
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnion control of tautomeric equilibria: Fe–H vs. N–H influenced by NH⋯F hydrogen bonding†
Geoffrey M.
Chambers
,
Samantha I.
Johnson
 ,
Simone
Raugei
and
R. Morris
Bullock
*
,
Simone
Raugei
and
R. Morris
Bullock
*
Center for Molecular Electrocatalysis, Pacific Northwest National Laboratory, Richland, Washington 99352, USA. E-mail: morris.bullock@pnnl.gov
First published on 23rd November 2018
Abstract
Counterions can play an active role in chemical reactivity, modulating reaction pathways, energetics and selectivity. We investigated the tautomeric equilibrium resulting from protonation of Fe(PEtNMePEt)(CO)3 (PEtNMePEt = (Et2PCH2)2NMe) at Fe or N. Protonation of Fe(PEtNMePEt)(CO)3 by [(Et2O)2H]+[B(C6F5)4]− occurs at the metal to give the iron hydride [Fe(PEtNMePEt)(CO)3H]+[B(C6F5)4]−. In contrast, treatment with HBF4·OEt2 gives protonation at the iron and at the pendant amine. Both the FeH and NH tautomers were characterized by single crystal X-ray diffraction. Addition of excess BF4− to the equilibrium mixture leads to the NH tautomer being exclusively observed, due to NH⋯F hydrogen bonding. A quantum chemical analysis of the bonding properties of these systems provided a quantification of hydrogen bonding of the NH to BF4− and to OTf−. Treatment of Fe(PEtNMePEt)(CO)3 with excess HOTf gives a dicationic complex where both the iron and nitrogen are protonated. Isomerization of the dicationic complex was studied by NOESY NMR spectroscopy.
Introduction
Tautomerization has been extensively studied, the archetypal example being the keto–enol equilibrium of ketones and aldehydes.1–3 Hydrogen bonding and other non-covalent interactions can profoundly influence tautomeric equilibria, and consequentially, affect the selectivity of catalytic reactions.4 Tautomers play a key role in the structure and reactivity of transition metal complexes. Tautomeric equilibria between dihydrogen and dihydride complexes (eqn (1)) were identified early in the history of H2 complexes.5–11 Subsequent studies examined more deeply the properties of elongated dihydrogen complexes,12,13 and the relationship between dihydrogen and dihydride tautomers.14,15 Tautomeric equilibria between M(η2-SiH4) complexes and M(H)(SiH3) complexes have also been observed.16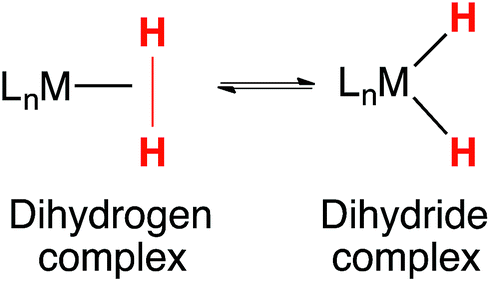 | (1) |
Proton transfers play a prominent role in catalysis. Studies on the kinetic and thermodynamic acidity of metal hydrides17–19 and dihydrogen complexes20 provide a foundation for understanding catalytic reactions that require proton transfers. Pendant amines in diphosphine ligands can function as proton relays,21 leading to electrocatalysts for production of H2 (ref. 22 and 23) and oxidation of H2.24,25 Studies of the intramolecular26 and intermolecular27 proton transfers in these systems has provided insight into the mechanisms of catalysis. Proton transfer between a pendant amine and a metal is often rapid, a feature that enhances catalytic rates. It has seldom been possible to separately observe or characterize the two tautomers.
Ion pairing can influence the thermodynamics and kinetics of transition metal chemistry, particularly those involving proton transfer reactions of metal hydrides.28–31 Bifurcated hydrogen bonding of BF4− to two NH bonds was observed in a crystal structure of a di-iron complex.32 Rauchfuss and co-workers found that excess BF4− shifted the equilibrium between Fe–H and N–H tautomers in a synthetic [FeFe]-hydrogenase model complex, increasing the amount of the ammonium tautomer.33 The role of counterions on reactivity has been studied computationally,34 with much of the work focusing on the influence of counterions on transition states and how they lower kinetic barriers35 or change the regioselectivity in catalysis.36,37 Poli, Shubina and co-workers reported computations showing how specific hydrogen bonding interactions between Mo dihydride complexes and BF4− could be altered by solvent choice, which ultimately determined the thermodynamic equilibrium between Mo dihydrides and dihydrogen complexes.30 Dub and Gordon suggested that hydrogen bonding between BF4− and H2 ligands may influence enantioselective ketone hydrogenations.38 Computations from Heinze and co-workers showed that non-covalent interactions between NO2-substituted gold tetraarylporphyrins and PF6− counterion change the electronic structure from a metal-centered to a ligand-centered radical.39
Hydrogen bonding involving metal hydrides is especially interesting, as the M–H bond can engage in two different types of hydrogen bonding, as discussed in reviews by Shubina and co-workers31,40–42 Crabtree43 and Brammer.44 Intramolecular Ir–H⋯H–N interactions were discovered independently and reported in 1994 by Morris45 and by Crabtree.46,47 In these examples, the M-H bond serves as the weak base (hydrogen bond acceptor). The protic–hydridic attraction (Hδ+⋯Hδ−) in the “dihydrogen bond”48 has been studied in many examples since these interactions were recognized. Intermolecular M–H⋯H–O interactions of metal hydrides have been characterized by extensive spectroscopic studies,49 particularly in the interaction of metal hydrides with acidic alcohols. Studies of these dihydrogen bonds provide insights into details of hydrogen bonding preceding proton transfer reactions.50–52 A Ru–H⋯H–N interaction is cleaved in reaction with CO2, leading to a ruthenium formate complex.53 In iron electrocatalysts for oxidation of H2, an Fe–H⋯H–N dihydrogen bond was characterized by neutron diffraction, providing precise structural characterization.54 NMR spectroscopy can provide evidence for dihydrogen bond formation; Manor and Rauchfuss found that the dihydrogen bond formed by addition of HNMe3+ to a hydride bridging Fe and Ni led to a change of about 2 ppm in the 1H NMR chemical shift.55
Metal hydrides exhibit versatile reactivity patterns; in addition to the hydrogen bonds described above, metal hydrides engage in hydrogen bonding where the M–H bond is the weak acid, or hydrogen bond donor. Early examples were reported for hydrogen bonding between cationic metal hydrides and the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds of phosphine oxides, such as Ph3PO.56,57 Subsequent detailed studies showed that neutral metal hydrides engage in intermolecular hydrogen bonding with phosphine oxides or amines.58 Hydrogen bonding has been found between dihydrogen ligands and BF4− (ref. 59) or OTf− anions.60
O bonds of phosphine oxides, such as Ph3PO.56,57 Subsequent detailed studies showed that neutral metal hydrides engage in intermolecular hydrogen bonding with phosphine oxides or amines.58 Hydrogen bonding has been found between dihydrogen ligands and BF4− (ref. 59) or OTf− anions.60
We report here that protonation of Fe(PEtNMePEt)(CO)3 (PEtNMePEt = (Et2PCH2)2NMe) can occur at either the N, or the Fe, or both. Singly protonated N–H and Fe–H tautomers were isolated, and their structures were characterized by X-ray diffraction. The preference for protonation at N or Fe is strongly influenced by the counterion; addition of BF4− to Fe–H tautomer converts it to the N–H tautomer. Computational studies provide insights into the N–H⋯F hydrogen bonding that strongly influences the tautomeric equilibria. The doubly protonated complex resulting from protonation at both N and Fe has been characterized by spectroscopic and crystallographic studies, and is shown to exist as an interconverting mixture of isomers.
