
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAlder-ene reactions driven by high steric strain and bond angle distortion to form benzocyclobutenes†
Saswata
Gupta
a,
Yongjia
Lin
b,
Yuanzhi
Xia
 *b,
Donald J.
Wink
a and
Daesung
Lee
*a
*b,
Donald J.
Wink
a and
Daesung
Lee
*a
aDepartment of Chemistry, University of Illinois at Chicago, 845 West Taylor Street, Chicago, Illinois 60607, USA. E-mail: dsunglee@uic.edu
bCollege of Chemistry and Materials Engineering, Wenzhou University, Wenzhou, Zhejiang Province 325035, P. R. China. E-mail: xyz@wzu.edu.cn
First published on 19th December 2018
Abstract
A unique aryne-based Alder-ene reaction to form benzocyclobutene is described. In this process, the thermodynamic barrier to form a four-membered ring is compensated by the relief of the strain energy of an aryne intermediate. On the other hand, the driving force to overcome the high kinetic barrier is provided by the gearing effect of the bulky substituent at the ortho-position of the ene-donor alkene. To maximize the steric strain by the ortho-substituent, a structural element for internal hydrogen bonding is installed, which plays a crucial role for both the hexadehydro Diels–Alder and the Alder-ene reactions. DFT calculations show that the bulky hydrogen bonding element lowers the activation barrier for the Alder-ene reaction by destabilizing the intermediate, which is due to the severe bond angle distortion. The preferred formation of cis-isomers can also be explained by the extent of bond angle distortion.
A variety of chemical transformations have been developed relying on the high reactivity of arynes, which is generally manifested through a strained alkyne, 1,2-diradical, or 1,2-zwitterionic character depending on the reacting counterpart.1 In the presence of silver salt as a catalyst,2 yet another mode of reactivity was revealed, which is best formulated as a 1,2-dicarbene/carbenoid. Among many reactions involving arynes, the Alder-ene reaction has been shown to be one of the most efficient processes wherein arynes behave as a powerful ene-acceptor.3 Our interest in aryne-mediated Alder-ene reactions prompted us to explore both intramolecular and intermolecular Alder-ene reactions of arynes4 generated via the hexadehydro Diels–Alder reaction.5
In our endeavor to further expand the scope of aryne-based Alder-ene reactions, we were intrigued by the possibility of an intramolecular Alder-ene reaction to form a four-membered ring, which is expected to be extremely difficult, and thus has not been reported to the best of our knowledge (Scheme 1). The paucity of four-membered ring formation via a thermal Alder-ene reaction is due to not only the ring-strain of the incipient ring but also the geometrical constraints for the cyclic transition state. In the case of an aryne as an ene-acceptor, however, the relief of its high strain energy may provide a sufficient driving force for the reaction to proceed to form benzocyclobutenes.6 This reagent-free access to compounds containing a benzocyclobutene substructure would be meritorious since they are known as important building blocks for natural products,7 pharmaceutical ingredients8 and advanced materials.9
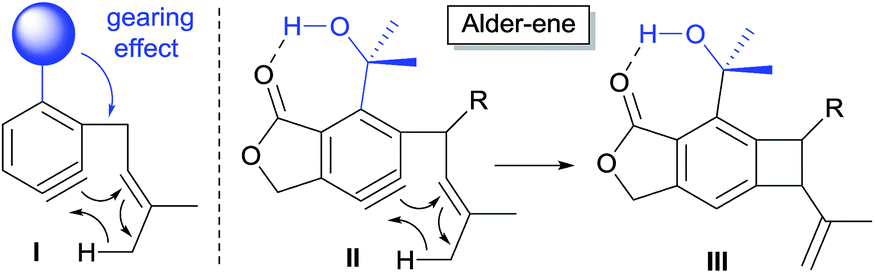 | ||
| Scheme 1 Aryne-mediated Alder-ene driven by hydrogen bonding and steric pressure. | ||
The calculated Gibbs free energy for the Alder-ene reaction suggests that the product formation has a sufficient thermodynamic driving force (ca. 50–63 kcal mol−1). To overcome the expected kinetic barrier, installation of a structural element (I) is expected to be crucial, whereby the steric bulk of the substituent will gear the ene-donor alkene via angle distortion to attain an appropriate transition state.10 In the actual substrate (II) the ortho-isopropanol moiety is expected to serve as an effective gearing element through internal hydrogen bonding. In this hydrogen-bonded conformation, the steric pressure exerted by the gem-dimethyl group against the ene-donor moiety would promote its interaction with the aryne moiety more effectively, which would facilitate the formation of benzocyclobutene (III).
