
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLanthanide nanoparticles for high sensitivity multiparameter single cell analysis†
Jothirmayanantham
Pichaandi‡
a,
Guangyao
Zhao‡
a,
Alexandre
Bouzekri
b,
Elsa
Lu
a,
Olga
Ornatsky
b,
Vladimir
Baranov
b,
Mark
Nitz
 *a and
Mitchell A.
Winnik
*a
*a and
Mitchell A.
Winnik
*a
aDepartment of Chemistry, University of Toronto, 80 St George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: mnitz@chem.utoronto.ca; mwinnik@chem.utoronto.ca
bFluidigm Canada Inc., 1380 Rodick Road, Markham, Ontario L3R 4G5, Canada
First published on 23rd January 2019
Abstract
Mass cytometry (MC) is a high throughput multiparameter analytical technique for determining biomarker expression in cells. In MC, antibodies (Abs) are tagged with heavy metal isotopes via conjugation to metal chelating polymers (MCPs). To improve the sensitivity of MC towards low abundance biomarkers, we are developing nanoparticle (NP)-based reagents as mass tags for Abs. We examine the use of silica-coated NaHoF4 NPs (d ∼ 12 nm) decorated with PEG5k conjugated to thiol-modified primary or secondary Abs for MC assays. We compare the sensitivity of NP–Ab conjugates to MCP–Ab conjugates towards seven biomarkers with varying expression levels across six cell lines. We also perform a multi-parameter assay using a cocktail of both NP- and MCP-based reagents to detect seven cellular markers in peripheral blood mononuclear cells (PBMCs). In the case of highly abundant markers, signal enhancements from NP–Ab conjugates offer minimal advantages over MCP–Ab conjugates, which already give strong signals. In the case of biomarkers with lower abundance, the level of signal enhancements depended on the nature of the biomarker being detected, or on the type of detection method used. When comparing the indirect detection of CD14 on THP-1 cells using NPs or MCPs conjugated to secondary Abs, the NP reagents offered little signal enhancements compared to the MCP reagents. However, in the case of direct CD14 detection on THP-1 or U937 cells using NPs or MCPs conjugated to primary Abs, a 30- or 450-fold signal enhancement was seen from the NP-based reagent. In the experiments where both NP–Ab and MCP–Ab conjugates were used together to stain PBMCs, we found that the presence of the NP–Ab conjugates did not affect the function of MCP–Ab conjugates, and the NP–Ab conjugates showed minimal non-specific interaction with cells without the target biomarker (CD14). Furthermore, these NP–Ab conjugates could be used to identify rare CD14+ monocytes from the PBMC mixture with a 20-fold signal increase when compared to the use of only MCP–Ab conjugates. Collectively, the strong signal amplification obtained from NP reagents demonstrate the potential of these reagents to be used in conjunction with MCP-reagents to detect rare cellular markers or cell types that may otherwise be overlooked when using MCP-reagents alone.
Introduction
Modern medical research requires highly sensitive, multiplexed assays of cellular biomarkers to interrogate the complex biology of underlying diseases.1–3 Many cellular markers associated with pathogenesis are expressed at low levels making detection difficult.4 Flow cytometry requires exceptionally bright fluorophores for the analysis of low abundance biomarkers, but their use is limited by the spectral overlap when they are used in highly multiplexed experiments.5Mass cytometry (MC) was designed to overcome the multiplexing limitations of flow cytometry by employing heavy metal isotopes as tags, which results in no background signal, and at the same time enables the measurement of over 40 markers per cell.6 MC combines cytometric injection of cells with inductively coupled plasma ionization and time-of-flight mass spectrometry detection.7 Since the classic contribution of Nolan and coworkers8 in 2011 demonstrating the ability of mass cytometry to provide a uniquely detailed view of cell differentiation in the human hematopoietic system, more than 300 papers have been published that take advantage of this technique.9 In a commentary in Science on the Nolan group contribution, Benoist and Hacohen10 suggest that “Mass cytometry is poised to revolutionize our studies of disorders in the human immune system by probing multiple critical parameters in parallel, across a broad range of cells and pathways.” Recent issues of J. Immunol. Methods11and Cytometry Part B12 devoted to MC provide an overview of the current state of the art.
Unfortunately, with currently available reagents, MC lacks the sensitivity observed in flow cytometry with the brightest fluorophores (e.g., phycoerythrin), and this complicates the detection of low-abundance cellular biomarkers by MC.13 Current heavy metal isotope tags (mass tags) used in MC are metal-chelating polymers (MCPs) that carry 20–50 metal atoms per tag, corresponding to 50 to 200 metal atoms per antibody–MCP conjugate.14–16 For detecting low-abundance biomarkers, mass cytometry requires ‘brighter’ mass tags which can be obtained by substantially increasing the number of metal atoms per tag.13
There have been two approaches so far to increase the number of metal atoms per tag. One approach is to conjugate Abs to semiconducting polymer-micelle dots carrying chelators for lanthanide ions.17 On average, these polymer-micelle dots contain ca. 1000 to 2000 lanthanide ions. With these reagents, researchers observed an increase in sensitivity by a factor of 4 to 6 over a commercial MCP reagent, depending on the marker studied.
The second approach is to increase the number of metals per tag by using inorganic nanoparticles. These nanoparticles, depending on their size, carry hundreds or thousands of metal atoms per tag. We are aware of two publications that used a commercially available CD3-CdSe quantum dot (QD) conjugate as part of a MC panel.18,19 These QDs have been shown to contain about 800 Cd atoms (1.49 × 10−13 μg Cd per QD)20 of which 21.4% is 112Cd and 28.75% is 114Cd. Thus the number of each isotope per Ab is comparable to the number of lanthanide isotope on a MCP reagent. Note that Cd isotopes have masses in the low sensitivity range of the MC detector21 and are not appropriate in this size range as high sensitivity reagents.
In an attempt to improve sensitivity in MC measurements, Schultz and coworkers employed commercially available streptavidin coated silver nanoparticles of two different diameters (d), 10 and 40 nm, to identify the marker CD25 in peripheral blood mononuclear cells (PBMC).22 With the d = 40 nm silver NPs, they achieved an enhancement in sensitivity of only 2 to 3 times over a MCP reagent. This increase is surprisingly small, as a 40 nm silver NP contains about 2 million silver atoms, of which 51.8% are the 107Ag isotope. Depending on the transmission coefficient of Ag for their instrument (on the order of 10−5), each 40 nm NP should generate at least 20 to 30 counts of signal. These reports on enhancing the sensitivity of the MC signal did not seem to fulfill the theoretical potential of nanoparticle reagents.
There are two reported examples of MC-based single cell assays that provide valuable information about NP–cell interactions and about the advantages of NP reagents for MC. Irvine and coworkers23 administered gold NPs intratracheally to a mouse model to understand, at the cellular level, the biodistribution of the NPs for drug delivery and imaging applications. Comparing BODIPY-labelled NPs, detected by flow cytometry, with unlabeled NPs detected by MC they found significantly greater sensitivity using MC. By flow cytometry, they found gold NPs associated with only 12 to 24% of the cells. In contrast, MC showed that the NPs were present in more than 96% of the cells, and the gold NPs were concentrated in the macrophages. This experiment shows the power of MC for the study of NPs in biological samples compared to other common analysis techniques.
Muller and coworkers24 used MC to study the toxicity of 10 nm silver NPs to bacterial cells. Increasing NP concentrations led to significant toxicity, and, interestingly, there was a heterogeneous distribution of silver NPs among the bacterial cells.