Results and discussion
Synthesis and characterization of Fe(diphosphine)(CO)3 complexes
The iron complex Fe(PEtNMePEt)(CO)3 (Fe0) is prepared by heating toluene solutions of Fe(COT)(CO)3 (COT = cyclooctatetraene) and PEtNMePEt to 110 °C (eqn (2)). It is purified by chromatography and isolated in 78% yield as a pale yellow, mildly air-sensitive microcrystalline crystalline solid. The aniline derivative Fe(PEtNPhPEt)(CO)3 (PEtNPhPEt = (Et2PCH2)2NPh) was prepared in a similar manner. The solid state structure of Fe0 was determined by single crystal X-ray diffraction (Fig. 1). The iron adopts a trigonal bipyramidal geometry with the diphosphine spanning an axial and equatorial site.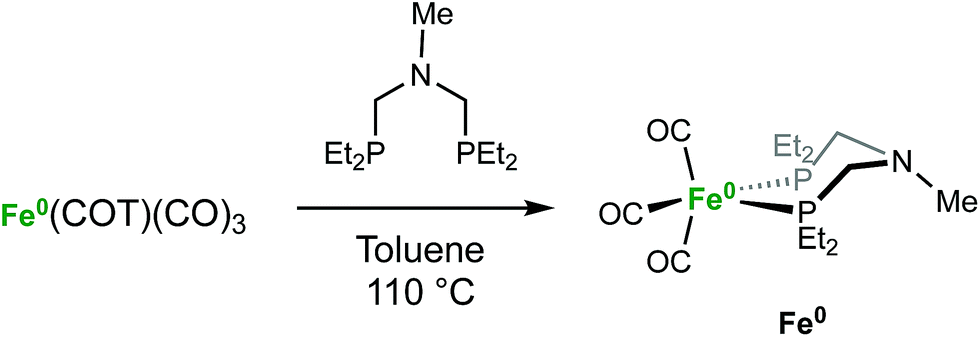 | (2) |
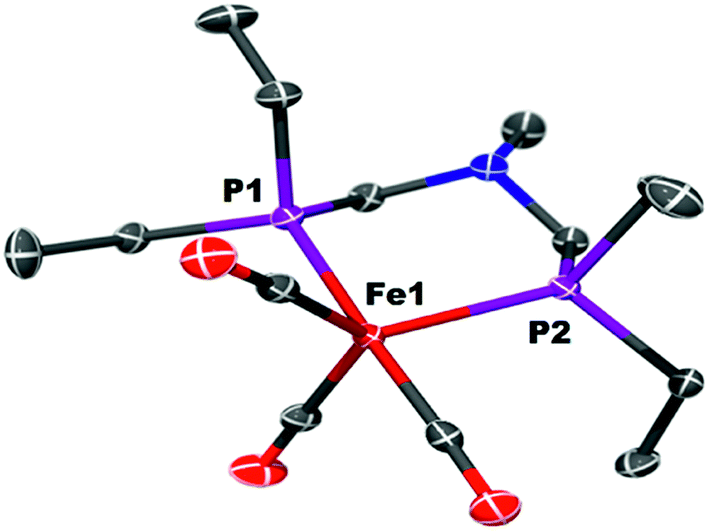 | ||
| Fig. 1 Solid state structure of Fe(PEtNMePEt)(CO)3 (Fe0). Thermal ellipsoids shown at the 50% probability level. Hydrogen atoms are omitted. | ||
Only one phosphorus resonance is observed in the 31P{1H} NMR spectra of Fe0 at room temperature, indicating that the structures are fluxional in solution. The 1H NMR spectrum of Fe0 shows a single resonance for the N–Me protons and the PCH2N methylene groups, consistent with rapid inversion of the nitrogen center. The IR spectrum of Fe0 in CH2Cl2 solution has three bands (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) CO = 1976, 1900, 1873 cm−1).
CO = 1976, 1900, 1873 cm−1).
Protonation and tautomerization studies
Protonation of Fe0 with [(Et2O)2H]+[B(C6F5)4]− occurs at the metal to give a cationic iron hydride complex (Scheme 1). After crystallization from Et2O/n-pentane, the hydrides [Fe(PEtNMePEt)(CO)3H]+[B(C6F5)4]− ([FeH]+[B(C6F5)4]−) and [Fe(PEtNPhPEt)(CO)3H]+[B(C6F5)4]− were isolated as white solids. The structure of [FeH]+[B(C6F5)4]− was determined by X-ray crystallography (Fig. 2). The metal hydride resonance of [FeH]+ is observed in the 1H NMR spectrum as a triplet at δ −9.27 (t, 2JPH = 45 Hz). Treatment of solutions of the iron hydrides with NEt3 gives the corresponding neutral Fe(0) compounds, demonstrating reversible protonation.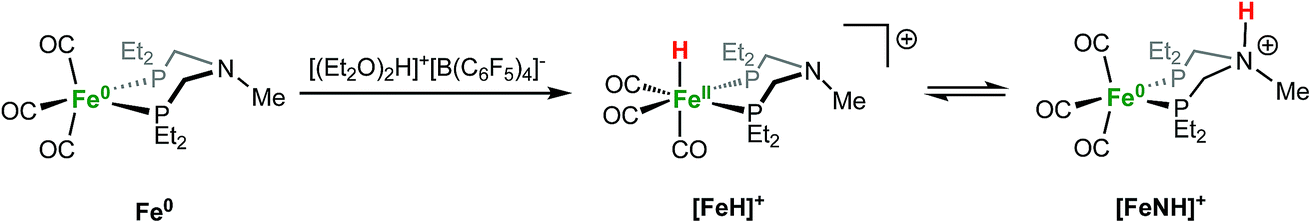 | ||
| Scheme 1 | ||
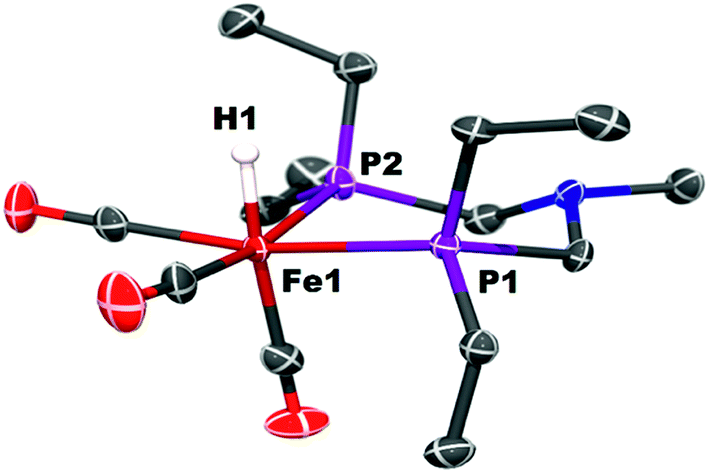 | ||
| Fig. 2 Solid state structure of [Fe(PEtNMePEt)(CO)3H]+[B(C6F5)4]−, [FeH]+. Thermal ellipsoids shown at the 50% probability level. All other hydrogen atoms except the Fe–H are omitted. | ||
Protonation of Fe0 with HBF4·OEt2 in CH2Cl2 gives protonation at both the Fe and the pendant amine. Monitoring the reaction by IR spectroscopy (Fig. 3) shows CO bands at 1992, 1920, and 1894 cm−1 assigned to the amine-protonated isomer [FeNH]+, along with bands for [FeH]+. The shift in the CO bands to higher energy in [FeNH]+ relative to Fe0 is consistent with the overall positive charge of [FeNH]+, and the relatively lower energy of the CO bands compared to [FeH]+ is consistent with the difference in formal oxidation state between the two tautomers.