Having these concerns in mind, we designed propiolate-based triyne substrates 1a–h and ynamide-based tetrayne 1i and triyne 1j (Table 1). These substrates contain a substituent that will impose a variable degree of steric interactions with the prenyl group at the transition state of the Alder-ene reaction. A prenyl group was chosen as the ene-donor based on its most favorable capacity for Alder-ene reactions.11 When triyne 1a bearing a trimethylsilyl group (SiMe3, TMS) was heated at 130 °C (toluene), complete conversion was reached after 30 h and the isolated product was identified as the Alder-ene product 2a in 45% yield (entry 1). Unexpectedly, the predominant diastereomer of 2a (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr) was confirmed to have a cis-relationship between the silyloxy and isopropenyl groups. Under the same conditions, triyne 1b containing a triethylsilyl group (SiEt3, TES) showed similar reactivity, affording product 2b in 48% yield (entry 2). The reaction of 1c containing a tert-butyldimethylsilyl group (SiMe2tBu, TBS) was much slower, resulting in 25% conversion after 30 h to produce 2c in 19% yield (entry 3). Triyne 1d containing a triisipropylsilyl group (SiiPr3, TIPS) remained intact even after 48 h (entry 4), which seems to be the consequence of extreme steric bulk of the TIPS group that prohibits the formation of an aryne intermediate. Gratifyingly, triyne 1e containing an isopropanol moiety (CMe2OH) afforded Alder-ene product 2e as a single diastereomer (for nOe data showing the cis-relationship see s32 and s33 of the ESI†) in 65% yield with full conversion within 14 h (entry 5). The 1H NMR of product 2e shows a characteristic sharp singlet at 6.2 ppm suggesting a hydrogen-bonded hydroxyl group. To compare the role of hydrogen bonding, similar substrates without a free hydroxyl group such as 1f and 1g were tested. While the TBS ether 1f showed little conversion (<5%) (entry 7), tbutyl-containing substrate 1g afforded 2g (5:1 dr) in 45% yield at 75% conversion after 40 h (entry 6 and 7). Triyne 1h containing an unsubstituted propiolate moiety did not provide an Alder-ene product although full conversion was achieved within 12 h (entry 8). As expected this is the consequence of relatively facile formation of an aryne intermediate but the Alder-ene reaction did not occur due to the lack of the necessary gearing effect. By the same token, the failure of a tetrayne 1i in producing 2i is the consequence of a small steric bulk of the 1-hexynyl substituent (entry 9). The important role of the hydrogen bonding was further demonstrated by a carbonyl-free triyne 1j containing a CMe2OH substituent, which provided 2j in only 28% yield although full conversion was achieved within 14 h (entry 10).
