
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighlighting spin selectivity properties of chiral electrode surfaces from redox potential modulation of an achiral probe under an applied magnetic field†
Tiziana
Benincori
 a,
Serena
Arnaboldi
*b,
Mirko
Magni
*b,
Sara
Grecchi
b,
Roberto
Cirilli
*c,
Claudio
Fontanesi
*d and
Patrizia Romana
Mussini
*b
a,
Serena
Arnaboldi
*b,
Mirko
Magni
*b,
Sara
Grecchi
b,
Roberto
Cirilli
*c,
Claudio
Fontanesi
*d and
Patrizia Romana
Mussini
*b
aDipartimento di Scienza e Alta Tecnologia, Università degli Studi dell'Insubria, Via Valleggio 11, 22100, Como, Italy
bDipartimento di Chimica, Università degli Studi di Milano, via Golgi 19, 20133 Milano, Italy. E-mail: serena.arrnaboldi@unimi.it; mirko.magni@unimi.it; patrizia.mussini@unimi.it
cIstituto Superiore di Sanità, Centro Nazionale per il Controllo e la Valutazione dei Farmaci, Viale Regina Elena 299, 00161, Roma, Italy
dDipartimento di Ingegneria “Enzo Ferrari”, Università degli Studi di Modena, Via Vivarelli 10, 41125, Modena, Italy. E-mail: claudio.fontanesi@unimore.it
First published on 4th January 2019
Abstract
Impressive spin-related effects are observed in cyclic voltammetry (CV) experiments performed under an applied magnetic field on a non-ferromagnetic electrode modified with a thin electroactive oligothiophene film, either “inherently chiral” or featuring chiral pendants with stereogenic centres. When flipping the magnet’s north/south orientation, the CV peaks of two achiral, chemically reversible Fe(III)/Fe(II) redox couples in aqueous or organic solution undergo impressive potential shifts (up to nearly 0.5 V depending on protocol conditions), specularly by changing the film’s (R)- or (S)-configuration. The magnitude of the potential shift decreases upon increasing both the polymer film thickness and the distance between the permanent magnet and the electrode surface. Such unprecedented spin-related redox potential modulation, obtained in the absence of a magnetic electrode acting as a spin injector, provides striking evidence (as well as an attractive evaluation criterion) of the spin selectivity properties of chiral thin films.
Introduction
The study of the mutual influence between chirality, electron spin and magnetism is a well-documented field of research.1–7 Implications span from pure fundamental research, to chemical applications of both analytical and synthetic character, to multidisciplinary purposes such as the development of computing oriented electronics.8,9 In this context the interrelated disciplines of magnetoelectrochemistry, spintronics and Spin Dependent Electrochemistry (SDE) play a crucial role. Actually, magnetic effects in electrochemical systems have been investigated since the XIX century, particularly focusing on Lorentz and Kelvin force effects.10,11 However, the large majority of the so far available magnetoelectrochemistry studies deal with achiral systems. For instance, the application of an external magnetic field is able to induce conformational variations (i.e. tilt angles) in self-assembled monolayers (SAMs).12 A magnetic field can also influence the reaction path in electrochemical redox mechanisms involving open-shell intermediate species,13–15 the morphology of molecular crystals16 and crystallization processes.17,18 The first meeting point between chirality and magnetoelectrochemistry has been achieved with magnetohydrodynamic (MHD) convection: as a result of the Lorentz force acting on ionic currents, micro-MHD vortices (themselves chiral, being clockwise and anticlockwise) are generated at the electrode surface. In the past two decades magnetohydrodynamics has been exploited in magnetoelectrodeposition/polymerization, to prepare films from achiral starting materials (metal salts or organic monomers, respectively) exhibiting macro- and microscopic chiral structures that resulted in enantioselectivity towards chiral electroactive analytes.19–22In this context, dealing with magnetic fields, it is important to distinguish between “true” and “false” chirality,23–29 an intriguing problem rationalized several decades ago by Barron.23–26 He stated that several time-dependent mirror physical systems are not truly chiral since they are interconverted by combined space and time reversal, while true chirality implies enantiomer interconversion by simple space inversion.23–27 Importantly, only when interacting with a truly chiral system the energy of enantiomeric probes can be different (corresponding to diastereomeric situations), while no loss of degeneration in energy levels can happen in a falsely chiral system; however, asymmetry could be obtained for processes out of thermodynamic equilibrium.27 Consistent with Barron's rationale, a magnetic field alone cannot produce true chirality, as already stated by Lord Kelvin in 1904,28 not even, in spite of an early assumption by Pierre Curie,28 upon addition of a collinear, uniform and constant electric field, a combination which, as more recently demonstrated,30 cannot affect the enantiomer equidistribution in a racemate at equilibrium. Instead, according to Barron's approach, an electron in helical roto-translational motion with spin–orbit coupling (i.e. translating in a “ballistic” motion with its spin projection parallel or antiparallel to the direction of propagation) can be regarded as chiral, existing as two possible enantiomers, corresponding to the α and β spin configurations, which do not coincide upon space and time inversion. Such peculiar “chiral actor” is the object of spintronics, the fascinating field of modern physics which deals with the active manipulation of spin degrees of freedom of charge carriers.31 In fact, a general spintronic device is based on the generation of carrier spin polarization (SP) in a suitable active material and spin polarized detection at the output.32 Important practical implications for the everyday life regard the magnetic information storage technology based on the giant magnetoresistance effect,33 resulting in a magnetic field-oriented modulation of electric resistance of the device. So far spintronics has been mainly developed exploiting metal, oxide or inorganic semiconductor materials; however, molecular spintronics is regarded as an attractive target on account of the wide range, flexibility, processability and low cost, typical of molecular materials.34
The combination of spintronics with magnetoelectrochemistry, particularly involving truly chiral molecular spin selectors, was promoted by the discovery of the Chiral Induced Spin Selectivity (CISS) effect by Ron Naaman and coworkers, observing SP in photo-ejected electrons transmitted through a thin layer of enantiopure material adsorbed on gold, acting as an electron spin filter.35 Such a CISS effect was subsequently exploited in electrochemical experiments, giving rise to spin dependent electrochemistry, SDE, in which electrons are generated, rather than by X-ray photoemission, by a low electric potential difference in electrochemical cells including a ferromagnetic electrode and a chiral molecular layer.29,36–41 The process thus involves interaction/transfer of a truly chiral probe, the electron with spin–orbit coupling, with/through a truly chiral phase, the chiral molecular film acting as a spin filter.
