. D. Scholz and T. C. Stadtman, Biochemistry, 1993, 32, 12555–12559 CrossRef CAS PubMed.
- R. K. Shrimali, R. D. Irons, B. A. Carlson, Y. Sano, V. N. Gladyshev, J. M. Park and D. L. Hatfield, J. Biol. Chem., 2008, 283, 20181–20185 CrossRef CAS PubMed.
- D. L. Hatfield, P. A. Tsuji, B. A. Carlson and V. N. Gladyshev, Trends Biochem. Sci., 2014, 39, 112–120 CrossRef CAS PubMed.
- C. M. Weekley, J. B. Aitken, S. Vogt, L. A. Finney, D. J. Paterson, M. D. de Jonge, D. L. Howard, P. K. Witting, I. F. Musgrave and H. H. Harris, J. Am. Chem. Soc., 2011, 133, 18272–18279 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- B. W. Tresca, R. J. Hansen, C. V. Chau, B. P. Hay, L. N. Zakharov, M. M. Haley and D. W. Johnson, J. Am. Chem. Soc., 2015, 137, 14959–14967 CrossRef CAS PubMed.
- S. K. Dey and G. Das, Chem. Commun., 2011, 47, 4983–4985 RSC.
- R. J. Batchelor, F. W. B. Einstein, I. D. Gay, C. H. W. Jones and R. D. Sharma, Inorg. Chem., 1993, 32, 4378–4383 CrossRef CAS.
- M. D. Hartle, D. J. Meininger, L. N. Zakharov, Z. J. Tonzetich and M. D. Pluth, Dalton Trans., 2015, 44, 19782–19785 RSC.
- P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC.
- D. Brynn Hibbert and P. Thordarson, Chem. Commun., 2016, 52, 12792–12805 RSC.
- F. G. Bordwell, Acc. Chem. Res., 2002, 21, 456–463 CrossRef.
- B. W. Tresca, L. N. Zakharov, C. N. Carroll, D. W. Johnson and M. M. Haley, Chem. Commun., 2013, 49, 7240–7242 RSC.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- A. Shahi and E. Arunan, J. Chem. Sci., 2016, 128, 1571–1577 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic details, NMR spectra, representative titrations. CCDC 1846890–1846892. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc03968b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2019 |