
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nickel-catalyzed cyanation of aryl halides and triflates using acetonitrile via C–CN bond cleavage assisted by 1,4-bis(trimethylsilyl)-2,3,5,6-tetramethyl-1,4-dihydropyrazine†
Yohei
Ueda
 ,
Nagataka
Tsujimoto
,
Taiga
Yurino
,
Hayato
Tsurugi
* and
Kazushi
Mashima
*
,
Nagataka
Tsujimoto
,
Taiga
Yurino
,
Hayato
Tsurugi
* and
Kazushi
Mashima
*
Department of Chemistry, Graduate School of Engineering Science, Osaka University, Toyonaka, Osaka 560-8531, Japan. E-mail: tsurugi@chem.es.osaka-u.ac.jp; mashima@chem.es.osaka-u.ac.jp
First published on 26th November 2018
Abstract
We developed a non-toxic cyanation reaction of various aryl halides and triflates in acetonitrile using a catalyst system of [Ni(MeCN)6](BF4)2, 1,10-phenanthroline, and 1,4-bis(trimethylsilyl)-2,3,5,6-tetramethyl-1,4-dihydropyrazine (Si–Me4-DHP). Si–Me4-DHP was found to function as a reductant for generating nickel(0) species and a silylation reagent to achieve the catalytic cyanation via C–CN bond cleavage.
Nitriles are highly versatile functional compounds often used to prepare the corresponding amines, amides, and carboxylic acids.1 Several useful synthetic protocols have been established as not only stoichiometric reactions, such as the Sandmayer reaction and Rosenmund-von Braun reaction using CuCN,2 but also metal-catalyzed cyanation reactions of aryl halides or aryl triflates using toxic cyanide reagents,1f,3 such as NaCN,4 KCN,5 CuCN,6 Zn(CN)2,7 and Me3SiCN.8 Recent efforts have been aimed at developing non-toxic cyanation reactions using alkylnitriles,5h,9 despite the relatively high bond dissociation energy of the C–CN bond (e.g. 133 kcal mol−1 for the C–CN bond of acetonitrile) compared with that of the typical C(sp3)–C(sp3) bond (ca. 83 kcal mol−1).10 In fact, pivotal studies using acetonitrile as a cyano source were recently achieved at high temperature (120–160 °C): Cheng et al. used phosphine complexes of palladium and nickel for the catalytic cyanation of ortho-mono and ortho-disubstituted aryl halides with acetonitrile in the presence of zinc powder at elevated temperature (160 °C) (Fig. 1a),9d and Shen et al. applied a catalyst system of copper(II) nitrate with 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) for the cyanation of aryl iodides in acetonitrile at high temperature (150 °C) (Fig. 1b).9e,f Quite recently, Morandi et al. demonstrated that a combination of bis(acetylacetonato)nickel(II) with Xantphos became a catalyst for the cyanation of aryl chlorides and aryl triflates with n-butyronitrile as a cyano source in the presence of highly reactive Lewis acids such as tri(isobutyl)aluminum at high temperature (120 °C) (Fig. 1c).9g
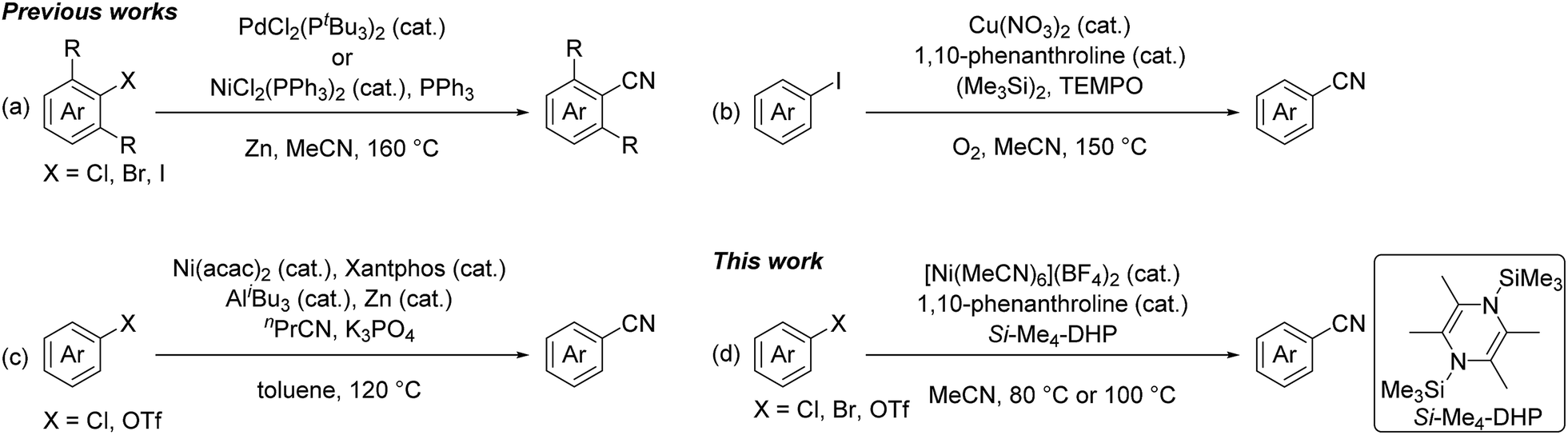 | ||
| Fig. 1 Recent catalytic cyanation reactions using alkylnitriles. | ||
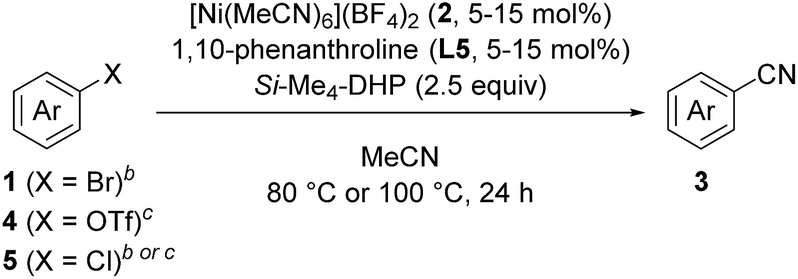
In this context, we have continuously focused our efforts on finding a catalyst system active for such cyanation using acetonitrile under milder reaction conditions. Herein, we report a nickel-catalyzed cyanation of aryl halides and aryl triflates with acetonitrile at 80 °C or 100 °C in the presence of an organosilicon compound, 1,4-bis(trimethylsilyl)-2,3,5,6-tetramethyl-1,4-dihydropyrazine (Si–Me4-DHP), which served as both an effective reductant and a silylating reagent for nickel species (Fig. 1d).11 It is noteworthy that C–CN bond cleavage of acetonitrile was achieved by α-methyl elimination from an [N-(trimethylsilyl)iminoacyl]nickel intermediate, which was generated by insertion of acetonitrile into Ni–SiMe3 species.