In this frame, a key issue is the availability of molecular layers of high electroactivity and enantioselectivity providing top level spin filters. Actually, the development of efficient, stable and robust chiral electrode surfaces able to discriminate enantiomers of electroactive probes is a general-scope hot topic in electrochemistry and electroanalysis. As recently reviewed by Mussini and coworkers,42,43 among the huge amount of materials and approaches that have been proposed in the literature, a breakthrough, leading to unprecedented, outstanding chirality manifestations, was recently obtained by the exploitation of “inherently chiral functional molecular materials” (ICFMMs).44–46 In ICFMMs both chirality and key functional properties (like electroactivity) originate from the same source, like a conjugated thiophene-based backbone with a tailored torsion resulting from the insertion of atropisomeric cores or helicoidal elements.42,43 A further essential feature is regioregularity, ensuring propagation as well as amplification of the properties of single chiral units into helical/foldamer macro- or supramolecular structures. Thus, charge carriers entirely originate and move along a twisted left-handed or right-handed chiral pathway, a powerful asset for magnetoelectrochemistry/spintronics applications (much more than bio-based chiral macromolecules with helical secondary structures but not electroactive, or chiral electroactive molecules with low macro- or supramolecular order).42,43 Another quite convenient feature for CISS studies is that the aforementioned ICFMMs are thiophene-based, and oligothiophene chains have already shown good ability as spin charge carrier transporters (with a spin diffusion length up to 200 nm at room temperature).32 In this frame we present voltammetry experiments under an applied magnetic field at a non-ferromagnetic electrode modified with a thin electroactive oligothiophene film, either “inherently chiral” or featuring chiral pendants with stereogenic centres. Unprecedented spin-related modulation of redox potentials of reversible achiral probes, observed in the absence of a magnetic electrode acting as spin injector, provides striking evidence as well as an attractive evaluation criterion of the spin selectivity properties of chiral thin films.
Results and discussion
Chiral films obtained by electrooligomerization of (R)- or (S)-BT2T4 monomers44 (Fig. 1a, synthesized as a racemate and resolved according to the procedure described in the SI.1.1†), successfully tested as chiral selectors for electroactive chiral molecular probes in former experiments,45,46 were evaluated as spin selectors, as a prototype for thiophene-based ICFMMs. Their performance was also compared with that of chiral thiophene-based enantiopure films (c-PEDOT) obtained from electropolymerization of (2R,3R)-2,3-dimethyl-2,3-dihydrothieno[3,4-b][1,4]dioxine (c-EDOT) monomers, including two stereocentres localized outside the main conjugated backbone (Fig. 1b, synthesized according to the procedure described in the SI.1.1†). To rule out any false positive results, achiral PEDOT (electrodeposited from commercially available 2,3-dihydrothieno[3,4-b][1,4]dioxine monomers), was tested with the same protocol. Our proposed strategy is a variation of the SDE protocol, where, in general, a permanent magnet is placed in close contact to a ferromagnetic Ni electrode that acts as an electron spin injector for the adjacent chiral interface obtained by proper functionalization of the metal surface.36,38,40,41 Instead, the setup adopted in the present work involves a non-ferromagnetic material as the electron collector (source/drain) for the working electrode (indium tin oxide coated glass, ITO). A magnetic field is applied using an external magnet, placed at a distance of ca. 2.6 mm from the ITO|film interface, considering that the thickness of the bare ITO electrode and cuvette were ca. 1.0 mm and ca. 1.6 mm, respectively (Fig. 1c); this implies no direct spin injection into the film by the electrode substrate, resembling recently proposed solid-state architectures.3 In particular, cyclic voltammetry measurements were carried out employing as a working electrode (WE) a slice (0.8 × 4.5 cm2) of ITO coated glass functionalized with a semiconducting, chiral, and enantiopure organic thin film (geometric area ca. 1 cm2), electrodeposited by potentiodynamic oxidation at 0.2 V s−1 (one CV cycle) of (a) (R)- or (S)-BT2T4 and (b) c-EDOT, in acetonitrile (ACN) with 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) supporting electrolyte (see the SI.1.2† for more details). For the sake of comparison achiral PEDOT films were also electrodeposited under the same conditions. No external magnetic field was applied during the electrodeposition procedure. As schematically depicted in Fig. 1c, the hybrid ITO|chiral film WE, and a Pt wire and an aqueous saturated calomel electrode (SCE) used as the counter (CE) and reference (RE), respectively, were immersed in (i) an equimolar aqueous solution of K3[Fe(CN)6] and K4[Fe(CN)6] (reversible achiral redox couple, in the following indicated as Fe(III)/Fe(II)), each of them at 2.5 mM concentration, with 400 mM KCl as the supporting electrolyte, or in (ii) a 2 mM solution of ferrocene in ACN with 0.1 M TBAPF6 as the supporting electrolyte.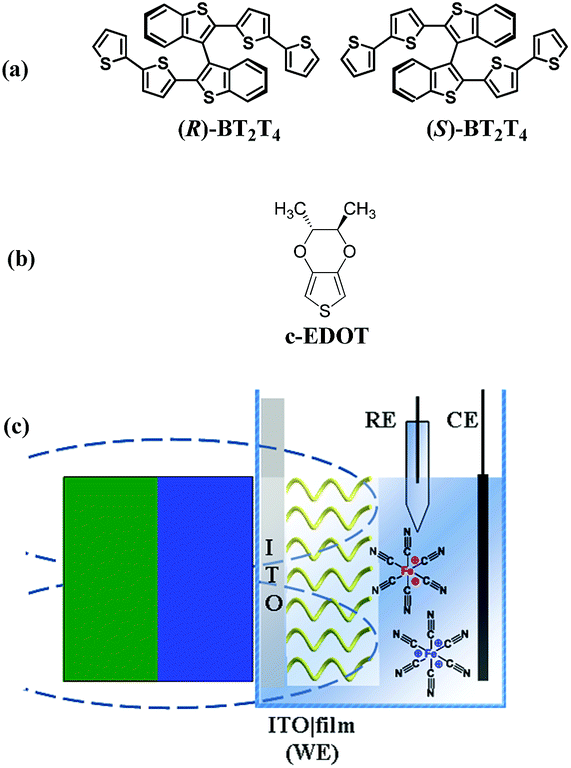 | ||
| Fig. 1 Chemical structures of thiophene-based monomers used in this work: (R)- and (S)-BT2T4 (a) and c-EDOT (b). Schematic representation of the magnetoelectrochemistry experimental setup (c). | ||
During CV tests with the achiral redox couple a magnetic field was applied, perpendicular to the electrode surface (SI.1.3†), by placing a permanent magnet (nickel-coated NdFeB B88X0 Grade N42 K&J Magnet, Inc.; magnetic field strength at the surface = 6353 Gauss). Change in the orientation of the magnetic field was obtained by mechanically flipping the magnet along its magnetic axis (north versus south magnetic pole).
