
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCargo shuttling by electrochemical switching of core–shell microgels obtained by a facile one-shot polymerization†
Olga
Mergel
 ab,
Sabine
Schneider
a,
Rahul
Tiwari‡
c,
Philipp T.
Kühn
b,
Damla
Keskin
b,
Marc C. A.
Stuart
d,
Sebastian
Schöttner
e,
Martinus
de Kanter
f,
Michael
Noyong
g,
Tobias
Caumanns
h,
Joachim
Mayer
h,
Christoph
Janzen
i,
Ulrich
Simon
g,
Markus
Gallei
e,
Dominik
Wöll
a,
Patrick
van Rijn
b and
Felix A.
Plamper
*aj
ab,
Sabine
Schneider
a,
Rahul
Tiwari‡
c,
Philipp T.
Kühn
b,
Damla
Keskin
b,
Marc C. A.
Stuart
d,
Sebastian
Schöttner
e,
Martinus
de Kanter
f,
Michael
Noyong
g,
Tobias
Caumanns
h,
Joachim
Mayer
h,
Christoph
Janzen
i,
Ulrich
Simon
g,
Markus
Gallei
e,
Dominik
Wöll
a,
Patrick
van Rijn
b and
Felix A.
Plamper
*aj
aInstitute of Physical Chemistry, RWTH Aachen University, Landoltweg 2, 52056 Aachen, Germany
bDepartment of Biomedical Engineering-FB40, University of Groningen, University Medical Center Groningen, A. Deusinglaan 1, Groningen, 9713 AV, The Netherlands
cDWI – Leibniz Institute for Interactive Materials, RWTH Aachen University, Forckenbeckstraße 50, 52056 Aachen, Germany
dGroningen Biomolecular Sciences and Biotechnology Institute, Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands
eErnst-Berl-Institute for Chemical Engineering and Macromolecular Chemistry, Technische Universität Darmstadt, Alarich-Weiss-Straße 4, D-64287 Darmstadt, Germany
fChair for Laser Technology LLT, RWTH Aachen University, Steinbachstr. 15, 52074 Aachen, Germany
gInstitute of Inorganic Chemistry, JARA-SOFT, RWTH Aachen University, Landoltweg 1, 52056 Aachen, Germany
hGFE Central Facility for Electron Microscopy, RWTH Aachen University, Ahornstraße 55, D-52074 Aachen, Germany
iFraunhofer Institute for Laser Technology (ILT), Steinbachstr. 15, 52074 Aachen, Germany
jInstitute of Physical Chemistry, TU Bergakademie Freiberg, Leipziger Straße 29, 09599 Freiberg, Germany. E-mail: felix.plamper@chemie.tu-freiberg.de; Fax: +49-3731-39-3588; Tel: +49-3731-39-2139
First published on 13th December 2018
Abstract
Controlling and understanding the electrochemical properties of electroactive polymeric colloids is a highly topical but still a rather unexplored field of research. This is especially true when considering more complex particle architectures like stimuli-responsive microgels, which would entail different kinetic constraints for charge transport within one particle. We synthesize and electrochemically address dual stimuli responsive core–shell microgels, where the temperature-responsiveness modulates not only the internal structure, but also the microgel electroactivity both on an internal and on a global scale. In detail, a facile one-step precipitation polymerization results in architecturally advanced poly(N-isopropylacrylamide-co-vinylferrocene) P(NIPAM-co-VFc) microgels with a ferrocene (Fc)-enriched (collapsed/hard) core and a NIPAM-rich shell. While the remaining Fc units in the shell are electrochemically accessible, the electrochemical activity of Fc in the core is limited due to the restricted mobility of redox active sites and therefore restricted electron transfer in the compact core domain. Still, prolonged electrochemical action and/or chemical oxidation enable a reversible adjustment of the internal microgel structure from core–shell microgels with a dense core to completely oxidized microgels with a highly swollen core and a denser corona. The combination of thermo-sensitive and redox-responsive units being part of the network allows for efficient amplification of the redox response on the overall microgel dimension, which is mainly governed by the shell. Further, it allows for an electrochemical switching of polarity (hydrophilicity/hydrophobicity) of the microgel, enabling an electrochemically triggered uptake and release of active guest molecules. Hence, bactericidal drugs can be released to effectively kill bacteria. In addition, good biocompatibility of the microgels in cell tests suggests suitability of the new microgel system for future biomedical applications.
Introduction
In recent years, stimuli-responsive systems,1 which can undergo reversible physical or chemical changes upon an internal/chemical or external/physical trigger,2 have gained significant research interest due to their powerful use in e.g. drug delivery3–5 and controlled release systems6,7 and their wide field in potential applications not only in biomedical technologies, but also separation and purification technologies,8 and sensor interfaces.9–11 While internal switching is accompanied by an addition of chemicals (pH, ionic strength, solvent composition), the external switching is possible by application of a magnetic/electric field,12 irradiation, or a change in temperature.13 The advantage of an electrochemical stimulus is the change in composition without any addition of chemicals. Hence the electrochemical stimulus is simple and clean and can be widely applied in biological systems and materials science.10,14–17 In this respect, nanoelectrochemistry has moved into the focus. One aspect is the understanding of electrochemical processes in a single colloidal entity, which can be even of more complex architecture (like for gold nanorods or gold-core/silver-shell nanoparticles).18,19 Less common is the use of polymeric nanoparticles.20 Though redox-active polymers have been known for decades, utilization of an electrochemical stimulus21–23 to switch polymer properties is a rather undiscovered field compared to the vast majority of reports dealing with stimuli such as temperature, light and pH to induce changes in the polymer chain conformation.13,24 This particularly holds for microgels,25 which are crosslinked polymeric particles in the colloidal size range, often with an ability to undergo a volume phase transition (VPT) upon response to the environmental changes.9,11,26 Recent progress in use of “smart” multi-responsive microgels as central component of advanced, functional colloidal materials for controlled drug delivery14 systems and sensors27–31 illustrates the importance and the need of further research in this direction.9 In many cases, temperature-responsive poly(N-isopropylacrylamide) (PNIPAM) acts as a microgel scaffold, which exhibits a VPTT (volume phase transition temperature) in aqueous environment within a narrow temperature range around 32 °C, close to body temperature.26,32In contrast, redox-active gels33 and microgel dispersions are less common. Most instances deal with chemical oxidation of e.g. poly(ferrocenylsilane) (PFS) moieties34–36 or emphasize non-aqueous systems.24 Vancso and co-workers took advantage of redox-switchable PFS in polyelectrolyte multilayer capsules for changing the permeability.37,38 Also vesicle permeability was switched via redox-responsive self-assembly of amphiphilic block copolymers and polyoxometalates.39 Redox active colloids were used for redox flow batteries.40 Injectable conducting hydrogels41 with self-healing properties (based on host–guest self-assembly)39,42 are promising for cardiac tissue repair.43 Alternatively, electrode surfaces were modified with an active layer,44 where the local electrochemistry influences the phase transition of electroactive gel layers45 and vice versa. Hence, the electrochemistry can be affected by the temperature induced phase transition of thin films/brushes, which are attached to electrodes.46–50 However, the electrochemical manipulation of the microgel state and of the swelling/collapse behavior of these dispersed particles has almost been neglected.13 Recently we demonstrated a microgel size manipulation by purely electrochemical means.51 Here the change in size is based on a host/guest interplay: the valency of guest counterions is switched electrochemically and the dimensions of the host microgels are influenced by the difference in complexation behavior of the differently charged counterions. In this case, the molar ratio of host microgel to multivalent counterion is decisive for the electrochemical switching ability. Hence, it would be beneficial to alleviate the need of adjustment of the host and guest quantities. As a remedy, the permanent/covalent attachment of the redox-active groups to the polymer network can be suggested. At the same time, an electrochemical response in size and polarity is expected at any dilution. As a possible avenue to such electrochemically-active systems, ferrocene units (Fc) can be incorporated into the network,52,53 yielding organometallic polymers. Organometallic polymers have attracted considerable attention, as they combine the processing characteristics of organic polymers with the properties common for transition metals, and thus impart unique electrical, optical, magnetic and catalytic properties to these polymers.54–59 Polymers bearing a ferrocene moiety have been subject of research for years and attracted attention due to their variety of promising properties, such as redox, magnetic, photophysical and semiconductive features.24,60–65 Water-soluble polyvinylferrocene (PVFc) derivatives were used to explore redox-controlled drug delivery agents as various water-soluble ferrocenium salts have been shown to display anticancer activity.66,67 Also supramolecular systems based on ferrocene in combination with cyclodextrins were extensively studied with regard to an electrochemical response.10,64 Again, the host–guest interplay takes the crucial role in the electrochemically triggered assembly and disassembly of these supramolecular structures. When now favorably combining different stimuli-sensitive moieties into one microgel,24 the core–shell architecture offers some advantages. This is also seen when considering the diverse and widespread applicability of core–shell particles consisting of inorganic cores and organic shells and vice versa,68–71 combining more than one functional property. These entities attracted great interest with focus on coating, electronics, biomedical applications, optics, catalysis, separation and diagnostics.70,71 Advantageous of core–shell particles is the possibility to encase the core material in a shell with different compositions to protect the core from extraneous chemical and physical changes.68 At the same time, colloidal stability might be imposed. However, the synthesis of these functional core–shell particles is complex and requires often several steps.
In our study, we describe a one batch precipitation polymerization to obtain core/shell particles with a poly(vinylferrocene)-rich core and a poly(N-isopropylacrylamide)-rich shell based on the different reactivities of the two monomers.60,72 The hydrophobic vinylferrocene (VFc)34,64,73,74 can be polymerized in water using solubilizing β-cyclodextrin (CD), which forms an inclusion compound with ferrocene units.75 As shown by Shishido et al./Ritter et al., linear copolymers containing NIPAM are accessible in the same manner.76,77 Beside the temperature-responsive behavior of PNIPAM, the incorporation of the redox active monomer VFc makes the system addressable for an additional stimulus, which can be triggered by both, chemical and electrochemical means. The accompanied change from a neutral to a positively charged species results in a significant increase in the microgel volume (Scheme 1). This is due to the polyelectrolyte effect, which can be traced back to contributions of the osmotic pressure of confined counterions and repulsion of the positive charges in the oxidized state (Fc+). Usually, the switching of polarity induces a drastic change in solubility and many solvents become inappropriate for one of the two states.24,78,79 In our case, we can overcome this limitation by introducing a more solubilizing shell around the ferrocene core, which offers the opportunity to conduct experiments both in water and organic solvents. This is due to the high NIPAM-content in the shell, as PNIPAM as such is well soluble in (cold) water as well as in organic solvents like THF. At the same time, the morphological changes upon an electrochemical trigger may become relevant for different fields of application. To date, electrochemically-induced polarity changes of colloidal/micellar systems were hardly in the focus, specifically for microgels.80 This offers possible applications in controlled drug release devices upon entrapment of negatively charged, or hydrophobic drugs and controlled release upon switching to the neutral Fc, or charged Fc+ state electrochemically14,81 (e.g. in the vicinity of implants). Hence, this study addresses the preparation, the (electrochemical) characterization of the microgels alongside with redox-induced uptake/release of guest molecules into the microgel network (for bactericidal action) and with biocompatibility tests.