Lanthanide nanoparticles of the general form NaLnF4 are particularly attractive as potential high sensitivity reagents for MC-based single cell assays. A number of research groups have reported the synthesis of lanthanide-doped NaYF4 NPs for optical upconversion applications and NaGdF4 NPs as contrast agents for magnetic resonance imaging applications.25–28 Thus effective synthesis protocols are available,29–31 and we have developed syntheses of spherical NaLnF4 NPs of uniform size for a range of Ln elements.32 A bigger challenge is to coat them to make them compatible with cell suspensions in physiological media. The coating has to provide colloidal stability in phosphate-containing media and contain “stealth” polymers such as polyethylene glycol (PEG) to minimize or eliminate non-specific interactions with cells.33 Elimination of non-specific binding to cells is particularly problematic.
We are interested in using NPs to detect low-abundance biomarkers (<1000 per cell), and typical cell staining conditions require 106 Abs per cell. Thus even 0.1% non-specific binding would appear as a false positive signal. The demands for suppressing non-specific binding are far more stringent for MC than for other applications that involve treating cells with NPs. We have examined ligand exchange of the as-synthesized, oleate-coated NaLnF4 NPs with multidentate ligands based on PEG chains end-labeled with two or four phosphonate groups.34,35 We also explored coating 30 nm NaYF4:Yb,Er upconverting NPs with lipid bilayers containing a PEGylated lipid.36 Low to modest levels of non-specific interactions with cells were found with these highly stable bilayer lipid coated NPs. For example, using THP-1 cells and KG1a cells treated with a dosage of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 NPs per cell led to non-specific binding of between 3 to 10 NPs per cell. These d ≈ 30 nm NaYF4 NPs generated on the order of 4 to 5 counts per NP in the yttrium channel. This resulted in a background signal reaching 30 to 40 counts. This low level is still above the target we hope to achieve in next-generation experiments with Ab-labeled NPs. We address two major challenges in this paper. The first is to develop a surface modification process which will exhibit very minimal non-specific binding. The second is to conjugate Abs to the NPs in a way that will lead to a substantial enhancement in signal when compared to that of the current MCP based reagents.
000 NPs per cell led to non-specific binding of between 3 to 10 NPs per cell. These d ≈ 30 nm NaYF4 NPs generated on the order of 4 to 5 counts per NP in the yttrium channel. This resulted in a background signal reaching 30 to 40 counts. This low level is still above the target we hope to achieve in next-generation experiments with Ab-labeled NPs. We address two major challenges in this paper. The first is to develop a surface modification process which will exhibit very minimal non-specific binding. The second is to conjugate Abs to the NPs in a way that will lead to a substantial enhancement in signal when compared to that of the current MCP based reagents.
Here we describe experiments in which we coated d = 12 nm NaHoF4 NPs with a thin shell of silica which was then functionalized with heterobifunctional PEGs. The –COOH groups at the distal ends of the PEGs serve as sites to attach antibodies. We tested these NPs for their ability to detect biomarkers on a variety of cell lines. Labeling a goat anti-mouse (GAM) secondary antibody with the PEGylated NPs enables experiments to determine specific and non-specific binding, in the presence and absence of a mouse primary antibody. Labeling biomarkers with a broad range of expression levels showed a substantial enhancement of sensitivity for these NP reagents compared to the same GAM antibody sample labeled with a commercial MCP. In experiments labeling a primary antibody (anti-CD14) with the silica-coated NaHoF4 NPs we observe more than a 450-fold enhancement in signal of U937 cells compared to the corresponding MCP reagent.
Results and discussion
Nanoparticle synthesis
Our primary goal is to develop NP reagents able to identify low abundance cellular markers by MC. We chose NaHoF4 nanoparticles because Ho is monoisotopic and its atomic mass (165) corresponds to the maximum sensitivity of our (CyTOF®-2) mass cytometry instrument. The d = 12 nm NaHoF4 NPs we use are close to the size of an antibody and contain sufficient Ho (ca. 12000 atoms per NP) to register to 2–3 counts upon passage through the mass cytometry instrument. Oleic acid stabilized NaHoF4 NPs (d = 12 ± 0.6 nm) were synthesized by optimizing the conditions for the solvothermal coprecipitation method originally reported for NaYF4 NPs by Zhang and coworkers29 (see details in ESI†). A transmission electron microscopy image (TEM) of these NPs is presented in Fig. 1A. Particles with a narrow size distribution are essential for mass cytometry applications, because one needs to minimize the variation in metal atom content among NPs. These NPs were then coated with a silica shell through a reverse microemulsion process.37 We found that the silica shell increased the diameter of the NaHoF4 NPs to 21.3 ± 1.1 nm, which corresponds to a shell thickness of ca. 5 nm (Fig. 1B). The narrow size distribution after the silica coating confirms that the NPs were uniformly coated with a silica layer.
 | ||
| Fig. 1 TEM images of (A) oleate coated NaHoF4 NPs, d = 12.0 ± 0.6 nm, (B) NaHoF4@SiO2 NPs, after coating with a silica shell, d = 21.3 ± 1.1 nm. The scale bars are 100 nm. | ||
In order to provide colloidal stability in aqueous media and suppress non-specific binding to cells, the NPs were functionalized with PEG chains through a two-step process. The surface of the silica@NaHoF4 NPs was modified by hydrolyzing mercaptopropyl trimethoxysilane (MPTMS) in ethanol at 85 °C to introduce thiol groups on the surface of the silica. The thiol-functionalized silica-coated NaHoF4 NPs could be dispersed in water. The particles oxidized on storage and thus were treated immediately with a heterobifunctional maleimide-PEG5k-COOH (M ≈ 5000 g mol−1), leading to colloidally stable species. Characterization by dynamic light scattering (DLS) measurements gave a z-average hydrodynamic diameter (dz) of 100 nm. As shown in Fig. S1 (ESI†), these NPs maintained the same hydrodynamic diameter in PBS buffer as in water. They showed no sign of aggregation in PBS buffer as monitored by DLS over 24 h, but we did not monitor colloidal stability in PBS buffer over longer times. The NPs could be stored in water for several months and the dz of the NPs did not change over time (data not shown). The PEG-chain content of the NPs was determined using an Ellman's assay for thiol groups, before and after treatment of a NP sample with the maleimide-PEG5k-COOH reagent. In this way, we calculate approximately 830 PEG chains per silica-coated NP (details are provided in ESI†). This corresponds to a density of 0.4 PEG chains per nm2.
Antibody conjugation
A schematic of the antibody conjugation process is presented in Fig. 2. First, the COOH groups on the surface of NP-PEG-COOH were activated by reacting with EDC and sulfo-NHS. These NHS esters were then condensed with N-(2-aminoethyl)maleimide to introduce maleimide groups. In parallel, an antibody sample (goat anti-mouse Ab (GAM), or an anti-CD14 Ab) was treated with Traut's reagent to introduce reactive thiol groups. The maleimide-activated NPs and the thiolated antibodies were prepared synchronously and conjugated immediately, using an excess of Abs (ca. 45 Abs per NP), to minimize spontaneous hydrolysis of the maleimide groups and oxidation of the thiols. The NP–Ab conjugates were purified from unbound antibodies by sedimentation (16000 × g, 20 min) and the pellet was washed with PBS buffer. The conjugates were stored at 4 °C in PBS buffer containing 0.5% (w/v) BSA and maintained colloidal stability over the 7 days that they were monitored.