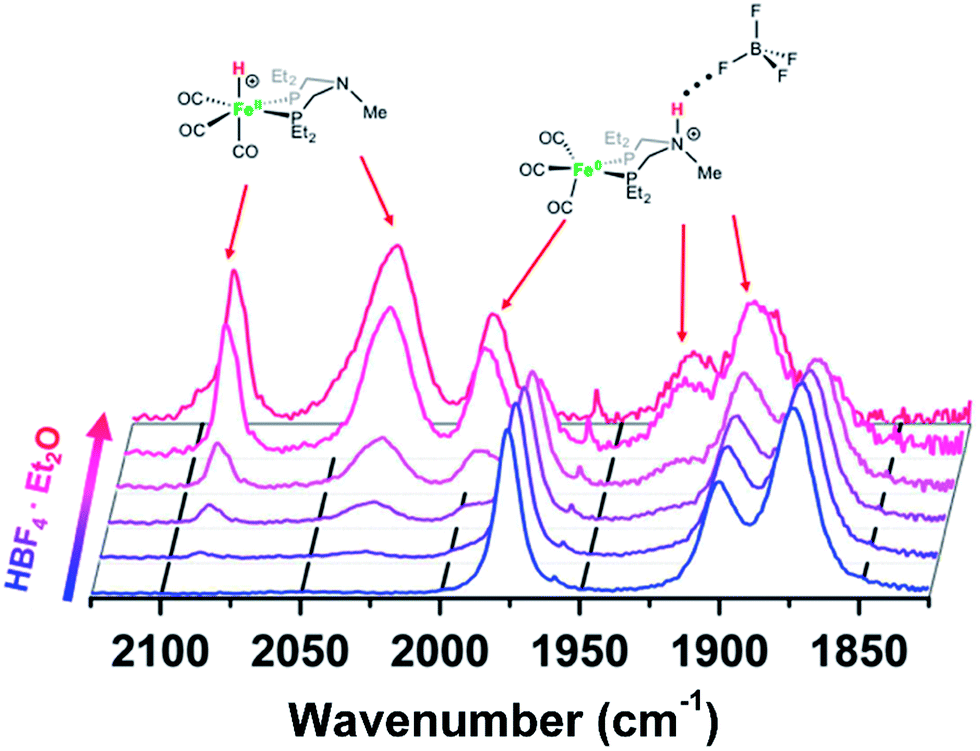 | ||
| Fig. 3 IR spectra (CO region) of Fe0 treated with varying amounts of HBF4·OEt2 in CH2Cl2. Blue = Fe0 in CH2Cl2 solution before addition of acid. Red = spectrum after 1 equiv. of HBF4·OEt2. | ||
Crystals of the protonated amine tautomer, [FeNH]+BF4−, were obtained from the reaction mixture from MeCN/Et2O solutions, and its structure was determined by X-ray crystallography (Fig. 4). The NH⋯F distance was found to be 1.93(1) Å, consistent with the presence of NH⋯F hydrogen bonding of the protonated amine to an F of the BF4− anion. Structural studies of organic compounds with protonated amines with BF4− counterions show NH⋯F hydrogen bonding.61,62 Infrared spectroscopy (KBr) of the crystallized [FeNH]+BF4− gave CO bands similar to those assigned to [FeNH]+ in the solution spectra shown in Fig. 3. The triflate derivate was also prepared by protonation of Fe0 with HOTf, and [FeNH]+OTf− was also characterized crystallographically (Fig. 5).
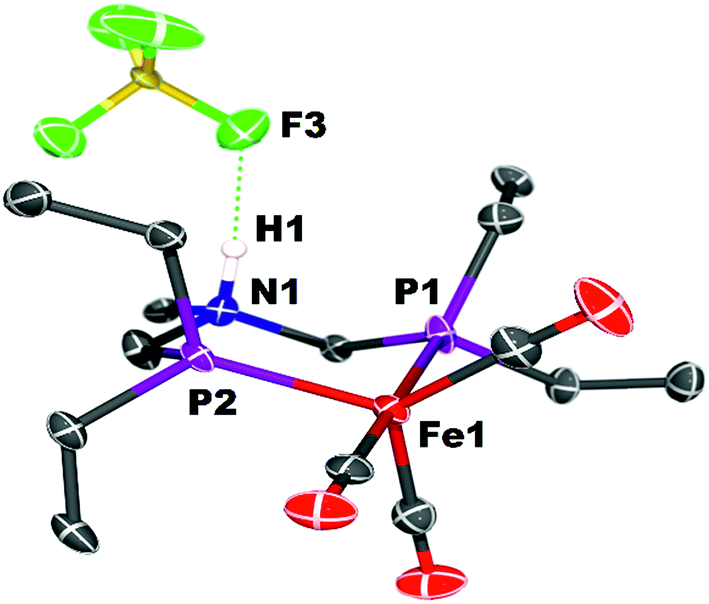 | ||
| Fig. 4 Solid state structure of [FeNH]+BF4−. Thermal ellipsoids shown at the 50% probability level. Hydrogen atoms other than the NH are omitted. | ||
 | ||
| Fig. 5 Solid state structure of [FeNH]+OTf−. Thermal ellipsoids shown at the 50% probability level. All hydrogen atoms have been omitted except for the ammonium hydrogen. | ||
To determine the influence of the anion BF4− on the equilibrium between [FeH]+ and [FeNH]+, solutions of [FeH]+[B(C6F5)4]− were treated with [Et4N]+BF4−. Treatment of CH2Cl2 solutions of [FeH]+[B(C6F5)4]− with less than 1 equiv. of [Et4N]+BF4− gives IR spectra indicating a preference for [FeH]+. As more [Et4N]+BF4− is added, [FeNH]+ becomes the preferred tautomer. At high concentrations of [Et4N]+BF4−, the Fe–H isomer is no longer detected (Fig. 6). These results suggest that hydrogen bonding of N–H to BF4− dramatically influences the equilibrium because of the formation of an NH⋯F hydrogen bond. When monitored by 1H spectroscopy, isolated single crystals of [FeNH]+BF4− in CD2Cl2/THF-d8 (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) solution treated with K+[B(C6F5)4]− give [FeH]+, as determined by 1H NMR spectroscopy, concomitant with precipitation of KBF4 (Fig. S11†). Similarly, the hydride resonance in CD2Cl2 solutions of [FeH]+[B(C6F5)4]− is no longer observed following treatment with [Et4N]+BF4− (Fig. S12†).
:5) solution treated with K+[B(C6F5)4]− give [FeH]+, as determined by 1H NMR spectroscopy, concomitant with precipitation of KBF4 (Fig. S11†). Similarly, the hydride resonance in CD2Cl2 solutions of [FeH]+[B(C6F5)4]− is no longer observed following treatment with [Et4N]+BF4− (Fig. S12†).
 | ||
| Fig. 6 IR spectra of a solution (≈10 mM) of [FeH]+[B(C6F5)4]− in CH2Cl2 treated with increasing amounts of [Et4N]+BF4−. Blue = [FeH]+[B(C6F5)4]−. Green = [FeH]+[B(C6F5)4]− with ∼1 equiv. of [Et4N]+BF4−. Red = [FeH]+[B(C6F5)4]− with ∼5 equiv. of [Et4N]+BF4−. | ||
Recognizing the precedents cited above for hydrogen bonding involving metal hydrides, we considered whether hydrogen bonding of the Fe–H bond of [FeH]+ to BF4− might provide some stabilization. As a more direct probe of that possible interaction, we examined a related complex that does not have a pendant amine in the diphosphine ligand. Addition of 94 mM [Et4N]+BF4− to a CD2Cl2 solution of [FeH(depp)(CO)3]+[B(C6F5)4]− (depp = 1,3-bis(diethylphosphino)propane) led to a shift of the 1H NMR resonance of the hydride from δ −9.028 (triplet, JPH = 44.8 Hz) to δ −9.045. A further shift to δ −9.066 was observed following addition of 280 mM [Et4N]+BF4− to the solution. We interpret these relatively small 1H NMR spectroscopic shifts to indicate weak, if any, hydrogen bonding of the Fe–H to the BF4− anion.
