
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA cheap metal for a challenging task: nickel-catalyzed highly diastereo- and enantioselective hydrogenation of tetrasubstituted fluorinated enamides†
Yu-Qing
Guan
a,
Zhengyu
Han
a,
Xiuxiu
Li
a,
Cai
You
a,
Xuefeng
Tan
b,
Hui
Lv
 *acd and
Xumu
Zhang
b
*acd and
Xumu
Zhang
b
aKey Laboratory of Biomedical Polymers of Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, P. R. China. E-mail: huilv@whu.edu.cn
bDepartment of Chemistry, Shenzhen Grubbs Institute, Southern University of Science and Technology, Shenzhen, Guangdong 518055, P. R. China
cEngineering Research Center of Organosilicon Compounds & Materials, Ministry of Education, College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, 430072, China
dBeijing National Laboratory for Molecular Sciences, CAS Key Laboratory of Molecular Recognition and Function, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China
First published on 16th October 2018
Abstract
Nickel-catalyzed asymmetric hydrogenation of challenging tetrasubstituted fluorinated enamides has been achieved, affording chiral α-fluoro-β-amino esters in high yields with excellent diastereo- and enantioselectivities (up to 98% yield, >99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, up to >99% ee). Deuterium-labeling experiments and control experiments were conducted to probe the mechanism, and the results indicated that the acidity of the solvent plays a critical role in the control of diastereoselectivity by trapping the adduct of nickel hydride to C
:1 dr, up to >99% ee). Deuterium-labeling experiments and control experiments were conducted to probe the mechanism, and the results indicated that the acidity of the solvent plays a critical role in the control of diastereoselectivity by trapping the adduct of nickel hydride to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds via protonolysis, giving the hydrogenation product with stereospecific syn-selectivity. This protocol provides efficient access to chiral α-fluoro-β-amino esters which have important potential applications in organic synthesis and medicinal chemistry.
C bonds via protonolysis, giving the hydrogenation product with stereospecific syn-selectivity. This protocol provides efficient access to chiral α-fluoro-β-amino esters which have important potential applications in organic synthesis and medicinal chemistry.
Highly efficient construction of chiral compounds in an environmentally friendly and cost-effective manner is highly significant in organic synthesis and the pharmaceutical industry.1 Asymmetric hydrogenation of tetrasubstituted alkenes provides ideal access to chiral compounds containing two contiguous stereocenters due to its perfect atom economy and efficiency.2 Although great effort has been made so far, only a few successful examples of asymmetric hydrogenation of tetrasubstituted olefins have been reported via catalysts based on Rh, Ru, Ir, and Pd with high costs and deleterious environmental impact.2 In contrast, catalysts based on cheap, earth-abundant first-row transition metals offer potential advantages in terms of cost and sustainability.3 Consequently, replacing precious metals with cheap, earth-abundant first-row transition metals in asymmetric hydrogenation of tetrasubstituted alkenes is desirable.4 In the past few years, first-row transition metal catalyzed asymmetric hydrogenation has experienced rapid development, such as iron-, cobalt- and nickel-catalyzed asymmetric (transfer) hydrogenation of C
C, CO, and CN bonds.3–5 However, first-row transition metal catalyzed asymmetric hydrogenation of tetrasubstituted alkenes has not been achieved.
Owing to the unique ability of fluorine to modulate the physical and biological properties of organic molecules,6 introduction of fluorine atoms to nitrogen-containing molecules has emerged as an attractive strategy in drug discovery,7 and a series of synthetic molecules containing chiral vicinal fluoroamine motifs, such as MK-0731,8a clofarabine,8b and sitafloxacin8c (Fig. 1a), have been reported to exhibit important bioactivities. However, asymmetric synthetic routes to β-fluoroamines with a vicinal stereogenic center are still very limited,9–11 and most of them involve the formation of C–F bonds and C–N bonds. As an alternative approach, transition metal catalyzed asymmetric hydrogenation of tetrasubstituted fluorinated enamides, one of the most powerful transformations available in synthetic chemistry,12 is unexploited. There are several reasons that may account for the absence of asymmetric hydrogenation in this transformation. Firstly, the combined effect of both the steric hindrance and electron-deficient nature of tetrasubstituted fluorinated alkenes greatly decreases the coordination ability of the alkene to the metal, which leads to the low activity in asymmetric hydrogenation.12 Secondly, the competition between defluorination and hydrogenation makes this reaction more challenging.13 Especially, the reaction might be further challenged by relatively low catalytic activity when a nickel complex is employed as the catalyst. Moreover, the mechanism of Ni-catalyzed hydrogenation is distinct from the rhodium dihydride and ruthenium monohydride pathways.14 For example, hydrogenation of acrylates involves the formation of O–nickel enolate through 1,4-conjugate addition of Ni–H to acrylate or Ni–H insertion to CC bonds and subsequent isomerization (Fig. 1b),5q,s and it is extremely difficult to afford stereospecific syn-addition products by the protonation. Given that the protonolysis of the carbon–metal bond may occur with retention of configuration at carbon under suitable conditions,15 we envisioned that if we could inhibit the formation of O–nickel species or accelerate the protonolysis of C–nickel species by acidic solvents or acids, syn hydrogenation products would be obtained stereospecifically (Fig. 1c). Herein, we report the first example of nickel-catalyzed asymmetric hydrogenation of tetrasubstituted fluorinated enamides, giving α-fluoro-β-amino esters with excellent diastereo- and enantioselectivities.