We started by searching for the best nickel catalyst system for the cyanation of 4-bromoanisole (1a) in acetonitrile, and the results are summarized in Table 1. The reaction mixture of 1a and Si–Me4-DHP (2.5 equiv.) in the presence of catalytic amounts of [Ni(MeCN)6](BF4)2 (2, 5 mol%) and 2,2′-bipyridine (L1, 5 mol%) in acetonitrile was heated at 80 °C for 24 h to give p-anisonitrile (3a) in 33% yield (entry 1). We further examined other nitrogen-based bidentate ligands: 4,4′-disubstituted-2,2′-bipyridine L2–L4 afforded 3a in low yields (entries 2–4), whereas 1,10-phenanthroline (L5) drastically improved the yield of 3a up to 91% (entry 5). Other 1,10-phenanthroline-based ligands bearing substituents at 4,7-positions, such as methyl (L6), methoxy (L7), and phenyl (L8), were less effective than L5, giving 3a in 58%, 30%, and 59% yields, respectively (entries 6–8). Accordingly, we selected L5 as the best ligand. Furthermore, we found that the catalytic activity was quite sensitive to the amounts of L5: 2 equiv. of L5 slightly decreased the yield (86%) of 3a and 3 equiv. of L5 significantly suppressed the activity to 54% yield (entries 9 and 10), the latter of which was consistent with the observation that an isolated dicationic tris(1,10-phenanthroline)nickel complex, [Ni(L5)3](BF4)2, afforded 3a in 61% yield (entry 11). Upon varying the reaction temperature at 60 °C and 100 °C, we obtained 3a in lower yields (75% and 86% yields, respectively) compared with the reaction conducted at 80 °C, giving 3a in 91% yield (entries 12 and 13 vs. entry 5). The addition of L5 was essential: the catalyst system without L5 gave 3a in a low yield (10%) (entry 14). The selection of the reducing reagents is the most significant factor for the cyanation reaction: no cyanation was observed when applying typical metal-based reducing reagents, such as zinc and manganese powder, as well as a typical organic reductant, tetrakis(dimethylamino)ethylene (TDAE) (entries 15–17), and the reaction without Si–Me4-DHP did not produce 3a (entry 18), indicating that Si–Me4-DHP was indispensable for the catalytic cyanation reaction (vide infra). As a result, we selected the mixture of catalytic amounts of 2 (5 mol%) and L5 (5 mol%) in combination with an excess amount of Si–Me4-DHP (2.5 equiv.) as the best catalyst system.12

|
|
|||
|---|---|---|---|
| Entry | Ligand (x mol%) | Reductant | Yieldb (%) |
| a Reaction conditions: 1a, 0.10 mmol (0.025 M). b 1H NMR yield using 1,3,5-trimethoxybenzene as an internal standard. c [Ni(L5)3](BF4)2 was used instead of 2. d 60 °C. e 100 °C. | |||
| 1 | L1 (5) | Si–Me4-DHP | 33 |
| 2 | L2 (5) | Si–Me4-DHP | 19 |
| 3 | L3 (5) | Si–Me4-DHP | 17 |
