
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSize-dependent synthesis and catalytic activities of trimetallic PdAgCu mesoporous nanospheres in ethanol electrooxidation†
Hao
Lv
a,
Lizhi
Sun
a,
Lu
Zou
a,
Dongdong
Xu
 *a,
Huiqin
Yao
b and
Ben
Liu
*a
*a,
Huiqin
Yao
b and
Ben
Liu
*a
aJiangsu Key Laboratory of New Power Batteries, Jiangsu Collaborative Innovation Center of Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing, Jiangsu 210023, China. E-mail: ddxu@njnu.edu.cn; ben.liu@njnu.edu.cn
bSchool of Basic Medical Sciences, Ningxia Medical University, Yinchuan 750004, China
First published on 11th December 2018
Abstract
Mesoporous noble metal nanocrystals have exhibited significant potential in electrocatalysis. However, it remains a big challenge to controllably synthesize sub-100 nm multimetallic mesoporous nanospheres (MNSs) with precisely tunable sizes and to further understand their size-dependent electrocatalytic performances. In this manuscript, a one-pot solution-phase strategy was developed for the formation of nanosized trimetallic PdAgCu MNSs with cylindrically open mesoporous nanochannels and continuous frameworks. The resultant Pd-based MNSs were precisely tailorable not only in terms of size (from 21 to 104 nm), but also in terms of elemental ratios and compositions (PdAgCu, PdAgPt, PdAgFe, PdPtCu, and PdCuRu). This system thus provided a facile yet straightforward means to evaluate the size effect of trimetallic MNSs in electrocatalysis. As an example, trimetallic PdAgCu MNSs with an average size of 36 nm exhibited the best activity of 4.64 A mgPd−1 in the electrocatalytic ethanol oxidation reaction, 1.1–1.7 fold higher than that of MNSs with smaller or larger sizes and 5.9 fold higher than that of commercial Pd black catalyst. By means of kinetic studies, the size-dependent electrocatalytic performance can be ascribed to the optimization and balance between electron transfer and mass transfer processes inside PdAgCu MNSs. We expect that the size effect of multimetallic MNS nanocatalysts presented here may provide a general synthetic methodology for rational design of size-dependent nanocatalysts for a broad range of applications.
1. Introduction
Noble metal nanocrystals have attracted fast-growing research interest in hydrogen storage, surface plasmon response, sensors, and catalysis, due to their physicochemical properties.1–7 Among the multiple nanostructures of nanocrystals, mesoporous nanospheres (MNSs) with three-dimensional (3D) interconnected nanochannels have received special attention in (electro)catalysis, owing to their abundant edge/corner sites, accessible mesopores, and high surface area.8–15 Furthermore, noble metal MNSs with cylindrically open nanochannels and sub-100 nm sizes would further enlarge the accessibility to the reactants, facilitate reactant/product transfer, accelerate the reaction kinetics, and thus enhance the (electro)catalytic performance.16–20 During the past few decades, dozens of synthetic methods and techniques, including the hard-templating method,21–23 soft-templating method,14,24–27 and solution-phase approach,28,29 have been employed to fabricate various nanosized MNSs with monometallic or multimetallic compositions. However, in most cases, successful synthesis of nanosized noble metal MNSs (<100 nm) consisted of spherical mesopores and uncontinuous frameworks,30–32 which kinetically inhibited the mass transfer inside mesopores and electron transfer on mesoporous frameworks during the (electro)catalysis. Therefore, it is still highly desired to develop an effective and general method to prepare nanosized noble metal MNSs with open/interconnected mesopores and continuous frameworks.Apart from the nanostructures, the sizes of noble metal nanocatalysts also strongly influence their (electro)catalytic performance (denoted as the size effect).33–40 Taking noble metal MNSs for catalysing the ethanol oxidation reaction (EOR) as an example, it is still inconclusive how (or why) the sizes of sub-100 nm noble metal MNSs affected the electrocatalytic performance. These uncertainties and conflicts possibly originated from the opposite size effects in electron transfer and mass transfer of the MNSs during electrocatalysis.41,42 On the one hand, the larger MNSs with continuously mesoporous frameworks efficiently facilitated electron transfer on the nanocatalysts, kinetically accelerated the electrooxidation of ethanol, and thus enhanced their electrocatalytic performance. On the other hand, the smaller MNSs structurally accelerated the transport of the reactants, intermediates and products (mass transfer) inside the mesoporous nanostructures, and accordingly achieved higher electrocatalytic performance. These opposite size effects in electron and mass transfer have not been well resolved so far and could be mainly attributed to the poor controllability of the precise formation of size-tuned noble metal MNSs, especially with open mesochannels and continuous frameworks.23,43 Therefore, in view of both fundamental interests and practical applications, a general synthetic method for controllable synthesis of size-tuned multimetallic MNSs with cylindrically open nanochannels and continuous frameworks must be urgently developed to better understand their size–performance relationship towards EOR electrocatalysis.