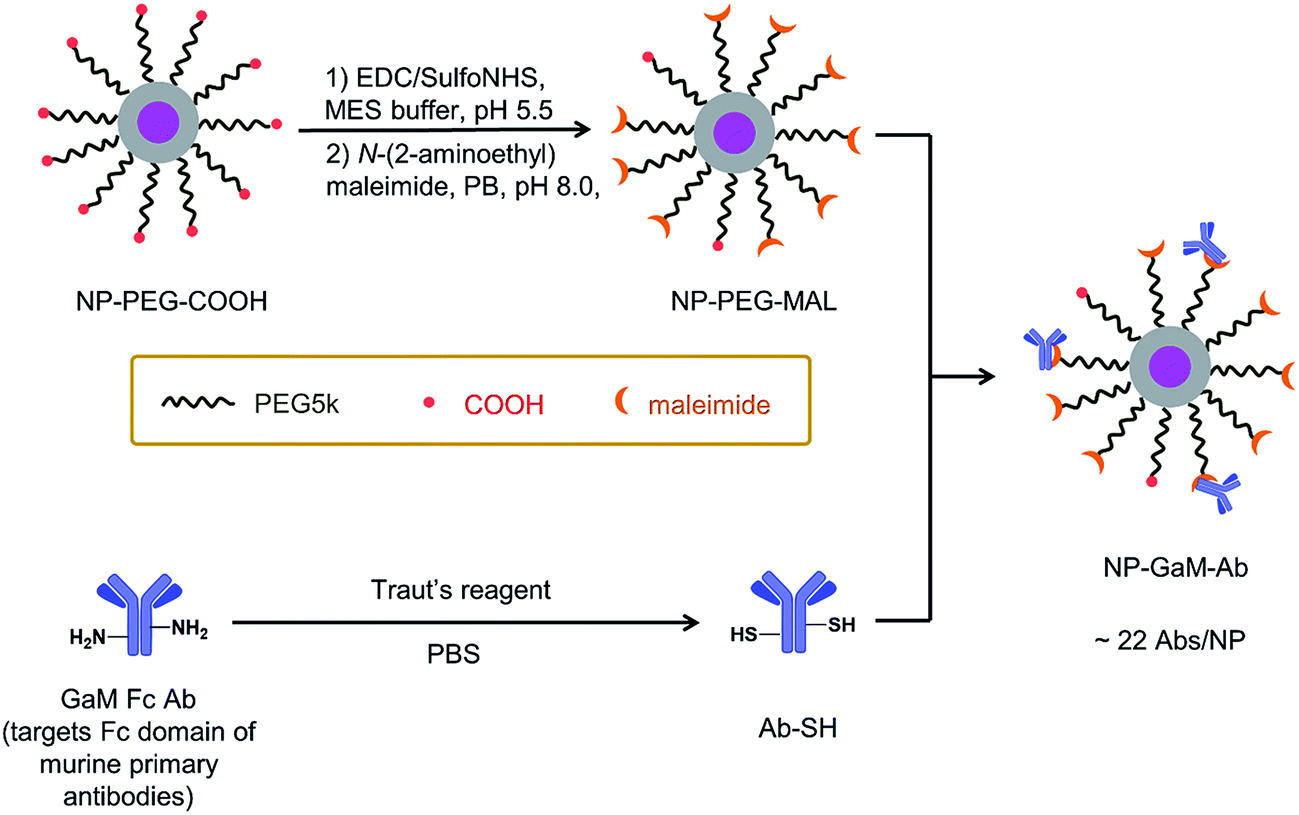 | ||
| Fig. 2 Preparation of the NaHoF4 NP–GAM–Ab conjugate via thiol-maleimide coupling. The COOH groups on the NP surface were first activated by (1) EDC and sulfo-NHS in MES buffer at pH 5.5, then the sulfo-NHS esters were displaced with (2) N-(2-aminoethyl)maleimide to introduce maleimide groups to the NPs. At the same time, the GAM Fc Ab was reacted with Traut's reagent in PBS at pH 8.0 to introduce thiol groups onto the Ab. The maleimide-activated NPs were then reacted with the thiolated Abs (ca. 45 Abs per NP) for 16 h in 50 mM PB, 75 mM NaCl, 2 mM EDTA, pH 7.2 solution to form the Ab–NP conjugates. The NPs were purified by sedimentation at 16000 × g for 20 minutes. After the centrifugation step, a Bradford assay of the supernatant led to a value of ca. 22 Abs per NP. | ||
The NP–GAM conjugates were examined by TEM and by DLS in PBS buffer. As seen in Fig. S2,† we obtained a monomodal CONTIN plot, and the sample was characterized by dz = 173 nm. From the TEM image in Fig. S2† one can see that the NPs retained their shape and size after the antibody conjugation process, but they appear to stick together upon drying on the grid. We used an indirect approach to estimate the number of GAM Abs per NP. From the known amount of Ab in the conjugation reaction, we subtracted the amount of unbound antibody in the supernatant after particle sedimentation (determined by a Bradford protein assay, Fig. S3†), which gave an average number of 22 GAM Ab per NP.
Mass cytometry assays based upon GAM
The NP–GAM Ab conjugate as a secondary antibody was examined in MC functional assays, where performance is defined by specific binding and enhanced sensitivity. Suspensions of cells of human cell lines related to acute myeloid leukemia (HL60 and KG1a), B lymphoma (Ramos), acute T cell leukemia (Jurkat) and acute monocytic leukemia (THP-1 and U937) were stained with murine anti-human primary antibodies directed to the cellular markers of interest. The cell-bound primary antibodies were then stained with the NP–GAM–Ab conjugate at varied titers. The stained cells were analyzed by MC, yielding a signal intensity histogram of 165Ho vs. cell number that indicated the binding between the cells and the NP–GAM–Ab. The 165Ho signal distribution observed represents the total binding (TB) of the NP–GAM–Ab to the cells. For simplicity, we will compare the arithmetic means of the signal distributions. Specific binding (SB) is defined as (total binding) − (non-specific binding) (NSB). NSB was determined by the 165Ho signal observed in a parallel experiment omitting the primary antibody. In all of the experiments reported here, the arithmetic mean of the NSB < 0.2% TB, and thus NSB makes a negligible contribution to the measured TB signals. As a consequence, we will consider the TB to be equivalent to the SB signals. SB/NSB values are used to determine the sensitivity of the NP conjugates.Analysis of CD4 on Jurkat-T cells
It is challenging to measure CD4 on Jurkat T cells with MCP-Ab reagents due to low abundance of CD4. We examined four different concentrations of NP–GAM–Ab (130, 660, 3300 and 16700 NPs per cell). Each experiment was carried out in parallel on Jurkat cells that were treated with anti-CD4 primary Abs (TB) and those not treated with the primary Ab (NSB). Also in parallel, two identical cell samples, in the presence and absence of the primary Ab, were incubated with 0.5 μg MCP(Tm)–GAM as a comparison control. Each experiment involved treating 2 × 106 primary stained or unstained live cells for 2 h at 4 °C in 100 μL PBS buffer containing 0.5 wt% bovine serum albumin (BSA) with the respective secondary conjugates. These experiments were carried out at 4 °C to minimize endocytosis of the NPs by the cells. Note that for an average of 20 Abs per NP, the range of antibody concentrations used (0.034 μg mL−1 to 3.34 μg mL−1) is significantly below the MCP(Tm)–GAM conjugate concentration (5 μg mL−1).