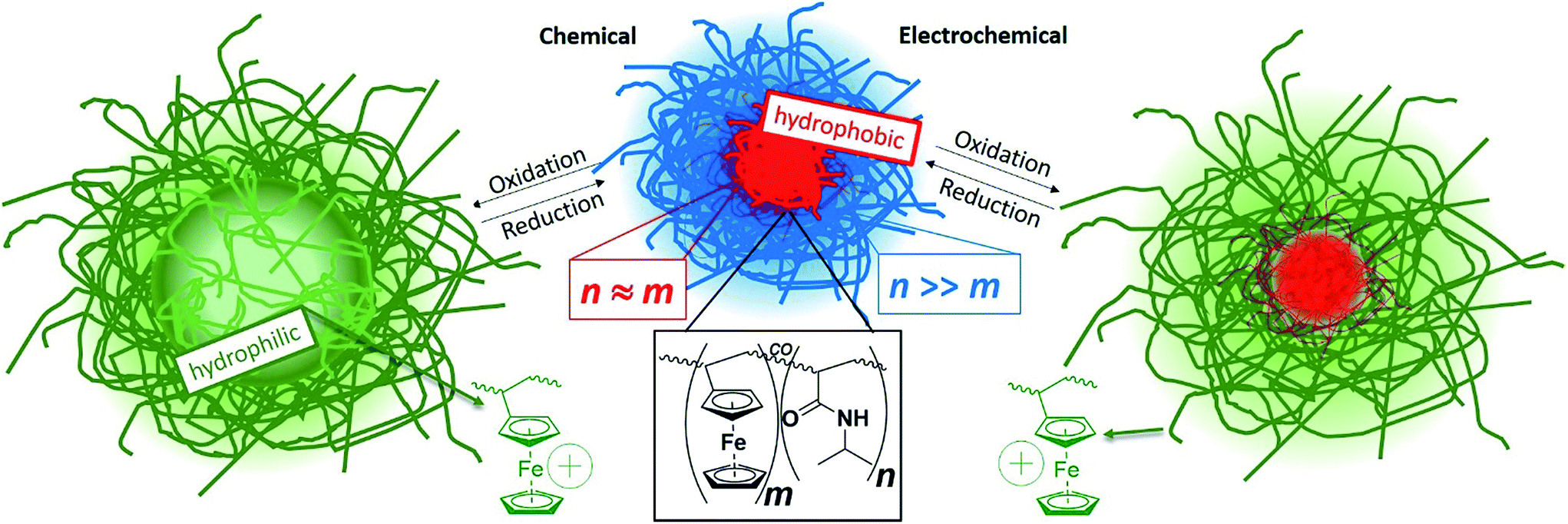 | ||
| Scheme 1 Schematic illustration of reversible volume and polarity modulation of core–shell microgels (core contains mainly ferrocene units while shell contains mainly N-isopropyl acrylamide), allowing the transition of a solid core to a highly swollen (hollowish) domain. | ||
Experimental section
Materials
Vinylferrocene (97%, VFc), methyl-β-cyclodextrin (CD), 2,2′-azobis(2-methylpropionamidine)dihydrochloride (97% AAPH, V 50), triclosan and iron(III) chloride hexahydrate (98%, FeCl3·6H2O) were purchased from Sigma-Aldrich. The crosslinker N,N′-methylenebis(acrylamide) (BIS) and the surfactant N-cetyl-N,N,N-trimethylammonium bromide (CTAB) were purchased from AppliChem, the monomer N-isopropylacrylamide (NIPAM) from Acros Organics. The redox-responsive salt potassium hexacyanoferrate(III) (K3[Fe(CN)6]) was purchased from Merck and the polyelectrolyte poly(styrene sulfonate) sodium salt (PSS) Mw = 3610 g mol−1, PDI < 1.2 was purchased from PSS Polymer Standards Service GmbH. All chemicals were used as received without any further purification. Deionized water (18.2 MΩ) from Millipore Milli-Q-purification system was distilled twice and used in all experiments.Synthesis of P(NIPAM-co-VFc) core–shell microgels
In a three-necked 250 mL flask equipped with a flat anchor-shaped mechanical stirrer, a reflux condenser and a nitrogen in- and outlet 0.642 g of the CD was dissolved in 95 mL bidestilled water to solubilize 0.104 g (0.5 mmol, 4 mol%) of the hydrophobic VFc under nitrogen stream. The solution was heated to 65 °C and 1.347 g (12 mmol, 91 mol%) of NIPAM, 0.108 g (0.7 mmol, 5 mol%) of BIS and 0.004 g (12.5% of the cmc = 0.92 mM) of CTAB were added to the reaction mixture and degassed with N2 over 1 h. The reaction was started by injecting the degassed initiator solution of 0.054 g (2 mM) V 50 in 5 mL bidestilled water to the reaction mixture. 10 min later, opalescence appeared and the reaction was continued for further 4 h at 65 °C and 200 rpm under nitrogen atmosphere. The reaction mixture was cooled down to room temperature and stirred overnight. The microgel dispersion was purified by ultracentrifugation (3 times at 40.000 rpm) and redispersion of the sediment in bidestilled water, leading to a removal of the CD. The product, a yellowish P(NIPAM-co-VFc) microgel (CS1), was freeze-dried after purification for further use. To verify the reproducibility, the polymerization (CS2) was repeated and conversion rate of the two co-monomers was followed by a kinetic study (see below). Comparison of the temperature dependent hydrodynamic radius (ESI Fig. S7†) shows very similar results for both batches, indicating a rather reproducible polymerization behavior, leading also to a very similar electrochemical addressability. We use mainly sample CS2, except were otherwise stated.General electrochemical techniques
Most electrochemical measurements were conducted on the CH Instruments Electrochemical Workstation Potentiostat CHI760D (Austin, Texas, USA). The experiments were performed in a conventional three-electrode setup in a water-jacketed cell connected to a thermostat (Thermo Scientific Haake A28). Electrolysis experiments were performed at 23 °C, or 30 °C while cyclic voltammetry experiments have been performed ranging the temperature between 20 °C to 50 °C. Two kinds of working electrodes have been used. For cyclic voltammetry, a platinum disk electrode, 4 mm disk diameter, and for bulk electrolysis experiments a platinum gauze electrode (35 mm × 30 mm) were used as working electrodes. Further, the counter electrode was chosen corresponding to the experiment. A platinum wire (50 mm) served as counter electrode for bulk electrolysis, which was immersed in 0.1 M KCl in H2O separated by a diaphragm from the remaining compartment and a spirally platinum electrode, 23 cm, for cyclic voltammetry measurements. An Ag/AgCl electrode stored in 1 M KCl served as aqueous reference electrode in aqueous solutions. All potentials in the text and figures are referenced to Ag/AgCl. The solutions were purged with Ar for 10 min to remove dissolved oxygen.Cryogenic transmission electron microscopy (cryo-TEM)
For morphological observation, cryo-TEM samples were prepared by rapid vitrification in liquid ethane from aqueous dispersion (0.05 wt%) using plasma-treated lacey carbon grids or Quantifoil (3.5/1) in a vitrobot system. Images were acquired on a LIBRA 120 operating at 120 keV or on a Tecnai T20 operating at 200 keV with a slow scan CCD under low-dose conditions.Dynamic light scattering (DLS)
Most experiments were performed on an ALV setup equipped with a 633 nm HeNe laser (JDS Uniphase, 35 mV), a goniometer (ALV, CGS-8F), digital hardware correlator (ALV 5000), two avalanche photo diodes (Perkin Elmer, SPCM-CD2969), a light scattering electronics (ALV, LSE-5003), an external programmable thermostat (Julabo F32) and an index-match-bath filled with toluene. Angle- and temperature-dependent measurements were recorded in pseudo-cross correlation mode varying the scattering angle from 35° to 80° at 5° intervals and variation of temperature in the range of 10 °C to 50 °C at 2 K intervals and measurement time of 60 s. The microgel samples were highly diluted to avoid multiple scattering and measured in 0.1 mM CTAB solution to ensure colloidal stability at high temperatures. For data evaluation, the first cumulant from second order cumulant fit was plotted against the squared length of the scattering vector q.2 The data were fitted with a homogeneous linear regression, whereas the diffusion coefficient was extracted from the slope and the hydrodynamic radius Rh calculated by using the Stokes–Einstein equation. Once, the size distributions obtained from ALV DLS setup were compared with a backscattering probe (NANO-flex from Particle Metrix), which can be directly employed in the electrochemical cell. In both cases, similar results were obtained (ESI Fig. S11†).Triclosan loading and release of/from P(NIPAM-co-VFc) microgel
5 mL of a predissolved P(NIPAM-co-VFc) microgel suspension (3.2 mg mL−1) were added to 20 μL of a triclosan stock solution in THF (prepared by dissolving 600 mg Triclosan in 400 μL THF) and stirred overnight. The resulting solution with microgel/triclosan was dialyzed (MWCO 3500 kDa) against water for 2 d. Half of the triclosan loaded microgel suspension was mixed with the oxidizing agent FeCl3 in a microgel/FeCl3 mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (see ESI†) by addition of 13 μL of the FeCl3 stock solution with a concentration of 173 mg mL−1. The mixture was stirred for 1 h and both suspensions, the triclosan loaded P(NIPAM-co-VFc) before and after oxidation were freeze dried for further use.
:3 (see ESI†) by addition of 13 μL of the FeCl3 stock solution with a concentration of 173 mg mL−1. The mixture was stirred for 1 h and both suspensions, the triclosan loaded P(NIPAM-co-VFc) before and after oxidation were freeze dried for further use.
Cultering and harvesting of bacterial strain S. aureus ATCC 12600
The strain was grown overnight at 37 °C on an agar plate from a frozen stock that was stored in DMSO at −80 °C. For experiments, one colony was transferred to inoculate 10 mL of tryptone soy broth (TSB, OXOID, Basingstoke, U.K.) at 37 °C for 24 h in ambient air. This preculture was diluted 1:20 in 200 mL of TSB and grown under static conditions for 16 h at 37 °C. Bacterial cultures were harvested by centrifugation at 5000g for 5 min, washed with potassium phosphate buffered saline (PBS, 5 mM K2HPO4, 5 mM KH2PO4, and 150 mM NaCl, pH 7.0), sonicated for 30 s at 30 W (Vibra Cell model VCX130; Sonics and Materials Inc., Newtown, Connecticut, USA, equipped with ice/water cooling) in order to break possible aggregates and subsequently suspended in TSB to a concentration of 6 × 105 bacteria per mL after quantification by use of the Bürker-Türk counting chamber.