We herein demonstrate a facile one-pot solution-phase synthetic approach to engineer the nanostructure of uniform and monodisperse sub-100 nm trimetallic PdAgCu MNSs and further investigate their size effect in the electrocatalytic EOR. Trimetallic PdAgCu MNSs are synthesized with dioctadecyldimethylammonium chloride (DODAC) as the surfactant in mixed solvents of H2O/ethanol at room temperature (25 °C). Through carefully controlling the synthetic conditions, the MNSs exhibit precise tunability of size (from 21 to 104 nm), elemental ratio and composition (e.g., PdAgCu, PdAgPt, PdAgFe, PdPtCu, and PdCuRu). All the MNSs possess multiple features of cylindrically open mesochannels, continuous crystalline frameworks and trimetallic elemental compositions, and this synthetic system thus offers a general and ideal platform to clearly evaluate the role of the size effect of the MNSs in EOR electrocatalysis.
2. Results and discussion
2.1 Synthesis and structural characterization
Fig. 1a schematically illustrates our design principle for size-tuned synthesis of trimetallic Pd-based MNSs via kinetic control based on a surfactant-directing cooperative assembly route. The key features of our approach to prepare trimetallic Pd-based MNSs with different sizes include the utilization of amphiphilic DODAC as the surfactant template and precise control over the reduction kinetics of metal precursors. On the one hand, the amphiphilic surfactant DODAC, which structurally has two hydrophobic long-chain tails (C18) and one hydrophilic quaternary ammonium head (Fig. S1†), drives the self-assembly into cylindrical micelles in a mixed solution of H2O/ethanol.44,45 After mixing with metal precursors (for example, H2PdCl4, AgNO3, and Cu(NO3)2 for PdAgCu MNSs), the electrostatic and coordination interactions and subsequent co-reduction by ascorbic acid (AA) cooperatively direct the assembly and in situ crystallization growth along cylindrical micelles of DODAC into trimetallic PdAgCu MNSs with cylindrically open nanochannels and continuous mesoporous frameworks. When octadecyltrimethylammonium chloride (C18TAC) or N-(2-carboxyethyl)-N,N-dimethyloctadecan-1-ammonium chloride (C18TAOC) with one hydrophobic alkyl tail, or Pluronic F127 was utilized as the surfactant template,46 the MNSs with a very broad size distribution of 20–60 nm or non-mesoporous nanostructures were obtained under the same synthetic conditions (Fig. S2†). These results implied our hypothesis of the key role of DODAC for the synthesis of uniform MNSs with cylindrically open mesopores in the mixed solvents with the higher ethanol content (>15% by volume) (discussed later). | ||
| Fig. 1 Synthesis and structural characterization of trimetallic PdAgCu MNSs with precisely controlled sizes. (a) Schematic illustrating the formation of size-tuned PdAgCu MNSs using DODAC as the surfactant template through kinetic control. Typical TEM images of PdAgCu MNSs with different sizes synthesized at pH of (b) 5.76, (c) 6.03, (d) 6.34, (e) 7.92, (f) 9.33 and (g) 10.61. | ||
