
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simple electrochemical reduction of nitrones to amines†
Eduardo
Rodrigo
and
Siegfried R.
Waldvogel
 *
*
Institut für Organische Chemie, Johannes-Gutenberg-Universität Mainz, Duesbergweg 10–14, 55128 Mainz, Germany. E-mail: waldvogel@uni-mainz.de
First published on 11th December 2018
Abstract
The use of electricity allows the reduction of nitrones containing aromatic and heteroaromatic rings to the corresponding amines. The main advantage of this protocol relies on the fact that only electrons are needed, avoiding the use of different chemicals and harsh procedures, something that reinforces some green aspects. The conversion tolerates different moieties and a large variety of functional groups. In addition, this electrolysis can be performed on a simple undivided beaker-type cell with constant current conditions.
Introduction
The role of nitrones in organic chemistry is very well stablished, fundamentally due to its synthetic versatility.1 This is caused principally by the electrophilicity of the double C–N bond, which allows them to act as dipoles in 1,3-dipolar additions in order to afford nitrogen-containing heterocycles, such as isoxazolidines.2 Another reaction of interest is their reduction, which has primarily been guided to the synthesis of hydroxylamines.1,3 However, nitrones can also be reduced and transformed into the corresponding amines. In fact, the reduction of open nitrones to amines needs two independent steps or at least two different reagents for both reductions.4 The first step, which allows the formation of the imine through a deoxygenation process, is normally performed using a metal,4,5 even though there are also examples using different organic reagents.6 However, many of these methods are limited by harsh procedures, side reactions or lack of chemoselectivity, whereas the second one to yield the final amine has been often carried out through hydrogenation or the use of hydrides.7 Therefore, the reduction of a nitrone into the corresponding amine using one single reagent remains elusive. This is an important aspect, since, for instance, amines are usually synthesized in the pharmaceutical industry through reductive aminations,8 and in some cases, anilines are more expensive than the corresponding aryl nitroderivatives.9Recently, the field of organic electrochemistry has experienced a renaissance,10 a fact that has permitted the opening of new paths in organic synthesis, as well as the development of novel concepts in the field.11 The use of electricity as “single reagent” allows the avoidance of big amounts of oxidizers or reducing agents, something that offers many advantages and benefits from both an ecological and an economic perspective, providing the development of sustainable procedures.
We envisioned that we could carry out the direct reduction of nitrones to amines using electrochemistry. The cleavage of N–O bonds with electricity has been described and it is known for some organic moieties, such as nitrile N-oxides or oximes,12 whereas there are some electrochemical examples of reductive processes with imines, although guided to the synthesis of diamines.13,14
Herein we present an electro-reduction methodology of nitrones for the synthesis of secondary amines. This approach allows the reduction of experimental steps to the minimum and the avoidance of two different reagents for such a process (Fig. 1), being the first time that this double reduction is performed with one single reagent, something which has not been described yet with any other classical chemical. In addition, the green aspects of the transformation are also important, since only electric current is utilized, something very significant in terms of atom economy and from an ecological point of view.