Killing of planktonic staphylococci by triclosan-loaded microgels
To determine the Minimal Inhibitory Concentration (MIC) and Minimal Bactericidal Concentration (MBC), 100 μL of each P(NIPAM-co-VFc), triclosan-loaded P(NIPAM-co-VFc), the triclosan-loaded and oxidized P(NIPAM-co-VFc+) – all with a microgel concentration of c = 4.2 mg mL−1 – and the oxidation agent FeCl3 (c = 1.1 mg mL−1) in TSB solution and serial dilution thereof were mixed with 20 μL of a S. aureus ATCC12600 suspension in TSB (6 × 108 bacteria per mL) in order to obtain a final concentration of 1 × 105 bacteria per mL and the mixture was incubated for 24 h at 37 °C. The MIC values represent the lowest concentration at which bacterial growth was absent. Subsequently, the MBC values were determined by plating aliquots (10 μL) of suspensions with concentrations yielding no visible growth of bacteria on TSB agar plates and the lowest concentration at which colony formation remained absent was taken as the MBC. The experiments have been performed in triplicates.Cell culture L929 cells
Mouse adipose derived fibroblasts (L929) were incubated at 37 °C, 5% CO2 at maximum humidity. DMEM (dulbecco's modified eagle medium) high glucose (completed with 10% FBS (fetal bovine serum), 1% P/S (penicillin, streptomycin) and 0.1% AA2P (ascorbic acid 2-phosphate) in DMEM-HG) was used as growth medium. Cells were harvested from culture flasks using trypsin for 3–5 min at 37 °C. After culturing until ∼80% confluence 30% of the cells were seeded in a new daughter flask. Cells were counted using a Neubauer chamber (hemocytometer).Cytotoxicity assay (XTT)
L929 cells were used to determine the in vitro cytotoxicity of P(NIPAM-co-VFc) following the ISO 10993-5 protocol. A direct cytotoxicity assay was performed by adding P(NIPAM-co-VFc) to mouse adipose derived fibroblasts (L929). Cells were grown and harvested before seeding in a 96-wells plate with a density of 104 cells per well. Cells were incubated for 24 hours to ensure sufficient adhesion and were washed with PBS prior to addition of the microgels compounds. Cells were incubated for 24 and 120 hours together with microgels dispersed in growth medium. For the 120 hour incubation, the medium was exchanged every 48 hours with new microgel containing medium. A mixture of XTT reagent (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide) and activation reagent PMS (N-methyl dibenzopyrazine methyl sulfate) in a volume ratio of 1:0.02 (according to protocol) were added to the cells and incubated for 3 hours prior to analysis using absorbance measurements at λ = 485 nm and λ = 690 nm. The absorbance at 485.0 nm is used for quantifying the metabolic activity observed as the reduction of XTT, while measuring at 690 nm provides the nonspecific absorbance. Statistical analysis was performed using GraphPad (GraphPad Software, La Jolla California USA). All experimental data are presented as average standard deviations over minimum triplicate experiments. Differences were analyzed using One-Way ANOVA, or as indicated and considered to be statistically significant when p < 0.05 (*) of p < 0.0005 (**).
Results
P(NIPAM-co-VFc) microgels responsive to multiple stimuli were obtained by a precipitation polymerization utilizing a one batch synthesis approach in water. To solubilize the hydrophobic vinylferrocene monomer, methyl-β-cyclodextrin was added during the synthesis. The incorporated overall amount of iron (Fe: 1.2–1.9 wt%) in the final microgel was examined by elementary analysis (Table S1, ESI†) and is in good agreement with the amount adjusted during the synthesis (6 wt% or 4 mol% vinylferrocene corresponding to 1.7 wt% iron). However, a kinetic study revealed a substantial difference in monomer conversion rate (Fig. 1). This implies a significant difference in composition between central and peripheral parts of the microgel leading to a structured microgel. At the same time, premature abortion of the precipitation polymerization can allow a tuning of the structure and properties of the microgel (Fig. S3–S5, ESI†). Approaching full conversion, a uniform core–shell-type architecture could be indeed detected by cryogenic transmission electron microscopy (cryo-TEM; Fig. 1). This can be explained by a central region occupied predominantly by VFc (faster conversion, see Fig. 1), while NIPAM is located preferentially in the shell, as indicated by the cryo-TEM image (Fig. 1; VFc gives good contrast due to the electron-rich iron; see also Scanning Transmission Electron Microscopy STEM with Energy Dispersive X-ray spectroscopy EDX in Fig. S24/S25 ESI†).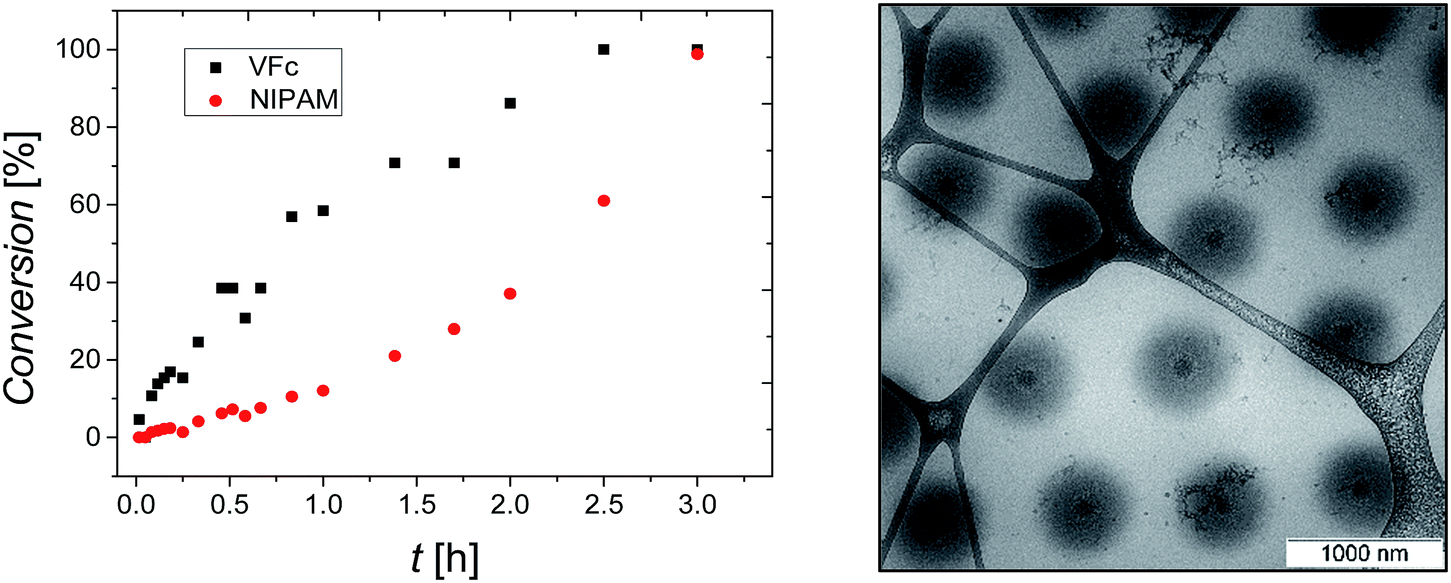 | ||
| Fig. 1 Conversion of VFc and NIPAM during the radical polymerization of the P(NIPAM-co-VFc) microgel in H2O (left) and cryo-TEM image of P(NIPAM-co-VFc) microgel (reduced/pristine state Fc) in H2O vitrified from a dispersion at 20 °C (CS1, right). | ||
A possible explanation for this exceptional compartmentalization can be sought in the linear region of conversions within the first hour of polymerization (Fig. 1): the composition of the produced polymer changes only slightly, providing a hydrophobic VFc-rich core. After this initial state, already ∼60% of the VFc was consumed, while ∼90% of the majority component NIPAM is still unreacted, providing a considerable amount of material for the shell synthesis with much lower VFc content. Another explanation for the rather sharp differentiation between core and shell can be found by the gradual shift of the local LCST with radially decreasing VFc content, giving a specific local composition at which the polymer becomes insoluble at the specified temperature used for vitrification before cryo-TEM imaging. This is in line with the properties of a rather homogeneous linear polymer with similar composition, which would turn insoluble at approximately 20 °C.76 Hence, the size of one domain (the core) can be easily adjusted by temperature (also indicated by comparison of Fig. 1 and 5 taken at different temperatures), which is in contrast to most thermoresponsive core–shell microgels. As discussed below, such a variable hydrophobic domain holds promise for uptake and release applications.