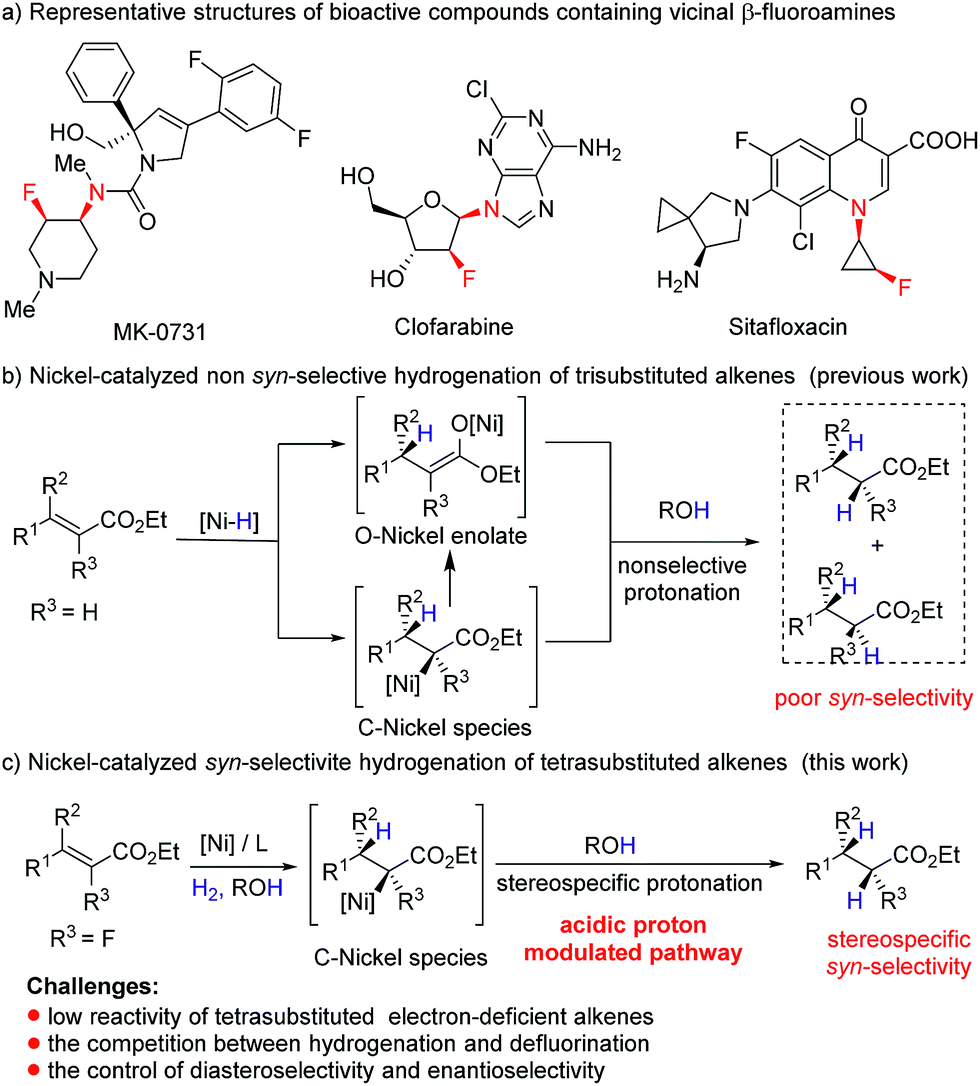 | ||
| Fig. 1 Ni-catalyzed asymmetric hydrogenation of olefins. | ||
Initially, asymmetric hydrogenation of (E)-ethyl-3-acet-amido-2-fluoro-3-phenylacrylate (E)-1a was investigated using 5 mol% Ni(OAc)2 and 5.5 mol% ligand under 70 atm of H2 at 80 °C in CF3CH2OH for 24 h. A variety of diphosphine ligands (Fig. 2) developed by our group and some commercially available chiral ligands were examined (Table 1). When (RC,SP)-DuanPhos and (R)-BINAP were employed for this reaction, both of them showed excellent reactivities and enantioselectivities, but an undesired defluorination product was observed (Table 1, entries 1–2). (S,S)-Me-DuPhos and (S,S)-Ph-BPE exhibited low catalytic activity for this transformation albeit with good enantioselectivities (Table 1, entries 3–4). To our delight, full conversion and excellent ee were obtained by using (S)-Binapine as the ligand (Table 1, entry 5, >99% conversion and >99% ee). Notably, no defluorination or diastereoisomer products were detected in this process, which indicated that the Ni(OAc)2/(S)-Binapine catalytic system is efficient and effective for the asymmetric hydrogenation of (E)-1a. Subsequently, the solvent effect was investigated, and the results revealed that solvents play critical roles in this reaction, affecting the catalytic activity and selectivity greatly. When toluene, tetrahydrofuran (THF), ethyl acetate (EtOAc) and dichloromethane (CH2Cl2) were used as solvents, the reactions were totally inhibited or only trace products were detected (Table 1, entries 7–9). When the reactions were conducted in MeOH or EtOH, the defluorination product was observed with poor yields, affording 3a as the major product (Table 1, entries 10–11). The effect of H2 pressure for this reaction was also evaluated. Full conversion with an unchanged ee value was obtained when the reaction was carried out in CF3CH2OH under a lower hydrogen pressure of 50 atm (Table 1, entry 12). When the temperature decreased to 50 °C, the reaction proceeded with 32% conversion (Table 1, entry 13). Therefore, the optimized reaction conditions involved the use of Ni(OAc)2/(S)-Binapine as a catalyst under 50 atm of H2 pressure in CF3CH2OH at 80 °C for 24 h.