CV curves of the achiral Fe(III)/Fe(II) redox probe on the bare ITO surface do not show any change as a function of the magnetic field orientation (Fig. 2a) with a half-wave potential (E1/2) of 0.22 V vs. SCE, E1/2 being defined as the average between peak potentials of forward and backward scan; such a chemically reversible signal is virtually identical to that obtained in the absence of the magnetic field (not shown). No difference is also observed when recording CV curves for the same achiral probe on the ITO electrode modified with an enantiopure oligo-BT2T4 film of either (R)- or (S)-configuration, in the absence of an applied magnetic field (Fig. 2b). Perturbing this last setup by the external magnetic field perpendicular to the WE surface, working e.g. on the enantiopure oligo-(S)-BT2T4 film (Fig. 2c), an impressive ∼0.47 V splitting is observed for the E1/2 of the achiral Fe(III)/Fe(II) couple upon flipping the magnetic field orientation, north vs. south and vice versa (see also the ESI, Table SI.2.1.1†).
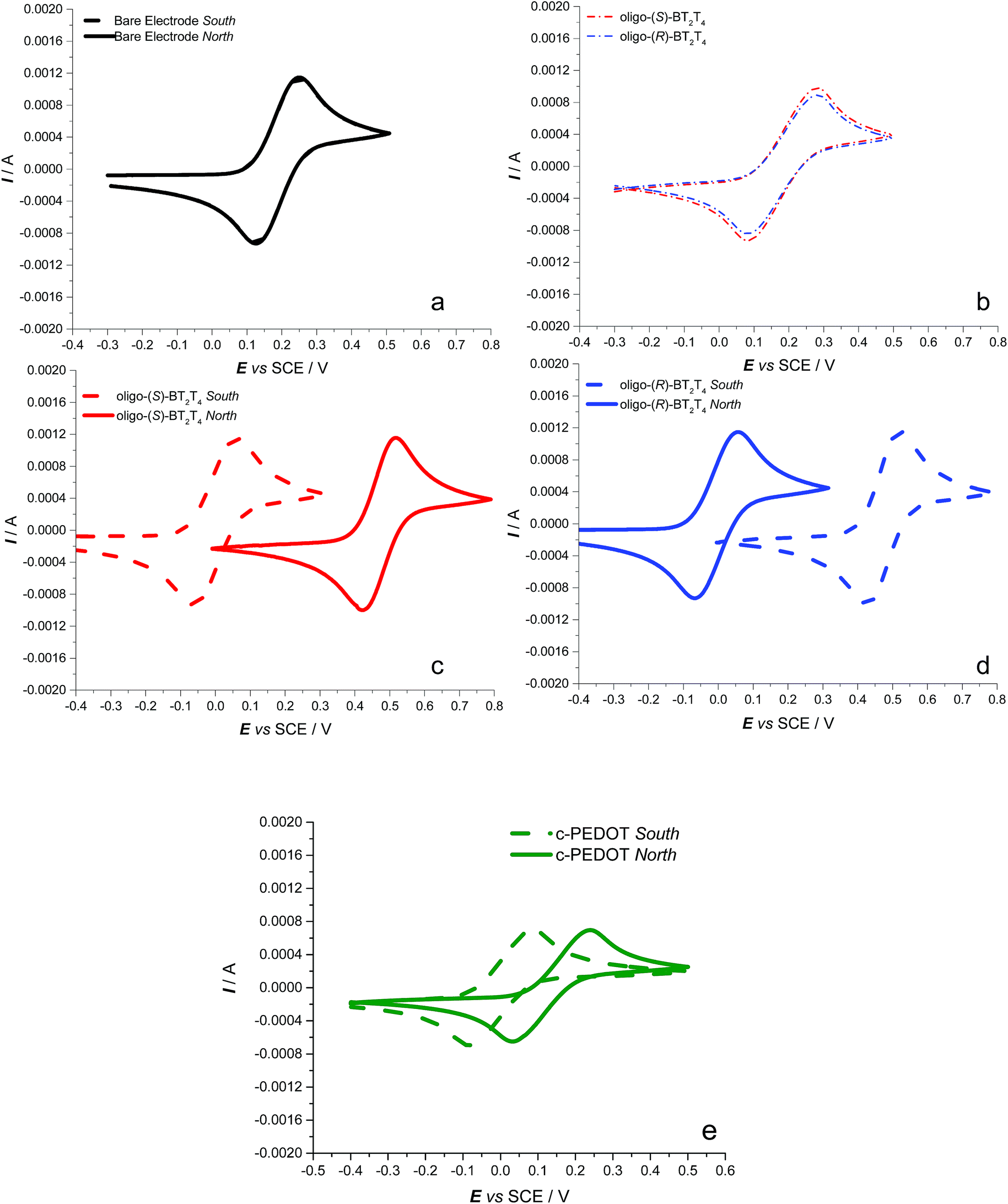 | ||
| Fig. 2 CV patterns recorded at 0.05 V s−1 for the Fe(III)/Fe(II) achiral couple: (a) on a bare ITO electrode, as a function of the magnetic field orientation (solid line for the north pole towards the electrode, dashed line for the south pole); (b) on hybrid ITO|oligo-BT2T4 electrodes (blue line for the (R)-configuration, red for (S)-) without an external magnet; (c) and (d) at the hybrid ITO|oligo-BT2T4 interface, as a function of the applied magnetic field orientation (solid vs. dashed lines, the same color legend as before for the film configuration); (e) at the hybrid ITO|c-PEDOT interface (the same legend for the magnetic field orientation). | ||
Specular results are obtained working on the enantiomeric oligo-(R)-BT2T4 electrode (Fig. 2d, Table SI.2.1†). Consistent with former studies about spin coherence conservation,32 when increasing the film thickness (by a higher number of electrodeposition cycles) the shift in the Fe(III)/Fe(II) peak potential upon magnet flipping decreases, becoming negligible for thickness around 50 nm (Fig. SI.2.2†).