On the other hand, kinetic control over the co-reduction rate of metal precursors was utilized to tune the crystallization growth and the resultant sizes of PdAgCu MNSs. The reaction conditions, including pH, solvents, and surfactant and precursor concentrations, were tuned to precisely control the reduction kinetics and the resultant MNSs. We first changed the pH of the reaction solution carefully to tailor the nanostructures and sizes of trimetallic PdAgCu MNSs. The higher pH would kinetically decrease the reduction rate of metal precursors by AA, thus resulting in thermodynamically stable MNSs with a larger size. By contrast, kinetically stable MNSs with a smaller size were synthesized in the lower pH range. Typical low-magnification transmission electron microscopy (TEM) images (Fig. 1b–g and 3) showed that all the MNSs synthesized in the pH range of 5.75–10.61 were homogeneously dispersed and pretty uniform with obvious spherical morphology and mesoporous nanochannels over the whole framework. Amongst them, at a low pH of 5.76, the as-obtained MNSs had partially disordered mesopores. By contrast, when the pH was higher than 6.03, mesoporous nanochannels became more ordered and cylindrically open (Fig. 1c–g, see more TEM images in Fig. S3†). Meanwhile, an obvious pH-dependent increase in MNS size was clearly observed as expected (Fig. 2a). As the pH changed from 5.76 to 6.03, 6.34, 7.13, 7.92, 9.33 and 10.61, the initially smallest size of 21.3 nm gradually increased to 35.7, 48.8, 58.3, 71.4, 93.3 and 103.8 nm (denoted as MNS-21, MNS-36, MNS-49, MNS-58, MNS-71, MNS-93 and MNS-104 hereafter), respectively. All the MNSs possessed a narrow size distribution, indicating precise controllability of MNS size by tuning the pH of the reaction solution. However, in the lower (<5.23) or higher pH (>11.12) range, no mesoporous nanostructure or MNSs with a disordered nanoparticle structure were obtained (Fig. S4 and S5†), due to the quicker/slower reduction rates or changed micelle structures of DODAC.
 | ||
| Fig. 2 Size control diagrams. Controlling the size of PdAgCu MNSs by (a) pH, (b) ethanol content and (c) DODAC concentration. | ||
The sizes of PdAgCu MNSs were also tuned by carefully changing the ethanol content in the mixed solvents (Fig. 2b). We emphasize that the mixed solvents of ethanol and H2O cooperatively controlled the cylindrical micelles of DODAC and reduction kinetics of metal precursors. In comparison to H2O, ethanol with weaker solvent polarity could efficiently stabilize the cylindrical micelles or even change the assembled features, thus resulting in the formation of more uniform MNSs. When using H2O as the sole solvent or with small amounts of ethanol (<1 mL in 10 mL of total solvent), DODAC self-assembled into vesicular and cylindrical “dual” micelles simultaneously,47,48 thus resulting in the formation of hollow MNSs (Fig. S6†), as reported in our previous studies.18 By contrast, upon increasing the ethanol content to 1.5 mL, uniform MNSs were obtained (Fig. S7a and S7b†). As shown in Fig. 2b, with increasing ethanol content from 1.5 to 3.0 mL (totally 10 mL of solvent), a nearly linear increase in the size of MNSs was achieved from 44.6 nm to 88.0 nm (see Fig. S7† for TEM images), possibly originating from the gradually decreased reduction rates and more stable cylindrical micelles at the high ethanol content. However, at higher ethanol content (>4 mL), the cylindrical micelle of DODAC would be disassembled and only nanoparticles were formed (Fig. S8†). Furthermore, we further tried to tune the sizes of MNSs by changing the surfactant concentration in the solution (Fig. 2c). A higher surfactant concentration caused more concentrated micelles and eventually formed the slightly larger MNSs (Fig. S9†). Besides, a slightly increased size of PdAgCu MNSs was seen when increasing the concentration of metal precursors (Fig. S10†). The above results definitely implied the decisive role of synthetic conditions (pH, ethanol content and concentration) on the formation of size-tuned PdAgCu MNSs.