 | ||
| Fig. 2 Structures of the phosphine ligands for hydrogenation of (E)-ethyl-3-acetamido-2-fluoro-3-phenylacrylate (E)-1a. | ||
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Solvent | Yieldb [%] |
2a:3ab |
eec | |
| 2a + 3a | 4a | |||||
|
a Conditions: Ni(OAc)2/(S)-Binapine/(E)-1a 1:1.1:20, in 0.5 mL of solvent.
b The conversion and ratio were determined using 1H NMR analysis.
c The ee values of 2a were determined using HPLC analysis with a chiral stationary phase.
d 50 atm of H2.
e 50 °C.
|
||||||
| 1 | L1 | TFE | 72 | 28 | >99:1 |
97 |
| 2 | L2 | TFE | 88 | 12 | >99:1 |
95 |
| 3 | L3 | TFE | <10 | 0 | >99:1 |
99 |
| 4 | L4 | TFE | <10 | 0 | >99:1 |
−72 |
| 5 | L5 | TFE | >99 | 0 | >99:1 |
>99 |
| 6 | L5 | Toluene | NR | NR | — | — |
| 7 | L5 | THF | Trace | — | — | — |
| 8 | L5 | EtOAc | Trace | — | — | — |
| 9 | L5 | CH2Cl2 | Trace | — | — | — |
| 10 | L5 | MeOH | 42 | 5 | 1:2 |
>99 |
| 11 | L5 | EtOH | 25 | 20 | <1:20 |
— |
| 12d | L5 | TFE | >99 | 0 | >99:1 |
>99 |
| 13d,e | L5 | TFE | 32 | 0 | >99:1 |
>99 |
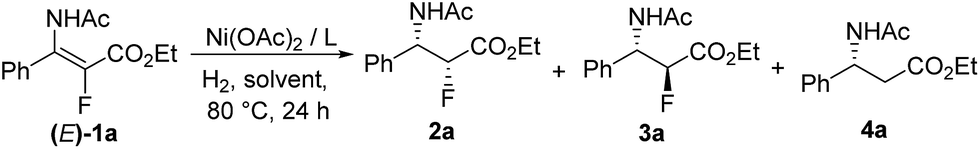
With the optimized reaction conditions in hand, a series of β-enamido-α-fluoro esters were examined to evaluate the substrate scope and generality of this catalytic reaction. As shown in Scheme 1, compounds (E)-1 with different ester groups were found to be good substrates to give the desired β-amido-α-fluoro esters in excellent yields with outstanding stereocontrol (2a and 2b). A wide range of β-aryl-β-enamido-α-fluoro esters with electron-rich or -poor aryl groups were examined. High yields and excellent enantioselectivities were observed in most cases, regardless of the substitution position (2c–2k). Moreover, excellent enantioselectivity was obtained with the substrate containing the naphthyl group (2l). In addition, when the acetyl group, the protection group of the