A search of the Cambridge Structural Database was carried out for cationic Fe hydrides with two or more Fe–P bonds and a BF4− counterion. The structure63 of {FeH[P(CH2CH2PPh2)3]}+BF4− reveals a Fe–H⋯F separation of 2.91 Å, and an Fe–H⋯F separation of 3.40 Å was found64 in [FeH(CO)3(dppf)]+BF4− (dppf = 1,1′-bis(diphenylphosphino)ferrocene). The location of the hydride ligand is usually subject to uncertainty when determined by X-ray diffraction, so that will potentially make these comparisons less precise. Compared to the sum of the van der Waals radii of H and F (about 2.67 Å), these structures indicate that any Fe–H⋯F interactions are, at most, weak. The shortest H⋯F distance in the cationic molybdenum hydride, [Cp*MoH(PMe3)3]+PF6−, is 3.33 Å.65 While that distance is longer than would normally be considered a hydrogen bond, a computational study provided evidence for a weak interaction.
Computational studies of the influence of the anion
Structures of [FeH]+, [FeNH]+, [FeNH]+BF4−, and [FeNH]+OTf− were optimized computationally, and the bonding properties were analyzed in the natural bond orbital (NBO)66 theory framework, as discussed below. Both anions have a similar overall effect on the structure of [FeNH]+. Without any anion, the N–H bond of [FeNH]+ tucks in towards the Fe, with an Fe⋯H distance of 2.51 Å (Fig. S32†). When the counterion is added, the N–H bond moves away from the Fe. For comparison, the Fe⋯H distance is 3.59 Å in [FeNH]+BF4− and 3.69 Å in [FeNH]+OTf−. The computed N⋯F distance of 2.60 Å in [FeNH]+BF4− is nearly identical to the computed N⋯O distance of 2.59 Å in [FeNH]+OTf− (Fig. 7). However, the N–H bond length in [FeNH]+BF4− (1.07 Å) is shorter than in [FeNH]+OTf− (1.10 Å), suggesting a stronger N–H⋯X interaction in the latter.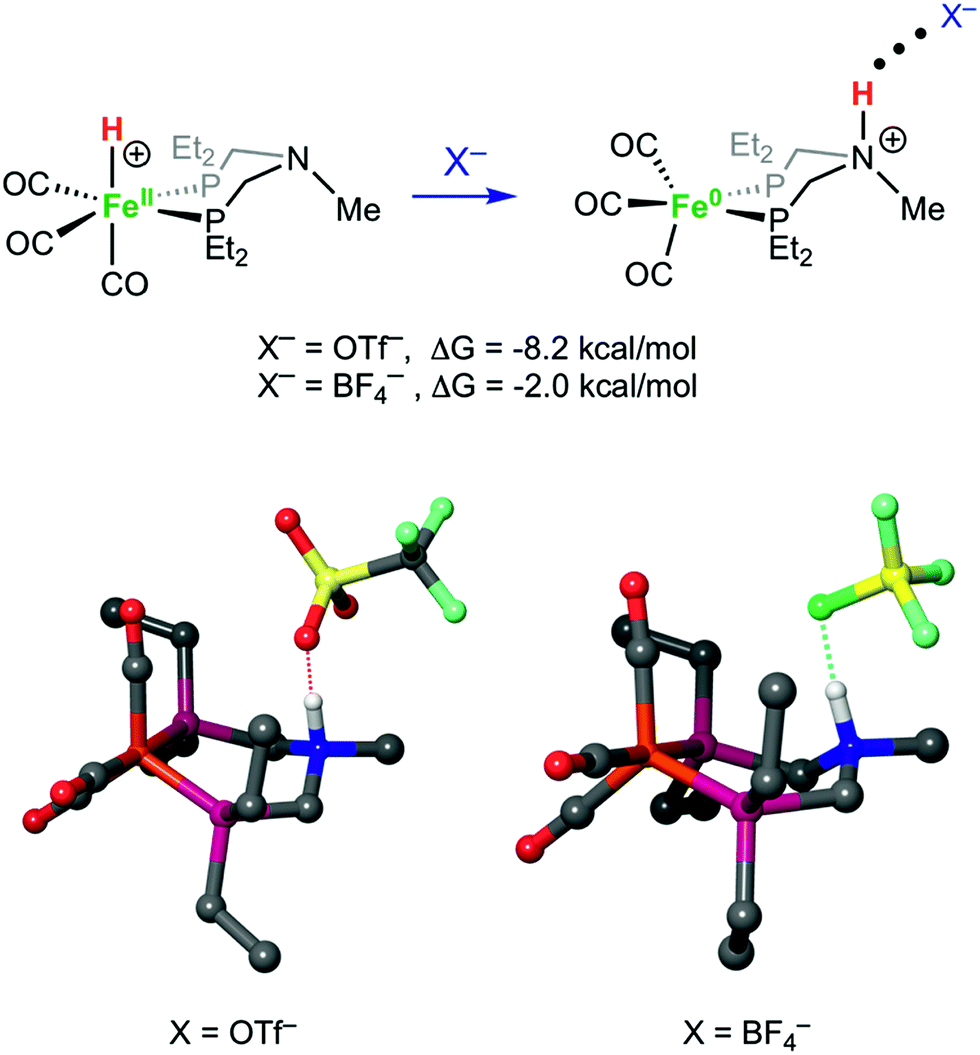 | ||
| Fig. 7 The tautomer with a protonated amine ligand is favored with a OTf− or BF4− counterion, as shown by DFT calculations. The computed structures of [FeNH]+OTf− and [FeNH]+BF4− are shown in the lower part of the figure. Hydrogen atoms have been omitted, except for the protonated ammonium. | ||
Our calculations clearly show that anions can provide energetic stabilization of the N-protonated ligand. The calculated [FeH]+ → [FeNH]+ isomerization free energy of ΔG0 = −2.5 kcal mol−1 shifts to −2.0 kcal mol−1 and −8.2 kcal mol−1, when BF4− or OTf− are added. The nearly isoergic formation of [FeNH]+BF4− is consistent with the experimental observation of both tautomers being present at a 1:1 ratio of the iron complex to BF4−.