Thermo- and redox-response
In contrast to the more gradual increase in core size, the reversible collapse of the remaining non-collapsed PNIPAM shell can be triggered within a narrow temperature region. This volume phase transition temperature VPPT of approximately 32 °C is typical for NIPAM microgels26 (Fig. S2, ESI†). The pronounced VPPT is another indication that the collapse is mainly caused by the shell, which is predominantly occupied by PNIPAM. Otherwise, linear copolymers composed of NIPAM and VFc with similar molar ratios as used for the microgel synthesis exhibit a decrease of the lower critical solution temperature (LCST ∼20 °C) compared to pure NIPAM.76The reversible redox response of the incorporated Fc units was demonstrated by cyclic voltammetry (CV) in organic media, which is a good solvent for the whole PNIPAM network (red curve in Fig. 2 and S6, ESI†). Taking the dimensions of the microgels into account, a direct electron transfer (via electron tunneling) into the core of the microgel is very unlikely (tunneling distance of ∼200 nm is much too large for typical tunneling distances of 1–2 nm).84 Hence, we assume that we primarily address VFc units in the outer parts of the microgels, when conducting electrochemical experiments on short time scale (several seconds), though electron self-exchange/hopping between ferrocene/ferrocenium units could lead to some redistribution of charges within the colloid. A comparison between monomer and microgels is in line with this assumption (see ESI† for a more detailed derivation): a microgel sample containing the same amount of ferrocene exhibits considerably lower currents than the corresponding monomer solution (dark red curve in Fig. 2), indicating a reduced addressability of the (inner) microgel-bound ferrocene units (i.e. a hindered electron self-exchange/hopping), which is in line with previous discussions on redox-active colloids.85,86
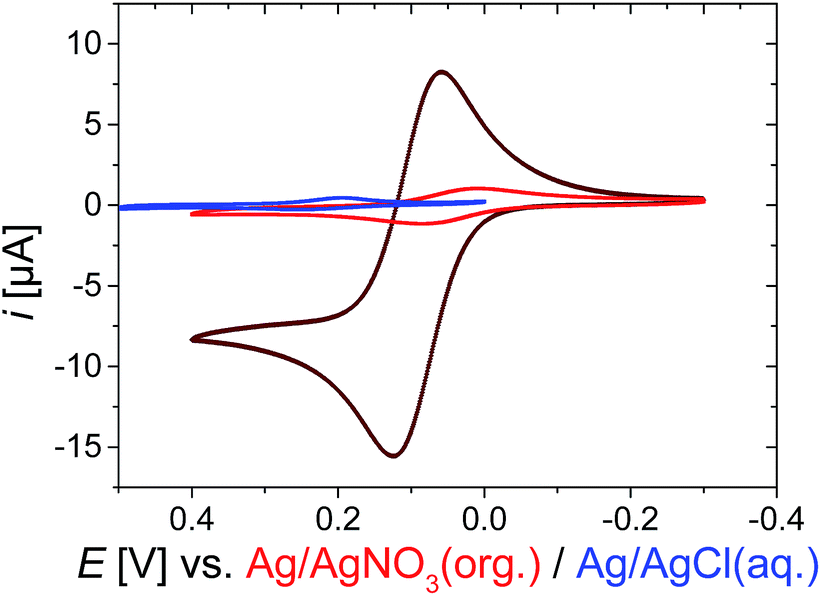 | ||
| Fig. 2 Cyclic voltammogram CV of 1 mM Fc as monomer (dark red curve) or incorporated into the microgel P(NIPAM-co-VFc) (red curve; 3 g L−1, 0.3 wt%), both organic medium in 0.1 M TBAP (tetrabutylammonium perchlorate) in CH3CN, or in aqueous medium in 0.1 M KCl; at room temperature, scan rate v = 5 mV s−1 (see ESI† for enlarged CVs). | ||
Compared to organic solvents, the electrochemical addressability of the microgel is further reduced in aqueous media, indicating an additional internal blockade for charge transport (blue curve in Fig. 2). Nevertheless, on longer time scales, 15% of Fc can be oxidized within 3 h bulk electrolysis at 23 °C (Fig. S9†), leading already to substantial changes of the microgel properties (see below). The bulk electrolysis cannot be assigned to first-order kinetics, indicating different kinetic regimes (Fig. S9†). Electrolysis of “conventional” electroactive matter would provide an exponential decay in current under stirring due to proceeding conversion of the reactants.87 In our case, the exponential decay can only be anticipated for certain time scales, indicating a non-uniform accessibility of the redox-active sites. Though CV currents show a maximum at 34 °C (Fig. S20†), an increased temperature of 30 °C leads to a reduced bulk switching speed compared to the one at 23 °C. Then, 21% of Fc (which refers approximately to the Fc content in the shell) can be electrochemically oxidized within 2 d during bulk electrolysis, while 100% can be oxidized within approximately 5 d (Fig. S10, ESI†). Here, electron self-exchange/hopping between the VFc moieties toward the core can eventually lead to a rather full oxidation/reduction of the microgel by electrochemical means. Hence, the electrochemical addressability of the whole colloid can be adjusted by the time scales of the electrochemical experiments, while the colloidal properties are mainly governed by PNIPAM in the shell. The increased timescale of the switching process compared to a triggered microgel size modulation by a counterion switching approach (∼0.5 to 1 h for full electrochemical conversion)51 can be partly explained by the immobilization of the redox unit directly linked to the microgel network and an additional shielding of the redox units by the NIPAM shell. Our observations of maximum current in CV at 34 °C also indicate that electron self-exchange/hopping is supported by a decreased distance between redox sites88 in the shell as long as certain dynamics are assured in the network for charge exchange (Fig. S20, ESI†).89,90 Both requirements are fulfilled at intermediate temperatures for the (thin) shell, exhibiting higher currents at short time scales (CV), but requiring a considerably longer time scale for oxidizing the large collapsed core at 30 °C. Finally, all faradaic signals vanish far above the VPTT,91 which is similar to the processes seen for the microgels modulated by the counterion switching approach:92 diminished internal mobility93 of redox-active sites prevents charge transfer/hopping. This effect is also resembled in a temperature-dependent electrochemical impedance spectroscopy study, where the charge-transfer resistance is increased considerably above 36 °C (Fig. S23, ESI†). Thus, the disappearing faradaic charge transfer at elevated temperatures in H2O can be comprehended as an encapsulation of the redox responsive moieties due to the collapsing PNIPAM shell (in contrast, good – organic – solvents allow addressability at all temperatures: see e.g. Fig. S22, ESI†). This explains also the reduced electrochemical addressability in water (Fig. 2) and the prolonged timescales (∼5 d) for a full electrochemical oxidation due to the hindered charge exchange between the more mobile, directly addressable exterior VFc units and the glassy microgel interior, which is compacted by hydrophobic interactions. We conclude that we have rather different redox kinetics within a single particle during electrochemical stimulation. In all cases, the electron transfer/hopping kinetics are strongly coupled to swelling of the matrix network, which in turn is influenced by the overall oxidation state and therefore by the electron transfer itself.
The influence of redox sensitivity on the thermo-response of the microgels and vice versa was studied by dynamic light scattering (DLS). Electrochemical oxidation of the neutral Fc moieties leads to a change in hydrophilicity resulting in a partially positively charged microgel. Such a switching to a polyelectrolyte endows an increase in osmotic pressure inside the gel. Hence, swelling occurs in addition to the repulsion of the positively charged Fc+ units (ferrocenium ion). The VPTT shifts from originally 32 °C to elevated temperatures (∼35 °C) upon oxidation.77 The fact that the observable VPTT hardly changes compared to pure PNIPAM is again explained by the major contribution of the VFc-poor shell on the overall hydrodynamic size.
A systematic study of several oxidants to investigate, whether full oxidation of a ferrocene containing polymer can be achieved, yielded that ferric chloride and L-ascorbic acid are suitable oxidants and reducing agents, respectively, in aqueous solutions.54 Hence, the oxidation and reduction was carried out both electrochemically (by electrolysis) and chemically (by addition of chemicals FeCl3 (pH = 4) and L-ascorbic acid, respectively, with a molar ratio of 3:1 in relation to the Fc units inside the microgel, see Fig. 3).54 The microgel swelling behaviors induced by electrochemical electrolysis and chemically by addition of FeCl3 are in very good agreement (see Fig. S8†).
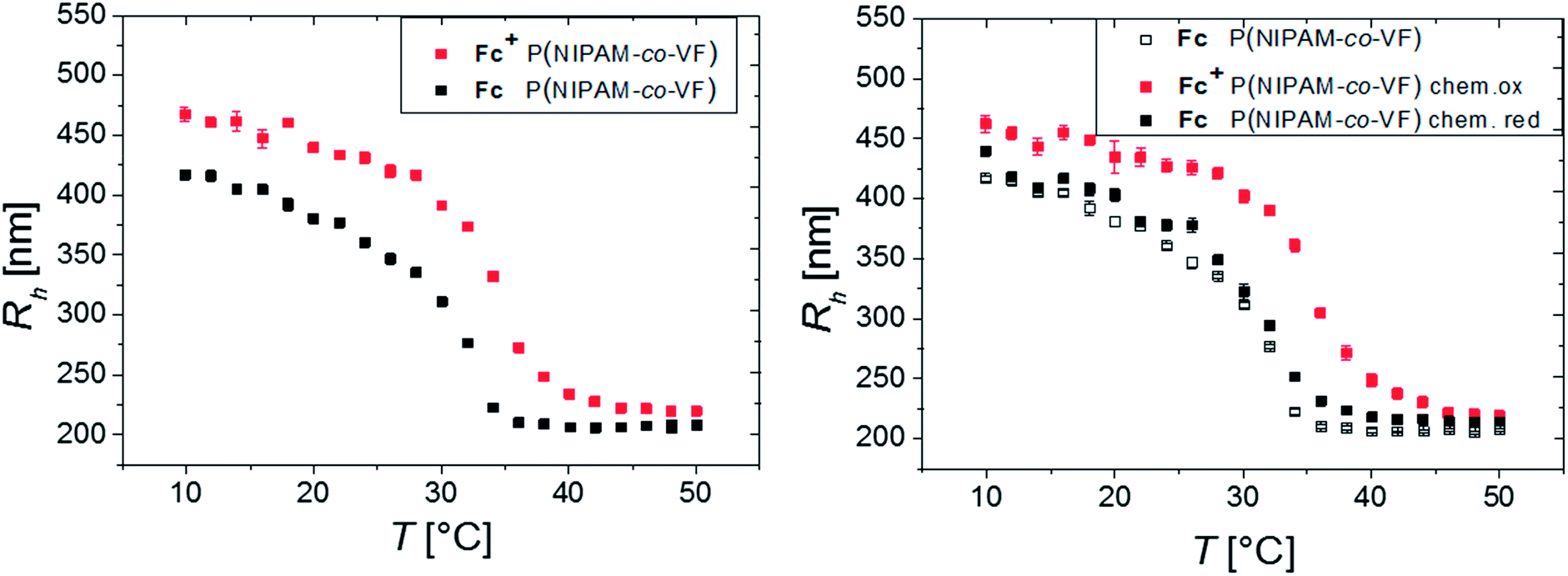 | ||
| Fig. 3 Hydrodynamic radius against temperature of the electrochemically fully oxidized state Fc+ (red) and reduced (non-switched) state Fc (black) of P(NIPAM-co-VFc) microgel in H2O (left) and chemically oxidized state Fc+ (red) by addition of FeCl3 and the chemically reduced state Fc (black) by addition of L-ascorbic acid in H2O and the reduced (pristine) P(NIPAM-co-VFc) microgel (open symbols, right). | ||
The biggest change in hydrodynamic radius Rh of the microgel is located at intermediate temperatures, between the volume phase transition temperatures of the neutral Fc and the oxidized Fc+ state at 34 °C. Consequently, the temperature response enhances the swelling ratio accessible by an electrochemical trigger.
Therefore, the combination of several stimuli and domains is beneficial and favorable from various aspects: protection of the core material from possible degradation, solubilizing the hydrophobic core in water and most importantly an increase of the electrochemical swelling response at intermediate temperatures. Moreover, a recent study on multi-stimuli responsive block copolymer micelles revealed the individual addressability of redox and temperature as external triggers.94 Thus, a temperature of 34 °C was chosen to analyze the oxidation process stepwise (Fig. 4) by DLS revealing a gradual swelling of the microgel with increasing transfer of charges and consequently increasing number of ferrocenium moieties (Fc+) inside the gel. The size distribution stays narrow during oxidation, giving hardly any sign for bimodality. Hence, the size change occurs simultaneously in a gradual way for all microgels (implying even some limited charge exchange between microgels in bulk dispersion) and not instantaneously in a digital way (full oxidation) for only those microgels, which have impacted and experienced charge transfer at the electrode. This is in line with the observation discussed above.