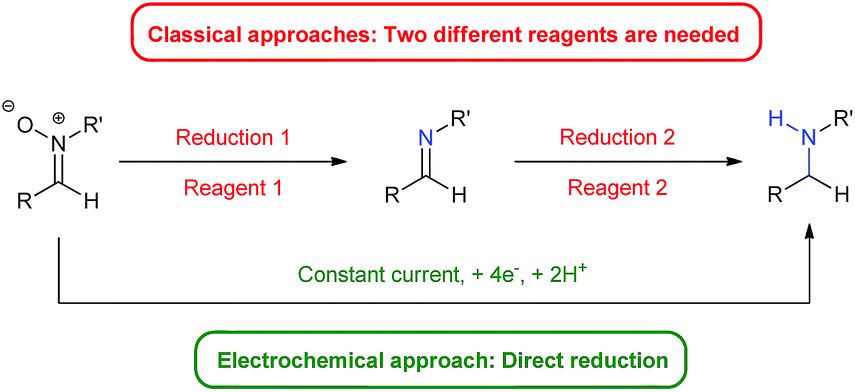 | ||
| Fig. 1 Classical approach vs. electrochemical approach. | ||
Results and discussion
The initial experiments were carried out to adjust the required quantity of charge and to determine the reaction medium. These results are summarized in Table 1.|
|
||||
|---|---|---|---|---|
| Entrya | Solvent | Additivec | Conv. | Yield (2a)d |
| a 0.15 mmol of nitrone were dissolved in 5 mL of solvent. b With Q = 4F, only 70% was achieved; with Q = 5F, 83%. c 0.3 mmol of additive were used. d Yields were determined by NMR using 1,3,5-trimethoxybenzene as internal standard. | ||||
| 1 | CH3CNb | — | >95% | 11% |
| 2 | DMF | — | >95% | 10% |
| 3 | EtOH | — | 94% | 24% |
| 4 | CH3CN/EtOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
— | >95% | 0% |
| 5 | CH3CN/H2O (1:1) |
— | >95% | 29% |
| 6 | CH3CN/H2O (1:1) |
NEt3 | >95% | 42% |
| 7 | CH3CN/H2O (1:1) |
NaOH | >95% | 38% |
| 8 | CH3CN/H2O (1:1) |
Na2CO3 | >95% | 44% |
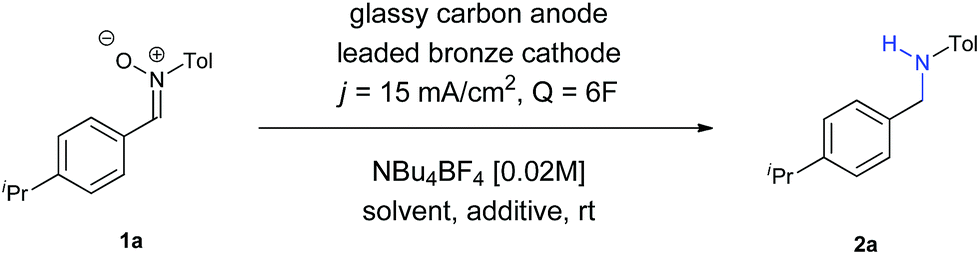
Firstly, we evaluated the quantity of charge needed for completion of the reaction, in CH3CN as solvent, with a glassy carbon anode, a leaded bronze cathode (CuSn7Pb15) and NBu4BF4 as supporting electrolyte, at a current density of 15 mA cm−2. A minimum theoretical amount of 4F was not enough, with some remaining nitrone in the reaction crude. Increasing the charge up to 6F yielded the amine, although the yield was very low (Entry 1). The use of DMF did not afford any improvement (Entry 2), while the utilization of a protic solvent such as EtOH (Entry 3) raised the yield. An equimolecular mixture of CH3CN and EtOH gave a complex mixture with no formation of the amine (Entry 4), whereas the use of CH3CN with another protic solvent such as water was found as optimal (Entry 5). In an electrochemical process for a radical addition of imines to acrylates, the use of triethylamine proved to be efficient to increase the adduct yields.15 When using NEt3 as additive the yield increased up to 42% (Entry 6). Therefore, other bases such as NaOH and Na2CO3 were tested (Entries 7 and 8, respectively), being the last one the one who gave better results, raising the yield up to 44%.