:1 dr) was confirmed to have a cis-relationship between the silyloxy and isopropenyl groups. Under the same conditions, triyne 1b containing a triethylsilyl group (SiEt3, TES) showed similar reactivity, affording product 2b in 48% yield (entry 2). The reaction of 1c containing a tert-butyldimethylsilyl group (SiMe2tBu, TBS) was much slower, resulting in 25% conversion after 30 h to produce 2c in 19% yield (entry 3). Triyne 1d containing a triisipropylsilyl group (SiiPr3, TIPS) remained intact even after 48 h (entry 4), which seems to be the consequence of extreme steric bulk of the TIPS group that prohibits the formation of an aryne intermediate. Gratifyingly, triyne 1e containing an isopropanol moiety (CMe2OH) afforded Alder-ene product 2e as a single diastereomer (for nOe data showing the cis-relationship see s32 and s33 of the ESI†) in 65% yield with full conversion within 14 h (entry 5). The 1H NMR of product 2e shows a characteristic sharp singlet at 6.2 ppm suggesting a hydrogen-bonded hydroxyl group. To compare the role of hydrogen bonding, similar substrates without a free hydroxyl group such as 1f and 1g were tested. While the TBS ether 1f showed little conversion (<5%) (entry 7), tbutyl-containing substrate 1g afforded 2g (5:1 dr) in 45% yield at 75% conversion after 40 h (entry 6 and 7). Triyne 1h containing an unsubstituted propiolate moiety did not provide an Alder-ene product although full conversion was achieved within 12 h (entry 8). As expected this is the consequence of relatively facile formation of an aryne intermediate but the Alder-ene reaction did not occur due to the lack of the necessary gearing effect. By the same token, the failure of a tetrayne 1i in producing 2i is the consequence of a small steric bulk of the 1-hexynyl substituent (entry 9). The important role of the hydrogen bonding was further demonstrated by a carbonyl-free triyne 1j containing a CMe2OH substituent, which provided 2j in only 28% yield although full conversion was achieved within 14 h (entry 10).
| a 20–25 mg scale of 1. b Determined by 1H NMR of crude mixtures. c Isolated yield. d Diastereomeric ratios. e 52% yield with 75% conversion at 150 °C, 40 h. f Determined by 1H NMR with an internal standard. g Starting material intact at 130 °C but decomposed at 170 °C. h No product observed with decomposition of the starting materials. i Heating at 90 °C. |
|---|

|
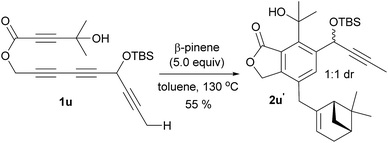
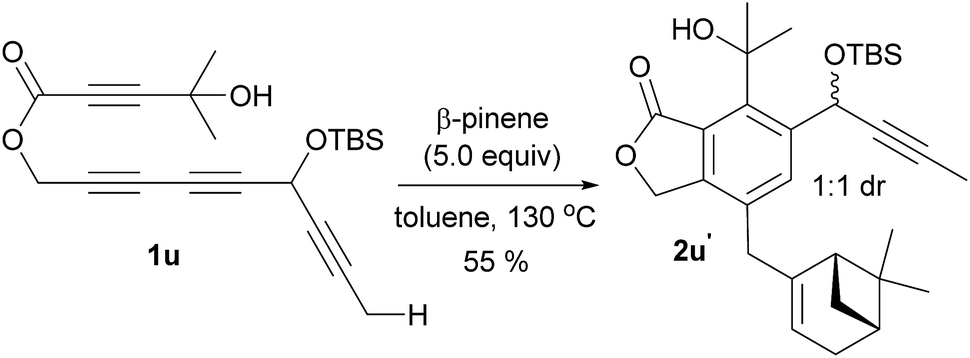
Having identified the crucial elements for the 4-membered-ring forming Alder-ene reaction of aryne species, we next examined the structural requirement of the ene-donor moiety (Table 2). Triyne 1k, identical to 1e but devoid of a silyloxy group, afforded 2k in 72% yield (entry 1). In contrast, a free hydroxyl-containing substrate 1l afforded product 2l in significantly lower yield (30%) (entry 2). We surmise that an alternative hydrogen bonding by the secondary hydroxyl group may perturb the hydrogen bonding of the 3° hydroxyl group at the stage of the ene reaction. This hypothesis is supported by a new broad singlet at 5.6 ppm, which is significantly different from the characteristic signal in other products including 2e. This clearly suggests the beneficial effect of the hydrogen bonding between 3° hydroxyl groups and the lactone carbonyl for the Alder-ene reaction. A structural limitation was recognized from the failure of triyne 1m in generating Alder-ene product 2m (entry 3). We assume that this failure is caused by the severe steric interaction between the aryne moiety and the incipient quaternary carbon center at the transition state of the ene reaction. Unexpectedly, triyne 1n containing a trans-disubstituted ene-donor did not generate 2n instead the starting material was decomposed (entry 4). Triyne 1o containing a geraniol moiety afforded 2o in 67% yield (E/Z, 2.5:1) (entry 5). On the other