To clearly reveal the mesoporous nanostructures and trimetallic elemental compositions, PdAgCu MNS-58 with a medium size was characterized in detail as a typical example by a combination of TEM, X-ray diffraction (XRD) and X-ray photoelectron spectroscopy (XPS) techniques. Typical low-magnification TEM and high-angle annular dark-field scanning TEM (HAADF-STEM) images (Fig. 3a–c) showed that the MNSs were morphologically spherical and structurally mesoporous with high uniformity and purity. The average diameter of the MNSs was 58 nm with a narrow distribution of ±3 nm. The MNSs were composed of well-defined mesoporous structures with cylindrically open and interconnected mesochannels (see more TEM images in Fig. S11†), which were also indicated clearly by a high-magnification TEM image (Fig. 3d). The mesoporous framework was continuous and crystalline (not nanoparticle aggregates), which would facilitate the electron and mass transfer within the MNSs during the electrocatalysis. A higher-magnification TEM image showed that the MNSs had an average mesopore size of 3.7 nm and a framework thickness of 4.9 nm (Fig. 3e). The mesoporous nanostructure was also indicated by small-angle XRD, in which a typical peak at 0.99° corresponds to a mesoporous periodicity of 8.9 nm (Fig. S12a†). The well-matched XRD and TEM results further indicated the highly uniform mesostructures of PdAgCu MNSs. The high-resolution TEM image shown in Fig. 3f further presented clear lattice fringes with d-spacings of 0.229 and 0.230 nm, indexed to the (111) planes of face-centered cubic (fcc) nanocrystals. The corresponding selected-area electron diffraction (SAED) pattern (inset in Fig. 3f) displayed a series of continuous rings, assigned to the (111), (200), (220) and (311) planes, suggesting an alloyed polycrystalline feature (also Fig. S13†). The formation of PdAgCu nanoalloys (rather than their phase-separated counterparts) was also evidenced from the wide-angle XRD pattern, in which a single set of diffraction peaks were located in between standard peaks of fcc Ag (04-0783), Pd (46-1043), and Cu (04-0836) (Fig. S12b†). The elemental compositions and distributions of PdAgCu MNSs were further revealed by STEM mapping (Fig. 3g and h). The homogeneous distribution of Pd, Ag and Cu throughout the MNSs without any elemental segregation further evidenced the formation of PdAgCu nanoalloys. Similar results were also obtained by an STEM line scan (Fig. 3i). The elemental ratio of Pd, Ag and Cu in the MNSs was determined to be 62.