| 4 | L4 (5) | Si–Me4-DHP | 14 |
| 5 | L5 (5) | Si–Me4-DHP | 91 |
| 6 | L6 (5) | Si–Me4-DHP | 58 |
| 7 | L7 (5) | Si–Me4-DHP | 30 |
| 8 | L8 (5) | Si–Me4-DHP | 59 |
| 9 | L5 (10) | Si–Me4-DHP | 86 |
| 10 | L5 (15) | Si–Me4-DHP | 54 |
| 11c | — | Si–Me4-DHP | 61 |
| 12d | L5 (5) | Si–Me4-DHP | 75 |
| 13e | L5 (5) | Si–Me4-DHP | 86 |
| 14 | — | Si–Me4-DHP | 10 |
| 15 | L5 (5) | Zn | 0 |
| 16 | L5 (5) | Mn | 0 |
| 17 | L5 (5) | TDAE | 0 |
| 18 | L5 (5) | — | 0 |
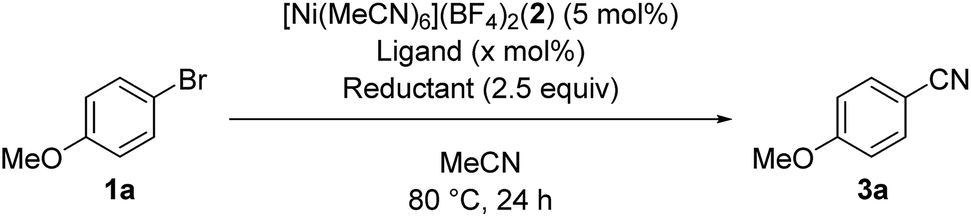
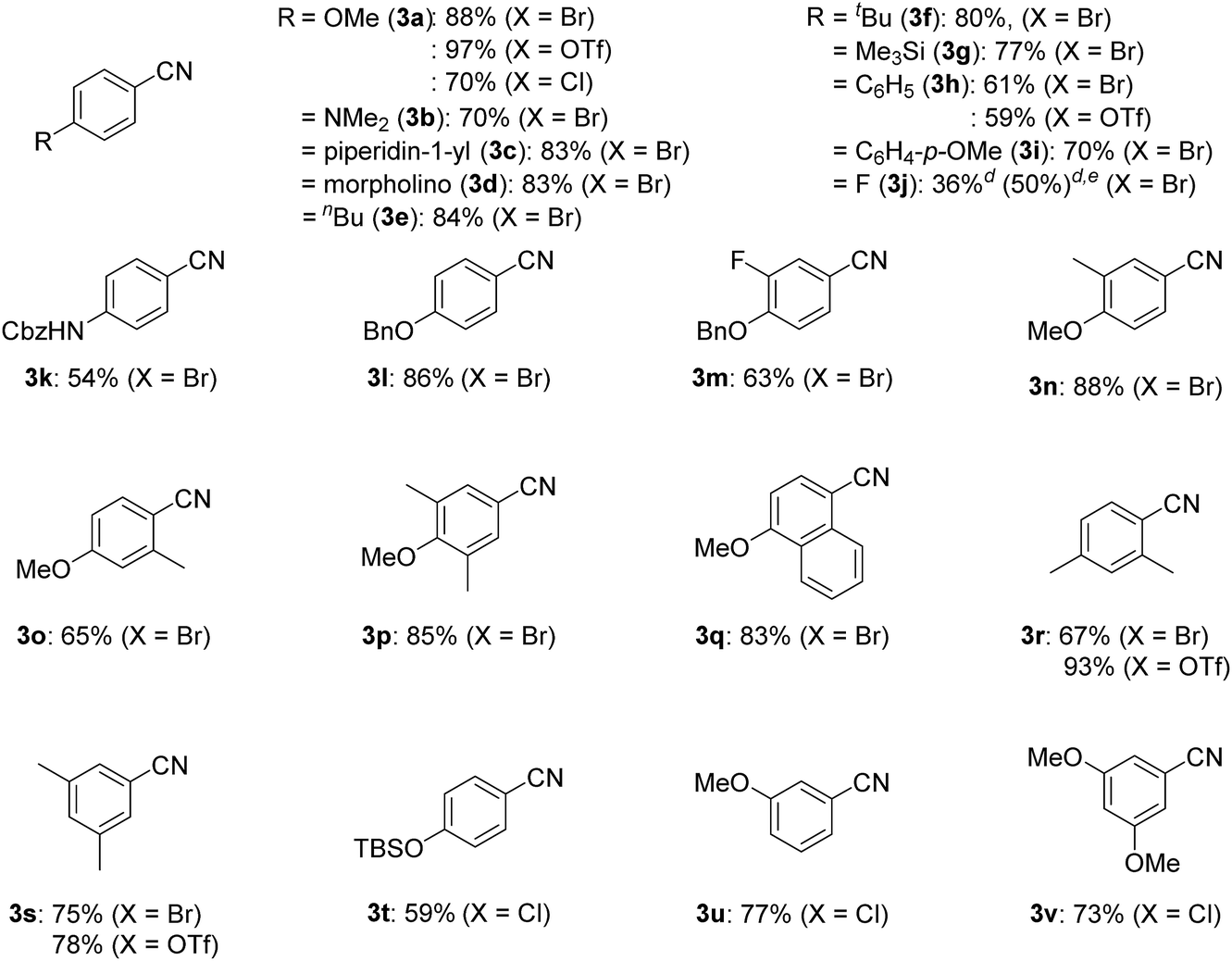
With the best catalyst system in hand, we examined the substrate scope of this nickel-catalyzed cyanation reaction (Table 2). Aryl bromides with electron-donating groups such as methoxy (1a), dialkylamino (1b–1d), and alkyl groups (1e and 1f) at the para-position gave the corresponding aryl nitriles 3a–3f in excellent to good yields (70–88%). Additionally, aryl bromides with electronically neutral para-substituents such as trimethylsilyl (1g), phenyl (1h), and p-methoxyphenyl (1i) were efficiently transformed into the corresponding aryl nitriles 3g–3i in good yields (61–77%). On the other hand, aryl bromides with electron-withdrawing groups significantly decreased the yield of the corresponding aryl nitriles though the aryl bromides were completely consumed; 4-fluorobromobenzene (1j) afforded 4-fluorobenzonitrile (3j) in 36% yield together with unidentified byproducts, while aryl bromides with cyano, methoxycarbonyl, nitro, and trifluoromethyl groups resulted in a complex mixture. When the amount of Si–Me4-DHP was reduced to 1.1 equiv. for the cyanation of 1j, 3j was obtained in 50% yield together with 4-fluoroacetophenone (14% yield) as the main byproduct after acidic work-up. Typical protecting groups such as benzyloxycarbonyl (Cbz) and benzyl (Bn) groups were well tolerated to give the corresponding aryl nitriles in 54–86% yields (3k–3m). Notably, this nickel catalyst system was less sensitive to the steric hindrance around the bromide group: 4-methoxybromoarenes 1n–1p, which respectively contained 3-Me, 2-Me, and 3,5-Me2 substituents, afforded the corresponding arylnitriles 3n–3p in 65–88% yields. In addition, the cyanation of a polycyclic compound, 1-bromo-4-methoxynaphthalene (1q), gave 3q in 83% yield. We further investigated the catalytic cyanation of several aryl triflates (4a, 4h, 4r, and 4s), which were effectively converted to the corresponding aryl nitriles 3a, 3h, 3r, and 3s in 59–97% yields by increasing the catalytic loadings (15 mol%) and temperature (100 °C). Moreover, the cyanation of aryl chlorides (5a and 5t–5v) afforded 3a and 3t–3v in 59–77% yields, whereas the cyanation of 4-iodoanisole resulted in complex mixtures.