In Fig. 3, we compare results for the four different concentrations of NP–GAM–Ab and plot both the NSB and TB signals. At the lowest dose (130 NPs per cell), the NSB mean intensity (counts) was only 0.7. In analyzing mass cytometry data, we treat signal at the level of 1 to 2 counts as background. This result is consistent with no detectable NSB. In contrast, the SB signal had a mean intensity of 150 counts. Nevertheless, there is still a considerable population of cells in the SB signal histogram showing a mean value of 0. This low value indicates a cell population showing no detectable 165Ho signal because of the relatively low abundance of the CD4 antigen. When the NP dose was increased from 130 NPs per cell to 660 and 3300 NPs per cell, the mean value of the NSB signal was comparably low but the SB mean intensity increased, shifting the entire population to higher intensities such that a negligible number of cells was below the detection limit at the 3300 NPs per cell dosage. At the highest dose (16700 NPs per cell), the NSB mean intensity increased to 14, whereas the SB mean intensity increased to 8000, yielding a SB/NSB ratio of 570. More importantly, the whole population of cells exhibited a detectable 165Ho signal in the SB experiment. The low level of non-specific binding observed in these experiments shows the potential of using NPs to detect low abundance biomarkers by MC.
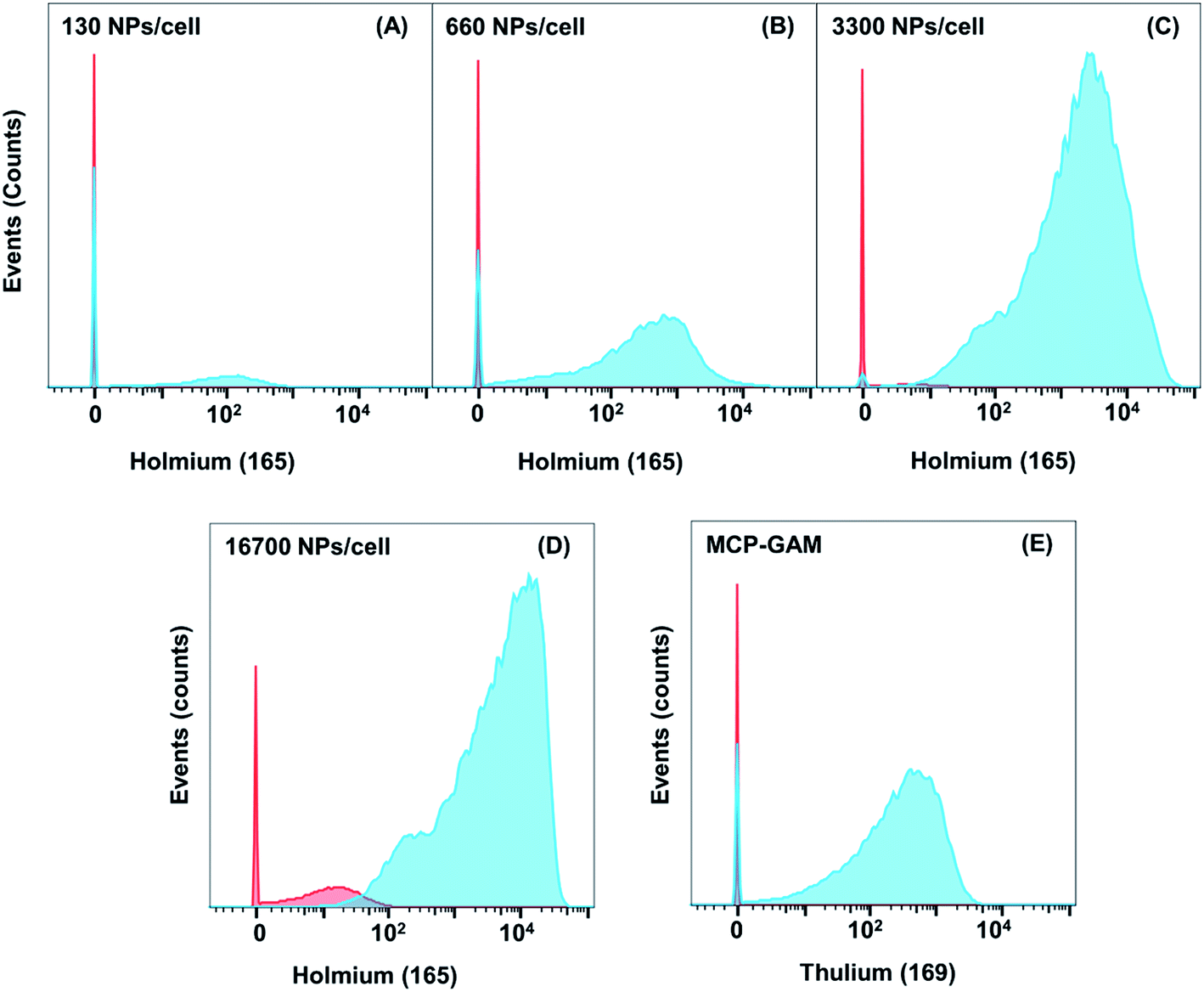 | ||
| Fig. 3 Histograms of the 165Ho signal obtained for the indirect detection of CD4 on Jurkat cells with NaHoF4 NP–GAM conjugates. These histograms are compared to the histogram obtained for a Tm-labeled MCP–GAM conjugate (ca. 3.8 × 106 MCPs per cell). The data are plotted as histograms in FlowJo format. The red histograms refer to NSB and the cyan histograms refer to TB, which is negligibly different from SB. For non-specific binding studies, the Jurkat cells were not incubated with CD4 primary antibody prior to incubation with NPs or MCPs. | ||
Because of the quantitative nature of mass spectrometry, the mean intensity of the mass cytometry histogram can be translated to the average number of NPs bound to each cell. In our experiments, the transmission coefficient of 165Ho (tHo), normalized to the signal of the 4-element calibration beads, is 2.57 × 10−4. The mean number of holmium atoms per NP were determined to be 12400 (NHo/NP) using ICP-MS measurements. In the SB experiment with the highest NP dose (16700 NPs per cell), the mean intensity IHo was 8000, which corresponds to an average of 2500 NPs per cell.
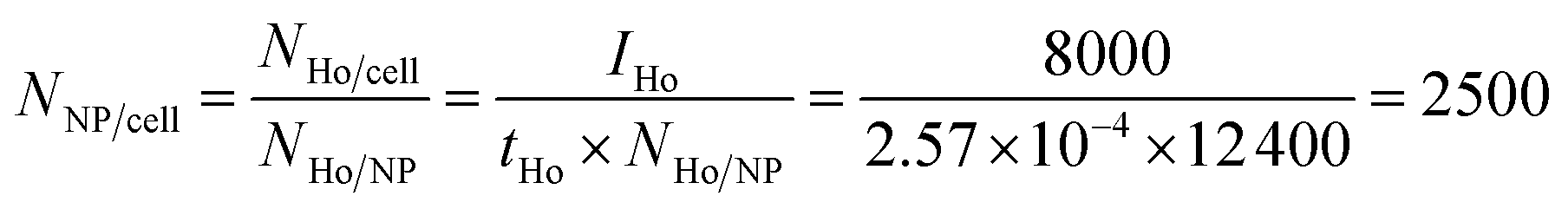 | (1) |
From this calculation, we learn that in the SB experiment, approximately 15% of the 16700 NPs per cell was specifically bound to the cells. In contrast, in the NSB experiment, the number of NPs per cell was calculated to be 14, which suggests only 14/16700 = 0.08% of the NPs were bound non-specifically to the cell surface. The numbers of NPs specifically and non-specifically bound to cells at each dosage are presented in Table 1.