To further confirm the role of the magnetic field in the observed phenomenon as well as evaluate the impact of its strength on the observed potential shift, a systematic series of CV experiments were carried out by placing different sheet spacers of increasing thickness between the cuvette and the permanent magnet. As expected, the potential difference observed by flipping the permanent magnet exhibited a sharp, regular decrease with increasing distance of the magnet from the ITO|oligo-(S)-BT2T4 interface (Fig. 3 inset, Table SI.2.3†). This is coherent with the complex, non-linear dependence of the strength of the magnetic field on the distance. The same behavior was observed when a different achiral probe, the ferricinium|ferrocene redox couple, was employed with the same set-up previously described, working in acetonitrile solvent (Fig. SI.2.4.1, SI.2.4.2 and Table SI.2.4†). In order to give more generality to the above results, we performed the same experiments with a constant protocol with the chiral thin film c-PEDOT, obtained by electrodeposition of c-EDOT, the relevant structure is shown in Fig. 1b. The latter monomer is still characterized by a C2 symmetry axis but it presents two identical homotopic stereocentres instead of a stereogenic axis. Fig. 2e shows the relevant CV patterns as a function of the magnetic field orientation. Also in this case a splitting was found for E1/2 of the achiral Fe(III)/Fe(II) probe, although smaller than in the previous case (about 0.15 V, Table SI.2.1†). This is in line with the fact that on one hand ICFMMs exhibit more prominent chirality manifestations;42–46 on the other hand it has been shown that monomers with chirality originating from localized stereocentres, but C2-symmetric and undergoing regioregular polymerization, can result in helical packing of predominantly planar chains, with high, although labile, chirality manifestations.47
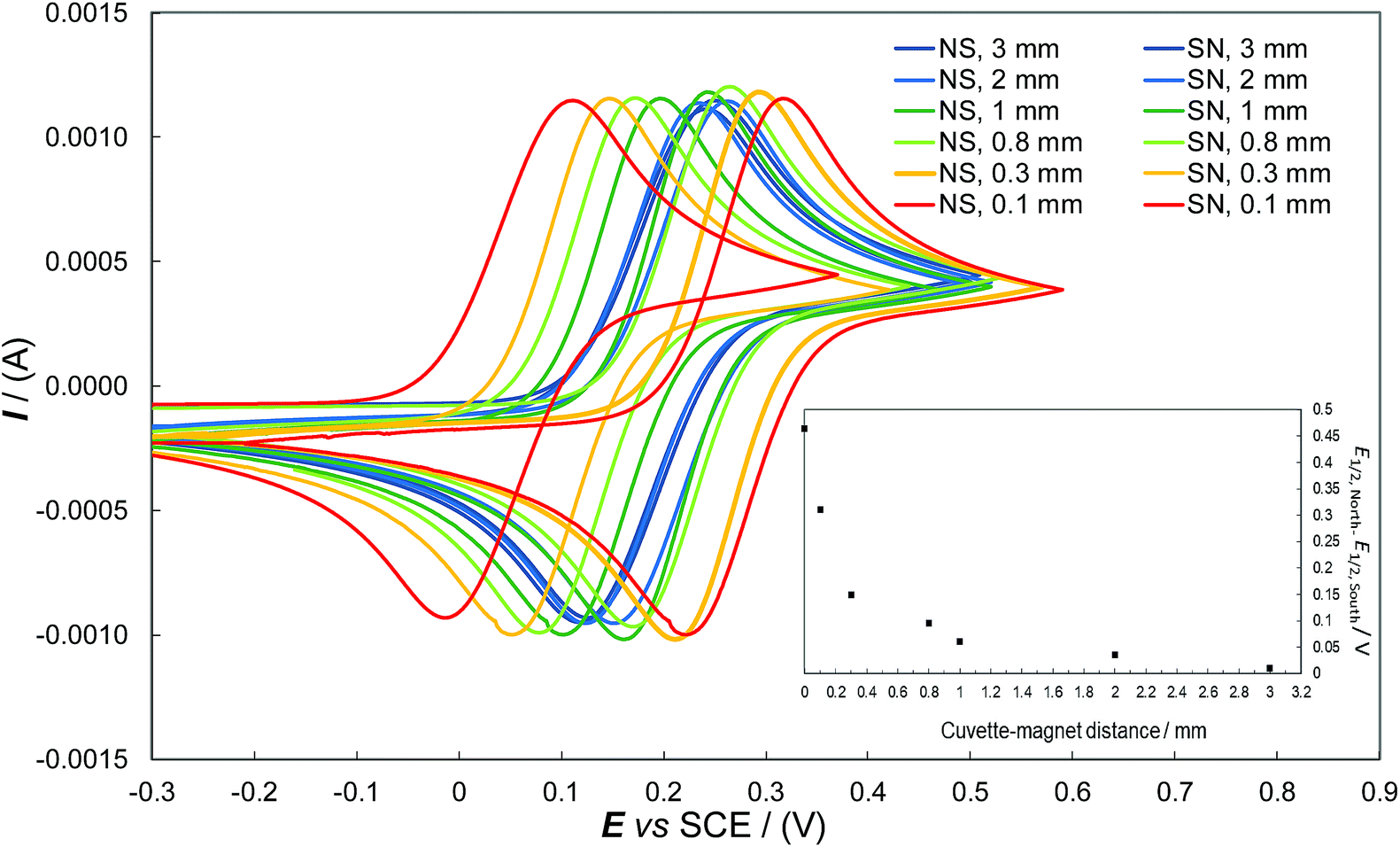 | ||
| Fig. 3 CV patterns recorded at 0.05 V s−1 for the Fe(III)/Fe(II) achiral couple in aqueous solution at the hybrid ITO|oligo-(S)-BT2T4 interface, as a function of the applied magnetic field orientation (north-south vs. south-north) and of the magnetic field strength. | ||
In order to verify that the observed huge potential shift of the redox couple is actually determined by the combination of the effects described above, measurements with achiral PEDOT, under the same conditions, with a constant protocol, were performed. In this case no peak shift for the Fe(III)/Fe(II) couple was observed by flipping the magnet’s orientation (Fig. SI.2.5†).