hand, triyne 1p containing a nerol moiety afforded a single isomer 2p in 62% yield, where only the methyl group reacted to become a methylidene moiety (entry 6). This is consistent with the previous observations that only the distal allylic C–H bonds on the trans-position of ene-donor alkenes can participate in the intramolecular Alder-ene reaction.3a Despite the possibility of forming an 8-membered ring, the exclusive formation of 4-membered ene product 2p from a nerol-derived substrate 1p suggests that a kinetic preference to form a 4-membered ring may override the thermodynamic preference to form an 8-membered ring. Triyne 1q tethered with a 3-methylcyclohexen-1-ol moiety afforded cyclohexane-fused benzocyclobutene 2q in 56% yield (entry 7). The reaction of triyne 1r provided 2r where the gem-dimethyl bridge should control the cis-ring junction stereochemistry, which was confirmed by nOe experiments and X-ray diffraction analysis12 (entry 8). As expected, 2r′ containing a triethylsilyl group was obtained in 46% yield at higher temperature with a longer reaction time (150 °C, 40 h) compared to 2r which was obtained in 68% at 130 °C within 14 h. A cholesterol-derived triyne 1s afforded 2s in 42% yield (entry 9). The relatively low yield of 2s is assumed to be the consequence of a conformationally restricted ene-donor methylene unit within the decaline system. On the other hand, triyne 1t tethered with a cyclooctylidene moiety afforded benzocyclobutene 2t in 69% yield, where the ene-donor methylene conformation should be more flexible to achieve an appropriate transition state for the ene reaction (entry 10). Under the same conditions, triyne 1u bearing an alkyne ene-donor was consumed but the Alder-ene product 2u was not obtained (entry 11). In the presence of β-pinene (5 equiv.) however, 1u afforded product 2u′ in 76% yield via an intermolecular reaction.13 This suggests that the putative aryne intermediate was formed properly from 1r but the Alder-ene reaction between the aryne and the tethered propyne moiety failed. This is presumably because of the difficulty in accessing the required transition state with the alkyne moiety of linear geometry.
|
a Conditions: toluene, 130 °C, 14 h.
b Isolated yield.
c
cis-Diastereomer only.
d Decomposition or polymerization of the aryne intermediates.
e
E/Z-mixture (2.5:1).
f With R = SiEt3, 2r′ was obtained in 46% yield at 150 °C at 40 h.
g Intermolecular Alder-ene reaction with β-pinene (5 equiv.) of 1u provided 2u′ (see ref. 13).
|
|---|

|
Next, we investigated the effect of the tether of triynes on the efficiency of the Alder-ene reaction (Table 3). Triyne 3a with a benzylic substituent afforded 4a as a 1:1 mixture of diastereomers in 62% yield (entry 1). Imide-tethered triynes 3b and 3c containing a gem-dimethyl or a cyclohexylidene moiety provided 4b and 4c in 72 and 56% yield (entries 2 and 3). Triyne 3d with a 3-methyl cyclohexen-1-ol moiety afforded 4d in relatively low yield (42%). In the same vein, arene-tethered triynes 3e and 3f afforded 4e and 4f in 87 and 76% yield (entries 5 and 6). Triyne 3g with an enone tether generate indenone 4g in 56% yield (entry 7). A racemic chiral alcohol 3h with sterically differentiated methyl and tert-butyl groups did not lead to the relative stereochemical induction, thus affording 4h as a 1:1 diastereomer in 87% yield (entry 8).

The competition for intramolecular Alder-ene reactions to form a strained benzocyclobutene versus an intermolecular reaction was examined with triyne 1e (Scheme 2). With 5 equivalents of β-pinene, a 4:1 mixture of 2e and 2e′ was obtained but the ratio changed to 5:95 in neat β-pinene. An internal competition with triyne 3i provided 6-membered ring product 4i along with its derivative 4i′.14 This indicates that the formation of a 6-membered ring is much more favorable than that of a 4-membered ring via the ene reaction.