| Dosage/Jurkat cells (NPs per cell) | Mean NSB | NSBb (NPs per cell) | Mean SB-CD4 | SBb (NPs per cell) |
|---|---|---|---|---|
| a The values (counts) presented in the table refer to the mean values obtained from the histograms in Fig. 4 for different NP dosages and for the MCP conjugate for the indirect detection of CD4 antigen in Jurkat cells. b Conversion of signal (counts) to NPs/cell uses the transmission coefficient of 165Ho (2.57 × 10−4) as described in eqn (1). c One million MCP(Tm)–GAM–Ab conjugates. Because 169Tm and 160Gd are very similar in atomic mass to 165Ho, their transmission coefficients, normalized to the calibration beads, are very similar: 165Ho (2.57 × 10−4), 169Tm (2.4 × 10−4), 160Gd (2.25 × 10−4). | ||||
| 130 | 0.7 | 0.2 | 150 | 50 |
| 660 | 1 | 0.3 | 930 | 290 |
| 3300 | 3 | 1 | 4000 | 1260 |
| 16700 |
14 | 5 | 8000 | 2500 |
| 3.8 × 106 (MCPs per cell)c | 0.07 | — | 500 | — |
Fig. 3E presents corresponding results for Jurkat cells incubated with 0.5 μg MCP(Tm)–GAM as a comparison control. This corresponds to 1 million MCP(Tm)–GAM conjugates per cell (3.8 × 106 MCPs per cell). The mean 169Tm intensity for SB by the MCP–GAM–Ab was 500 counts, compared to a signal intensity of 8000 found for the NP reagent at 16700 NPs per cell (Table 1). One sees that the mean signal intensity ratio between the NP- and MCP-based tags was 8000/500 = 16, indicating that the NP mass tag signal is 16-times stronger on average at these titres than the signal generated by the MCP mass tag. The plots in Fig. 3 show that the shapes of the histograms for both MCP(Tm)–GAM and for NP–GAM are skewed to the left. The same anti-CD4 antibody clone (RPA-T4) was used in both sets of measurements. The skewed nature of the distributions may be due to the broad expression level of CD4 antigen on Jurkat cell model, or it may be related to the particular antibody clone used, which may not be optimal for surface antigen detection on this cell line. For the MCP(Tm)–Ab conjugate titre, we still see a fraction of cells with a mean signal of zero, indicating that the MCPs do not possess the sensitivity to show the whole population of Jurkat cells positive for CD4 antigen. In contrast, for the specific binding experiments with NaHoF4 NP–GAM (3300 NPs per cell and 16700 NPs per cell), a negligible fraction of cells shows a count of zero due to the improvement in detection sensitivity with this type of tagging reagent. It is also important to note that the MCP reagent had a mean NSB signal of zero. This is very typical of MCP reagents in mass cytometry, that there is often no detectable NSB background.
Detection of other biomarkers
We then examined the performance of NP–GAM–Ab conjugates by staining of 6 other markers on 5 different cell lines and compared the results to the performance of MCP–GAM–Ab reagents using same series of NPs/cell dosages as above. For the MCP reagents, we used concentrations that correspond to ca. 1 × 106 MCP(Tm)–GAM–Ab conjugates per cell. Histograms of signal intensity for the SB and NSB experiments at each of the doses examined are presented in Fig. S4.† The mean intensity values of all the SB and NSB experiments are summarized in Table S1.† The SB intensity values for all 7 biomarkers on the various cell lines are presented in a bar graph format in Fig. 4. Based on the differences in SB intensity values between the NP- and MCP-conjugates, we have arbitrarily divided the biomarkers into three groups.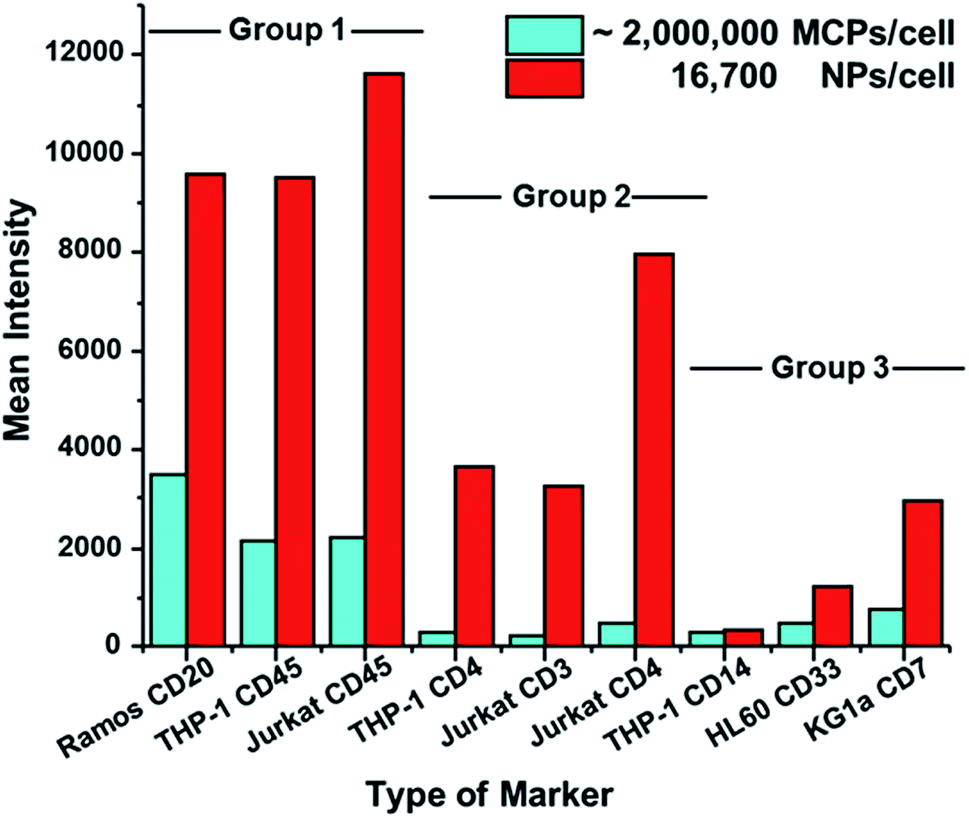 | ||
| Fig. 4 Bar graphs showing the difference in the specific binding (SB) signal between the MCP (Tm)–GAM and NaHoF4 NP–GAM conjugates. Depending on the signal difference between the MCPs and NPs the markers have been classified in to three groups. Group 1 has CD20, and CD45 markers which shows 2 to 3 times higher SB signal for NPs compared to MCPs. Group 2 comprises CD4, and CD3 markers which shows 15 to 16 times higher SB signal for NPs compared to MCPs. Group 3 comprises CD14, CD33 and CD7 markers, which show only 1 to 3 times higher signal for the NPs than their MCP counterparts. | ||
Group 1, which includes CD20 (clone – 2H7) on Ramos cells, CD45 (clone – HI30) on THP-1 cells and CD45 on Jurkat cells, represents a group of cellular markers that are highly expressed on their respective cell lines. The mean SB intensity values obtained by the use of the MCP tag on these markers exceeded 2000 counts. This is a very strong signal. The NP–GAM–Ab conjugates at 16700 NPs per cell gave an even stronger signal, reaching 10000 counts. For high-expression biomarkers, the NPs offer little or no advantage over MCP reagents for biomarker detection.