|
|
|---|
| a Isolated yields. b Reaction conditions for aryl bromides 1a–1s and aryl chloride 5a: 1 or 5, 0.40 mmol (0.027 M); 2 (5 mol%); L5 (5 mol%); 80 °C. c Reaction conditions for aryl triflates 4a, 4h, 4r, and 4s and aryl chlorides 5t–5v: 4 or 5, 0.40 mmol (0.027 M); 2 (15 mol%); L5 (15 mol%); 100 °C. d 1H NMR yield. e Si–Me4-DHP (1.1 equiv.) was used. |
|
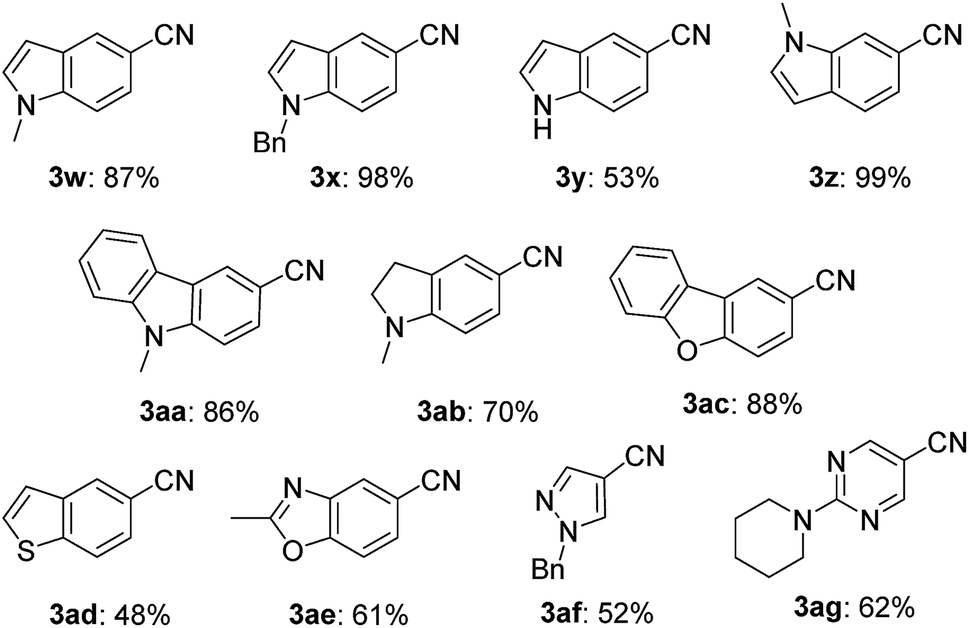
This nickel catalyst system was applicable to a wide range of heterocyclic compounds (Table 3): excellent to moderate yields were obtained for the cyanation of five- and six-membered heterocycles such as indoles (3w–3z) (53–99%), carbazole (3aa) (86%), indoline (3ab) (70%), dibenzofuran (3ac) (88%), benzothiophene (3ad) (48%), benzoxazole (3ae) (61%), pyrazole (3af) (52%), and pyrimidine (3ag) (62%). In all cases, no decomposition of the heterocycle motif was observed.