To rationalize such observations, we could first of all note that the probe’s reversible CV peaks barely undergo a potential shift, their morphology appearing practically unaffected by the magnet’s orientation, suggesting an effect of thermodynamic nature. Moreover, considering that no systematic variations in peak intensities were obtained, unlike previous SDE experiments,38,39 we think that the present phenomenon cannot be explained in terms of a different resistance, ΔR, for the transport of α and β electrons within the chiral film.38,39 In fact, a potential shift originating from an ohmic drop implies, according to E = IR, a distortion in the I vs. E voltammetric signal (the potential drop linearly increasing with the current); moreover, in the case of a chemically reversible signal, forward and backward peaks would be distorted and shifted in opposite directions, with E1/2 remaining practically constant. In contrast, the observed rigid shifts, resulting in neatly different E1/2, could be described as Iconfig,north(E) = Iconfig,south(E + constant) (with “config” standing for the (R)- or (S)-configuration of the chiral film; “north” or “south” standing for the pole close to the electrode surface; and const = 0.15 or 0.46 V, accounting for the constant potential shift observed for c-PEDOT and oligo-BT2T4, respectively).
Such considerations suggest to look for rationalization of our experimental observations in terms of spin-resolved electron energy levels (or, from the electrochemical perspective, of spin-resolved electrochemical potentials48) as a function of the orientation of the external achiral magnetic field as well as of the configuration of the built-in chiral one of the enantiopure film, as follows (by steps of increasing complexity referring to the cases reported in Fig. 2).
(a) Magnetic field only (Fig. 2a)
Application of an external magnetic field should result in a Zeeman splitting of the electron energy levels corresponding to the two possible α and β spin configurations (consistent with their being parallel or antiparallel to the magnetic field), with a small excess of electron population on the lower level, increasing with increasing energy gap and decreasing with temperature. A 180° rotation in the orientation of the magnetic field vector should result in a specular situation (e.g. lower β and higher α instead of lower α and higher β), energetically identical to the initial one in terms of energy levels and gap, as well as population ratio (Scheme 1, top right).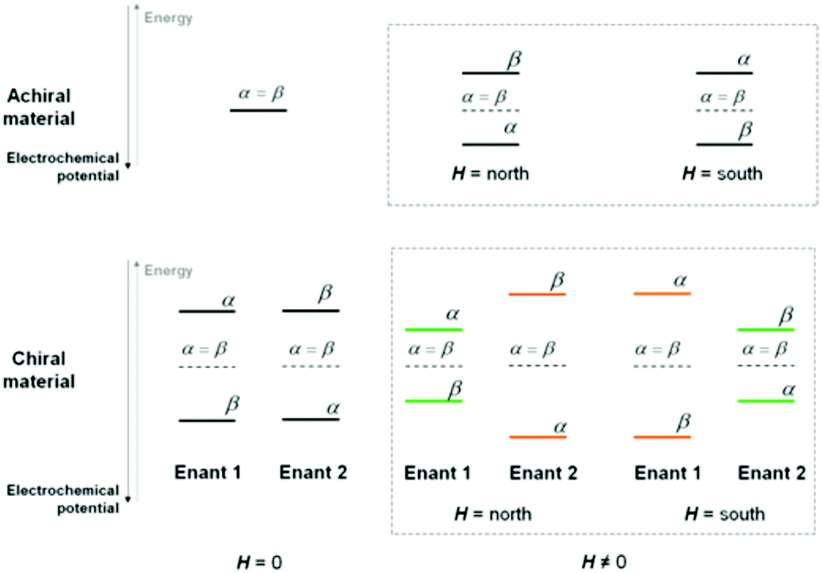 | ||
| Scheme 1 | ||
(b) Chiral film only (Fig. 2b)
A splitting of α and β electron energy levels should also occur within a chiral film phase, consistent with its molecular electrostatic potential49,50 (Scheme 1 bottom left); in this case specular and energetically identical situations must be obtained depending on the (R)- or (S)-film configuration. Incidentally, the splitting should be particularly remarkable in the case of inherently chiral selectors with high macro/supramolecular order.(c) Chiral film plus magnetic field (Fig. 2c and d, Scheme 1 bottom right)
Combining the above two effects, two couples of situations can arise (orange or green in the scheme), depending on the film configuration and magnet orientation (north-south, NS, or south-north, SN): (i) (R)-film/NS magnet and (S)-film/SN magnet and (ii) (R)-film/SN magnet and (S)-film/NS magnet. Importantly, one couple of situations (the orange cases) must correspond to the external applied magnetic field and internal “chiral field”49 in synergy, i.e. favouring the same α or β spin configuration while unfavouring the opposite β or α one. Instead, the other couple (the green cases) must correspond to the external applied magnetic field and internal “chiral field” in opposition, i.e. favouring/unfavouring opposite spin configurations (i.e. with the film favouring α and the magnet favouring β, or vice versa). The two “synergistic” situations are reciprocally specular and energetically equivalent (the only difference being the favoured spin configuration). However, they are energetically different from the two (reciprocally specular and equivalent) “opponent” situations. In particular the “synergistic” cases must correspond to a lower energy for the preferred spin and a higher energy for the unfavoured one, resulting in a higher energy gap between the two levels, and therefore a higher population ratio for the lower level with respect to the higher one. Such a situation can justify the observed half-wave potential shift.Notably, similar outstanding potential differences were formerly observed by some of us on oligo-BT2T4 surfaces “recognizing” the (R)- or (S)-enantiomer of redox probes in solution, without an applied magnetic field.44–46 Indeed, by analogy with the present case, one might now consider explaining such results in terms not only of probe–film molecular interactions but also of spin-related electron energy level splitting, resulting in the chiral film field being modulated by the presence of the (R)- or (S)-probe enantiomer. In any case, comparing the cited experiments with the present ones, our achiral redox probes combined with the external magnetic field appear as mimics of chiral redox probes in the absence of a magnetic field.