amino group, was changed to a benzoyl group, high yield and excellent enantioselectivity were also achieved (2m). Notably, when the aryl group was changed to an alkyl group, such as methyl, i-butyl, or n-amyl, the reaction also proceeded smoothly, affording β-amido-α-fluoro esters with good yields and high enantioselectivities (2n–2p). Moreover, in order to determine the absolute configuration of the products, X-ray analysis of 2b was conducted, and the configuration was determined as (2R,3S).16
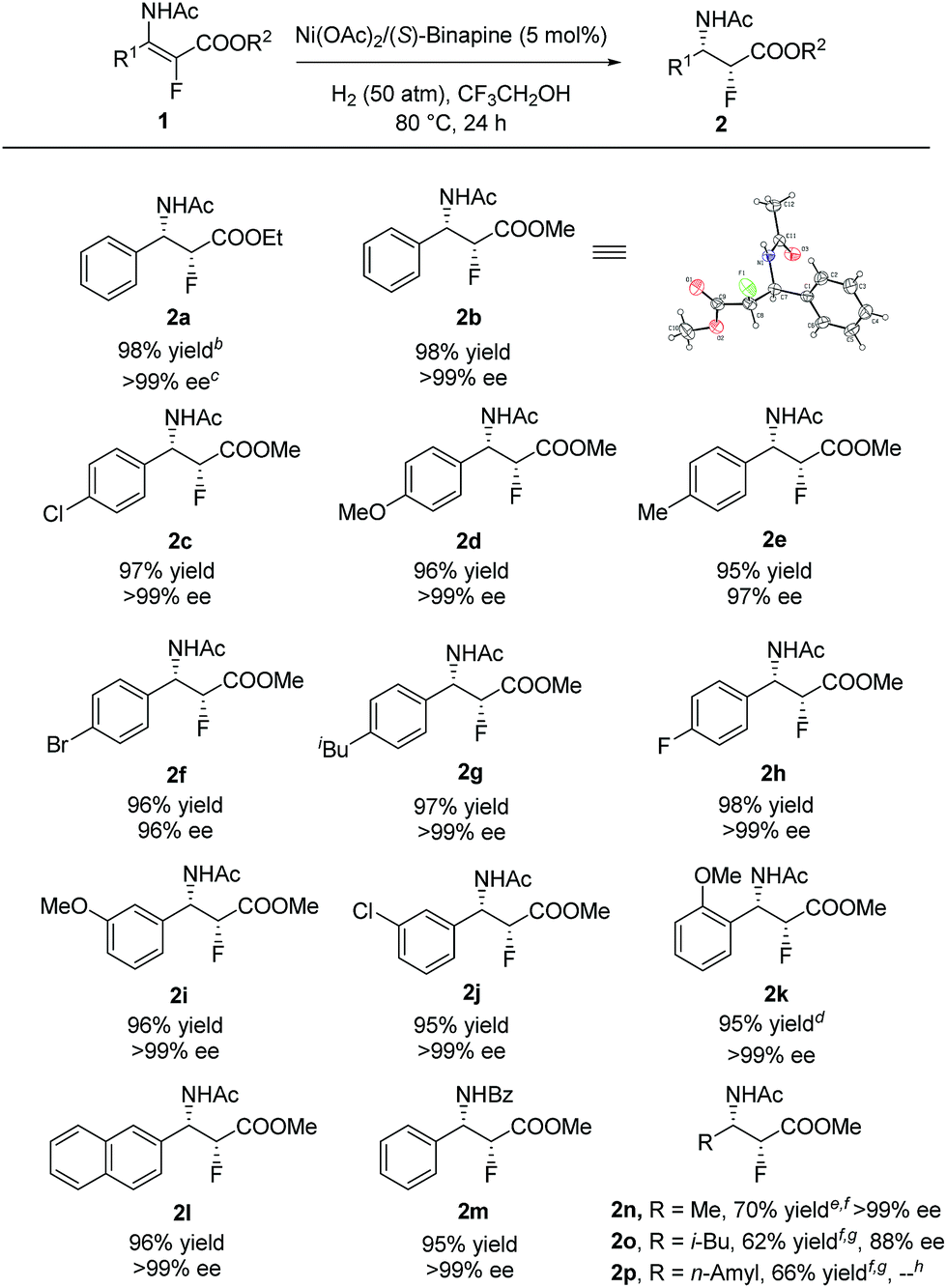 | ||
| Scheme 1 Substrate scope. aUnless otherwise mentioned, all reactions were carried out with a Ni(OAc)2/(S)-Binapine/substrate ratio of 1:1.1:20, in 0.5 mL of CF3CH2OH, at 80 °C, under hydrogen (50 atm) for 24 h. bYield of the isolated product. cDetermined by HPLC analysis using a chiral stationary phase. dUnder 70 atm of H2. eUnder 100 atm of H2. fDefluorination product was detected. gThe reaction was conducted with a Ni(OAc)2/(S)-Binapine/substrate ratio of 1:1.1:10 under 90 atm of H2 at 80 °C for 46 h. hThe ee was not possible to detect. | ||
To obtain insight into this catalytic system, a series of isotopic labeling studies were conducted. Firstly, when 1b was hydrogenated with 50 atm of D2 in CF3CH2OH at 80 °C, the deuterium atom was solely added at the β position (Scheme 2a). Then, when the experiment was carried out with 70 atm of H2 in CD3OD, the deuterium atoms were incorporated at the α position of the ester (Scheme 2b). Finally, when 2b was stirred in a mixture of 1:1 CD3OD/CF3CH2OH at 80 °C, the deuterium atoms were found at ester and amido groups, excluding the H/D exchange at the α position of the ester after the product was formed (Scheme 2c).17
 | ||
| Scheme 2 Deuterium labeling studies of asymmetric hydrogenation of 1b. | ||