 | ||
| Scheme 2 Inter vs. intramolecular Alder-ene reaction. | ||
To gain insight into the role of the hydrogen bonding and the unexpected cis-stereoselectivity, the Alder-ene reactions of aryne intermediates IN-1e and IN-1h were compared by DFT calculations (Scheme 3).15 The more facile Alder-ene reaction of IN-1e is due to a smaller angle distortion by ca. 10° for reaching the transition states TS-21e and TS-21e′, compared to ca. 18° for IN-1h to reach TS-21h. This difference then translates to an activation energy difference of more than 11 kcal mol−1 between these two systems. Although trans-isomer 2e′ is more stable than cis-isomer 2e by 1.3 kcal mol−1, the energy of TS-21e is lower than that of TS-21e′ by 1.2 kcal mol−1, which can be attributed to the slightly lower angle distortion in TS-21e, leading to the formation of cis-diastereomer 2e.16
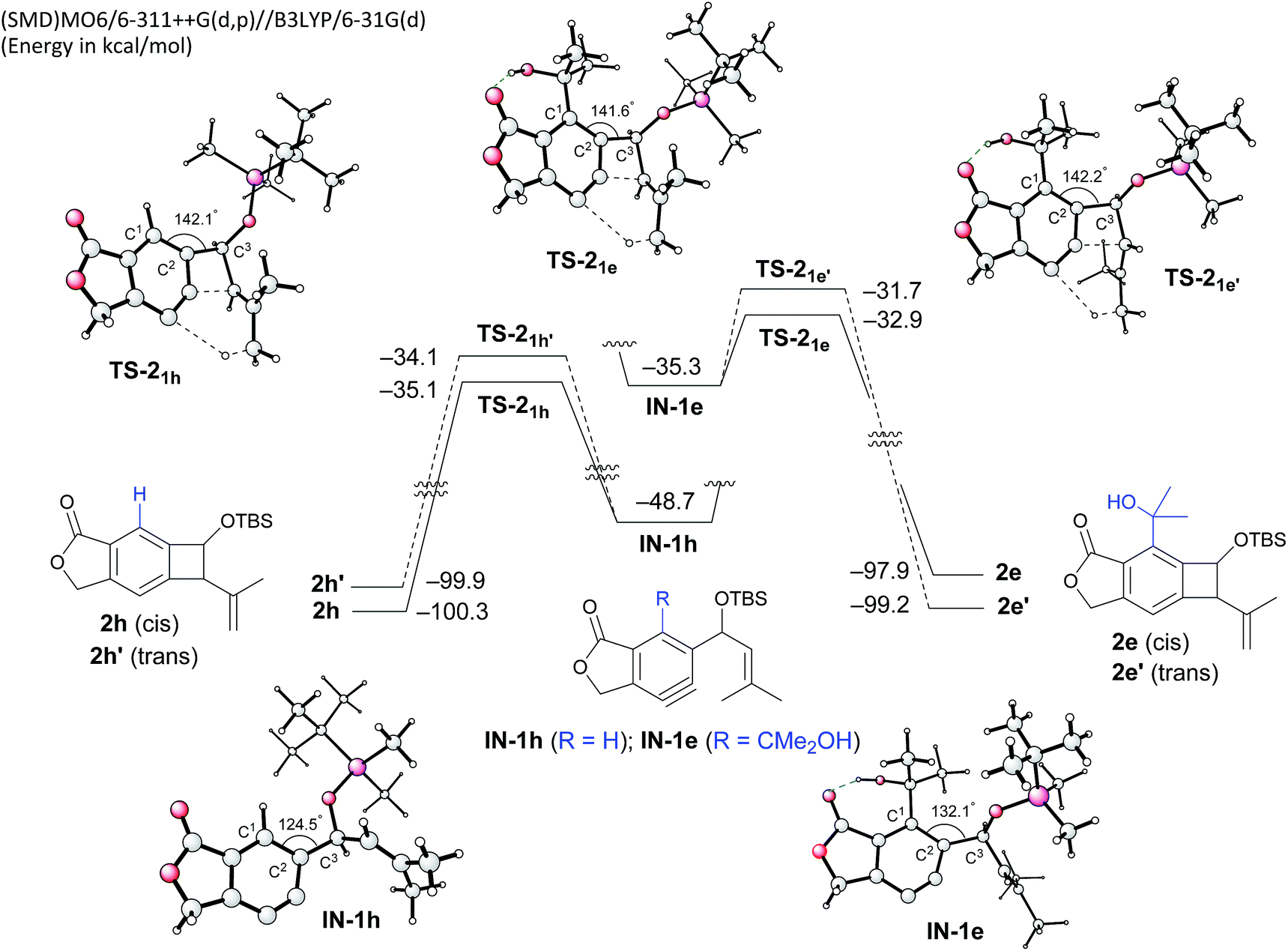 | ||
| Scheme 3 DFT-based analysis of the energy profiles of Alder-ene reactions and the origin of cis-selectivity. | ||
Finally, the benzocyclobutenes generated by Alder-ene reactions could be converted to various other molecular structures via ring expansion or cleavage of the 4-membered ring (Scheme 4). Oxidation of benzocyclobutenol 2l with MnO2 resulted in ring opening followed by two independent subsequent allylic oxidations, generating enal 5a in good yield. However, efficient oxidation of 2l to the corresponding ketone 5b could be achieved with Dess–Martin periodinane. One-carbon insertion into 5b afforded 5c.17 Treating silyl ether 2e with TBAF resulted in the ring-opened product 5d as a mixture (1.5:1) of a terminal and an internal alkene. Also, direct ring expansion of the cyclobutanol moiety of 2o cleanly afforded cyclooctenone 5e under basic conditions.
 | ||