Group 2 (Fig. 4) includes markers that are less abundant than those in Group 1, namely CD4 (clone – RPA-T4) on THP-1 cells and on Jurkat cells, and CD3 (clone – UCHT1) on Jurkat cells. The mean SB intensity values from the MCP conjugates in this group were between 200 and 500 counts, a factor of 2–10 lower than the values in Group 1. When the NaHoF4 NP–GAM–Ab conjugates were used to detect these markers, the mean SB intensity values were 10–15 times stronger than the values given by the MCP tags, showing a clear evidence of the improved sensitivity of NP tags in detecting relatively low abundance markers compared to MCP tags.
Group 3 includes CD7 (clone – 6B7) on KG1a cells, CD33 (clone – WM53) on HL60 cells and CD14 (clone – M5E2) on THP-1 cells, all of which are low abundance markers. Here, however, the mean SB intensities of the NP conjugate, compared to its MCP counterpart, was enhanced by only a factor of 1 to 3. For example, the NP conjugate at 16700 NPs per cell gave a mean signal of 3000 counts for CD7 on KG1a cells, whereas the MCP conjugate gave a mean signal of 800 counts. An even smaller difference was found for CD14 on THP-1 cells. This NP dosage yielded a mean signal of 340 counts whereas the MCP conjugate gave a mean signal of 290 counts. We have no definitive explanation for the lower increases in intensity that would be expected from the NaHoF4–NP–GAM Ab conjugates. Later assays using labeled primary antibodies (see below) show significant enhancements in sensitivity, suggesting that the staining conditions or particulars of the secondary antibody recognition complicate this analysis.
Detection of CD14 using a NaHoF4 NP–Anti CD14 Ab conjugate
We next turned our attention to the use of a monoclonal primary Ab for low expression biomarker detection. Here we examine CD14 expression in THP-1 and U937 cells with the NaHoF4 NPs conjugated to an anti-CD14 Ab. CD14 is a well-known low abundance marker. In most cases, the expression level is enhanced through the stimulation of cells. Our intention was to see whether we can detect CD14 expression in THP-1 and U937 cells without any kind of external stimulation to the cells. Conjugation of anti-CD14 Ab to the NaHoF4 NPs was carried out as described above for the NP–GAM conjugate. The Bradford assay also led to a value of ca. 20 Abs per NP. Ramos cells, which do not express CD14, were employed as a control to determine NSB. We incubated the THP-1, U937 and Ramos cells (2 million cells, PBS buffer + 0.5% BSA, 4 °C) with a series of NP dosages ranging from 200 NPs per cell to 200000 NPs per cell. Histograms for low doses of NP conjugates are presented in Fig. S5,† and histograms for higher doses are presented below in Fig. 5.
 | ||
| Fig. 5 Histograms of the signal obtained for the detection of CD14 marker in THP-1 cells and U937 cells using a primary anti-CD14 Ab. (A, B) Refer to THP-1 cells treated with two different titers of NaHoF4 NP–CD14 conjugates. (C, D) Refer to U937 cell treated with titres equivalent to the THP-1 cells. (E) Refers to THP-1 cells treated with a one million Gd-labeled MCP–CD14 conjugates per cell. (F) Refers to U937 cells treated with the same dose of MCP–CD14 conjugate. The blue curves represent the positive signal (specific binding) and the red curves represent the negative signal (NSB). The negative controls used for these measurements are Ramos cells, which do not express CD14. | ||
At the initial dosage of 200 NPs per cell, there was no signal observed for CD14 expression in both U937 and THP-1 cells. At 1000 NPs per cell for both cell lines (Fig. S5†), we found mean SB values of 15–20 counts, with a significant population of cells (∼80%) below the detection limit. As the staining concentration was increased to 5000, and 25000 NPs per cell (Fig. S5†) the histogram shifted to mean signal intensities of 110 and 410 counts (Table S3†), respectively for THP-1 cells and 80 and 290 counts for U937 cells. There is still a portion of cells which is below the detection limit at these staining concentrations (Fig. S5†). When the staining dosage was further increased to 75000 NPs per cell (Fig. 5), the mean value increased to 880 counts for THP-1 cells and 480 counts for U937 cells (Tables S3 and S4†). For the highest dosage of 200000 NPs per cell, the mean 165Ho signal was 1920 counts for THP-1 cells and 900 counts for U937 cells (Fig. 5).
To our surprise, the mean NSB on Ramos cells (CD14−) at the highest dosage (200000 NPs per cell) was just 15 counts, similar to the mean NSB signal (22 counts) for the NP–GAM–Ab conjugate at 16700 NPs per cell used to detect CD14 on THP-1 cells. Calculation of the mean number of nanoparticles in the 200000 NPs per cell experiments gives ca. 600 NPs per cell for the SB experiment with the primary CD14 Ab and only ca. 5 NP per cell in the NSB control. The NSB binding here represents only 0.0025% of the NPs applied. This is significantly below the NSB observed in the application of the NaHoF4–NP–GAM secondary antibody (0.18% at 16700 NPs per cell). GAM is a polyclonal antibody that consists of many different monoclonal IgGs, each of which will have a different non-specific binding characteristic. With this monoclonal CD14 (clone-M5E2) antibody, the IgG is characterized by low NSB. With a different clone or another monoclonal Ab, this may not be the case. It is also accepted that polyclonal antibodies in general have higher background.
Furthermore, when comparing the primary NP-conjugate to the primary MCP–160Gd–antiCD14 Ab conjugate, significant improvements in sensitivity were observed. For THP-1 cells stained with 1 million MCP–160Gd–Ab conjugates/cell, a mean value of ca. 66 counts was observed and for U937 cells, the mean signal was near the detection limit (3 counts) (Table S3†). Even though the mean value obtained for CD14 in THP-1 cells is far above the background, we observed that the histogram shows a substantial signal at zero counts. When we compare the performance of NaHoF4 NP–antiCD14 conjugates with the MCPs, we find that the total population of THP-1 cells has been shifted towards a mean value that is much higher than the background signal. When we compare the mean value of 66 obtained from the MCP-based reagent with the NP reagent, the NP reagents (200000 NPs per cell) performed 30 times better. For U937 cells the MCP–160Gd–antiCD14 Ab reagent yielded such a low signal (2 counts, essentially equal to background), that one would conclude that CD14 is not expressed on U937 cells. When we compare the mean intensities obtained from the NPs (200000 NPs per cell) and MCPs the NPs exhibit a signal at least 450 times higher than that of the MCPs. This result shows how nanoparticle–Ab conjugates can be a powerful tool to identify and detect low abundance markers and rare phenotypes.