The strength of hydrogen bonds between the anion and the protonated amine was calculated in the NBO framework.66 From an electronic standpoint, the formation of a hydrogen bond is accompanied by a sizeable charge transfer from the lone pair n(X) of the hydrogen bond acceptor to the σ*(N–H) anti-bonding orbital of the donor. The energy stabilization due to the hydrogen bond between [FeNH]+ and the counterion is given by the ratio of the off-diagonal components of the Fock matrix and the difference in energy between the two orbitals.66 This analysis also gives an estimate of the amount of charge transferred, qCT, from the donor orbital to the acceptor orbital using eqn (3), wherein ΔE(2)nσ* is the energy lowering, and the denominator represents the difference in orbital energies.66
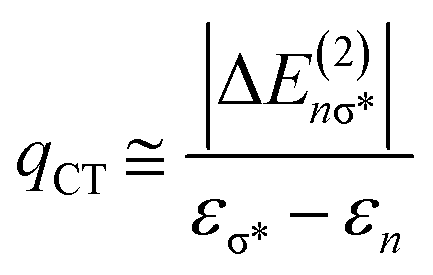 | (3) |
For comparison, the interaction between N–H and one Cl of dichloromethane solvent was calculated. The calculated ΔE(2)nσ* and qCT values are reported in Table 1. The magnitude of the charge transfer from both anions is comparable to values calculated for hydrogen bonding interactions between anions and water.67 The second-order stabilization energy between the complex and BF4− and OTf− is ΔE(2)nσ* = 14.8 and 20.3 kcal mol−1, respectively. The charge transfer and stabilization energy from both anions is orders of magnitude larger than the negligible stabilization energy from dichloromethane solvent. This result implies that the hydrogen bonding interaction is significant, and not merely the result of an interaction between an electronegative atom and the N–H bond. Further details of the contributing interactions are provided in the ESI.†
| Interaction | Total charge transfer, qCT | Stabilization energy, ΔE(2)nσ* (kcal mol−1) |
|---|---|---|
| N–H⋯ClCH2Cl | 0.0004 | 0.2 |
| N–H⋯FBF3 | 0.0220 | 14.8 |
| N–H⋯OTf | 0.0399 | 20.3 |
In contrast to the results with the protonated pendant amine, there is no donation from the lone pair of F to the Fe–H. This is not surprising, in view of the hydridic nature of the Fe–H, compared to the protic nature of the N–H. The atomic charge of the H of the ammonium was calculated to be +0.46, which contrasts greatly with the calculated value of −0.05 when bound to iron. Thus, the interaction between the hydride and the counterion will be repulsive, which is indicated by the energetics (ΔG = 7.9 kcal mol−1) shown in Fig. S33.† Indeed, the small stabilization due to the interaction between the iron hydride bonding orbital, σ(Fe–H), and the four B–F antibonding orbitals, σ*(B–F), does not compensate for the overall electrostatic repulsion. The delocalized nature of the σ(Fe–H)-to-σ*(B–F) interaction is indicated by the geometry, wherein the BF4− anion is close to the hydride, with three fluorine atoms oriented toward the hydride, rather than the directed interaction seen in the geometry of the protonated amine complex. It is important to note that the σ(Fe–H)-to-σ*(B–F) interaction is much smaller than the σ(N–H)-to-σ*(B–F) interaction. Summing all of the stabilization energies gives ΔE(2)σσ* = 3.0 kcal mol−1, for the Fe–H BF4− interaction, which is far less than the ΔE(2)σσ* = 15 kcal mol−1 exhibited by the interaction with between the N–H and BF4−.
Protonation at both Fe and N to give a dication
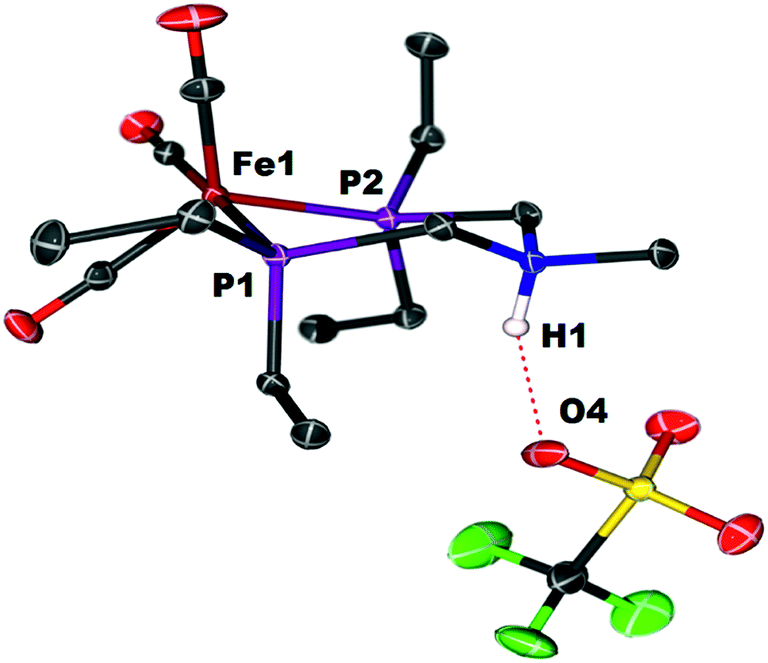
When Fe0 is treated with excess HBF4·OEt2 or HOTf, protonation at both Fe and N is observed, giving a dication (Scheme 2). The doubly protonated complex, [FeHNH]2+[OTf−]2, was characterized crystallographically (Fig. 8), showing one OTf− that is hydrogen bonded to the N–H, and one OTf− counterion that is not interacting. This dicationic complex exhibits a notably shorter NH⋯O distance (1.80(3) Å) relative to the monoprotonated complex [FeNH]+OTf− (1.89(3) Å), suggesting a stronger hydrogen bonding interaction. The IR spectra of the doubly protonated [FeHNH]2+[BF4−]2 and [FeHNH]2+[OTf−]2 complexes featureCO bands shifted to higher energy relative to [FeH]+; the difference is similar to that observed between Fe0 and [FeNH]+.
 | ||
| Scheme 2 | ||
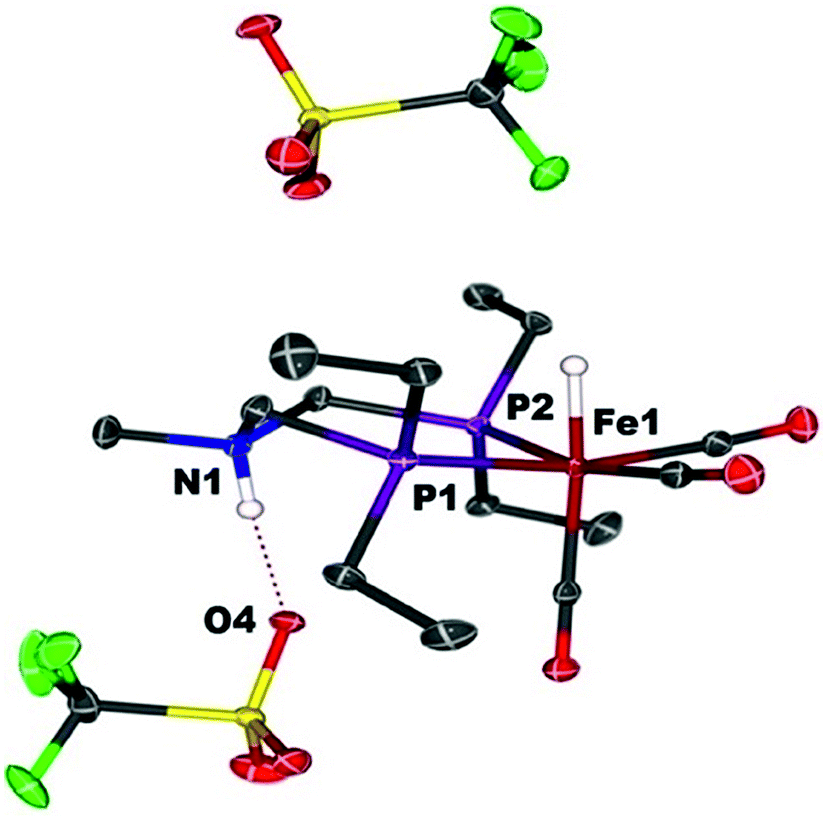 | ||
| Fig. 8 Solid state structure of [FeHNH]2+[OTf−]2. Thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms are omitted, except the N–H and Fe–H. | ||
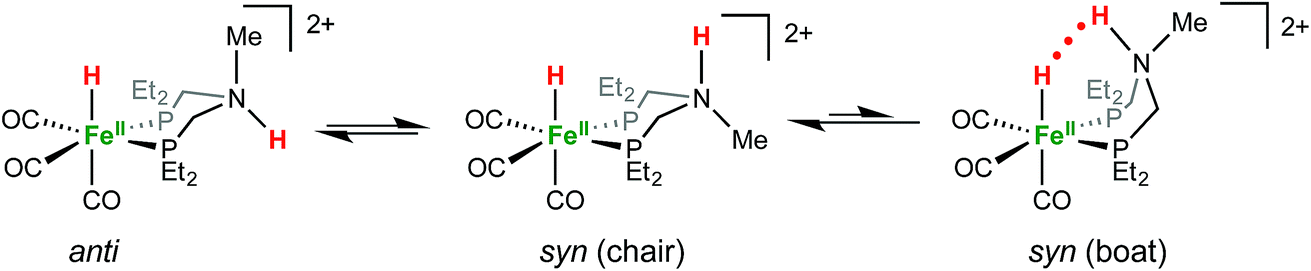
1H NMR spectra in CD3CN solution of crystals of [FeHNH]2+[OTf−]2 reveal multiple isomers. At 25 °C, two distinct species are indicated by two ammonium NH resonances (δ 9.40 and 8.89) and two hydride resonances (δ −9.62 and −9.71). These isomers are assigned to the syn (chair) and anti isomers, differing in the relative orientation of the FeH and NH (Scheme 3). The NOESY spectrum of [FeHNH]2+[OTf−]2 at 25 °C (Fig. S17†) shows an exchange correlation between the FeH and NH resonance of one isomer, proposed to be the syn isomer. An additional exchange correlation is observed between the NH resonances of the two isomers, consistent with interconversion between syn (chair) and anti isomers.