In order to ascertain the role of solvent in modulation of diastereoselectivity, we conducted this reaction in MeOH by adding different ratios of acetic acid (Table 2), and the results revealed that the yields and diastereoselectivities were gradually improved as the ratios of acetic acid increased. Joyfully, when 15 equivalents of acetic acid were added, only the syn hydrogenation product was obtained. These results suggested that the acidic proton plays a critical role in the control of diastereoselectivity.
|
|
||||
|---|---|---|---|---|
| Entry | HOAc (x equiv.) | Yield/% | Ratio | |
| 2a + 3a | 4a |
2a:3a |
||
|
a All reactions were carried out with a Ni(OAc)2/(S)-Binapine/substrate ratio of 1:1.1:20, in 0.5 mL of CF3CH2OH, at 80 °C, under hydrogen (70 atm) for 24 h.
|
||||
| 1 | — | 42 | 5 | 1:2 |
| 2 | 0.5 | 51 | <1 | 1:1 |
| 3 | 1.0 | 61 | <1 | 68:32 |
| 4 | 5.0 | >99 | <1 | 93:7 |
| 5 | 15.0 | >99 | 0 | >99:1 |

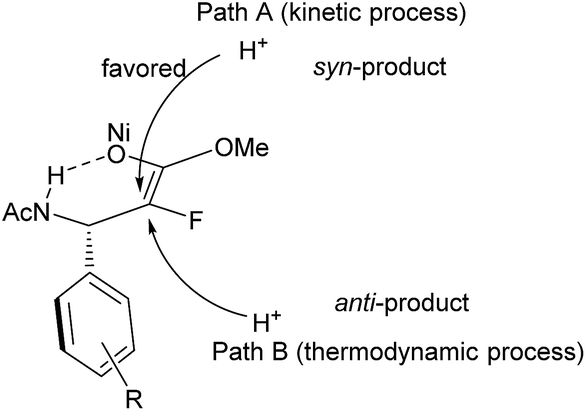
Considering that the reaction was conducted at relatively high temperature, and anti-selective product 3a was the major product in ethanol or methanol, we assumed that the protonation of nickel enolate was a thermodynamically controlled process, which was beneficial for the formation of the major product with anti-selectivity.18 Based on these results and analysis, we proposed the plausible reaction pathway (Fig. 3). The heterolytic cleavage of H2 gave the NiH complex5q,s followed by reaction with enamide to form nickel intermediate I. There were two possible reaction pathways from intermediate I. When the reaction was conducted in CF3CH2OH, the acidity of the solvent greatly accelerated the stereospecific protonolysis of nickel species I, which led to the inhibition of conversion of I to II, generating the syn hydrogenation product (path A). When a less acidic solvent (methanol) was used, the reaction rate of isomerization of intermediate I to II was faster than protonolysis of intermediate I, thus forming nickel enolate II preferentially; then II underwent protonation to afford anti/syn mixed products (path B). Both pathways coexisted in methanol; therefore the ratio of anti/syn is poor. The thermodynamic protonation is the predominant pathway in ethanol, which resulted in a high ratio of anti/syn.