At that point, we decided to test how the current density and the electrodes affected the outcome of the reaction (Table 2). To our surprise, an increase of the current density up to 30 mA cm−2 led to an increase of the yield (compare Entries 1–3), with larger values decreasing both the conversion and the yield (Entry 4). A plausible explanation for this fact is that, the higher the current density is, the faster the reaction is, and therefore, the intermediates and the radical species involved do not evolve into secondary or undesired products so easily. We have recently observed in our research group in the electrochemical reduction of nitrile oxides,16 which present an analogue structure to nitrones, that iminoxyl radicals dimerized to aldazine bis-N-oxides, evolving to aldehydes by loss of nitrogen. This issue seems to be eliminated at higher current densities, probably because the further reduction occurs faster when this value is increased. In addition, both the nitrone and the intermediate imine can be sensitive to hydrolysis in the reaction media, so if the reaction time is reduced, this circumstance is also minimized. Finally, it is also well known that when imines are reduced through the intermediate formation of radicals using metals,17 photocatalysis18 or metal-mediated electrochemistry,14 the diamine is obtained, a fact that could also contribute to decrease the final yield of the reaction.
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Anode | Cathode | Q (F) | j (mA cm−2) | Conv. | Yieldb |
| a 0.15 mmol of nitrone were dissolved in 5 mL of solvent. b Yields were determined by NMR using 1,3,5-trimethoxybenzene as internal standard. c Isolated yield: 60%. | ||||||
| 1 | Glassy carbon | Leaded bronze | 6 | 18 | >95% | 50% |
| 2 | Glassy carbon | Leaded bronze | 6 | 24 | >95% | 51% |
| 3 | Glassy carbon | Leaded bronze | 6 | 30 | 93% | 55% |
| 4 | Glassy carbon | Leaded bronze | 6 | 45 | 87% | 46% |
| 5 | Glassy carbon | Leaded bronze | 6.5 | 30 | <95% | 56% |
| 6 | Glassy carbon | Platinum | 6.5 | 30 | 80% | 19% |
| 7 | Glassy carbon | Lead | 6.5 | 30 | 95% | 65% |
| 8 | Glassy carbon | Lead | 7 | 30 | >95% | 67%c |
| 9 | Graphite | Lead | 6.5 | 30 | >95% | 9% |
| 10 | BDD | Lead | 6.5 | 30 | >95% | 15% |
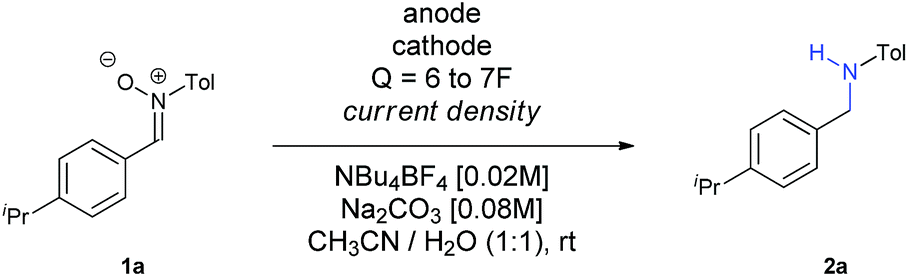
To get full conversion with j = 30 mA cm−2, 6.5F of charge were needed (Entry 5). Other metallic cathodes were tested, being Pb the best one (Entry 7) in comparison to Pt or leaded bronze, though another small increase of charge up to 7F was mandatory to get full conversion (Entry 8). Although the counter electrode doesn't always play an important role in electrochemical transformations, in this case the use of other carbon based electrodes as anodes dropped the yield dramatically (Entries 9 and 10), an issue that we observed recently in the electrochemical reduction of nitro derivatives in aqueous conditions.19 In this case, its function cannot be considered as secondary, since water seems to provide protons for the reduction and final formation of the amine, while it is also decomposed at the same time at the anode. Furthermore, the basic pH of the reaction media favours this process, since the oxidation potential of water is lower at higher pH values.
At this point, we started to evaluate the scope of the reaction. The results are summarized in Scheme 1.