 | ||
| Scheme 3 | ||
At −40 °C, a broad resonance observed at δ 7.11 is proposed to arise from a third isomer in which the FeH and NH are syn and the six-membered ligand ring adopts a boat conformation, bringing the FeH and NH in close proximity. The FeH and NH in the syn (boat) isomer are likely stabilized to some extent by dihydrogen bonding48 between the hydridic FeHδ− and the protic NHδ+.54 Computational analysis shows ΔE(2)σσ* = 2.9 kcal mol−1 in the presence of counterion. A more detailed discussion of this dihydrogen bonding is provided in the ESI.† The NOESY spectrum at −40 °C (Fig. S18†) shows an exchange correlation of the FeH only with the NH resonance of the syn (boat) isomer, presumably occurring through an unobserved Fe(H2)+ intermediate.54,68
Preparation, isolation and reactions of stable 17e− Fe cations
The cyclic voltammogram of Fe0 (Fig. 9) in fluorobenzene shows a reversible one-electron oxidation at −0.37 V vs. Cp2Fe0/+.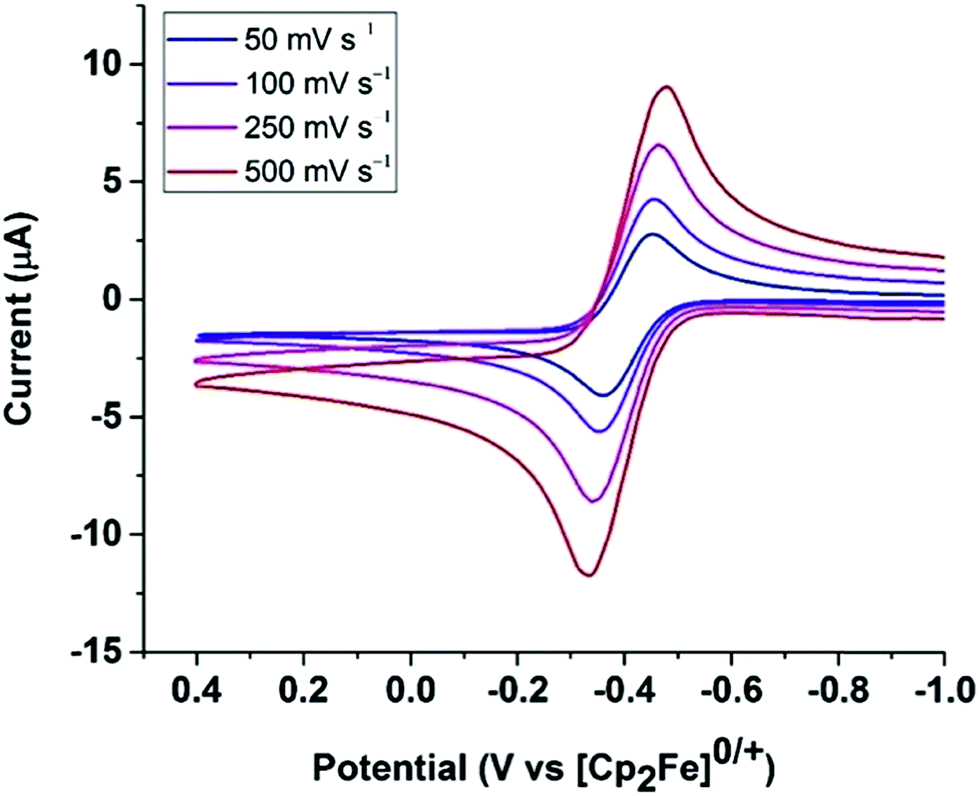 | ||
| Fig. 9 Cyclic voltammogram of Fe0 in PhF under Ar at various scan rates, showing a reversible oxidation at E1/2 = −0.37 V vs. [Cp2Fe]0/+. Conditions: [Bu4N][B(C6F5)4] (100 mM) as the supporting electrolyte, glassy carbon as the working electrode, a silver wire as a pseudo reference, and a Pt wire as the counter electrode. | ||
Oxidation of Fe0 by [Cp2Fe]+[BArF4]− (1 equiv.; ArF = 3,5-bis(trifluoromethyl)phenyl) resulted in an immediate color change of the solution from yellow to dark green. The paramagnetic complex [Fe(PEtNMePEt)(CO)3]+[BArF4]−, [FeI]+, was isolated as air-sensitive dark green crystals. X-Band EPR spectroscopy of [FeI]+ at 22 °C gives giso = 2.05 and a phosphorus hyperfine coupling Aiso = 63 MHz (see ESI†), similar to spectra reported for related complexes.64,69 The 17-electron iron center adopts a square pyramidal geometry, as indicated by X-ray crystallography (Fig. 10). The carbonyl stretching frequencies (CO = 2069, 2007, 1999 cm−1) shift to higher energies compared to 1, consistent with reduced backbonding in the cationic iron complex.