 | ||
| Fig. 3 Proposed mechanism. | ||
To demonstrate the potential utility of this methodology, asymmetric hydrogenation of (E)-1b was performed on a gram scale with 1 mol% catalyst loading, and the desired product (2b) was obtained with 90% yield and >99% ee (Scheme 3).
 | ||
| Scheme 3 Gram-scale reaction. | ||
Conclusions
In conclusion, a Ni(OAc)2/(S)-Binapine complex catalyzed asymmetric hydrogenation of tetrasubstituted β-enamido-α-fluoro esters has been achieved in high yields with excellent diastereo- and enantioselectivities (up to 98% yield, up to >99% ee, and up to >99:1 dr). More importantly, this investigation reveals the critical role of acidic solvent in modulating the reaction pathway and in the control of diastereoselectivity, which throws a new light on the reaction mechanism. This method features a broad substrate scope, excellent stereoselectivity and diastereoselectivity, and provides an efficient and concise route to α-fluoro-β-amino esters. Further investigations on nickel-catalyzed asymmetric hydrogenation are in progress in our lab.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (Grant No. 21871212, 21402145 and 21432007), the Natural Science Foundation of Hubei Province (2018CFB430), the Fundamental Research Funds for the Central Universities (2042017kf0177), the Important Sci-Tech Innovative Project of Hubei Province (2015ACA058) and the “111” Project of the Ministry of Education of China.Notes and references
- (a) W. S. Knowles, Acc. Chem. Res., 1983, 16, 106 CrossRef CAS; (b) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97 CrossRef CAS; (c) Q.-H. Fan, Y.-M. Li and A. S.-C. Chan, Chem. Rev., 2002, 102, 3385 CrossRef CAS; (d) W.-J. Tang and X.-M. Zhang, Chem. Rev., 2003, 103, 3029 CrossRef CAS; (e) I. D. Gridnev and T. Imamoto, Acc. Chem. Res., 2004, 37, 633 CrossRef CAS; (f) S. J. Roseblade and A. Pfaltz, Acc. Chem. Res., 2007, 40, 1402 CrossRef CAS; (g) Y.-G. Zhou, Acc. Chem. Res., 2007, 40, 1357 CrossRef CAS PubMed; (h) H. Shimizu, I. Nagasski, K. Matsumura, N. Sayo and T. Saito, Acc. Chem. Res., 2007, 40, 1385 CrossRef CAS; (i) W. Zhang, Y. Chi and X.-M. Zhang, Acc. Chem. Res., 2007, 40, 1278 CrossRef CAS; (j) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713 CrossRef CAS; (k) Y. Liu, Z. Wang and K.-L. Ding, Acta Chim. Sin., 2012, 70, 1464 CrossRef CAS; (l) J.-H. Xie and Q.-L. Zhou, Acta Chim. Sin., 2012, 70, 1427 CrossRef CAS; (m) B.-G. Zhao, Z.-B. Han and K.-L. Ding, Angew. Chem., Int. Ed., 2013, 52, 4744 CrossRef CAS; (n) Y.-M. He, Y. Feng and Q.-H. Fan, Acc. Chem. Res., 2014, 47, 2894 CrossRef CAS PubMed; (o) Z.-F. Zhang, N. A. Butt and W.-B. Zhang, Chem. Rev., 2016, 116, 14769 CrossRef CAS PubMed.
- S. Kraft, K. Ryan and R. B. Kargbo, J. Am. Chem. Soc., 2017, 139, 11630 CrossRef CAS PubMed.
- P. J. Chirik, Acc. Chem. Res., 2015, 48, 1687 CrossRef CAS.
- R. H. Morris, Acc. Chem. Res., 2015, 48, 1494 CrossRef CAS.