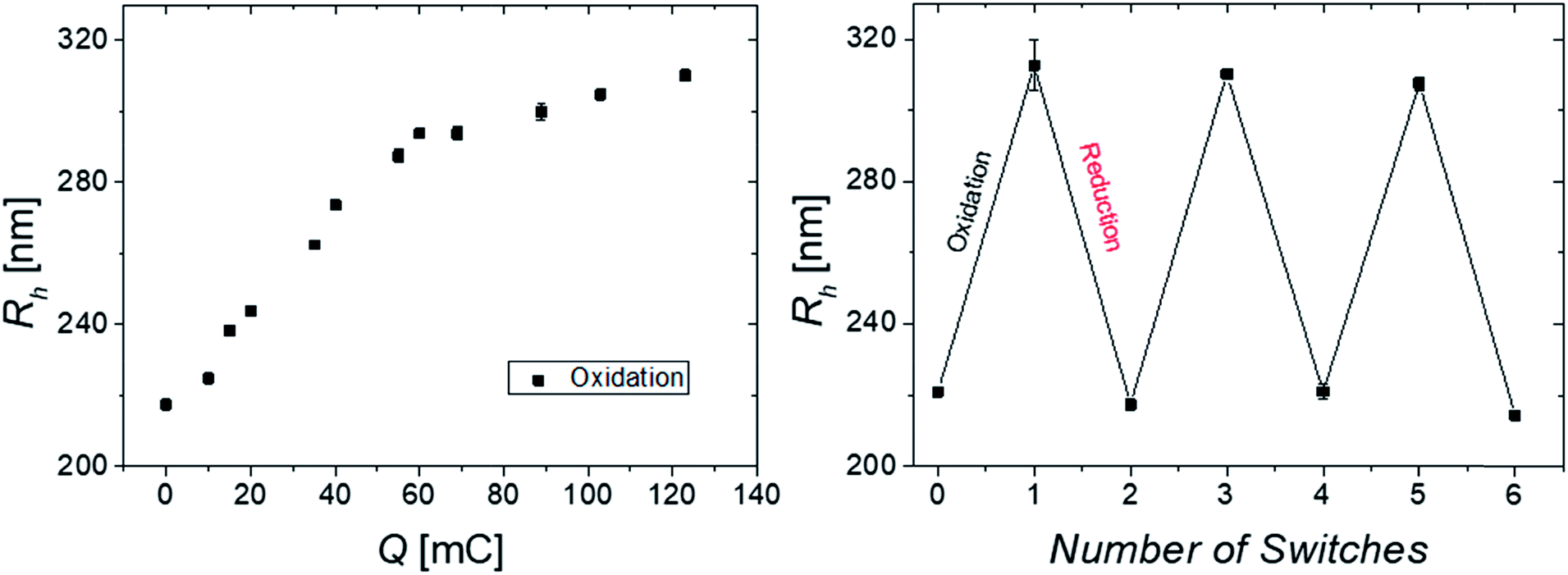 | ||
| Fig. 4 Hydrodynamic radius at 34 °C against the transferred charge during stepwise electrochemical oxidation (left) and hydrodynamic radius at 34 °C against the number of switches (right); initial solution of 2.1 mM Fc (95 mg/10 mL; ∼1 wt% P(NIPAM-co-VFc), CS1) in 0.1 M KCl; oxidation potential: 0.5 V, reduction potential: 0 V at 23 °C. | ||
In order to investigate the reversibility of the microgel size modulation triggered by electrochemical means, the oxidation and reduction was performed several times using the same sample. The fully reversible swelling/collapse of the microgel around ΔRh ∼ 90 nm upon oxidation/reduction is demonstrated in Fig. 4. The volume changes triggered by electrochemical means are around 40% in respect to possible volume changes upon temperature response (Fig. S2, ESI†). However only 15% of the redox-active species were switched from Fc to Fc+ (Fig. 4 and S9, ESI†), which leads to shortened response times of the colloidal suspension while still having maximum change in size (within 2 h). Any longer electrolysis does not provide significant changes in hydrodynamic radius Rh (see Fig. S10, ESI†). However, full electrochemical oxidation and chemical oxidation with FeCl3 reveal nearly the same final size (Fig. S8, ESI†) and similar reversibility (Fig. 3), whereas the swelling of the positively charged core seems to be restricted by the NIPAM shell.
Cryo TEM imaging method was used to analyze and visualize the microgel structure at both states at 34 °C before and after electrochemical, or chemical switching (Fig. 5).95 While the core–shell morphology is clearly seen before electrochemical oxidation, the core starts to disappear partially upon oxidation due to swelling (repulsion of Fc+ units). Here 50% of the Fc units are oxidized leading to a decrease in core radius from RC (Fc) = 102 nm ± 11 nm to RC (Fc+/50%) = 56 nm ± 4 nm (Fig. 5B). These results show that not only the shell network is electrochemically addressable, but also the core reacts (slowly) on the oxidation (the core units are partially oxidized in the more mobile regions, and/or the swollen shell facilitates partial core swelling due to network strain). Also, the swelling of the total core–shell microgel particles upon oxidation can be followed by cryo TEM from RCS (Fc) 311 nm ± 16 nm to RCS (Fc+/50%) 384 nm ± 47 nm (Fig. 5A and B), yielding in general bigger particle radii RCS compared to hydrodynamic radii Rh determined by DLS (which might be due to partial deformation of the microgels within the thin layers of vitrified water). In addition, the difference in radii ΔRCS = 70 nm is smaller compared to ΔRh, originating from the low contrast of the swollen polymeric NIPAM shell in the cryo TEM images.
 | ||
| Fig. 5 Cryo TEM images of the pristine reduced Fc microgel ((A), blue), electrochemically oxidized state Fc+ (50% of the Fc oxidized, (B), red) and chemically oxidized state Fc+ (100% of the Fc oxidized, (C), light green) of P(NIPAM-co-VFc) microgel in H2O vitrified from a dispersion at 34 °C; right: enlargements of marked microgels (with size comparison). | ||
Besides an electrochemical stimulus, the chemical oxidation allows a more rapid oxidation of the core. The full oxidation leads to microgels with an opposite structure compared to the pristine microgels. Instead of a core–shell architecture with dense core, full oxidation provides core–shell structures with a highly swollen core with a radius of RC (Fc+/100%) = 129 nm ± 17 nm (Fig. 5C) and a less swollen shell RCS (Fc+/100%) 386 nm ± 34 nm (hollowish microgel;96Fig. 5C). This structure corroborates again the high content of ferrocene in the core, leading to pronounced swelling due to a high charge density and high local osmotic pressure after oxidation. The chemically oxidized microgels were electrochemically reduced (Fig. S26, ESI†), then resembling the pristine microgels. This indicates that the electrochemical reduction is facile and even allows a reduction of the core. This can again be explained by the unfavourable high charge density and the gain in hydrophobic interactions upon core reduction, leading to an electron funneling toward the core (electron harvesting).
In sum, these core–shell microgels are unique, as other “one-shot” copolymer microgels with interior collapsed phases show often a “dirty snowball” structure.97E.g. poly(N-isopropylacrylamide-co-1-vinyl-3-alkylimidazolium bromide), P(NIPAM-co-VIMnBr) with long alkyl chains exhibit nanophase separated structures (8–12 nm in radius) based on hydrophobic domains, which are randomly distributed inside the microgel.98
Uptake and release of guest molecules
After structural investigation and insight into the redox properties of the microgels, we are interested in changes in polarity of these constructs for triggered uptake/release.99 While specific ion adsorption of polymer-modified electrodes has been demonstrated before,100 the shuttling of hydrophobic molecules was less regarded. In order to demonstrate the change in polarity of the microgel upon switching, uptake/release experiments were performed with hydrophobic fluorescence dyes and negatively charged counterions (for hexacyanoferrates and linear polyelectrolytes: see ESI†).As model compounds for e.g. hydrophobic drug molecules, polarity sensitive fluorescence dyes are suitable probes: the emission characteristics are altered in dependence of the polarity of their surroundings. During spectroelectrochemistry, we employ 4-(dicyanomethylene)-2-methyl-6-(4-dimethylaminostyryl)-4H-pyran, abbreviated as 4HP or DCM, which is sensitive both toward polarity (by a shift in the maximum of the emission spectrum) and toward microviscosity (by modulation of the fluorescence intensity). Due to the reduced sensitivity of the spectroelectrochemical setup for fluorescence experiments (platinum grid working electrode partly obstructs the passage of light), we cannot resolve the location of the weak fluorescence peak at ∼620 nm of the freely dissolved dye (i.e. in hydrophilic aqueous surroundings e.g. for the oxidized state; 4HP quantum yield in 0.1 M KCl only 1.4% – see ESI†). But as evidenced in Fig. 6 (purple curve, left hand side), the incorporation of the dye into a hydrophobic (reduced) domain with high microviscosity is indicated by the pronounced peak at 600 nm (excitation at 470 nm). This incorporation is corroborated by the temperature-dependent measurement in the same setup, which resembles very much literature data83 (see ESI Fig. S14†), and by the fluorescence analysis of the supernatant separated at the different redox states. This shows a 4HP uptake into the microgels in the reduced state, which is reversed in the oxidized state (see ESI†). This correlation between dye uptake and its fluorescence intensity increase was utilized to study the electrochemical uptake and release of hydrophobic substances for our microgels (see Fig. 6). Indeed, the fluorescence intensity is reduced upon oxidation at constant temperature, indicating the release of fluorescence probes. In a reversible fashion, the dye molecules can return into the microgels upon reduction. Taking into account the fluorescence quenching of 4HP by ferrocene by photoinduced electron transfer,101 the response in Fig. 6 is probably mainly related to the 4HP located in the shell with low ferrocene but high NIPAM content. That means that the average distance between ferrocene and 4HP is still so large that total quenching is suppressed in the shell, but still substantial differences in solubilization of at least part of this network can be invoked by the oxidation and reduction of the electroactive units. Taking further into account the reduced electrochemical reactivity of the core, these microgels act as a two-compartment reservoir for hydrophobic substances, allowing in sum the adjustment of advanced release profiles.