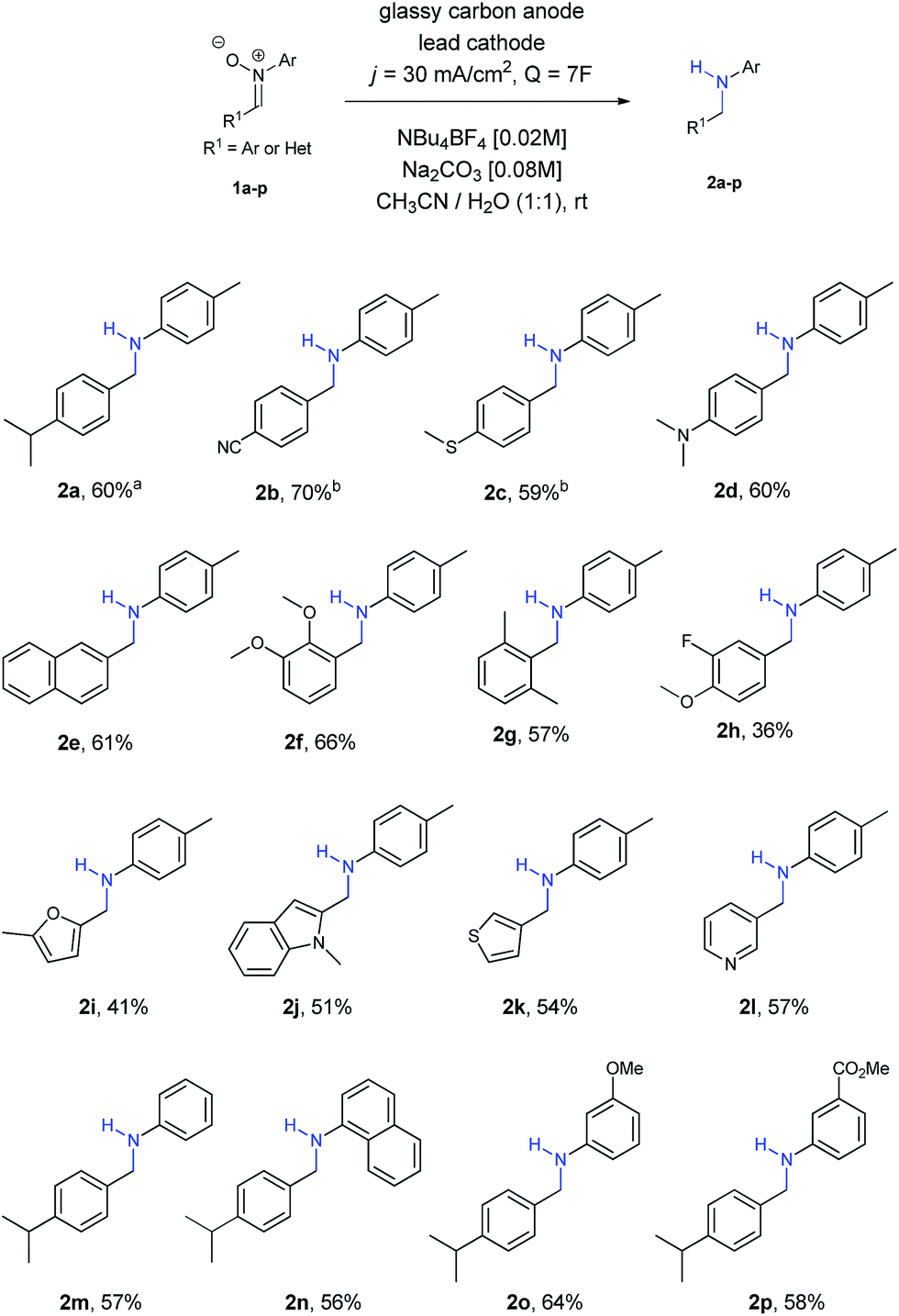 | ||
| Scheme 1 Scope of the reaction. a58% yield in a 25 mL beaker type-cell. b8F of charge were needed. | ||
Firstly, we observed that the reaction went smoothly, no matter the nature of the substituent or the substituted position at the ring, with both EWG and EDG, as well as ortho, meta and para positions (amines 2a–2g). Only in the case of nitrones 1b and 1c a small increase of charge (8F) was needed to achieve the final amines 2b and 2c, owing to solubility reasons. Regarding the use of halogens, only fluorine was tolerated for the reaction, affording amine 2h, although in lower yields than other functionalities.