- (a) R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282 RSC; (b) Y. Y. Li, S. L. Yu, W. Y. Shen and J. X. Gao, Acc. Chem. Res., 2015, 48, 2587 CrossRef CAS; (c) R. H. Morris, Acc. Chem. Res., 2015, 48, 1494 CrossRef CAS; (d) R. Bigler, R. Huber and A. Mezzetti, Angew. Chem., Int. Ed., 2015, 54, 5171 CrossRef CAS PubMed; (e) J. F. Sonnenberg, K. Y. Wan, P. E. Sues and R. H. Morris, ACS Catal., 2017, 7, 316 CrossRef CAS; (f) S. Monfette, Z. R. Turner, S. P. Semproni and P. J. Chirik, J. Am. Chem. Soc., 2012, 134, 4561 CrossRef CAS PubMed; (g) M. R. Friedfeld, M. Shevlin, J. M. Hoyt, S. W. Krska, M. T. Tudge and P. J. Chirik, Science, 2013, 342, 1076 CrossRef CAS PubMed; (h) M. R. Friedfeld, M. Shevlin, G. W. Margulieux, L. Campeau and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 3314 CrossRef CAS; (i) J. H. Chen, C. H. Chen, C. L. Ji and Z. Lu, Org. Lett., 2016, 18, 1594 CrossRef CAS; (j) G. R. Morello, H. Zhong, P. J. Chirik and K. H. Hopmann, Chem. Sci., 2018, 9, 4977 RSC; (k) J. Chen and Z. Lu, Org. Chem. Front., 2018, 5, 260 RSC; (l) Y. Hamada, Y. Koseki, T. Fujii, T. Maeda, T. Hibino and K. Makino, Chem. Commun., 2008, 6206 RSC; (m) P. Yang, H. Y. Xu and J. R. Zhou, Angew. Chem., Int. Ed., 2014, 53, 12210 CrossRef CAS; (n) S. Y. Guo, P. Yang and J. R. Zhou, Chem. Commun., 2015, 51, 12115 RSC; (o) H. Y. Xu, P. Yang, P. Chuanprasit, H. Hirao and J. R. Zhou, Angew. Chem., Int. Ed., 2015, 54, 5112 CrossRef CAS; (p) P. Yang, L. H. Lim, P. Chuanprasit, H. Hirao and J. R. Zhou, Angew. Chem., Int. Ed., 2016, 55, 12083 CrossRef CAS; (q) M. Shevlin, M. R. Friedfeld, H. Sheng, N. A. Pierson, J. M. Hoyt, L. Campeau and P. J. Chirik, J. Am. Chem. Soc., 2016, 138, 3562 CrossRef CAS; (r) S. Y. Guo and J. R. Zhou, Org. Lett., 2016, 18, 5344 CrossRef CAS; (s) W. Gao, H. Lv, T. Zhang, Y. Yang, L. W. Chung, Y.-D. Wu and X. Zhang, Chem. Sci., 2017, 8, 6419 RSC; (t) X. Li, C. You, S. Li, H. Lv and X. Zhang, Org. Lett., 2017, 19, 5130 CrossRef CAS; (u) M. R. Friedfeld, H. Zhong, R. T. Ruck, M. Shevlin and P. J. Chirik, Science, 2018, 360, 888 CrossRef CAS; (v) Z. Zhang, N. A. Butt, M. Zhou, D. Liu and W. Zhang, Chin. J. Chem., 2018, 36, 443 CrossRef CAS.
- (a) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315 CrossRef CAS; (b) J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432 CrossRef CAS; (c) K. Mikami, Y. Itoh and M. Yamanaka, Chem. Rev., 2004, 104, 1 CrossRef CAS.
- K. Mueller, C. Faeh and F. Diederich, Science, 2007, 317, 1881 CrossRef CAS.
- (a) C. D. Cox and R. M. Garbaccio, Adv. Anticancer Agents Med. Chem., 2013, 1, 269 CAS; (b) A. D. Ajavon, P. L. Bonate and D. R. Taft, Eur. J. Pharm. Sci., 2010, 40, 209 CrossRef CAS; (c) R. S. Obach, F. Lombardo and N. J. Waters, Drug Metab. Dispos., 2008, 36, 1385 CrossRef CAS.
- (a) N. Shibata, E. Suzuki and Y. Takeuchi, J. Am. Chem. Soc., 2000, 122, 10728 CrossRef CAS; (b) D. Cahard, C. Audouard, J.-C. Plaquevent and N. Roques, Org. Lett., 2000, 2, 3699 CrossRef CAS; (c) M. Marigo, D. Fielenbach, A. Braunton, A. Kjærsgaard and K. A. Jørgensen, Angew. Chem., Int. Ed., 2005, 44, 3703 CrossRef CAS PubMed; (d) D. D. Steiner, N. Mase and C. F. Barbas III, Angew. Chem., Int. Ed., 2005, 44, 3706 CrossRef CAS PubMed; (e) T. D. Beeson and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 8826 CrossRef CAS PubMed; (f) M. L. Schulte and C. W. Lindsley, Org. Lett., 2011, 13, 5684 CrossRef CAS PubMed; (g) C. Appayee and S. E. Brenner-Moyer, Org. Lett., 2010, 12, 3356 CrossRef CAS; (h) Y.-Y. Huang, X. Yang, Z. Chen, F. Verpoort and N. Shibata, Chem.–Eur. J., 2015, 21, 8664 CrossRef CAS PubMed.