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :27.9:9.7 by weight percentage (wt%) (Fig. S14†), in good agreement with that from X-ray photoelectron spectroscopy (XPS) (Fig. S15†). The real atomic ratio in the obtained PdAgCu MNS-58 was very close to the feed ratio (6:3:1) and the atomic conversion efficiency was calculated to be more than 80%, highlighting the high yield of our synthesis method (also see the ESI†). The surface electron states of Pg, Ag, and Cu in trimetallic PdAgCu MNSs were also indicated, in which both slightly positive shifts and partially oxidized species (Pd2+, Ag+ and Cu2+) in high-resolution XPS further implied the formation of alloyed PdAgCu MNSs.49–51
:27.9:9.7 by weight percentage (wt%) (Fig. S14†), in good agreement with that from X-ray photoelectron spectroscopy (XPS) (Fig. S15†). The real atomic ratio in the obtained PdAgCu MNS-58 was very close to the feed ratio (6:3:1) and the atomic conversion efficiency was calculated to be more than 80%, highlighting the high yield of our synthesis method (also see the ESI†). The surface electron states of Pg, Ag, and Cu in trimetallic PdAgCu MNSs were also indicated, in which both slightly positive shifts and partially oxidized species (Pd2+, Ag+ and Cu2+) in high-resolution XPS further implied the formation of alloyed PdAgCu MNSs.49–51
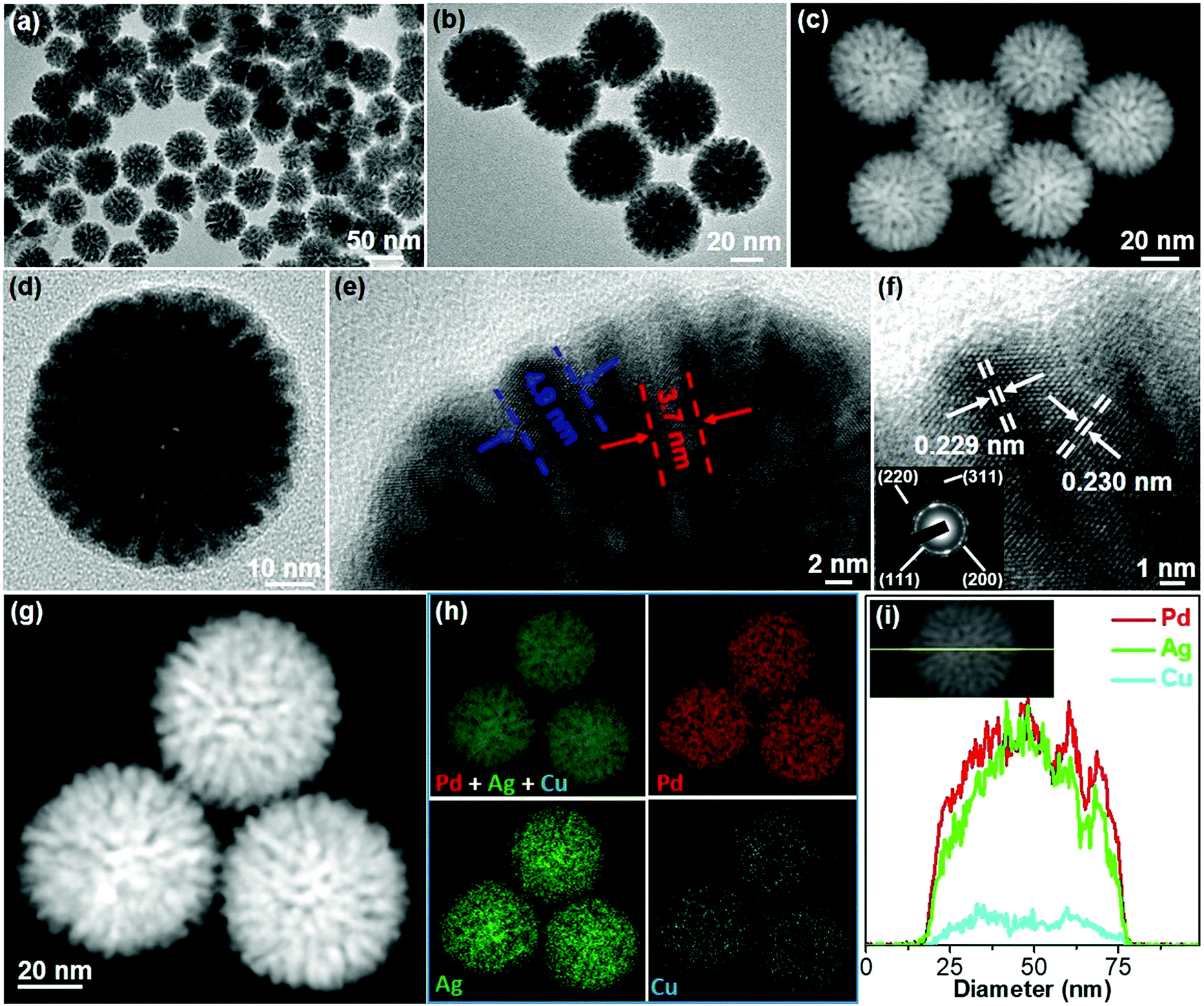 | ||
| Fig. 3 Structural and compositional characterization of PdAgCu MNS-58. (a and b) Low-magnification TEM and (c) HAADF-STEM, (d and e) high-magnification and (f) high-resolution TEM images and the corresponding SAED pattern (inset in (f)) of trimetallic PdAgCu MNS-58 synthesized at a pH of 7.13. (g) High-magnification HAADF-STEM image of PdAgCu MNSs, and (h) the corresponding STEM mapping and (i) line scan analysis of Pd, Ag and Cu in trimetallic PdAgCu MNS-58. | ||
To evaluate the universality of our synthetic strategy, various trimetallic Pd-based MNSs with different elemental ratios and compositions were synthesized using in situ co-reduction routes. Also taking PdAgCu MNS-58 as an example, we first tuned the composition ratios by changing the Ag and Cu contents. All the MNSs had average sizes in the range of 50–60 nm with cylindrically open mesochannels and continuous frameworks, further indicating the good controllability. Specifically, increasing Ag ratios structurally resulted in PdAgCu MNSs with thicker frameworks and finally nearly solid nanospheres without mesochannels (Fig. S16†). This may have originated from the easier crystalline nucleation of Ag, compared to Pd and Cu in the PdAgCu system.18,44 By contrast, a thinner framework was obtained at higher Cu content (Fig. S17†), also implying the tunability of mesoporous nanochannels and frameworks of PdAgCu MNSs. Lastly, we further tuned the elemental compositions in trimetallic Pd-based MNSs (Fig. 4). The 5d metal of Pt, 4d metals of Ru and Ag, and 3d metals of Fe and Cu were successfully alloyed with Pd