 | ||
| Fig. 10 Solid state structure of [FeI]+[BArF4]−. Thermal ellipsoids are shown at the 15% probability level. X-ray diffraction data were collected at 230 K because of fracture of the crystal at lower temperatures. Two molecules were present in the asymmetric unit; both are similar, and one molecule was chosen arbitrarily to be represented here. | ||
Solutions of [FeH]+BF4− treated with TEMPO (2,2,6,6-tetramethylpiperidin-1-oxyl) immediately become dark green, converting the hydride to [FeI]+, as confirmed by IR spectroscopy (eqn (4)). Rauchfuss and co-workers reported similar reaction proceeding through a proton-coupled electron transfer reaction in studies of [FeFe]-hydrogenase model complexes with pendant amines, in which the protonated amine form is strongly favored.70
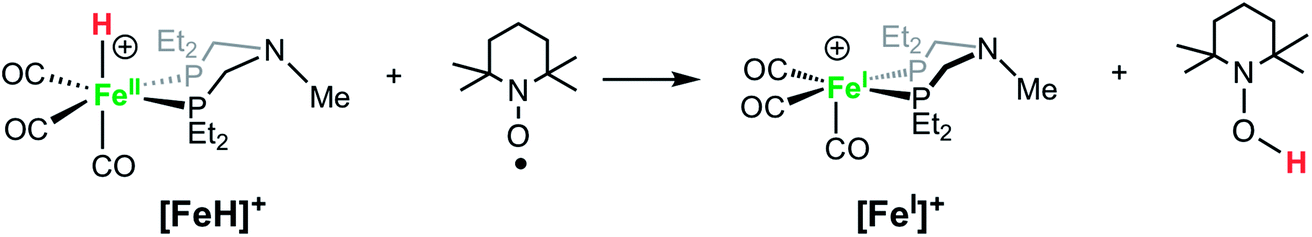 | (4) |
Reactions of FeI complexes with H2 are seldom reported, but Peters and co-workers reported a FeI complex that reacts with H2 to generate a rare FeI(H2) complex.71 We found that dark green solutions of [FeI]+ react slowly with H2 (1 atm) to give pale yellow solutions of the hydride [FeH]+, but the reaction is slow, occurring over a few days. When monitored by 1H NMR spectroscopy, the broad resonances of the paramagnetic complex [FeI]+ decrease, and new resonances appear, corresponding to the iron hydride [FeH]+, but unidentified side products are observed. The yield of [FeH]+ was determined to be approximately 60% by integration against an internal standard. Camara and Rauchfuss reported72 that the reaction of a [FeFe]-hydrogenase complex with H2 is accelerated by a one-electron oxidation, even though that oxidant is not capable of oxidizing the complex. We carried out the reaction of [FeI]+ with H2 with added [Cp2Fe]+, but the reaction rate and yield were similar to that observed with no oxidant added. The very slow reaction with H2 and the low yield of the iron hydride preclude using [FeI]+ as a competent electrocatalyst for H2 oxidation.
Conclusion
Protonation of the iron diphosphine complex Fe(PEtNMePEt)(CO)3 occurs at Fe and the amine, generating tautomers. The iron hydride complex [Fe(PEtNMePEt)(CO)3H]+[B(C6F5)4]− is converted to the N–H tautomer, with a protonated amine, by addition of excess [Et4N]+BF4−. Both isomers were characterized by spectroscopy and X-ray crystallography. Control of the tautomeric equilibrium occurs because of the favorable formation of an NH⋯F hydrogen bonding interaction between the protonated amine and the BF4− anion. Quantum chemical calculations quantified the strength of hydrogen bonding and its influence on the equilibrium. Protonation of Fe(PEtNMePEt)(CO)3 with an excess of HOTf gives a doubly protonated dication with Fe–H and N–H bonds. The dication exists as interconverting isomers in solution, as determined by 2D NMR spectroscopic studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported as part of the Center for Molecular Electrocatalysis, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences. Pacific Northwest National Laboratory (PNNL) is operated by Battelle for the U.S. DOE. We thank Dr Eric Walter for the EPR spectra. EPR experiments were performed using the Environmental Molecular Sciences Laboratory (EMSL), a National Scientific User Facility sponsored by the DOE's Office of Biological and Environmental Research and located at PNNL. Computations were performed using the Cascade supercomputer at EMSL.References
- Y. Chiang, A. J. Kresge, Y. S. Tang and J. Wirz, J. Am. Chem. Soc., 1984, 106, 460–462 CrossRef.
- A. J. Kresge, Acc. Chem. Res., 1990, 23, 43–48 CrossRef.
- Z. Rappoport and S. E. Biali, Acc. Chem. Res., 1988, 21, 442–449 CrossRef.
- H. J. Davis and R. J. Phipps, Chem. Sci., 2017, 8, 864–877 RSC.
- G. J. Kubas, R. R. Ryan and D. A. Wrobleski, J. Am. Chem. Soc., 1986, 108, 1339–1341 CrossRef.
- G. J. Kubas, C. J. Unkefer, B. I. Swanson and E. Fukushima, J. Am. Chem. Soc., 1986, 108, 7000–7009 CrossRef.
- M. S. Chinn and D. M. Heinekey, J. Am. Chem. Soc., 1987, 109, 5865–5867 CrossRef.
- M. S. Chinn and D. M. Heinekey, J. Am. Chem. Soc., 1990, 112, 5166–5175 CrossRef.
- X.-L. Luo and R. H. Crabtree, J. Am. Chem. Soc., 1990, 112, 6912–6918 CrossRef.
- K. A. Earl, G. Jia, P. A. Maltby and R. H. Morris, J. Am. Chem. Soc., 1991, 113, 3027–3039 CrossRef.
- G. J. Kubas, Chem. Rev., 2007, 107, 4152–4205 CrossRef PubMed.
- J. K. Law, H. Mellows and D. M. Heinekey, J. Am. Chem. Soc., 2001, 123, 2085–2086 CrossRef PubMed.
- J. K. Law, H. Mellows and D. M. Heinekey, J. Am. Chem. Soc., 2002, 124, 1024–1030 CrossRef PubMed.
- D. M. Heinekey, A. Lledos and J. M. Lluch, Chem. Soc. Rev., 2004, 33, 175–182 RSC.
- R. H. Morris, Coord. Chem. Rev., 2008, 252, 2381–2394 CrossRef.
- X.-L. Luo, G. J. Kubas, C. J. Burns, J. C. Bryan and C. J. Unkefer, J. Am. Chem. Soc., 1995, 117, 1159–1160 CrossRef.
- E. J. Moore, J. M. Sullivan and J. R. Norton, J. Am. Chem. Soc., 1986, 108, 2257–2263 CrossRef PubMed.
- R. T. Edidin, J. M. Sullivan and J. R. Norton, J. Am. Chem. Soc., 1987, 109, 3945–3953 CrossRef.
- S. S. Kristjánsdóttir and J. R. Norton, in Transition Metal Hydrides, ed. A. Dedieu, VCH, New York, 1991, ch. 9, pp. 309–359 Search PubMed.
- R. H. Morris, Chem. Rev., 2016, 116, 8588–8654 CrossRef PubMed.
- D. L. DuBois, Inorg. Chem., 2014, 53, 3935–3960 CrossRef PubMed.
- A. J. P. Cardenas, B. Ginovska, N. Kumar, J. Hou, S. Raugei, M. L. Helm, A. M. Appel, R. M. Bullock and M. O'Hagan, Angew. Chem., Int. Ed., 2016, 55, 13509–13513 CrossRef PubMed.
- S. Raugei, M. L. Helm, S. Hammes-Schiffer, A. M. Appel, M. O'Hagan, E. S. Wiedner and R. M. Bullock, Inorg. Chem., 2016, 55, 445–460 CrossRef PubMed.
- T. Liu, D. L. DuBois and R. M. Bullock, Nat. Chem., 2013, 5, 228–233 CrossRef PubMed.
- R. M. Bullock and M. L. Helm, Acc. Chem. Res., 2015, 48, 2017–2026 CrossRef PubMed.
- M. O'Hagan, W. J. Shaw, S. Raugei, S. Chen, J. Y. Yang, U. J. Kilgore, D. L. DuBois and R. M. Bullock, J. Am. Chem. Soc., 2011, 133, 14301–14312 CrossRef PubMed.
- M. O'Hagan, M. H. Ho, J. Y. Yang, A. M. Appel, M. Rakowski DuBois, S. Raugei, W. J. Shaw, D. L. DuBois and R. M. Bullock, J. Am. Chem. Soc., 2012, 134, 19409–19424 CrossRef PubMed.
- A. Macchioni, Chem. Rev., 2005, 105, 2039–2074 CrossRef PubMed.
- M. G. Basallote, M. Besora, C. E. Castillo, M. J. Fernandez-Trujillo, A. Lledos, F. Maseras and M. A. Manez, J. Am. Chem. Soc., 2007, 129, 6608–6618 CrossRef PubMed.