This statement is strongly supported by the observation that exactly the same feature has been found for circular dichroism, CD, spectra, which result from different absorption of the L- and R-circularly polarized light components by a chiral molecular probe, are specular according to the sample (R)- or (S)-configuration, and very sharp for inherently chiral chromophores.51,52 Although achiral chromophores are CD inactive, some of them yield increasingly sharp CD spectra under increasing magnetic field strength, specularly inverting the magnetic field orientation.53 Notably, both ferrocyanide and ferrocene, involved in the redox couples considered in the present work, have been long known to give magnetic dichroism.54,55 Also a very preliminary test on the temperature dependence of the spin-related potential splitting effect might be compared with magnetic CD features (Fig. SI.2.6†). Indeed optical and electronic dichroism are strictly connected, as recently pointed out.50
In summary, an achiral redox couple combined with an external magnetic field enables us to highlight the spin filter properties of a chiral film. In particular, with a constant protocol, the magnitude of the potential shift could be exploited as an analytical signal to highlight/compare spin filter properties of chiral thin layers, with the shift sign accounting for film configuration. In this sense the protocol reported here looks as a solution-based equivalent of magnetoresistance determination in all-solid devices (except for the distinction between electron transfer and electron transport, respectively).
An intriguing issue concerns classification of the observed phenomenon within the frame of “true/false chirality” mentioned in the Introduction. Notwithstanding the non-stationary character of the CV experiment, the observation of very large rigid shifts in reversible peak potentials with a constant peak morphology, for different redox probes and different films, points to a thermodynamic effect, which would imply true chirality. Actually, the electron translating in a ballistic (i.e. rototranslational) motion from the probe to electrode (oxidation) or from the electrode to probe (reduction), with its spin projection parallel or antiparallel to the direction of propagation through the truly chiral spin filtering interface, looks as a truly chiral system, with a loss of degeneration for the α and β electron energies (consistent with Naaman's CISS effect34). However, the resulting imbalance of the normal 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 α:β spin ratio in the chiral film phase is not perceivable from the CV signal, which is the same for the two possible film configurations since they correspond to specular, energetically equivalent situations (although one film configuration should correspond to the electron transfer process involving an excess of α electrons and the other an excess of β electrons). In this context the addition of a magnetic field, although in itself not producing true chirality, cooperates in evidencing the above spin filter effect by modifying the couple of energetically equivalent combinations into two couples of energetically different ones, according to its orientation once the chirality of the interphase is fixed. Such a “double splitting” effect by the magnetic field could gradually decrease with increasing magnet distance or film thickness, consistent with the above reported observations.
:50 α:β spin ratio in the chiral film phase is not perceivable from the CV signal, which is the same for the two possible film configurations since they correspond to specular, energetically equivalent situations (although one film configuration should correspond to the electron transfer process involving an excess of α electrons and the other an excess of β electrons). In this context the addition of a magnetic field, although in itself not producing true chirality, cooperates in evidencing the above spin filter effect by modifying the couple of energetically equivalent combinations into two couples of energetically different ones, according to its orientation once the chirality of the interphase is fixed. Such a “double splitting” effect by the magnetic field could gradually decrease with increasing magnet distance or film thickness, consistent with the above reported observations.