Different nitrones with heterocycles were also studied (1i–1l), as well as nitrones with different substituents at the ring directly bonded with the nitrogen atom (1m–1p). Amines 2i–2l containing heterocycles and aryl amines 2m–2p were obtained with moderate to good yields in all the examples.
Aliphatic nitrones were also studied, however their low stability in aqueous solutions20 was a major issue and only complex mixtures were observed after isolation. In the case of aliphatic substituents bonded to the nitrogen atom, the reaction was possible, but yields were extremely low and not comparable with the aryl ones.21
In order to prove the usefulness of the process, nitrone 1a was reduced into its corresponding amine 2a in a 25 mL beaker-type cell, with a very similar yield (58%) under the same reaction conditions. The experimental simplicity of the reaction, in which just a two-electrode arrangement in a beaker-type cell under constant current conditions is used, it also important to be pointed out.
A plausible mechanism for the described transformation is proposed in Scheme 2. Four electrons are needed for the reduction. The first two electrons yield the imine after cleavage of the N–O bond. The double bond of the imine is subsequently reduced with two more electrons to afford the corresponding amine after protonation. In order to reinforce this mechanistic proposal, we have performed two experiments. In the first one, we have carried out the reduction of 1a under the optimized conditions and stopped the reaction at Q = 3F, observing the presence of a small amount of imine (ca. 7%) along with 1a and 2a in the crude. Secondly, we took commercial N-benzylideneaniline and applied Q = 0, 1, 2 and 3F under the optimized conditions, recording all the NMR spectra. We could see the formation of the corresponding amine from this imine and some byproducts which also explain the loss of yield and Faraday efficiency (see ESI† for more details).
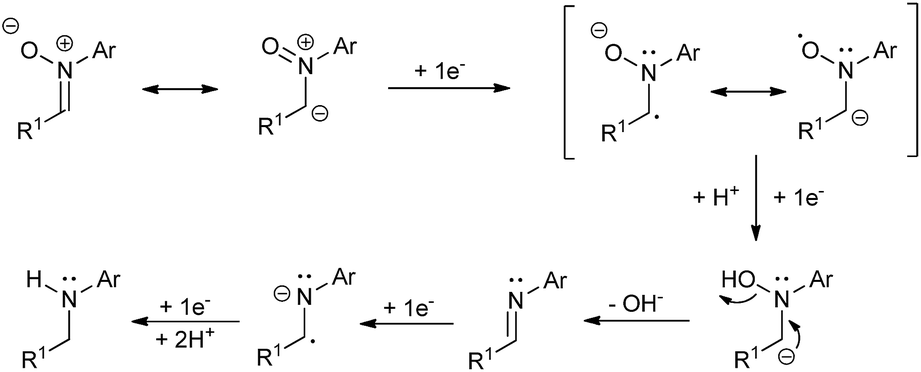 | ||
| Scheme 2 Proposed mechanism for the nitrone reduction. | ||
Conclusions
In summary, we have described an electrochemical method for the reduction of nitrones to amines. A broad scope of aromatic and heteroaromatic amines, containing a large variety of functional groups, was accessible using this methodology. The utilization of electric current allows for the first time the avoidance of two different reagents for this reduction, something that resulted mandatory if this transformation was performed. Furthermore, the simplicity of the method, consisting on an undivided cell under constant current conditions, may open new paths and approaches in terms of amine synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the DFG (Wa 1278/17-1) for financial support. Support by the Advanced Lab of Electrochemistry and Electrosynthesis – ELYSION (Carl Zeiss Stiftung) is gratefully acknowledged. The authors highly appreciate the financial support by the Center for INnovative and Emerging MAterials (CINEMA).Notes and references
- I. A. Grigoriev, in Nitrile Oxides, Nitrones and Nitronates in Organic Synthesis: Novel Strategies in Synthesis, ed. H. Feuer, Wiley, New Jersey, 2nd edn, 2008, pp. 129–434 Search PubMed.