to form trimetallic MNSs with cylindrically open mesopores and continuous frameworks. All the MNSs (PdAgPt, PdAgFe, PdPtCu, and PdCuRu) composed of highly uniform sizes (50–60 nm) and well-distributed compositions. Similarly, without Ag, denser and thinner mesoporous frameworks were obtained, for example PdPtCu and PdCuRu MNSs. This facile yet powerful method is thus expected to offer us dozens of multimetallic Pd-based MNSs.
 | ||
| Fig. 4 Typical examples of various trimetallic Pd-based MNSs. (a, c, e and g) TEM images and (b, d, f and h) the corresponding HAADF-STEM mapping of (a and b) PdAgPt, (c and d) PdAgFe, (e and f) PdPtCu, and (g and h) PdCuRu MNSs. | ||
2.2 Electrocatalytic performance
Successful synthesis of trimetallic PdAgCu MNSs displayed great potential in evaluating their size-dependent electrocatalytic activities. Here, we carefully compared the performance of the MNSs with different sizes in the range of 21 to 104 nm (synthesized at different pH, Fig. 1b–g and 3) in the electrocatalytic EOR under alkaline conditions (1.0 M KOH), and commercial Pd black (PdB) was also tested as a control. Cyclic voltammograms (CVs) were first collected in N2-saturated 1.0 M KOH at a scan rate (ν) of 50 mV s−1 (Fig. 5a). The obvious cathode peaks in the range of −0.5 to 0 V (vs. SCE (saturated calomel electrode)) for all the Pd-based nanocatalysts were clearly seen in the negative sweeps, suggesting that oxidized PdO was reduced to metallic Pd. The intensity of the CVs was strongly related to the Pd-based nanocatalysts, evidencing the different electrochemically active surface areas (ECSAs). We further calculated the ECSA according to the equation ECSA = QPdO/(0.405 (mC cm−2) × mPd), where QPdO and 0.405 mC cm−2 are the charge of the peak area and stranded charge for the reduction of PdO, respectively, and mPd is the mass loading (in grams) of Pd nanocatalysts on the electrode.18,50 As expected, a slight decrease in ECSA was seen with the increase of MNS sizes (Fig. 5b). The initially largest ECSA of 50.6 m2 gPd−1 for MNS-21 gradually decreased to 49.8 m2 gPd−1 for MNS-36, 46.5 m2 gPd−1 for MNS-49, 41.2 m2 gPd−1 for MNS-58, 38.1 m2 gPd−1 for MNS-71, 36.9 m2 gPd−1 for MNS-93, and 35.4 m2 gPd−1 for MNS-104, respectively. By contrast, commercial PdB had the lowest ECSA of 12.0 m2 gPd−1. The decreased ECSAs with increasing sizes of the MNSs corresponded partially to the slower mass transfer on the nanocatalysts with the longer mesoporous nanochannels. | ||
| Fig. 5 Electrochemical EOR performance. (a) CV curves and (b) summarized ECSAs of trimetallic PdAgCu MNSs with different sizes and commercial PdB obtained in 1.0 M KOH. (c) CV curves, (d) summarized mass activities, (e) Tafel plots, and (f) i–t curves of PdAgCu MNSs and PdB collected in 1.0 M KOH and 1.0 M ethanol. | ||