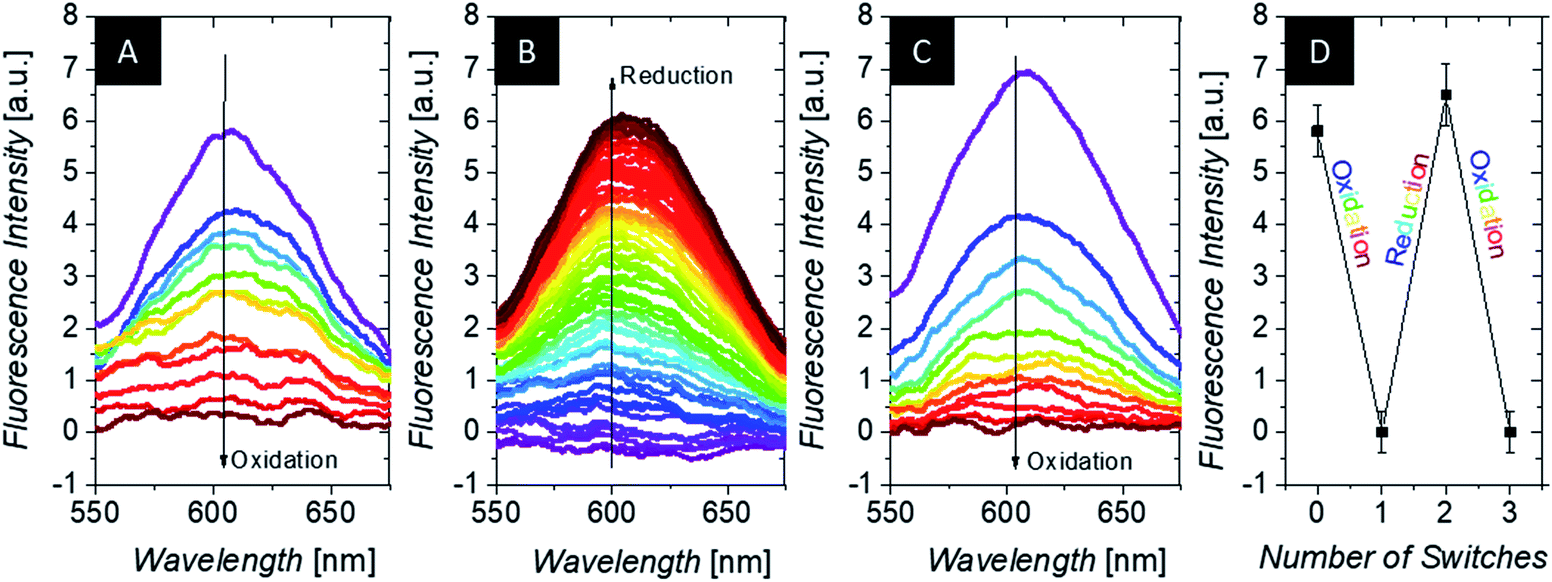 | ||
| Fig. 6 Change of 4HP fluorescence upon electrochemical oxidation at 0.5 V (A), subsequent reduction at 0 V (B), re-oxidation at 0.5 V (C); all potentials against Ag/AgCl) and the summary of the repetitive switching (D) of 1 mL of a 3 g L−1 microgel CS1 dispersion (in 0.1 M KCl and 1 × 10−5 M 4HP; the colored curves represent measurements with intervals of 10 min between them; all spectra were recorded with λ = 470 nm excitation and corrected for residual background). | ||
Hence, the system under investigation is promising for the stimulated uptake/release of active components. This can be utilized in corrosion protection as well as in biomedical context (e.g. coupled with smart implant devices containing biosensor feedback loops for automatized controlled release of bioactive substances). For the latter purpose, we also assessed the biocompatibility of the microgels.
Cytotoxicity
In order to access the possibility of future applications in the bio-medical field, the cytotoxic effect of P(NIPAM-co-VFc) microgels towards L292 mouse fibroblasts was tested. Cytotoxicity was determined for three concentrations of microgel particles using a XTT cell viability assay after 24 h (Fig. 7) and 5 d (ESI†) in comparison to cells not exposed to the microgel. Although differences are significantly different for some concentrations of both, the oxidized and reduced form of the microgel, the normalized absorbance is always above 80% and thus not considered cytotoxic. Surprisingly, this is even true for completely oxidized microgels, being cationic particles. One reason for the enhanced biocompatibility can be found in the peculiar microgel structure. As the polarity is changing predominantly in the microgel interior and the polar Fc+ groups are surrounded by a protecting PNIPAM shell, the zeta potential ζ (Fc) = 1.0 mV ± 4.0 mV is not changing considerably upon oxidation ζ (Fc+) = 3.3 mV ± 4.5 mV. At the same time, the detrimental interaction with negatively charged cell components is probably minimized. Hence, the zeta potential is in line with the negligible changes in biocompatibility of the microgel particles upon electrochemical switching. This assures their possible future use in e.g. medical context after adjusting the optimal temperature-dependent properties to the specific biomedical need (according to Fig. S20,† the maximum temperature for electrochemically-induced release is close to 36 °C for our system).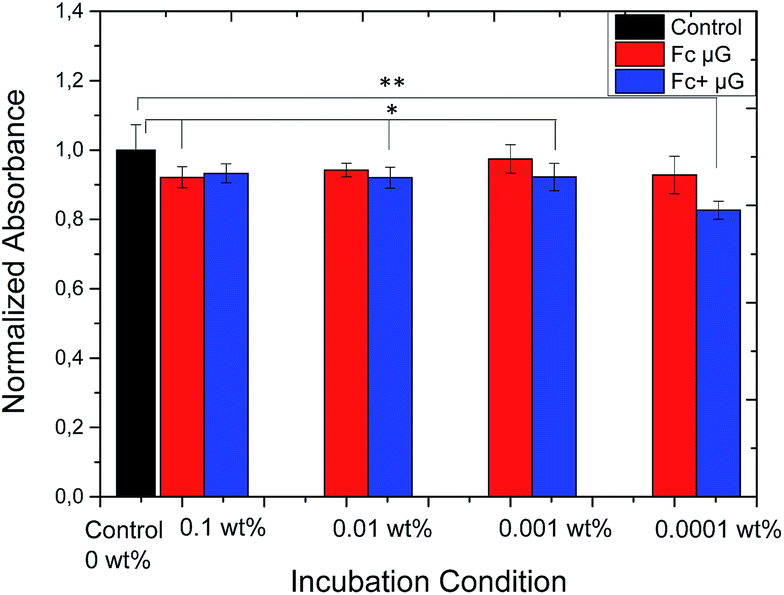 | ||
| Fig. 7 Normalized absorbance (cell viability of L929 mouse fibroblasts) at varying concentrations of P(NIPAM-co-VFc) microgel reduced (pristine) Fc and electrochemically oxidized Fc+ state at 37 °C after 1 d, * assigns statistical significant difference p < 0.05 and **p < 0.0005. | ||
Staphylococcal killing by triclosan loaded microgels
In order to demonstrate the applicability of this system, the model drug triclosan was encapsulated into the hydrophobic part of the nanocontainer P(NIPAM-co-VFc) and the killing efficiency of triclosan-loaded nanocarriers, Minimal Inhibitory (MIC) and Minimal Bactericidal Concentrations (MBC) for planktonic S. aureus ATCC12600 in planktonic growth mode were determined, as listed in Table 1.| MIC [μg mL−1] | MBC [μg mL−1] | |
|---|---|---|
| P(NIPAM-co-VFc) | 27 | 219 |
| P(NIPAM-co-VFc) + triclosan | 6.5 | 104 |
| P(NIPAM-co-VFc+) + triclosan | 3.3 | 13 |
Although already the unloaded microgel P(NIPAM-co-VFc) is able to kill the model microbe S. aureus ATCC12600, which is attributed to the Fe(II) as previously reported in literature,102 loading with triclosan increases the killing efficiency. It is expected that the drug can diffuse from the microgel to a certain extent and kill bacteria based on the deformability and porosity of the microgel. At culture conditions (37 °C) the temperature responsive microgel is present in its deswollen state (see Fig. 3) Then, the distribution of hydrophobic triclosan is determined by the Nernst partitioning law, probably allowing major entrapment of the drug. The partitioning is shifted to free triclosan by both temperature and oxidation state, leading to a substational release of triclosan upon oxidation (though the microgel size has only increased from the complete collapsed state by 50 nm at 37 °C, Fig. 3). This mechanism is in contrast to the sponge-like release of more hydrophilic cargoes by an outward flow of the solvent induced by the collapsing microgel, as described earlier.103 However, the triggered release of the model drug from the interior of the nanocarrier upon chemical oxidation is most effective in terms of bactericidal killing efficiency. A remarkable decrease in MBC from 104 μg mL−1 to 13 μg mL−1, one order of magnitude could be achieved upon the triggered release of triclosan (Table 1 and Fig. 8B and C). Thereby the oxidation agent FeCl3 didn't show any influence on the bacterial growth of S. aureus ATCC12600 under the used concentrations (see ESI, Fig. S18†).