|
|
|---|
| a Reaction conditions: 1, 0.40 mmol (0.027 M). Isolated yield for all the products. |
|

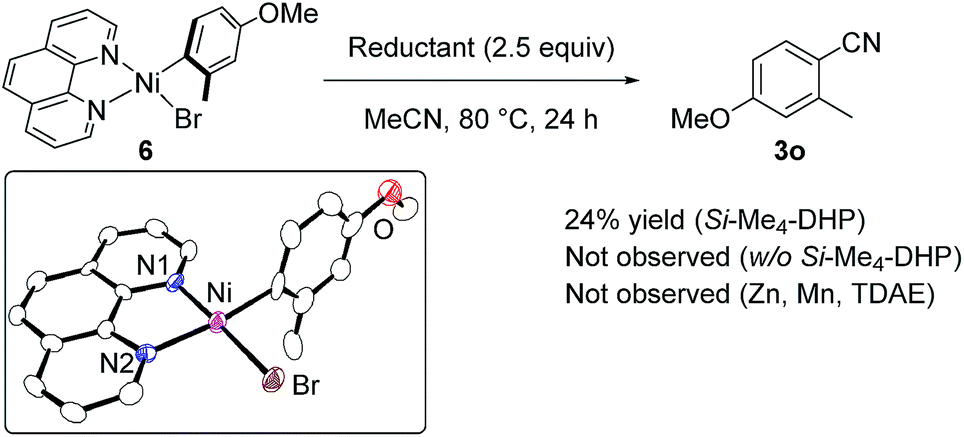
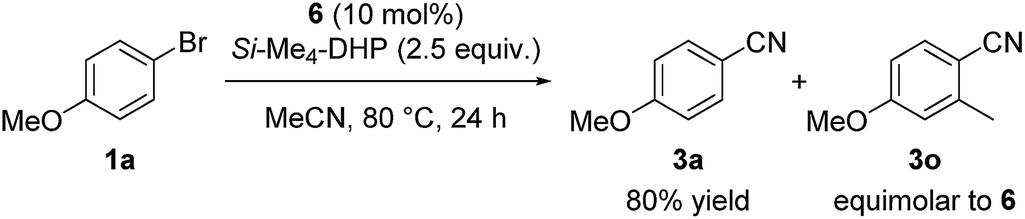
Regarding the reaction mechanism, we conducted some controlled experiments. The reaction of Ni(cod)2 with 4-methoxy-2-methylbromobenzene (1o) in the presence of 1 equiv. of L5 afforded a nickel(II) σ-aryl complex, Ni(C6H3-4-OMe-2-Me)Br(L5) (6), in 76% yield,13 which was characterized by spectral data as well as X-ray diffraction study of its single crystal. We then monitored the solution of 6 in acetonitrile with or without Si–Me4-DHP at 80 °C for 24 h: the reaction of 6 with acetonitrile in the presence of Si–Me4-DHP proceeded to give 3n in a substantial yield (24%), whereas no reaction of 6 with acetonitrile was observed in the absence of Si–Me4-DHP or in the presence of other reducing reagents (eqn (1)), clearly indicating that complex 6 was a key intermediate and Si–Me4-DHP played a key role for giving 3o. In fact, cyanation of 1a in the presence of 10 mol% of 6 produced 3a in 80% yield with stoichiometric amounts of 3o from 6 (eqn (2)). It is noteworthy that Si–Me4-DHP was consumed in the presence of Ni(cod)2 (10 mol%) at room temperature to produce (Me3Si)2 and 2,3,5,6-tetramethylpyrazine (Me4-pyrazine).14 We thus assume that Si–Me4-DHP acts as a silylation reagent of nickel species.
 | (1) |
 | (2) |
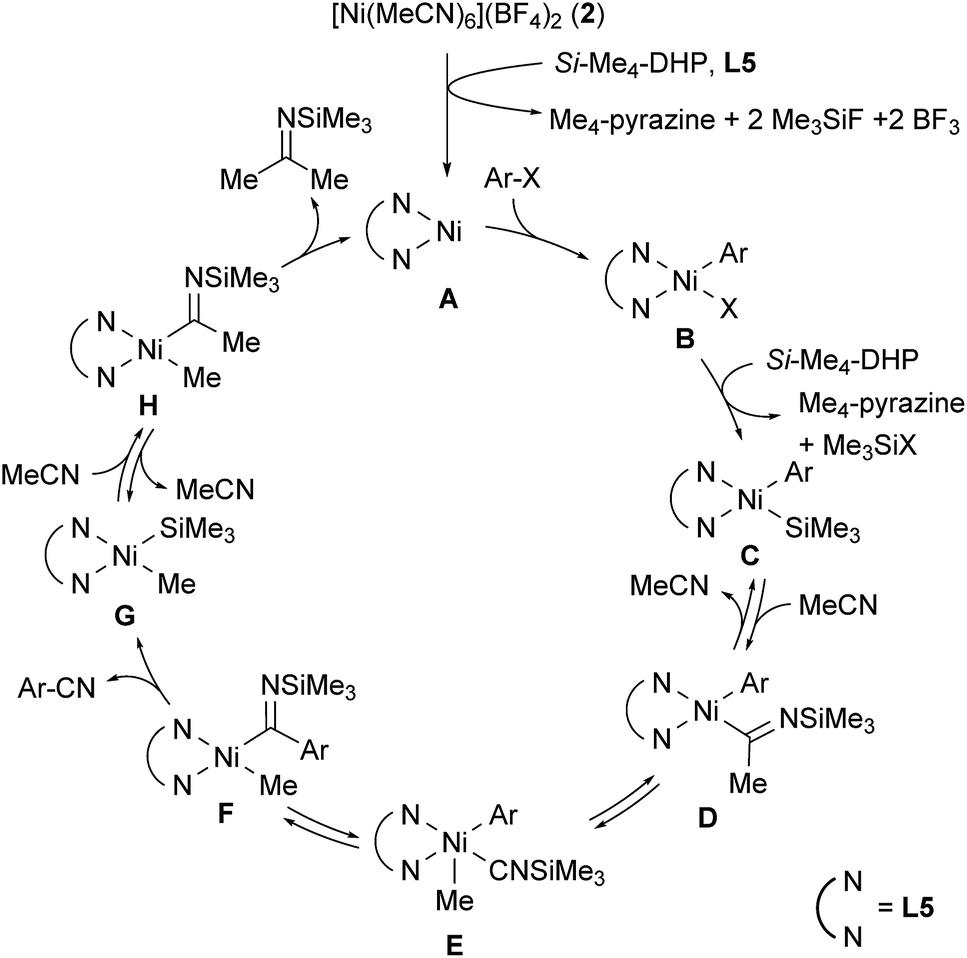
Scheme 1 shows a plausible catalytic cycle for the nickel-catalyzed cyanation reaction with acetonitrile as the sole cyano source based on the results of the controlled experiments shown in eqn (1) and (2). The initial step is the reduction of 2 by Si–Me4-DHP in the presence of L5, producing a 1,10-phenanthroline-coordinated Ni(0) species A.11b Subsequently, oxidative addition of aryl halides or aryl triflates (ArX) to A gives a Ni(II) species, Ni(Ar)X(L5) (B), X of which is silylated by the reaction with Si–Me4-DHP to afford Ni(Ar) (SiMe3) (L5) (C) with a release of Me3SiX and Me4-pyrazine. Insertion of acetonitrile into the Ni–Si bond of species C forms N-(trimethylsilyl)iminoacyl species D.15 α-Methyl elimination from D generates isonitrile species E, and the subsequent 1,1-insertion of CNSiMe3 into the nickel-aryl moiety results in the formation of F. β-Silyl elimination of F spontaneously induces a liberation of aryl nitriles accompanied by the generation of Ni(Me) (SiMe3) (L5) (G).16 Finally, the second insertion of acetonitrile into the Ni–Si bond of G followed by reductive elimination of N-(trimethylsilyl)propan-2-imine from H regenerates A. Notably, this is consistent with the formation of 4-fluoroacetophenone after acidic work-up when aryl bromide 1j with an electron-withdrawing substituent is used as a substrate; reductive elimination from species D or F is competitive with the formation of G and 3j.