- M. Berthet, T. Cheviet, G. Dujardin, I. Parrot and J. Martinez, Chem. Rev., 2016, 116, 15235 CrossRef CAS PubMed.
- W. R. Bowman and R. J. Marmon, in Comprehensive Organic Functional Group Transformations, ed. A. R. Katrisky, O. Meth-Cohn, C. W. Rees and S. V. Ley, Elsevier Science, Oxford, 1995, vol. 2, p. 344 Search PubMed.
- G. Radivoy, F. Alonso and M. Yus, Synthesis, 2001, 33, 427 CrossRef.
- (a) D. Konwar, R. C. Boruah and J. S. Sandhu, Synthesis, 1990, 22, 337 CrossRef; (b) R. Balicki, Chem. Ber., 1990, 123, 647 CrossRef CAS; (c) P. Ilankumaran and S. Chandrasekaran, Tetrahedron Lett., 1995, 36, 4881 CrossRef CAS; (d) N. Somasundaram and C. Srinivasan, Tetrahedron Lett., 1998, 39, 3547 CrossRef CAS; (e) A. Jeevanadram, C. Cartwright and Y. C. Ling, Synth. Commun., 2000, 30, 3153 CrossRef; (f) A. Saini, S. Kumar and J. S. Sandhu, Synlett, 2006, 17, 395 Search PubMed.
- (a) A. G. Hortmann, J. Y. Koo and C. C. Yu, J. Org. Chem., 1978, 43, 2289 CrossRef CAS; (b) N. Tokitoh and R. Okazaki, Chem. Lett., 1985, 14, 1517 CrossRef; (c) Y. Kawamura, Y. Iwano, Y. Shimizu, Y. Tokai and T. Horie, Chem. Lett., 1994, 23, 707 CrossRef; (d) J. R. Hwu, W. N. Tseng, H. V. Patel, F. F. Wong, D. N. Horng, B. R. Liaw and L. C. Lin, J. Org. Chem., 1999, 64, 2211 CrossRef CAS; (e) M. T. Kumar, S. Jegannathan and S. Dharaniya, Int. J. Chem. Sci., 2012, 10, 2027 CAS.
- (a) R. W. Layer, Chem. Rev., 1963, 63, 489 CrossRef CAS; (b) E. W. Baxter and A. B. Reitz, in Organic Reactions, ed. Larry E. Overman, John Wiley and Sons, New York, 2001, vol. 59 Search PubMed; (c) R. P. Tripathi, S. S. Verma, J. Pandey and V. K. Tiwari, Curr. Org. Chem., 2008, 12, 1093 CrossRef CAS; (d) T. C. Nugent and M. El-Shalzy, Adv. Synth. Catal., 2010, 352, 753 CrossRef CAS.
- S. Warren and P. Wyatt, in Organic Synthesis: The Disconnection Approach, Wiley-Blackwell, Oxford, 2nd edn, 2008, pp. 53–60 Search PubMed.
- For example, the price of 100 g of m-toluidine (99%) in Sigma-Aldrich (November 2018) is 28.60€, while the price of 100 g of m-nitrotoluene (99%) in the same company is only 15.90€. For the ortho derivatives, the price of 100 g of o-toluidine (≥99%) is 41.10€, whereas the price of 100 mL of o-nitrotoluene (≥99%) goes down to 23.30€.
- (a) S. R. Waldvogel and B. Janza, Angew. Chem., Int. Ed., 2014, 53, 7122 CrossRef CAS PubMed; (b) S. R. Waldvogel and S. Möhle, Angew. Chem., Int. Ed., 2015, 54, 6398 CrossRef CAS PubMed; (c) S. R. Waldvogel and M. Selt, Angew. Chem., Int. Ed., 2016, 55, 12578 CrossRef CAS PubMed; (d) M. Navarro, Curr. Opin. Electrochem., 2017, 2, 43 CrossRef CAS.