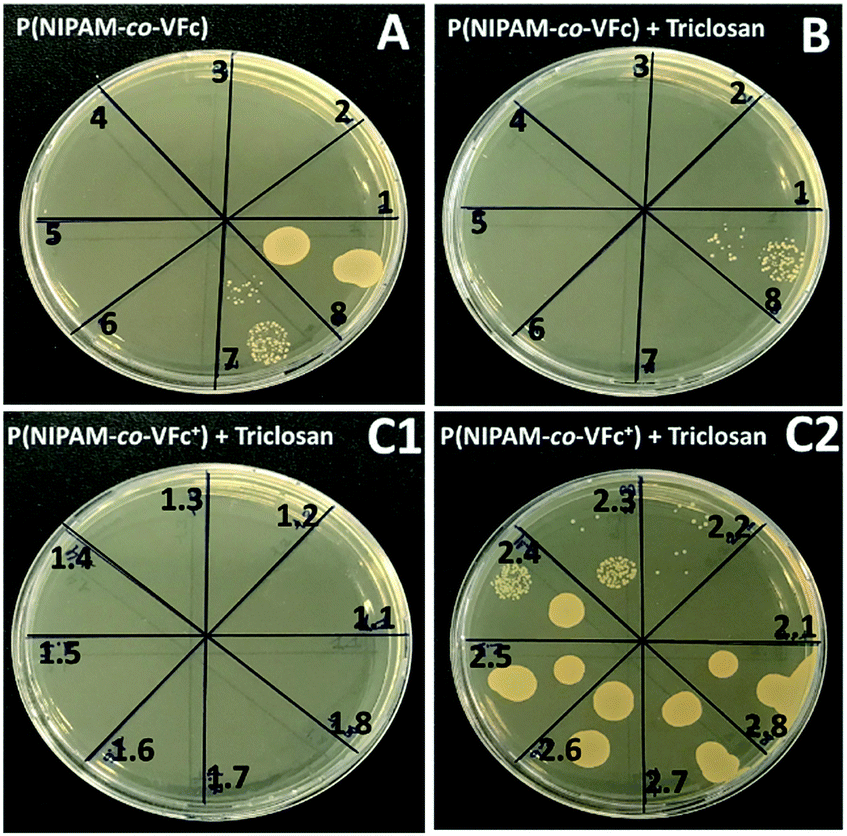 | ||
| Fig. 8 Representative images of TSB agar plates for determination of the MBC (Minimal Bactericidal Concentration) of S. aureus ATCC12600. (A) in presence of the unloaded P(NIPAM-co-VFc) microgel, MBC (section 6) = 219 μg mL−1; (B) triclosan loaded P(NIPAM-co-VFc) microgel, MBC (section 7) = 104 μg mL−1; (C1 and C2) triclosan loaded P(NIPAM-co-VFc+) microgel after oxidation with FeCl3, MBC (section 9, 2.1 of C2) = 13 μg mL−1. | ||
Conclusion
The use of electroactive microgels with complex architecture and double stimuli-sensitivity allows a fine-tuning of the (internal) electrochemical addressability and response times. These are directly coupled to an electrochemical switch in hydrophilicity/hydrophobicity, size or internal structure. Even more, it can combine different features into a colloid, which are often exclusive. As an example, oxidation provides a polycationic microgel, which in turn has a negligible effect on cytotoxicity due to the shielding of the major cationic sites in the interior of the particle. At the same time, the electrochemical addressability is still possible owing to the few (cationic) redox-active sites in the outer microgel part, which do not impart cytotoxicity. Then, network fluctuations facilitate the charge transport from the outer shell into the colloid interior. More specifically, our approach involves a method to generate architecturally advanced core–shell type microgels taking advantage of different reactivities of N-isopropylacrylamide (NIPAM) and vinylferrocene (VFc) and the intermediate solubilization of VFc with help of cyclodextrin. The resulting P(NIPAM-co-VFc) microgels can exhibit a remarkable electrochemically triggered swelling compared to the amount of oxidized ferrocene units. Further, the compact solid PVFc-rich core can turn into a highly swollen hydrophilic domain. The uptake/release of guest molecules is demonstrated upon switching polarity, which makes these microgels interesting for various biomedical applications, probably extending towards release of hydrophobic drugs or hydrophilic enzymes upon tailoring the properties suitable for the specific purpose.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Funding sources
German Research Foundation DFG (SFB 985).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank Michael Appold for his help during STEM sample preparation. They are also thankful for the continuous support of Walter Richtering and they appreciate the funding of the Deutsche Forschungsgemeinschaft DFG within SFB 985 “Functional Microgels und Microgel Systems” (project A6). Markus Gallei and Sebastian Schöttner acknowledge partial support in the frame of the LOEWE project iNAPO by the Hessen State Ministry of Higher Education, Research and the Arts. Olga Mergel gratefully acknowledges the Alexander von Humboldt Foundation for the Feodor Lynen Research Fellowship.References
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Müller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101 CrossRef PubMed.
- A. A. Steinschulte, A. Scotti, K. Rahimi, O. Nevskyi, A. Oppermann, S. Schneider, S. Bochenek, M. F. Schulte, K. Geisel, F. Jansen, A. Jung, S. Mallmann, R. Winter, W. Richtering, D. Wöll, R. Schweins, N. J. Warren and F. A. Plamper, Adv. Mater., 2017, 29, 1703495 CrossRef PubMed.
- S. Nayak, H. Lee, J. Chmielewski and L. A. Lyon, J. Am. Chem. Soc., 2004, 126, 10258–10259 CrossRef CAS PubMed.
- C. M. Nolan, C. D. Reyes, J. D. Debord, A. J. Garcia and L. A. Lyon, Biomacromolecules, 2005, 6, 2032–2039 CrossRef CAS PubMed.
- N. Murthy, Y. X. Thng, S. Schuck, M. C. Xu and J. M. Fréchet, J. Am. Chem. Soc., 2002, 124, 12398–12399 CrossRef CAS PubMed.
- M. V. Varma, A. M. Kaushal and S. Garg, J. Controlled Release, 2005, 103, 499–510 CrossRef CAS PubMed.
- L. Bromberg and V. Alakhov, J. Controlled Release, 2003, 88, 11–22 CrossRef CAS PubMed.
- L. Bromberg, M. Temchenko and T. A. Hatton, Langmuir, 2003, 19, 8675–8684 CrossRef CAS.
- M. Das, H. Zhang and E. Kumacheva, Annu. Rev. Mater. Res., 2006, 36, 117–142 CrossRef CAS.
- L. Peng, A. Feng, M. Huo and J. Yuan, Chem. Commun., 2014, 50, 13005–13014 RSC.
- A. Pich and W. Richtering, in Chemical Design of Responsive Microgels, ed. A. Pich and W. Richtering, Springer Berlin Heidelberg, 2011, vol. 234, ch. 70, pp. 1–37 Search PubMed.
- F. Kroener, A. Heerwig, W. Kaiser, M. Mertig and U. Rant, J. Am. Chem. Soc., 2017, 139, 16510–16513 CrossRef CAS PubMed.
- F. Plamper, in Porous Carbons – Hyperbranched Polymers – Polymer Solvation, ed. T. E. Long, B. Voit and O. Okay, Springer International Publishing, 2015, vol. 266, ch. 284, pp. 125–212 Search PubMed.
- S. Szunerits, F. Teodorescu and R. Boukherroub, Eur. Polym. J., 2016, 83, 467–477 CrossRef CAS.
- J. Kim, J. H. Kim and K. Ariga, Joule, 2017, 1, 1–30 CrossRef.
- K. Hernández-Burgos, Z. J. Barton and J. Rodríguez-López, Chem. Mater., 2017, 29, 8918–8931 CrossRef.
- H.-S. Yang, J. Jang, B.-S. Lee, T.-H. Kang, J.-J. Park and W.-R. Yu, Langmuir, 2017, 33, 9057–9065 CrossRef CAS PubMed.
- B. J. Plowman, N. P. Young, C. Batchelor-McAuley and R. G. Compton, Angew. Chem., Int. Ed., 2016, 55, 7002–7005 CrossRef CAS PubMed.
- L. R. Holt, B. J. Plowman, N. P. Young, K. Tschulik and R. G. Compton, Angew. Chem., Int. Ed., 2016, 55, 397–400 CrossRef CAS PubMed.
- X. F. Zhou, W. Cheng and R. G. Compton, Angew. Chem., Int. Ed., 2014, 53, 12587–12589 CAS.
- O. Mergel, P. T. Kühn, S. Schneider, U. Simon and F. A. Plamper, Electrochim. Acta, 2017, 232, 98–105 CrossRef CAS.
- F. A. Plamper, L. Murtomäki, A. Walther, K. Kontturi and H. Tenhu, Macromolecules, 2009, 42, 7254–7257 CrossRef CAS.
- O. Bertrand, A. Vlad, R. Hoogenboom and J.-F. Gohy, Polym. Chem., 2016, 7, 1088–1095 RSC.
- M. Mazurowski, M. Gallei, J. Li, H. Didzoleit, B. Stühn and M. Rehahn, Macromolecules, 2012, 45, 8970–8981 CrossRef CAS.
- F. A. Plamper and W. Richtering, Acc. Chem. Res., 2017, 50, 131–140 CrossRef CAS PubMed.
- R. Pelton, Adv. Colloid Interface Sci., 2000, 85, 1–33 CrossRef CAS PubMed.
- Q. M. Zhang, D. Berg, J. Duan, S. M. Mugo and M. J. Serpe, ACS Appl. Mater. Interfaces, 2016, 8, 27264–27269 CrossRef CAS PubMed.
- H.-z. Bu, S. R. Mikkelsen and A. M. English, Anal. Chem., 1995, 67, 4071–4076 CrossRef CAS.
- H.-z. Bu, A. M. English and S. R. Mikkelsen, Anal. Chem., 1996, 68, 3951–3957 CrossRef CAS.
- L. V. Sigolaeva, S. Y. Gladyr, O. Mergel, A. P. H. Gelissen, M. Noyong, U. Simon, D. V. Pergushov, I. N. Kurochkin, F. A. Plamper and W. Richtering, Anal. Chem., 2017, 89, 6091–6098 CrossRef CAS PubMed.
- L. V. Sigolaeva, O. Mergel, E. G. Evtushenko, S. Y. Gladyr, A. P. H. Gelissen, D. V. Pergushov, I. N. Kurochkin, F. A. Plamper and W. Richtering, Langmuir, 2015, 31, 13029–13039 CrossRef CAS PubMed.
- K. Özlem Nazli, C. W. Pester, A. Konradi, A. Böker and P. van Rijn, Chem.–Eur. J., 2013, 19, 5586–5594 CrossRef PubMed.
- F. Boujioui, O. Bertrand, B. Ernould, J. Brassinne, T. Janoschka, U. S. Schubert, A. Vlad and J.-F. Gohy, Polym. Chem., 2017, 8, 441–450 RSC.
- M. H. George and G. F. Hayes, J. Polym. Sci., Polym. Chem. Ed., 1975, 13, 1049–1070 CrossRef CAS.
- R. L. N. Hailes, A. M. Oliver, J. Gwyther, G. R. Whittell and I. Manners, Chem. Soc. Rev., 2016, 45, 5358–5407 RSC.
- R. M. Choueiri, A. Klinkova, S. Pearce, I. Manners and E. Kumacheva, Macromol. Rapid Commun., 2018, 39, 1700554 CrossRef PubMed.
- M. A. Hempenius, C. Cirmi, F. L. Savio, J. Song and G. J. Vancso, Macromol. Rapid Commun., 2010, 31, 772–783 CrossRef CAS PubMed.
- Y. Ma, W.-F. Dong, M. A. Hempenius, H. Möhwald and G. Julius Vancso, Nat. Mater., 2006, 5, 724 CrossRef CAS PubMed.
- D. Chong, J. Tan, J. Zhang, Y. Zhou, X. Wan and J. Zhang, Chem. Commun., 2018, 54, 7838–7841 RSC.
- Z. T. Gossage, K. Hernández-Burgos, J. S. Moore and J. Rodríguez-López, ChemElectroChem, 2018, 5, 3006–3013 CrossRef CAS.
- Y. Wu, B. Guo and P. X. Ma, ACS Macro Lett., 2014, 3, 1145–1150 CrossRef CAS.
- Z. Deng, Y. Guo, X. Zhao, P. X. Ma and B. Guo, Chem. Mater., 2018, 30, 1729–1742 CrossRef CAS.
- R. Dong, X. Zhao, B. Guo and P. X. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 17138–17150 CrossRef CAS PubMed.