 | ||
| Scheme 1 Plausible reaction mechanism. | ||
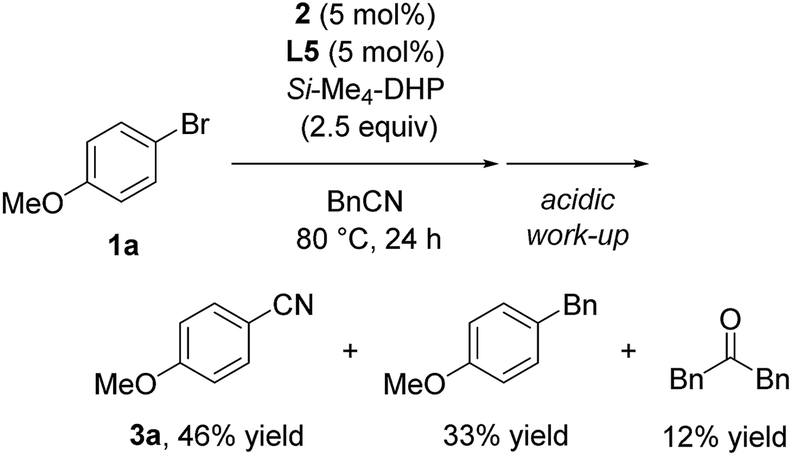
Furthermore, we conducted the catalytic reaction in benzylnitrile under the same catalytic conditions, and we obtained a mixture of 3a (46%), 1-benzyl-4-methoxybenzene (33%), and dibenzylketone (12%) (eqn (3)).17 Thus, the formation of 1-benzyl-4-methoxybenzene corresponded to reductive elimination from a similar intermediate, Ni(Ar) (Bn) (CNSiMe3) (L5) (E′ in SI), and the formation of dibenzylketone corresponded to the formation of acetone after hydrolysis of N-(trimethylsilyl)propan-2-imine derived during the reaction course.
 | (3) |
Conclusions
We developed a toxic cyanide-free cyanation of aryl halides and aryl triflates using a nickel catalyst system of [Ni(MeCN)6](BF4)2 and 1,10-phenanthroline along with a stoichiometric amount of an organosilicon reductant, Si–Me4-DHP, in acetonitrile. Noteworthy was that the mechanism of our catalytic system differs from the well-established direct C–CN bond cleavage involving Lewis acid-assisted oxidative addition to electron-rich low-valent group 10 metal complexes.9g,18,19 In this catalytic system, Si–Me4-DHP had dual functions to reduce the nickel(II) catalyst precursors but also to serve as a silylation reagent for nickel(II) σ-aryl species. Further application of Si–Me4-DHP as a silylating reagent to transition metals is ongoing in our laboratory.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank Mr Shunsuke Kato for the initial experimental results. Y. U. thanks the financial support by the JSPS Research Fellow-ships for Young Scientists. This work was supported by JSPS KAKENHI Grant No. JP26708012 (Grant-in-Aid for Young Scientist (A)) to H. T. and JP15H05808 (Precisely Designed Catalysis with Customized Scaffolding) to K. M.Notes and references
- (a) Z. Rappoport, Chemistry of the Cyano Group, Wiley, London, 1970 Search PubMed; (b) R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Wiley, New York, 1989 Search PubMed; (c) A. Kleeman, J. Engel, B. Kutschner and D. Reichert, Pharmaceutical Substances: Syntheses, Patents, Applications, Georg Theime, Stuttgart, 4th edn, 2001 Search PubMed; (d) S. I. Murahashi, Sci. Synth., 2004, 19, 345 Search PubMed; (e) L. H. Jones, N. W. Summerhill, N. A. Swain and J. E. Mills, MedChemComm, 2010, 1, 309 RSC; (f) P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049 RSC.
- (a) T. Sandmeyer, Ber. Dtsch. Chem. Ges., 1884, 17, 2650 CrossRef; (b) H. H. Hodgson, Chem. Rev., 1947, 40, 251 CrossRef CAS PubMed; (c) K. W. Rosenmund and E. Struck, Ber. Dtsch. Chem. Ges., 1919, 52, 1749 CrossRef.