It is also worth noting that the experimental outcome reported here is in general agreement with the “electrical magnetochirality anisotropy” results reported by Rikken, Avarvari and coworkers56 concerning single crystals of a bulk molecular conductor, as well as with impressive spin-polarization values observed in charge transmission through single chiral molecules by Mujica and coworkers.50 Moreover, the present enantioselective experimental results obtained in the absence of any ferromagnetic material are definitively consistent with a similar outcome obtained by electrochemically driving, and spin-filtering, the charge transmission through a chiral interface, when using a magnetless non-ferromagnetic (GaN) system.57
Conclusions
Electron transfer on non-ferromagnetic electrodes coated with chiral conductive thiophene-based films resulted in an impressive shift of the half-wave potential of two achiral Fe(III)/Fe(II) redox couples under an external magnetic field, either upon flipping the magnetic field orientation with a constant layer configuration or inverting the chiral layer configuration with a constant field orientation. Notably, unlike former CISS experiments, in the present case (a) the electrode support is non-ferromagnetic and, most importantly, (b) a wide potential shift as a function of the applied magnetic field is observed, rather than a current variation. Such an unprecedented phenomenon could be linked to the peculiar semiconductor films employed here, with chirality features amplified from regioregular units to helical/foldamer macro and/or supramolecular architectures. Actually, the findings reported here parallel our recent enantiorecognition results of chiral probes on inherently chiral films in terms of potential, rather than current, difference.Such spin-related redox potential modulation, obtained in the absence of a magnetic electrode acting as a spin injector, provides striking evidence and an attractive evaluation criterion of the spin selectivity properties of chiral thin films. Such a novel and appealing condensed state system, of easy preparation under ambient conditions, might also be exploited as a “potentiometric” spin-sensitive sensor or a “redox” spin injector, opening the way to innovative spintronic devices.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
C.F. gratefully acknowledges support from the University of Modena and Reggio Emilia (Department of Engineering ‘Enzo Ferrari’), through “Spin Dependent Electrochemistry”, FAR2016, and Prof. Ron Naaman, Weizmann Institute of Science, for deep discussion, ideas and encouragement concerning the application and development of SDE. P. R. M. gratefully acknowledges the support to inherent chirality research, including M. M.'s postdoc grant, by Fondazione Cariplo and Regione Lombardia (2016-0923 RST — Avviso congiunto FC-RL Sottomisura B) rafforzamento (Enhancing VINCE (Versatile INherently Chiral Electrochemistry)). The support of SmartMatLab Centre (cofunded by Regione Lombardia, Fondazione Cariplo and Università degli Studi di Milano, grant No. 2013-1766) and Dr Alessio Orbelli Biroli for profilometry measurements is also gratefully acknowledged. The authors thank Professor Francesco Sannicolò (University of Milano and Laboratori Alchemia S.r.l.) as well as Prof. Sergio Abbate, Prof. Giovanna Longhi and Dr Giuseppe Mazzeo (University of Brescia) for fruitful discussion and suggestions.Notes and references
- L. D. Barron, Nat. Mater., 2008, 7, 691 CrossRef CAS PubMed.
- R. A. Rosenberg, D. Mishra and R. Naaman, Angew. Chem., Int. Ed., 2015, 54, 7295 CrossRef CAS PubMed.
- O. B. Dor, S. Yochelis, S. P. Mathew, R. Naaman and Y. A. Paltiel, Nat. Commun., 2013, 4, 2256 CrossRef PubMed.
- O. B. Dor, S. Yochelis, A. Radko, K. Vankayala, E. Capua, A. Capua, S.-H. Yang, L. T. Baczewski, S. S. Papworth Parkin, R. Naaman and Y. Paltiel, Nat. Commun., 2017, 8, 14567 CrossRef PubMed.
- S. G. Kozlova, M. R. Ryzhikov, N. B. Kompankov and M. S. Zavakhina, J. Phys. Chem. B, 2016, 120, 7517 CrossRef CAS PubMed.
- J. R. Galán-Mascarós, Nat. Phys., 2015, 11, 7 Search PubMed.
- R. Sessoli, M.-E. Boulon, A. Caneschi, M. Mannini, L. Poggini, F. Wilhelm and A. Rogalev, Nat. Phys., 2015, 11, 69 Search PubMed.
- G. A. Printz, Science, 1998, 282, 1660 CrossRef.
- P. L. Popa, N. T. Kemp, H. Majjad, G. Dalmas, V. Faramarzi, C. Andreas, R. Hertel and B. Doudin, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 10433 CrossRef CAS PubMed.
- L. M. Monzon and J. M. D. Coey, Electrochem. Commun., 2014, 42, 38 CrossRef CAS.
- L. M. Monzon and J. M. D. Coey, Electrochem. Commun., 2014, 42, 42 CrossRef CAS.
- G. Saravanan and S. Ozeki, J. Phys. Chem. B, 2008, 112, 3 CrossRef CAS PubMed.
- M. R. Wasielewski, J. Org. Chem., 2006, 71, 5051 CrossRef CAS PubMed.
- J. Lee, H. Ye, S. Pan and A. Bard, J. Anal. Chem., 2008, 80, 7445 CAS.
- A. M. Scott, T. Miura, A. Butler Ricks, Z. E. X. Dance, E. M. Giacobbe, M. T. Colvin and M. R. Wasielewski, J. Am. Chem. Soc., 2009, 131, 17655 CrossRef CAS PubMed.
- J. Potticary, L. R. Terry, C. Bell, A. N. Papanikolopoulos, P. C. M. Christianen, H. Engelkamp, A. M. Collins, C. Fontanesi, G. Kociok-Köhn, S. Crampin, E. Da Como and S. R. Hall, Nat. Commun., 2016, 7, 11555 CrossRef CAS PubMed.
- N. Micali, H. Engelkamp, P. G. van Rhee, P. C. M. Christianen, L. Monsù Scolaro and J. C. Maan, Nat. Chem., 2012, 4, 201 CrossRef CAS PubMed.
- A. Nakamura, J. Ohtsuka, K.-i. Miyazono, A. Yamamura, K. Kubota, R. Hirose, N. Hirota, M. Ataka, Y. Sawano and M. Tanokura, Cryst. Growth Des., 2012, 12, 1141 CrossRef CAS.
- I. Mogi and K. Watanabe, Jpn. J. Appl. Phys., Part 1, 2005, 44, 1 CrossRef.
- I. Mogi and K. Watanabe, Sci. Technol. Adv. Mater., 2008, 9, 024210 CrossRef PubMed.
- I. Mogi and K. Watanabe, Int. J. Electrochem., 2011, 2011, 239637 Search PubMed.
- I. Mogi, R. Morimoto and R. Aogaki, Curr. Opin. Electrochem., 2018, 7, 1 CrossRef CAS.
- L. D. Barron, Chem. Phys. Lett., 1986, 123, 423 CrossRef CAS.