| Scheme 4 Reactions of benzocyclobutene derivatives. Conditions: (a) 10-camphorsulfonic acid, MeOH–CH2Cl2 (1:1), rt, 80%. (b) MnO2 (excess), CH2Cl2, rt, 72%. (c) Dess–Martin periodinane, CH2Cl2, rt, 82%. (d) TMSCHN2, BF3·Et2O, CH2Cl2, −20 °C, 55%. (e) TBAF, THF, rt, 86%. (f) NaH, THF, 50 °C, 81%. | ||
In summary, we have explored the Alder-ene reaction of arynes to form benzocyclobutenes, which was affected by installing a bulky substituent next to the ene-donor alkene moiety. It was found that internal hydrogen bonding is crucial not only to facilitate the hexadehydro Diels–Alder reaction to form an aryne intermediate but also to promote its Alder-ene reaction. DFT calculations revealed that the major role of the bulky hydrogen bonding element is to lower the activation barrier by destabilizing the intermediate, which is due to severe bond angle distortion. The transition state leading to the cis-isomer is more stable than the trans-isomer by 1.2 kcal mol−1, which is the consequence of slightly lower bond angle distortion than the transition state leading to the trans-isomer. This reagent-free method to form substituted benzocyclobutene scaffolds would be useful for natural product synthesis and transition metal-catalyzed transformations.18
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to NSF (CHE-1764141, D. L.) and NSFC (21873074 and 21572163, Y. X.) for financial support. The Mass Spectrometry Laboratory at UIUC is acknowledged.Notes and references
- General reviews on aryne chemistry: (a) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550–3577 CrossRef CAS PubMed; (b) C. M. Gampe and E. M. Carreira, Angew. Chem., Int. Ed., 2012, 51, 3766–3778 CrossRef CAS PubMed; (c) S. S. Bhojgude, A. Bhunia and A. T. Biju, Acc. Chem. Res., 2016, 49, 1658–1670 CrossRef CAS PubMed; (d) T. Roy and A. T. Biju, Chem. Commun., 2018, 54, 2580–2594 RSC.
- Review: R. Karmakar and D. Lee, Chem. Soc. Rev., 2016, 45, 4459–4470 RSC , and references therein.