- For representative reviews on stoichiometric or catalytic cyanation of aryl halides, see: (a) G. P. Ellis and T. M. Romney-Alexander, Chem. Rev., 1987, 87, 779 CrossRef CAS; (b) V. V. Grushin and H. Alper, Chem. Rev., 1994, 94, 1047 CrossRef CAS; (c) M. Tobisu and N. Chatani, Chem. Soc. Rev., 2008, 37, 300 RSC; (d) P. Anbarasan, H. Neumann and M. Beller, Chem. - Eur. J., 2011, 17, 4217 CrossRef CAS PubMed; (e) J. Kim, H. J. Kim and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 11948 CrossRef CAS PubMed; (f) Q. Wen, J. Jin, L. Zhang, Y. Luo, P. Lu and Y. Wang, Tetrahedron Lett., 2014, 55, 1271 CrossRef CAS; (g) Q. Wen, P. Lu and Y. Wang, RSC Adv., 2014, 4, 47806 RSC.
- Selected examples of using NaCN as cyano source: (a) L. Cassar, J. Organomet. Chem., 1973, 54, C57 CrossRef CAS; (b) L. Cassar, S. Ferrara and M. Foá, Adv. Chem. Ser., 1974, 132, 252 CrossRef CAS; (c) L. Cassar, M. Foá, F. Montanari and G. P. Marinelli, J. Organomet. Chem., 1979, 173, 335 CrossRef CAS; (d) T. Okano, M. Iwahara and J. Kiji, Synlett, 1998, 243 CrossRef CAS; (e) J. R. Dalton and S. L. Regen, J. Org. Chem., 1979, 44, 4443 CrossRef CAS; (f) J. Zanon, A. Klarpars and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 2890 CrossRef CAS PubMed; (g) A. V. Ushkov and V. V. Grushin, J. Am. Chem. Soc., 2011, 133, 10999 CrossRef CAS PubMed.
- Selected examples of using KCN as cyano source; (a) K. Takagi, T. Okamoto, Y. Sakakibara and S. Oka, Chem. Lett., 1973, 471 CrossRef CAS; (b) Y. Akita, M. Shimazaki and A. Ohta, Synthesis, 1981, 12, 974 CrossRef; (c) Y. Sakakibara, F. Okuda, A. Shimobayashi, K. Kirino, M. Sakai, N. Uchino and K. Takagi, Bull. Chem. Soc. Jpn., 1988, 61, 1985 CrossRef CAS; (d) B. A. Anderson, E. C. Bell, F. O. Ginah, N. K. Harn, L. M. Pagh and J. P. Wepsiec, J. Org. Chem., 1998, 63, 8224 CrossRef CAS; (e) M. Sundermeier, A. Zapf, M. Beller and J. Sans, Tetrahedron Lett., 2001, 42, 6707 CrossRef CAS; (f) M. Sundermeier, A. Zapf, S. Mutyala, W. Baumann, J. Sans, S. Weiss and M. Beller, Chem.–Eur. J., 2003, 9, 1828 CrossRef CAS PubMed; (g) C. Yang and J. M. Williams, Org. Lett., 2004, 6, 2837 CrossRef CAS PubMed; (h) H.-J. Cristau, A. Ouali, J.-F. Spindler and M. Taillefer, Chem.–Eur. J., 2005, 11, 2483 CrossRef CAS PubMed.
- A Selected example of using CuCN as cyano source: T. Sakamoto and K. Ohsawa, J. Chem. Soc., Perkin Trans. 1, 1999, 2323 RSC.
- Selected examples of using Zn(CN)2 as cyano source: (a) D. M. Tschaen, R. Desmond, A. O. King, M. C. Fortin, B. Pipik, S. King and T. R. Verhoeven, Synth. Commun., 1994, 24, 887 CrossRef CAS; (b) P. E. Maligres, M. S. Waters, F. Fleitz and D. Askin, Tetrahedron Lett., 1999, 40, 8193 CrossRef CAS; (c) M. Alterman and A. Hallberg, J. Org. Chem., 2000, 65, 7984 CrossRef CAS PubMed; (d) F. Jin and P. N. Confalone, Tetrahedron Lett., 2000, 41, 3271 CrossRef CAS; (e) J. Ramnauth, N. Bhardwaj, P. Renton, S. Rakhit and S. P. Maddaford, Synlett, 2003, 14, 2237 Search PubMed; (f) R. Chidambaram, Tetrahedron Lett., 2004, 45, 1441 CrossRef CAS; (g) R. S. Jensen, A. S. Gajare, K. Toyota, M. Yoshifuji and F. Ozawa, Tetrahedron Lett., 2005, 46, 8645 CrossRef CAS; (h) J. M. Veauthier, C. N. Carson, G. E. Collis, J. L. Kiplinger and K. D. John, Synthesis, 2005, 16, 2683 Search PubMed; (i) M. T. Martin, B. Liu, B. E. Cooley Jr and J. F. Eaddy, Tetrahedron Lett., 2007, 48, 2555 CrossRef CAS; (j) A. Littke, M. Soumeillant, R. F. Kaltenbach, R. J. Cherney, C. M. Tarby and S. Kiau, Org. Lett., 2007, 9, 1711 CrossRef CAS PubMed; (k) F. G. Buono, R. Chidambaram, R. H. Mueller and R. E. Waltermire, Org. Lett., 2008, 10, 5325 CrossRef CAS PubMed; (l) D. T. Cohen and S. L. Buchwald, Org. Lett., 2015, 17, 202 CrossRef CAS PubMed; (m) X. Zhang, A. Xia, H. Chen and Y. Liu, Org. Lett., 2017, 19, 2118 CrossRef CAS PubMed.