- D. Grieshaber, J. Vörös, T. Zambelli, V. Ball, P. Schaaf, J.-C. Voegel and F. Boulmedais, Langmuir, 2008, 24, 13668–13676 CrossRef CAS PubMed.
- T. Tatsuma, K. Takada, H. Matsui and N. Oyama, Macromolecules, 1994, 27, 6687–6689 CrossRef CAS.
- M. Mackiewicz, K. Marcisz, M. Strawski, J. Romanski, Z. Stojek and M. Karbarz, Electrochim. Acta, 2018, 268, 531–538 CrossRef CAS.
- J. A. Jaber and J. B. Schlenoff, Macromolecules, 2005, 38, 1300–1306 CrossRef CAS.
- T. A. García, C. A. Gervasi, M. J. Rodríguez Presa, J. I. Otamendi, S. E. Moya and O. Azzaroni, J. Phys. Chem. C, 2012, 116, 13944–13953 CrossRef.
- J. Liang, X. Yu, T. Yang, M. Li, L. Shen, Y. Jin and H. Liu, Phys. Chem. Chem. Phys., 2017, 19, 22472–22481 RSC.
- Y. Dou, J. Han, T. Wang, M. Wei, D. G. Evans and X. Duan, Langmuir, 2012, 28, 9535–9542 CrossRef CAS PubMed.
- O. Mergel, P. Wünnemann, U. Simon, A. Böker and F. A. Plamper, Chem. Mater., 2015, 27, 7306–7312 CrossRef CAS.
- H.-z. Bu, A. M. English and S. R. Mikkelsen, J. Phys. Chem. B, 1997, 101, 9593–9599 CrossRef CAS.
- X. Feng, K. Zhang, P. Chen, X. Sui, M. A. Hempenius, B. Liedberg and G. J. Vancso, Macromol. Rapid Commun., 2016, 37, 1939–1944 CrossRef CAS PubMed.
- M. I. Giannotti, H. Lv, Y. Ma, M. P. Steenvoorden, A. R. Overweg, M. Roerdink, M. A. Hempenius and G. J. Vancso, J. Inorg. Organomet. Polym. Mater., 2006, 15, 527–540 CrossRef.
- A. Alkan and F. R. Wurm, Macromol. Rapid Commun., 2016, 37, 1482–1493 CrossRef CAS PubMed.
- H. Gu, R. Ciganda, P. Castel, J. Ruiz and D. Astruc, Macromolecules, 2016, 49, 4763–4773 CrossRef CAS.
- D. Scheid, C. Lederle, S. Vowinkel, C. G. Schäfer, B. Stühn and M. Gallei, J. Mater. Chem. C, 2014, 2, 2583–2590 RSC.
- D. P. Puzzo, A. C. Arsenault, I. Manners and G. A. Ozin, Angew. Chem., Int. Ed., 2009, 48, 943–947 CrossRef CAS PubMed.
- G. Agrawal, M. P. Schürings, P. van Rijn and A. Pich, J. Mater. Chem. A, 2013, 1, 13244–13251 RSC.
- S. Schachschal, A. Balaceanu, C. Melian, D. E. Demco, T. Eckert, W. Richtering and A. Pich, Macromolecules, 2010, 43, 4331–4339 CrossRef CAS.
- I. Kawarazaki and A. Takasu, Macromol. Chem. Phys., 2016, 217, 2595–2600 CrossRef CAS.
- J. Izquierdo and C. Kranz, Eur. Polym. J., 2016, 83, 428–449 CrossRef CAS.
- R. L. Hailes, A. M. Oliver, J. Gwyther, G. R. Whittell and I. Manners, Chem. Soc. Rev., 2016, 45, 5358–5407 RSC.
- R. Pietschnig, Chem. Soc. Rev., 2016, 45, 5216–5231 RSC.
- B. Stein, D. Zopes, M. Schmudde, R. Schneider, A. Mohsen, C. Goroncy, S. Mathur and C. Graf, Faraday Discuss., 2015, 181, 85–102 RSC.
- P. Köpf-Maier, H. Köpf and E. W. Neuse, J. Cancer Res. Clin. Oncol., 1984, 108, 336–340 CrossRef.
- P. Köpf-Maier, H. Köpf and E. W. Neuse, Angew. Chem., Int. Ed., 1984, 23, 456–457 CrossRef.
- J. L. Antonio, L. Höfler, T. Lindfors and S. I. Córdoba de Torresi, ChemElectroChem, 2016, 3, 2146–2152 CrossRef CAS.
- H. J. Räder, T.-T.-T. Nguyen and K. Müllen, Macromolecules, 2014, 47, 1240–1248 CrossRef.
- E. M. White, J. Yatvin, J. B. Grubbs, J. A. Bilbrey and J. Locklin, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1084–1099 CrossRef CAS.
- R. Ghosh Chaudhuri and S. Paria, Chem. Rev., 2012, 112, 2373–2433 CrossRef CAS PubMed.
- A. Melle, A. Balaceanu, M. Kather, Y. Wu, E. Gau, W. Sun, X. Huang, X. Shi, M. Karperien and A. Pich, J. Mater. Chem. B, 2016, 4, 5127–5137 RSC.
- M. H. George and G. F. Hayes, J. Polym. Sci., Polym. Chem. Ed., 1976, 14, 475–488 CrossRef CAS.
- G. F. Hayes and M. H. George, in Organometallic Polymers, ed. J. E. Sheats and C. U. Pittman, Academic Press, 1978, pp. 13–24, DOI:10.1016/B978-0-12-160850-7.50006-X.
- M. Nakahata, Y. Takashima, H. Yamaguchi and A. Harada, Nat. Commun., 2011, 2, 511 CrossRef PubMed.
- H. Ritter, B. E. Mondrzik, M. Rehahn and M. Gallei, Beilstein J. Org. Chem., 2010, 6, 60 Search PubMed.
- N. Kuramoto and Y. Shishido, Polymer, 1998, 39, 669–673 CrossRef CAS.
- J. Elbert, M. Gallei, C. Rüttiger, A. Brunsen, H. Didzoleit, B. Stühn and M. Rehahn, Organometallics, 2013, 32, 5873–5878 CrossRef CAS.
- J. Elbert, F. Krohm, C. Rüttiger, S. Kienle, H. Didzoleit, B. N. Balzer, T. Hugel, B. Stühn, M. Gallei and A. Brunsen, Adv. Funct. Mater., 2014, 24, 1591–1601 CrossRef CAS.
- P. Anton, A. Laschewsky and M. D. Ward, Polym. Bull., 1995, 34, 331–335 CrossRef CAS.
- D. Scheid, M. von der Lühe and M. Gallei, Macromol. Rapid Commun., 2016, 37, 1573–1580 CrossRef CAS PubMed.
- T. Thavanesan, C. Herbert and F. A. Plamper, Langmuir, 2014, 30, 5609–5619 CrossRef CAS.
- J. Virtanen, H. Lemmetyinen and H. Tenhu, Polymer, 2001, 42, 9487–9493 CrossRef CAS.
- C. M. Hill, J. Kim and A. J. Bard, J. Am. Chem. Soc., 2015, 137, 11321–11326 CrossRef CAS PubMed.
- M. Burgess, J. S. Moore and J. Rodríguez-López, Acc. Chem. Res., 2016, 49, 2649–2657 CrossRef CAS PubMed.
- Z. T. Gossage, N. B. Schorr, K. Hernández-Burgos, J. Hui, B. H. Simpson, E. C. Montoto and J. Rodríguez-López, Langmuir, 2017, 33, 9455–9463 CrossRef CAS PubMed.
- J. J. Lingane, J. Am. Chem. Soc., 1945, 67, 1916–1922 CrossRef CAS.
- F. Pinaud, L. Russo, S. Pinet, I. Gosse, V. Ravaine and N. Sojic, J. Am. Chem. Soc., 2013, 135, 5517–5520 CrossRef CAS PubMed.
- K. Kaniewska, J. Romanski and M. Karbarz, RSC Adv., 2013, 3, 23816–23823 RSC.
- A. Heller, Curr. Opin. Chem. Biol., 2006, 10, 664–672 CrossRef CAS.
- C. Dähling, G. Lotze, H. Mori, D. V. Pergushov and F. A. Plamper, J. Phys. Chem. B, 2017, 121, 6739–6748 CrossRef PubMed.
- O. Mergel, A. P. H. Gelissen, P. Wünnemann, A. Böker, U. Simon and F. A. Plamper, J. Phys. Chem. C, 2014, 118, 26199–26211 CrossRef CAS.
- S. Maccarrone, O. Mergel, F. A. Plamper, O. Holderer and D. Richter, Macromolecules, 2016, 49, 1911–1917 CrossRef CAS.
- B. V. K. J. Schmidt, J. Elbert, C. Barner-Kowollik and M. Gallei, Macromol. Rapid Commun., 2014, 35, 708–714 CrossRef CAS PubMed.
- L. E. Franken, E. J. Boekema and M. C. A. Stuart, Adv. Sci., 2017, 4, 1600476 CrossRef PubMed.
- O. L. J. Virtanen, A. Mourran, P. T. Pinard and W. Richtering, Soft Matter, 2016, 12, 3919–3928 RSC.
- M. Keerl, J. S. Pedersen and W. Richtering, J. Am. Chem. Soc., 2009, 131, 3093–3097 CrossRef CAS PubMed.
- Q. Wu, C. Lv, Z. Zhang, Y. Li, J. Nie, J. Xu and B. Du, Langmuir, 2018, 34, 9203–9214 CrossRef CAS.
- S. Walta, D. V. Pergushov, A. Oppermann, A. A. Steinschulte, K. Geisel, L. V. Sigolaeva, F. A. Plamper, D. Wöll and W. Richtering, Polymer, 2017, 119, 50–58 CrossRef CAS.
- X. Su and T. A. Hatton, Phys. Chem. Chem. Phys., 2017, 19, 23570–23584 RSC.
- J. Yang, M. Li, L. Kang and W. Zhu, Sci. China: Chem., 2017, 60, 607–613 CrossRef CAS.
- E. V. Sorokina, T. P. Yudina, I. A. Bubnov and V. S. Danilov, Microbiology, 2013, 82, 439–444 CAS.
- H. Masoud and A. Alexeev, ACS Nano, 2012, 6, 212–219 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, elemental analysis, NMR data, detailed CV data and interpretation, chronoamperometry data, additional dynamic light scattering, uptake experiments, scanning transmission electron microscopy and cytotoxicity data. Experimental data for this article are available under request at https://hdl.handle.net/21.11102/b794f67f-caf8-11e8-a934-e41f1366df48. See DOI: 10.1039/c8sc04369h |
| ‡ School of Physical Sciences, Ingram Building, University of Kent, Canterbury, Kent, CT2 7NH, UK. |
| This journal is © The Royal Society of Chemistry 2019 |