- (a) T. T. Jayanth, M. Jeganmohan, M.-J. Cheng, S.-Y. Chu and C.-H. Cheng, J. Am. Chem. Soc., 2006, 128, 2232–2233 CrossRef CAS PubMed; (b) D. A. Candito, J. Panteleev and M. Lautens, J. Am. Chem. Soc., 2011, 133, 14200–14203 CrossRef CAS PubMed; (c) D. A. Candito, D. Dobrovolsky and M. Lautens, J. Am. Chem. Soc., 2012, 134, 15572–15580 CrossRef CAS PubMed; (d) Z. Chen, J. Liang, J. Yin, G.-A. Yu and S. H. Liu, Tetrahedron Lett., 2013, 54, 5785–5787 CrossRef CAS; (e) D. Niu and T. R. Hoye, Nat. Chem., 2014, 6, 34–40 CrossRef CAS PubMed; (f) P. Pérez and L. R. Domingo, Eur. J. Org. Chem., 2015, 2015, 2826–2834 CrossRef; (g) J. Zhang, D. Niu, V. A. Brinker and T. R. Hoye, Org. Lett., 2016, 18, 5596–5599 CrossRef CAS PubMed; (h) P. Trinchera, W. Sun, J. E. Smith, D. Palomas, R. Crespo-Otero and C. R. Jones, Org. Lett., 2017, 19, 4644–4647 CrossRef CAS PubMed.
- (a) R. Karmakar, P. Mamidipalli, S. Y. Yun and D. Lee, Org. Lett., 2013, 15, 1938–1941 CrossRef CAS PubMed; (b) S. Gupta, P. Xie, Y. Xia and D. Lee, Org. Lett., 2017, 19, 5162–5165 CrossRef CAS PubMed.
- (a) A. Z. Bradley and R. P. Johnson, J. Am. Chem. Soc., 1997, 119, 9917–9918 CrossRef CAS; (b) K. Miyawaki, R. Suzuki, T. Kawano and I. Ueda, Tetrahedron Lett., 1997, 38, 3943–3946 CrossRef CAS; (c) T. R. Hoye, B. Baire, D. Niu, P. H. Willoughby and B. P. Woods, Nature, 2012, 490, 208–212 CrossRef CAS PubMed; (d) Y. Hu, Y. Hu, Q. Hu, J. Ma, S. Lv, B. Liu and S. Wang, Chem.–Eur. J., 2017, 23, 4065–4072 CrossRef CAS PubMed. For reviews: (e) C. Holden and M. F. Greaney, Angew. Chem., Int. Ed., 2014, 53, 5746–5749 CrossRef CAS PubMed; (f) O. J. Diamond and T. B. Marder, Org. Chem. Front., 2017, 4, 891–910 RSC.
- Other methods for benzocyclobutene synthesis: (a) K. P. C. Vollhardt, Angew. Chem., Int. Ed., 1984, 23, 539–556 CrossRef; (b) M. Iwao, J. Org. Chem., 1990, 55, 3622–3627 CrossRef CAS; (c) I. S. Aidhen and J. R. Ahuja, Tetrahedron Lett., 1992, 33, 5431–5432 CrossRef CAS; (d) F. Toda, K. Tanaka, I. Sano and T. Isozaki, Angew. Chem., Int. Ed., 1994, 33, 1757–1758 CrossRef; (e) T. Matsumoto, H. Yamaguchi, T. Hamura, M. Tanabe, Y. Kuriyama and K. Suzuki, Tetrahedron Lett., 2000, 41, 8383–8387 CrossRef CAS; (f) M. Chaumontet, R. Piccardi, N. Audic, J. Hitce, J.-L. Peglion, E. Clot and O. Baudoin, J. Am. Chem. Soc., 2008, 130, 15157–15166 CrossRef CAS PubMed; (g) P. Álvarez-Bercedo, A. Flores-Gaspar, A. Correa and R. Martin, J. Am. Chem. Soc., 2010, 132, 466–467 CrossRef PubMed; (h) V. R. Yedulla, P. Pradhan, L. Yang and M. K. Lakshman, Eur. J. Org. Chem., 2015, 2015, 750–764 CrossRef CAS PubMed; (i) T. Yano, T. Kawasaki, T. Yuhki, N. Ishida and M. Murakami, Org. Lett., 2018, 20, 1224–1227 CrossRef CAS PubMed.