- Selected examples of using Me3SiCN as cyano source: (a) N. Chatani and T. Hanafusa, J. Org. Chem., 1986, 51, 4714 CrossRef CAS; (b) M. Sundermeier, S. Mutyala, A. Zapf, A. Spannenberg and M. Beller, J. Organomet. Chem., 2003, 684, 50 CrossRef CAS.
- Selected examples of using harmless organic nitrile compounds as cyano source are as follows. Acetone cyanohydrin; (a) M. Sundermeier, A. Zapf and M. Beller, Angew. Chem., Int. Ed., 2003, 42, 1661 ( Angew. Chem. , 2003 , 115 , 1700 ) CrossRef CAS PubMed; (b) T. Schareina, A. Zapf, A. Cotté, M. Gotta and M. Beller, Adv. Synth. Catal., 2011, 353, 777 CrossRef CAS; (c) F. Burg, J. Egger, J. Deutsch and N. Guimond, Org. Process Res. Dev., 2016, 20, 1540 CrossRef CASacetonitrile; (d) F.-H. Luo, C.-I. Chu and C.-H. Chen, Organometallics, 1998, 17, 1025 CrossRef CAS; (e) M. Zhao, W. Zhang and Z. Shen, J. Org. Chem., 2015, 80, 8868 CrossRef CAS PubMed; (f) Y. Zhu, M. Zhao, W. Lu, L. Li and Z. Shen, Org. Lett., 2015, 17, 2602 CrossRef CAS PubMedbutyronitrile; (g) P. Yu and B. Morandi, Angew. Chem., Int. Ed., 2017, 56, 15693 ( Angew. Chem. , 2017 , 129 , 15899 ) CrossRef CAS PubMedmalonitrile, aminonitrile, cyanoester; (h) Z. Jiang, Q. Huang, S. Chen, L. Long and X. Zhou, Adv. Synth. Catal., 2012, 354, 589 CrossRef CAS; (i) S. Zheng, C. Yu and Z. Shen, Org. Lett., 2012, 14, 3644 CrossRef CAS PubMed; (j) S.-L. Zhang and L. Huang, Org. Biomol. Chem., 2015, 13, 9963 RSC; (k) R. Takise, K. Itami and J. Yamaguchi, Org. Lett., 2016, 18, 4428 CrossRef CAS PubMed.
- (a) X. Kou, M. Zhao, X. Qiao, Y. Zhu, X. Tong and Z. Shen, Chem.–Eur. J., 2013, 19, 16880 CrossRef CAS PubMed; (b) A. A. Zavitsas, J. Phys. Chem. A, 2003, 107, 897 CrossRef CAS.
- (a) T. Saito, H. Nishiyama, H. Tanahashi, K. Kawakita, H. Tsurugi and K. Mashima, J. Am. Chem. Soc., 2014, 136, 5161 CrossRef CAS PubMed; (b) T. Yurino, Y. Ueda, Y. Shimizu, S. Tanaka, H. Nishiyama, H. Tsurugi, K. Sato and K. Mashima, Angew. Chem., Int. Ed., 2015, 54, 14437 CrossRef CAS PubMed.
- For effects of additional reaction parameters such as solvent and organosilicon reductants, see ESI.†.
- E. A. Standley and T. F. Jamison, J. Am. Chem. Soc., 2013, 135, 1585 CrossRef CAS PubMed.
- See ESI† for details..
- (a) H. Nakazawa, T. Kawasaki, K. Miyoshi, C. H. Suresh and N. Koga, Organometallics, 2004, 23, 117 CrossRef CAS; (b) F. L. Taw, P. S. White, R. G. Bergman and M. Brookhart, J. Am. Chem. Soc., 2002, 124, 4192 CrossRef CAS PubMed; (c) M. Tobisu, Y. Kita and N. Chatani, J. Am. Chem. Soc., 2006, 128, 8152 CrossRef CAS PubMed; (d) M. Tobisu, Y. Kita, Y. Ano and N. Chatani, J. Am. Chem. Soc., 2008, 130, 15982 CrossRef CAS PubMed; (e) S.-L. Zhang, L. Huang and W.-F. Bie, Organometallics, 2014, 33, 3030 CrossRef CAS.
- (a) C.-Y. Ho and L. He, Chem. Commun., 2012, 48, 1481 RSC; (b) Z. Chen, M. D. Leatherman, O. Daugulis and M. Brookhart, J. Am. Chem. Soc., 2017, 139, 16013 CrossRef CAS PubMed; (c) M. R. Elsby and S. A. Johnson, J. Am. Chem. Soc., 2017, 139, 9401 CrossRef CAS PubMed.
- For more details on the mass-balance of this reaction, see ESI.†.
- (a) J. J. Garcia, A. Arévalo, N. M. Brunkan and W. D. Jones, Organometallics, 2004, 23, 3997 CrossRef CAS; (b) Y. Nakao, A. Yada, S. Ebata and T. Hiyama, J. Am. Chem. Soc., 2007, 129, 2428 CrossRef CAS PubMed.
- A direct cleavage of the C–CN bond of acetonitrile by in situ generated Ni(0) center was excluded under our reaction condition (see ESI†)..
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for the synthesis of Si–Me4-DHP and 6, screening of reaction conditions, identification of the products, and crystal data for 6. CCDC 1852145. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04437f |
| This journal is © The Royal Society of Chemistry 2019 |