- For some recent reviews in organic electrochemistry, see: (a) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230 CrossRef CAS PubMed; (b) A. Wiebe, T. Gieshoff, S. Möhle, E. Rodrigo, M. Zirbes and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 5594 CrossRef CAS PubMed; (c) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018 CrossRef PubMed.
- O. Hammerich, in Organic Electrochemistry: Revised and Expanded, ed. O. Hammerich and B. Speiser, CRC Press, Boca Raton, 5th edn, 2016, pp. 599–600 Search PubMed.
- L. Horner and D. H. Skaletz, Liebigs Ann. Chem., 1975, 1210 CrossRef CAS.
- (a) T. Siu, W. Li and A. K. Yudin, J. Comb. Chem., 2001, 3, 554 CrossRef CAS PubMed; (b) K. Sahloul, L. Sun, A. Requet, Y. Chahine and M. Mellah, Chem.–Eur. J., 2012, 18, 11205 CrossRef CAS PubMed; (c) L. Sun, K. Sahloul and M. Mellah, ACS Catal., 2013, 3, 2568 CrossRef CAS.
- T. Shono, N. Kise, N. Kunimi and R. Nomura, Chem. Lett., 1991, 20, 2191 CrossRef.
- M. F. Hartmer and S. R. Waldvogel, Chem. Commun., 2015, 51, 16346 RSC.
- For some recent examples, see: (a) T. Tsukinoki, S. Nagashima, Y. Mitoma and M. Tashiro, Green Chem., 2000, 2, 117 RSC; (b) R. Annunziata, M. Benaglia, M. Caporale and L. Raimondi, Tetrahedron: Asymmetry, 2002, 13, 2727 CrossRef CAS; (c) T. Nishino, Y. Nishiyama and N. Sonoda, Heteroat. Chem., 2002, 13, 131 CrossRef CAS; (d) G. J. Mercer, M. Sturdy, D. R. Jensen and M. S. Sigman, Tetrahedron, 2005, 61, 6418 CrossRef CAS; (e) A. D. E. Prasad, R. A. Flowers and G. Hilmersson, Chem.–Eur. J., 2005, 11, 3279 CrossRef PubMed; (f) E. Vellemäe, O. Tšubrik, S. Mäeorg and U. Mäeorg, J. Chem. Res., 2006, 30, 149 CrossRef; (g) M. Norikazu, K. Tadashi, W. Makoto, M. Sei, M. Isao and S. Makoto, Chem. Lett., 2009, 38, 984 CrossRef; (h) S. Okamoto, J. Q. He, C. Ohno, Y. Oh-iwa and Y. Kawaguchi, Tetrahedron Lett., 2010, 51, 387 CrossRef CAS; (i) M. Soueidan, F. Hélion, J. L. Namy and J. Szymoniak, Tetrahedron Lett., 2011, 52, 1348 CrossRef CAS.
- S. Okamoto, R. Ariki, H. Tsujioka and A. Sudo, J. Org. Chem., 2017, 82, 9731 CrossRef CAS PubMed.
- E. Rodrigo and S. R. Waldvogel, Green Chem., 2018, 20, 2013 RSC.
- S. R. Sandler and W. Karo, in Organic functional group preparations, Academic Press, New York, vol. 12-III, 1972, p. 301, and references cited herein Search PubMed.
- In the case of R = t-Bu, the NMR yield was 15%, and for R = Pr, the NMR yield was lower than 10% (1,3,5-trimetoxybenzene was used as internal standard).
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, characterization data and copies of 1H and 13C NMR spectra. See DOI: 10.1039/c8sc04337j |
| This journal is © The Royal Society of Chemistry 2019 |