- (a) W. Oppolzer and K. Keller, J. Am. Chem. Soc., 1971, 93, 3836–3837 CrossRef CAS; (b) F. A. J. Kerdesky, R. J. Ardecky, M. V. Lakshmikantham and M. P. Cava, J. Am. Chem. Soc., 1981, 103, 1992–1996 CrossRef CAS; (c) M. Petit, G. Chouraqui, P. Phansavath, C. Aubert and M. Malacria, Org. Lett., 2002, 4, 1027–1029 CrossRef CAS; for reviews: (d) G. Mehta and S. Kotha, Tetrahedron, 2001, 57, 625–659 CrossRef CAS; (e) A. K. Sadana, R. K. Saini and W. E. Billups, Chem. Rev., 2003, 103, 1539–1602 CrossRef CAS PubMed.
- (a) S. Graf-Christophe, C. Kuehm-Caubere, P. Renard, B. Pfeiffer, A. Pierŕe, S. Léonce and P. Caubére, Bioorg. Med. Chem. Lett., 2000, 10, 2589–2591 CrossRef CAS; (b) A. Tsotinis, P. A. Afroudakis, P. J. Garratt, A. Bocianowska-Zbrog and D. Sugden, ChemMedChem, 2014, 9, 2238–2243 CrossRef CAS PubMed; (c) C. Zhang, F. Li, Y. Yu, A. Huang, P. He, M. Lei, J. Wang, Z. Huang, Z. Liu, J. Liu and Y. Wei, J. Med. Chem., 2017, 60, 3618–3625 CrossRef CAS PubMed.
- (a) G. Sakellariou, H. Ji, J. W. Mays, N. Hadjichristidis and D. Baskaran, Chem. Mater., 2007, 19, 6370–6372 CrossRef CAS; (b) Y. Cheng, J. Yang, Y. Jin, D. Deng and F. Xiao, Macromolecules, 2012, 45, 4085–4091 CrossRef CAS.
- (a) S. K. Ghosh and T. K. Sarkar, Tetrahedron Lett., 1986, 27, 525–528 CrossRef CAS; (b) K. Narasaka, Y. Hayashi and S. Shimada, Chem. Lett., 1988, 17, 1609–1612 CrossRef; a general review on gem-dimethyl effect: (c) M. E. Jung and G. Piizzi, Chem. Rev., 2005, 105, 1735–1766 CrossRef CAS PubMed.
- K. Mikami and M. Shimizu, Chem. Rev., 1992, 92, 1021–1050 CrossRef CAS.
- X-Ray crystallographic data of 2o (CCDC 1843923)†.
- In presence of 5.0 equiv. of β-pinene 1u formed 2u′ by an intermolecular Alder-ene reaction in 55% yield as a 1:1 mixture of diastereomers. This result clearly shows that the formation of aryne intermediate from this substrate is not a problem.
 .
. - The secondary product 4i′ is assumed to be generated from 4ivia a thermal benzylic alkoxy exchange reaction.
- All solvation energies; are relative to that of the starting material. The full detail can be found in the ESI.†.
- The reaction profile of substrate 1g lacking the hydrogen bonding element was also calculated (see ESI† for details).
- J.-H. Youn, J. Lee and J. K. Cha, Org. Lett., 2001, 3, 2935–2938 CrossRef CAS PubMed.
- (a) T. Xu and G. Dong, Angew. Chem., Int. Ed., 2012, 51, 7567–7571 CrossRef CAS PubMed; (b) N. Ishida, S. Sawano, Y. Masuda and M. Murakami, J. Am. Chem. Soc., 2012, 134, 17502–17504 CrossRef CAS PubMed; (c) N. Ishida, N. Ishikawa, S. Sawano, Y. Masuda and M. Murakami, Chem. Commun., 2015, 51, 1882–1885 RSC; (d) F. Juliá-Hernández, A. Ziadi, A. Nishimura and R. Martin, Angew. Chem., Int. Ed., 2015, 54, 9537–9541 CrossRef PubMed; (e) P.-H. Chen, J. Sieber, C. H. Senanayake and G. Dong, Chem. Sci., 2015, 6, 5440–5445 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedure and 1H and 13C spectra. CCDC 1843923. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04277b |
| This journal is © The Royal Society of Chemistry 2019 |