- P. A. Dub, N. V. Belkova, O. A. Filippov, J.-C. Daran, L. M. Epstein, A. Lledós, E. S. Shubina and R. Poli, Chem.–Eur. J., 2010, 16, 189–201 CrossRef PubMed.
- N. V. Belkova, L. M. Epstein, O. A. Filippov and E. S. Shubina, Chem. Rev., 2016, 116, 8545–8587 CrossRef PubMed.
- P. Das, J.-F. Capon, F. Gloaguen, F. Y. Petillon, P. Schollhammer and J. Talarmin, Inorg. Chem., 2004, 43, 8203–8205 CrossRef PubMed.
- M. E. Carroll, B. E. Barton, T. B. Rauchfuss and P. J. Carroll, J. Am. Chem. Soc., 2012, 134, 18843–18852 CrossRef PubMed.
- G. Jindal, H. K. Kisan and R. B. Sunoj, ACS Catal., 2015, 5, 480–503 CrossRef.
- M. C. Groenenboom and J. A. Keith, J. Phys. Chem. B, 2016, 120, 10797–10807 CrossRef PubMed.
- M. Arthuis, R. Beaud, V. Gandon and E. Roulland, Angew. Chem., Int. Ed., 2012, 51, 10510–10514 CrossRef PubMed.
- V. M. Lau, W. C. Pfalzgraff, T. E. Markland and M. W. Kanan, J. Am. Chem. Soc., 2017, 139, 4035–4041 CrossRef PubMed.
- P. A. Dub and J. C. Gordon, Dalton Trans., 2016, 45, 6756–6781 RSC.
- S. Preiß, J. Melomedov, A. Wünsche von Leupoldt and K. Heinze, Chem. Sci., 2016, 7, 596–610 RSC.
- N. V. Belkova, L. M. Epstein and E. S. Shubina, Eur. J. Inorg. Chem., 2010, 2010, 3555–3565 CrossRef.
- N. V. Belkova, E. S. Shubina and L. M. Epstein, Acc. Chem. Res., 2005, 38, 624–631 CrossRef PubMed.
- L. M. Epstein and E. S. Shubina, Coord. Chem. Rev., 2002, 231, 165–181 CrossRef.
- R. H. Crabtree, J. Organomet. Chem., 1998, 577, 111–115 CrossRef.
- L. Brammer, Dalton Trans., 2003, 3145–3157 RSC.
- A. J. Lough, S. Park, R. Ramachandran and R. H. Morris, J. Am. Chem. Soc., 1994, 116, 8356–8357 CrossRef CAS.
- E. Peris, J. C. Lee Jr and R. H. Crabtree, J. Chem. Soc., Chem. Commun., 1994, 2573 RSC.
- J. C. Lee Jr, E. Peris, A. L. Rheingold and R. H. Crabtree, J. Am. Chem. Soc., 1994, 116, 11014–11019 CrossRef.
- R. Custelcean and J. E. Jackson, Chem. Rev., 2001, 101, 1963–1980 CrossRef CAS PubMed.
- E. S. Shubina, N. V. Belkova, A. N. Krylov, E. V. Voronstov, L. M. Epstein, D. G. Gusev, M. Niedermann and H. Berke, J. Am. Chem. Soc., 1996, 118, 1105–1112 CrossRef CAS.
- N. V. Belkova, P. O. Revin, L. M. Epstein, E. V. Vorontsov, V. I. Bakhmutov, E. S. Shubina, E. Collange and R. Poli, J. Am. Chem. Soc., 2003, 125, 11106–11115 CrossRef CAS PubMed.
- N. V. Belkova, E. Collange, P. Dub, L. M. Epstein, D. A. Lemenovskii, A. Lledós, O. Maresca, F. Maseras, R. Poli, P. O. Revin, E. S. Shubina and E. V. Vorontsov, Chem.–Eur. J., 2005, 11, 873–888 CrossRef CAS PubMed.
- M. Baya, O. Maresca, R. Poli, Y. Coppel, F. Maseras, A. Lledos, N. V. Belkova, P. A. Dub, L. M. Epstein and E. S. Shubina, Inorg. Chem., 2006, 45, 10248–10262 CrossRef CAS PubMed.
- H. S. Chu, C. P. Lau, K. Y. Wong and W. T. Wong, Organometallics, 1998, 17, 2768–2777 CrossRef CAS.
- T. Liu, X. Wang, C. Hoffmann, D. L. DuBois and R. M. Bullock, Angew. Chem., Int. Ed., 2014, 53, 5300–5304 CrossRef CAS PubMed.
- B. C. Manor and T. B. Rauchfuss, J. Am. Chem. Soc., 2013, 135, 11895–11900 CrossRef CAS PubMed.
- L. M. Epstein, E. S. Shubina, A. N. Krylov, A. Z. Kreindlin and M. I. Rybinskaya, J. Organomet. Chem., 1993, 447, 277–280 CrossRef CAS.
- E. Peris and R. H. Crabtree, J. Chem. Soc., Chem. Commun., 1995, 2179–2180 RSC.
- N. V. Belkova, E. I. Gutsul, O. A. Filippov, V. A. Levina, D. A. Valyaev, L. M. Epstein, A. Lledos and E. S. Shubina, J. Am. Chem. Soc., 2006, 128, 3486–3487 CrossRef CAS PubMed.
- M. Schlaf, A. J. Lough, P. A. Maltby and R. H. Morris, Organometallics, 1996, 15, 2270–2278 CrossRef CAS.
- T. P. Fong, C. E. Forde, A. J. Lough, R. H. Morris, P. Rigo, E. Rocchini and T. Stephan, J. Chem. Soc., Dalton Trans., 1999, 4475–4486 RSC.
- K. Gotoh, R. Ishikawa and H. Ishida, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, o4016–o4017 CrossRef CAS.
- G. R. Juan Carlos, G. P. Carlos, N. Heinrich and F. P. Angelina, Eur. J. Inorg. Chem., 2004, 2004, 601–611 CrossRef.
- C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777–9780 CrossRef CAS PubMed.
- M. R. Ringenberg, F. Wittkamp, U.-P. Apfel and W. Kaim, Inorg. Chem., 2017, 56, 7501–7511 CrossRef CAS PubMed.
- M. Baya, P. A. Dub, J. Houghton, J.-C. Daran, N. V. Belkova, E. S. Shubina, L. M. Epstein, A. Lledós and R. Poli, Inorg. Chem., 2009, 48, 209–220 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- L. A. Curtiss, C. A. Melendres, A. E. Reed and F. Weinhold, J. Comput. Chem., 1986, 7, 294–305 CrossRef CAS.
- T. Liu, Q. Liao, M. O'Hagan, E. B. Hulley, D. L. DuBois and R. M. Bullock, Organometallics, 2015, 34, 2747–2764 CrossRef CAS.
- M. J. Therien and W. C. Trogler, J. Am. Chem. Soc., 1986, 108, 3697–3702 CrossRef CAS.
- M. T. Olsen, T. B. Rauchfuss and S. R. Wilson, J. Am. Chem. Soc., 2010, 132, 17733–17740 CrossRef CAS PubMed.
- Y. Lee, R. A. Kinney, B. M. Hoffman and J. C. Peters, J. Am. Chem. Soc., 2011, 133, 16366–16369 CrossRef CAS PubMed.
- J. M. Camara and T. B. Rauchfuss, J. Am. Chem. Soc., 2011, 133, 8098–8101 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic, electrochemical, crystallographic and computational details. CCDC 1810828 for [1]+[BArF4]−, 1810829 for [FeNH]+BF4−, 1810831Fe0, 1810832 for [FeH]+[B(C6F5)4]−, 1849473 for [FeHNH]2+[OTf−]2, 1849476 for [FeNH]+OTf−. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04239j |
| This journal is © The Royal Society of Chemistry 2019 |