Mass cytometry experiments to identify the CD14 population in PBMC
Based on the encouraging results described above, we next examined the interaction of NaHoF4 NP–CD14-conjugates with PBMCs. First, we note that mass and flow cytometry have been shown to provide comparable cellular subset and marker frequencies for PBMCs.38 The experiments reported here were carried out with three different objectives in mind: (i) to test whether the NP-based reagents could identify a specific phenotype in a mixture of cell types; (ii) to examine the level of non-specific interaction of NPs with a variety of cells; and (iii) to see if the NP reagent interfered with the detection of other biomarkers in a multiparameter assay with MCP-based reagents. In these experiments, the PBMC were treated sequentially with the CD14–NP conjugate followed, after washing, by a cocktail of six MCP–Ab conjugates targeting CD20, CD45, CD16, CD4, CD8a and CD3 surface markers. We studied a series of CD14–NP dosages, from 200 to 25000 NPs per cell. As a comparison control experiment, the NaHoF4 NP–antiCD14 conjugate was replaced with its MCP–160Gd–antiCD14 counterpart. The PBMC sample was gated using the CD20, CD45, CD16, CD4, CD14, CD8a and CD3 surface markers, and the gating strategy is shown in Fig. S6.† The classical monocytes (CD14+CD16−) show an increase in mean signal value for CD14 as the nanoparticle dosage was increased from 200 to 25000. The histograms and mean values for the dosages from 200 to 5000 NPs per cell are presented in Fig. S7 and Table S4.† The histograms for the NP-conjugate at 25000 NPs per cell and for the MCP reagents are presented in Fig. 6. At the highest dosage (25000 NPs per cell), we obtained a mean value of 2150 counts for the CD14+CD16− classical monocytes, whereas with the MCPs, we obtained 110 counts as the mean value (Table 2). It is clear that the NP conjugate is far more sensitive compared to the MCP conjugate. When we analyzed the CD4+ T-lymphocytes, the CD8+ T-lymphocytes and the B-lymphocytes for the 165Ho NP signal (Fig. 6, Table 2), the mean value was 4 to 5 counts, just slightly above the background. We see that the interaction of the NaHoF4 NP–CD14 conjugate with cells that do not express CD14 is minimal at 25000 NPs per cell.
 | ||
| Fig. 6 Histograms of the signals obtained from the NaHoF4 NP–antiCD14 conjugate (dosage 25000 NPs per cell) and the MCP–160Gd–antiCD14 conjugate obtained for six different cell phenotypes found in the PBMCs. (A) CD3+CD20− (T lymphocytes), (B) CD20+CD3− (B lymphocytes), (C) CD4+CD8a− (T lymphocytes), (D) CD4−CD8a+ (T lymphocytes), (E) CD14+CD16− (classical monocytes), (F) CD16+CD14− (NK/non-classical monocytes). The mean values for 165Ho and 160Gd for all six phenotypes are presented in Table 2. These data were obtained from a 7-plex experiment. For experiments with the NPs, the NPs were initially incubated with the cells (2 h at 4 °C), and after washing, followed by incubating with the six MCP–Ab conjugates. The cells were washed, fixed, and stained with the Ir intercalator prior to analysis by MC. For the control experiments with the MCP–160Gd–antiCD14 conjugate, the cells were stained with all seven MCP–Ab conjugates simultaneously. | ||
000 NPs per cell
| Phenotype (PBMC) | MCP–CD14 | NP–CD14 |
|---|---|---|
| CD3+CD20− (T-lymphocytes) | 2 | 3 |
| CD20+CD3− (B-lymphocytes) | 2 | 5 |
| CD4+CD8− (T-lymphocytes) | 2 | 4 |
| CD4−CD8+ (T-lymphocytes) | 1 | 2 |
| CD14+CD16− (classical monocytes) | 110 | 2160 |
| CD16+CD14− (NK/non-classical monocytes) | 0.5 | 3 |
The PBMC experiments also provide information on whether the NPs affect the ability of other MCP-based conjugates to bind to specific markers when used in a multiplexed assay. If the NPs interfered with antigen–antibody binding at the cell surface, the MCP-tagged reagents would show a lower signal for all markers in the presence of the NP conjugate compared to the control experiment in which the NPs were replaced with the MCP(Gd)–CD14 conjugate. The mean signals obtained for the MCP-tagged antibodies (CD20, CD4, CD8, CD3, CD16, CD45) determined in the presence of the NPs exhibit no significant statistical difference from those of the control sample. The mean values for all markers are presented in Table S5.† This result clearly establishes that the presence of the NPs did not hinder the marker recognition of other MCP–Ab conjugates.
Conclusions
We synthesized 12 nm NaHoF4 NPs coated with a thin silica shell to which functional PEG(5k) were attached. This construct was conjugated to either a GAM antibody or an anti-CD14 primary Ab using thiol maleimide chemistry. The GAM Ab conjugated NPs were tested for indirect detection of both abundant markers (CD20, CD45, CD33) and less abundant markers (CD3, CD4, CD14 and CD7) in different cell lines. We found that the NP–GAM conjugate showed an enhancement of ca. 15 times for the detection of CD4 and CD3 biomarkers in Jurkat cells when compared to the MCP reagent. With abundant markers like CD45 and CD20, the NP–GAM conjugates gave signals close to saturation of the instrument (∼10000 counts). The signal intensity when compared to MCP-based reagents was only 2 to 3 times higher for CD33 and CD7 markers. With CD14, there was no enhancement in signal from the NP–GAM conjugates compared to the MCPs. We do not have any explanation at this point to why less abundant markers like CD7, and CD14 perform poorly in this secondary Ab assay when compared to the other low-abundance markers CD4 and CD3.
In contrast, with the direct detection of CD14 marker using a primary NP–antiCD14 conjugate, we found large enhancements in signal compared to a corresponding MCP–antiCD14 conjugate. For THP-1 cells, the enhancement in signal for the NPs was more than 30 times higher. In the U937 cell line, which is similar to THP-1 macrophage cells, we found an even larger enhancement, more than two orders of magnitude higher (>400 times), compared to the MCP–antiCD14 reagent. It is clear that for this low expression biomarker, the NP reagent offers a significant advantage in terms of enhanced signal. Even at a very high dosage (200000 NPs per cell), the level of non-specific binding was remarkably low. We note that direct conjugation of NP to the primary CD14 Ab provides not only greater signal enhancement, but also lower NSB levels than experiments with NP–GAM as a secondary Ab.
A 7-plex assay carried out on PBMC samples showed that the presence of the NP–CD14 conjugate along with other MCP–Ab conjugates did not interfere with the ability of metal-tagged antibodies to recognize their target antigen. Additionally, the signal for the detection of classical CD14-positive monocytes showed a 20-fold increase for the NP conjugate compared to the control, where it was replaced with a MCP–antiCD14 conjugate. The NP reagent also exhibited a very small signal, only slightly above the background, for non-specific interaction with other PBMC cell types.
Experiments reported here using NaHoF4 NPs for mass cytometry single cell assays represent a very important first step in utilizing NPs as high sensitivity reagents to identify low abundance markers. Efforts are underway to extend the synthesis and coating protocols to other lanthanide NPs32 with the intent of creating a family of NP reagents for multiparameter MC assays.
Conflicts of interest
The University of Toronto authors declare no competing conflict of interest. Authors AB, OO, and VB are employees of Fluidigm Canada.Acknowledgements
The authors thank the NSERC Canada and Fluidigm Canada (Markham, ON), Ontario Centres of Excellence (OCE), and the Province of Ontario for their support of this research. J. P thanks MITACS Canada and Fluidigm Canada for a MITACS Accelerate fellowship.References
- T. Konry, I. Smolina, J. M. Yarmush, D. Irimia and M. L. Yarmush, Small, 2010, 7, 395–400 CrossRef PubMed.
- G. K. Behbehani, Clin. Lab. Med., 2017, 37, 945–964 CrossRef PubMed.