- L. D. Barron, J. Am. Chem. Soc., 1986, 108, 5539 CrossRef CAS.
- L. D. Barron, Rend. Fis. Acc. Lincei, 2013, 24, 179 CrossRef.
- L. D. Barron, Molecular light scattering and optical activity, Cambridge University Press, Cambridge, 2nd edn, 2004, p. 38 Search PubMed.
- B. L. Feringa and R. A. van Delden, Angew. Chem., Int. Ed., 1999, 38, 3418 CrossRef PubMed.
- P. Lazzeretti, J. Phys.: Condens. Matter, 2017, 29, 443001 CrossRef PubMed and references cited.
- K. Banerjee-Ghosh, O. Ben Dor, F. Tassinari, E. Capua, S. Yochelis, A. Capua, S.-H. Yang, S. S. P. Parkin, S. Sarkar, L. Kronik, L. T. Baczewski, R. Naaman and Y. Paltiel, Science, 2018, 360, 1331 CrossRef CAS PubMed.
- C. A. Mead, A. Moscowitz, H. Wynberg and F. Meuwese, Tetrahedron Lett., 1977, 12, 1063 CrossRef.
- I. Žutić, J. Fabian and S. Das Sarma, Rev. Mod. Phys., 2004, 76, 323 CrossRef.
- V. Dediu, M. Murgia, F. C. Matacotta, C. Taliani and S. Barbanera, Solid State Commun., 2002, 122, 181 CrossRef CAS.
- M. Baibich, J. M. Broto, A. Fert, F. Nguyen Van Dau, F. Petroff, P. Etienne, G. Creuzet, A. Friederich and J. Chazelas, Phys. Rev. Lett., 1988, 61, 2472 CrossRef CAS PubMed.
- R. Naaman and D. H. Waldeck, Annu. Rev. Phys. Chem., 2015, 66, 263 CrossRef CAS PubMed.
- K. Ray, S. P. Ananthavel, D. H. Waldeck and R. Naaman, Science, 1999, 283, 814 CrossRef CAS PubMed.
- D. Mishra, T. Z. Markus, R. Naaman, M. Kettner, B. Göhler, H. Zacharias, N. Friedman, M. Sheves and C. Fontanesi, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14872 CrossRef CAS PubMed.
- P. C. Mondal, N. Kantor-Uriel, S. P. Mathew, F. Tassinari, C. Fontanesi and R. Naaman, Adv. Mater., 2015, 27, 1924 CrossRef CAS PubMed.
- P. C. Mondal, C. Fontanesi, D. H. Waldeck and R. Naaman, Acc. Chem. Res., 2016, 49, 2560 CrossRef CAS PubMed.
- A. Kumar, E. Capua, K. Vankayala, C. Fontanesi and R. Naaman, Angew. Chem., Int. Ed., 2017, 56, 14587 CrossRef CAS PubMed.
- P. C. Mondal, W. Mtangi and C. Fontanesi, Small Methods, 2018, 2, 1700313 CrossRef.
- C. Fontanesi, Curr. Opin. Electrochem., 2018, 7, 36 CrossRef CAS.
- S. Arnaboldi, M. Magni and P. R. Mussini, Curr. Opin. Electrochem., 2018, 8, 60 CrossRef CAS.
- S. Arnaboldi, S. Grecchi, M. Magni and P. R. Mussini, Curr. Opin. Electrochem., 2018, 7, 188 CrossRef CAS.
- F. Sannicolò, S. Arnaboldi, T. Benincori, V. Bonometti, R. Cirilli, L. Dunsch, W. Kutner, G. Longhi, P. R. Mussini, M. Panigati, M. Pierini and S. Rizzo, Angew. Chem., Int. Ed., 2014, 53, 2623 CrossRef PubMed.
- S. Arnaboldi, T. Benincori, R. Cirilli, W. Kutner, M. Magni, P. R. Mussini, K. Noworyta and F. Sannicolò, Chem. Sci., 2015, 6, 1706 RSC.
- S. Arnaboldi, T. Benincori, R. Cirilli, S. Grecchi, L. Santagostini, F. Sannicolò and P. R. Mussini, Anal. Bioanal. Chem., 2016, 408, 7243 CrossRef CAS PubMed.
- B. M. W. Langeveld-Voss, R. A. J. Janssen and E. W. Meijer, J. Mol. Struct., 2000, 521, 285 CrossRef CAS.
- I. Žutić, J. Fabian and S. D. Sarma, Rev. Mod. Phys., 2004, 76, 323 CrossRef.
- V. V. Maslyuk, R. Gutierrez, A. Dianat, V. Mujica and G. Cuniberti, J. Phys. Chem. Lett., 2018, 9, 5453 CrossRef CAS PubMed.
- A. C. Aragonès, E. Medina, M. Ferrer-Huerta, N. Gimeno, M. Teixidó, J. L. Palma, N. Tao, J. M. Ugalde, E. Giralt, I. Díez and V. Mujica, Small, 2017, 13, 1602519 CrossRef PubMed.
- N. Berova, L. Di Bari and G. Pescitelli, Chem. Soc. Rev., 2007, 36, 914 RSC.
- J. T. Vázquez, Tetrahedron: Asymmetry, 2017, 28, 1199 CrossRef.
- B. Han, X. Gao, J. Lv and Z. Tang, Adv. Mater., 2018, 1801491 CrossRef PubMed.
- P. J. Stephens, R. L. Mowery and P. N. Schatz, J. Chem. Phys., 1971, 55, 224 CrossRef CAS.
- D. Nielson, D. Boone and H. Eyring, J. Phys. Chem., 1972, 76, 511 CrossRef CAS.
- F. Pop, P. Auban-Senzier, E. Canadell, G. L. J. A. Rikken and N. Avarvari, Nat. Commun., 2014, 5, 375.7 CrossRef.
- A. Kumar, E. Capua and C. Fontanesi, ACS Nano, 2018, 12, 3892 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04126a |
| This journal is © The Royal Society of Chemistry 2019 |