- (a) W. Kong, P. Feige, T. de Haro and C. Nevado, Angew. Chem., Int. Ed., 2013, 52, 2469 CrossRef CAS PubMed; (b) T. Honjo, R. J. Phipps, V. Rauniyar and F. D. Toste, Angew. Chem., Int. Ed., 2012, 51, 9684 CrossRef CAS PubMed; (c) R. J. Phipps, K. Hiramatsu and F. D. Toste, J. Am. Chem. Soc., 2012, 134, 8376 CrossRef CAS PubMed; (d) O. Lozano, G. Blessley, T. Martinez del Campo, A. L. Thompson, G. T. Giuffredi, M. Bettati, M. Walker, R. Borman and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 8105 CrossRef CAS; (e) D.-F. Lu, C.-L. Zhu, J. D. Sears and H. Xu, J. Am. Chem. Soc., 2016, 138, 11360 CrossRef CAS PubMed; (f) K. M. Mennie, S. M. Banik, E. C. Reichert and E. N. Jacobsen, J. Am. Chem. Soc., 2018, 140, 4797 CrossRef CAS PubMed.
- (a) D. Cahard, X. Xu, S. Couve-Bonnaire and X. Pannecoucke, Chem. Soc. Rev., 2010, 39, 558 RSC; (b) X. Yang, T. Wu, R. J. Phipps and F. D. Toste, Chem. Rev., 2015, 115, 826 CrossRef CAS; (c) Z. Jiao, J. J. Beiger, Y. Jin, S. Ge, J. S. Zhou and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 15980 CrossRef CAS.
- (a) M. Engman, J. S. Diesen, A. Paptchikhine and P. G. Andersson, J. Am. Chem. Soc., 2007, 129, 4536 CrossRef CAS; (b) P. Zhang, C. Wang, L. Zhou and J. Sun, Chin. J. Chem., 2012, 30, 2636 CrossRef CAS.
- D. M. Sedgwick and G. B. Hammond, J. Fluorine Chem., 2018, 207, 45 CrossRef CAS.
- (a) I. D. Gridnev, N. Higashi, K. Asakura and T. Imamoto, J. Am. Chem. Soc., 2000, 122, 7183 CrossRef CAS; (b) R. Giernoth, H. Heinrich, N. J. Adams, R. J. Deeth, J. Bargon and J. M. Brown, J. Am. Chem. Soc., 2000, 122, 12381 CrossRef CAS; (c) A. S. C. Chan, J. J. Pluth and J. Halpern, J. Am. Chem. Soc., 1980, 102, 5952 CrossRef CAS; (d) J. M. Brown and P. A. Chaloner, J. Chem. Soc., Chem. Commun., 1980, 344 RSC; (e) M. Kitamura, M. Tsukamoto, Y. Bessho, M. Yoshimura, U. Kobs, M. Widhalm and R. Noyori, J. Am. Chem. Soc., 2002, 124, 6649 CrossRef CAS.
- (a) H. Qian and R. A. Widenhoefer, J. Am. Chem. Soc., 2003, 125, 2056 CrossRef CAS PubMed; (b) H. Qian, T. Pei and R. A. Widenhoefer, Organometallics, 2005, 24, 287 CrossRef CAS.
- The X-ray crystal data for 2b have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1845389.†.
- The difference in the deuterium labelling ratio of the NHAc group in Scheme 2b and c may be attributed to the different purification procedures. In Scheme 2b, deuterium exchange between the NDAc and trace water in the eluent might occur in column chromatography; thus no NDAc was detected in NMR experiments.
- Although the kinetic protonation of nickel enolate can explain the formation of the syn-product in CF3CH2OH, it can't account for the experimental results in ethanol or methanol. In addition, it also can't explain why the syn/anti ratio of the product improved as the acidity
of solvent increased. Thus, it is excluded
.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and NMR spectra of the compounds. CCDC 1845389. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04002h |
| This journal is © The Royal Society of Chemistry 2019 |