- Q. Baca, A. Cosma, G. Nolan and B. Gaudilliere, Cytometry, Part B, 2017, 92, 10–11 CrossRef PubMed.
- P. H. Roos and N. Jakubowski, Bioanalysis, 2010, 2, 295–309 CrossRef CAS PubMed.
- P. K. Chattopadhyay, D. A. Price, T. F. Harper, M. R. Betts, J. Yu, E. Gostick, S. P. Perfetto, P. Goepfert, R. A. Koup, S. C. De Rosa, M. P. Bruchez and M. Roederer, Nat. Med., 2006, 12, 972–977 CrossRef CAS PubMed.
- D. R. Bandura, V. I. Baranov, O. I. Ornatsky, A. Antonov, R. Kinach, X. Lou, S. Pavlov, S. Vorobiev, J. E. Dick and S. D. Tanner, Anal. Chem., 2009, 81, 6813–6822 CrossRef CAS PubMed.
- E. W. Newell and Y. Cheng, Nat. Immunol., 2016, 17, 890–895 CrossRef CAS PubMed.
- S. C. Bendall, E. F. Simonds, P. Qiu, E.-a. D. Amir, P. O. Krutzik, R. Finck, R. V. Bruggner, R. Melamed, A. Trejo, O. I. Ornatsky, R. S. Balderas, S. K. Plevritis, K. Sachs, D. Pe'er, S. D. Tanner and G. P. Nolan, Science, 2011, 332, 687–696 CrossRef CAS PubMed.
- A. H. Rahman and M. R. G. O'Gorman, J. Immunol. Methods, 2018, 453, 1–2 CrossRef CAS PubMed.
- C. Benoist and N. Hacohen, Science, 2011, 332, 677–678 CrossRef CAS PubMed.
- J. Immunol. Methods, 2018, themed issue on Mass Cytometry: Methods and Applications Search PubMed.
- C. E. Wogsland, A. R. Greenplate, A. Kolstad, J. H. Myklebust, J. M. Irish and K. Huse, Cytometry, Part B, 2016, 92, 79–87 CrossRef PubMed.
- S. C. Bendall, G. P. Nolan, M. Roederer and P. K. Chattopadhyay, Trends Immunol., 2012, 33, 323–332 CrossRef CAS PubMed.
- D. Majonis, I. Herrera, O. Ornatsky, M. Schulze, X. Lou, M. Soleimani, M. Nitz and M. A. Winnik, Anal. Chem., 2010, 82, 8961–8969 CrossRef CAS PubMed.
- N. Illy, D. Majonis, I. Herrera, O. Ornatsky and M. A. Winnik, Biomacromolecules, 2012, 13, 2359–2369 CrossRef CAS PubMed.
- Z. B. Bjornson, G. P. Nolan and W. J. Fantl, Curr. Opin. Immunol., 2013, 25, 484–494 CrossRef CAS PubMed.
- X. Wu, Q. DeGottardi, I. C. Wu, J. Yu, L. Wu, F. Ye, C.-T. Kuo, W. W. Kwok and D. T. Chiu, Angew. Chem., Int. Ed., 2017, 56, 14908–14912 CrossRef CAS PubMed.
- A. Horowitz, D. M. Strauss-Albee, M. Leipold, J. Kubo, N. Nemat-Gorgani, O. C. Dogan, C. L. Dekker, S. Mackey, H. Maecker, G. E. Swan, M. M. Davis, P. J. Norman, L. A. Guethlein, M. Desai, P. Parham and C. A. Blish, Sci. Transl. Med., 2013, 5, 208ra145 Search PubMed (see Table S1 in ESI†).
- A. W. Kay, D. M. Strauss-Albee and C. A. Blish, in Natural Killer Cells: Methods and Protocols, Methods in Molecular Biology, ed. S. S. Somanchi, Humana Press, New York, 2016, ch. 2, vol. 1441, pp. 13–26 Search PubMed.
- F. T. Lee-Montiel and P. I. Imoukhuede, J. Mater. Chem. B, 2013, 1, 6434–6441 RSC.
- Z. B. Bjornson, G. P. Nolan and W. J. Fantl, Curr. Opin. Immunol., 2013, 25, 484–494 CrossRef CAS PubMed.
- A. R. Schulz, S. Stanislawiak, S. Baumgart, A. Grützkau and H. E. Mei, Cytometry, Part A, 2017, 91, 25–33 CrossRef CAS PubMed.
- Y.-S. S. Yang, P. U. Atukorale, K. D. Moynihan, A. Bekdemir, K. Rakhra, L. Tang, F. Stellacci and D. J. Irvine, Nat. Commun., 2017, 8, 14069 CrossRef CAS PubMed.
- Y. Guo, S. Baumgart, H.-J. Stärk, H. Harms and S. Müller, Front. Microb., 2017, 8, 1326 CrossRef PubMed.
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed.
- J. Zhou, Z. Liu and F. Li, Chem. Soc. Rev., 2012, 41, 1323–1349 RSC.
- N. J. J. Johnson, W. Oakden, G. J. Stanisz, R. Scott Prosser and F. C. J. M. van Veggel, Chem. Mater., 2011, 23, 3714–3722 CrossRef CAS.
- Z. Li and Y. Zhang, Nanotechnology, 2008, 19, 345606 CrossRef PubMed.
- J. Ryu, H.-Y. Park, K. Kim, H. Kim, J. H. Yoo, M. Kang, K. Im, R. Grailhe and R. Song, J. Phys. Chem. C, 2010, 114, 21077–21082 CrossRef CAS.
- C. Liu, Z. Gao, J. Zeng, Y. Hou, F. Fang, Y. Li, R. Qiao, L. Shen, H. Lei, W. Yang and M. Gao, ACS Nano, 2013, 7, 7227–7240 CrossRef CAS PubMed.
- L. Tong, E. Lu, J. Pichaandi, G. Zhao and M. A. Winnik, J. Phys. Chem. C, 2016, 120, 6269–6280 CrossRef CAS.
- U. Kostiv, V. Lobaz, J. Kučka, P. Švec, O. Sedláček, M. Hrubý, O. Janoušková, P. Francová, V. Kolářová, L. Šefc and D. Horák, Nanoscale, 2017, 9, 16680–16688 RSC.
- P. Cao, L. Tong, Y. Hou, G. Zhao, G. Guerin, M. A. Winnik and M. Nitz, Langmuir, 2012, 28, 12861–12870 CrossRef CAS PubMed.
- G. Zhao, L. Tong, P. Cao, M. Nitz and M. A. Winnik, Langmuir, 2014, 30, 6980–6989 CrossRef CAS PubMed.
- J. Pichaandi, L. Tong, A. Bouzekri, Q. Yu, O. Ornatsky, V. Baranov and M. A. Winnik, Chem. Mater., 2017, 29, 4980–4990 CrossRef CAS.
- F. Liu, Q. Zhao, H. You and Z. Wang, Nanoscale, 2013, 5, 1047–1053 RSC.
- K. J. Nicholas, A. R. Greenplate, D. K. Flaherty, B. K. Matlock, J. San Juan, R. M. Smith, J. M. Irish and S. A. Kalams, Cytometry, Part A, 2016, 89, 271–280 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional tables, figures and experimental details. See DOI: 10.1039/c8sc04407d |
| ‡ These authors contributed equally for this manuscript. |
| This journal is © The Royal Society of Chemistry 2019 |