Electrocatalytic activities in the EOR were further evaluated in 1.0 M KOH and 1.0 M ethanol and the results were normalized to the Pd mass in all Pd-based nanocatalysts. As shown in Fig. 5c, the shapes of all CVs were completely changed owing to ethanol electrooxidation. Two intense signals were seen from the CV curves, in which the broad one in the forward scan was ascribed to the electrochemical oxidation of ethanol into intermediates (CH3CO) and their decomposition (CO and CH3), while the sharp one in the backward scan was attributed to the further oxidation/removal of the above intermediates.52 The mass activities of PdAgCu MNSs with different sizes were then summarized based on the oxidation peaks in the forward scans (Fig. 5d). Overall, PdAgCu MNSs with different sizes displayed higher mass activities, which were approximately 4–6 fold higher than that of PdB. Meanwhile, it was easily noticed that the mass activity was strongly related to the size of PdAgCu MNSs. An obvious “volcano”-type EOR activity of PdAgCu MNSs versus the sizes was clearly observed, evidencing the typical size effect of the MNSs in EOR electrocatalysis. Among them, PdAgCu MNS-36 achieved the highest mass activity of 4.64 A mgPd−1, 1.1–1.7 fold higher than that of MNS-21 (the highest ECSA) and MNSs with the larger sizes (smaller ECSAs). Similarly, the same overall tendency was found in Tafel plots calculated from the CV curves, in which MNS-36 displayed the highest Tafel plot value of 193.4 mV dec−1 (Fig. 5e). Besides, better electrocatalytic stability was also achieved for MNS-36 (Fig. 5f). These results definitely evidenced the size-dependent electrocatalytic performance of the PdAgCu MNSs for the EOR.
PdAgCu MNSs with different sizes possessed similar intrinsic features, including cylindrically open nanochannels, continuous mesoporous frameworks and trimetallic compositions. Considering these structural and compositional merits that contributed equally to the enhanced electrocatalytic kinetics, PdAgCu MNSs thus behaved as the ideal nanocatalysts to fully understand their size-dependent electrocatalytic performance. Two key factors should be seriously considered for PdAgCu MNSs with different sizes: (i) electron transfer where the electrons rapidly relocate within continuous mesoporous frameworks for the electrooxidation of ethanol, and (ii) mass transfer, which transports the reactants, intermediates and products inside MNSs.35,43 It is easily understood that the larger sizes of the MNSs with continuous frameworks would accelerate the electron transfer, while the smaller sizes of the MNSs with open mesochannels facilitate the mass transfer during the electrocatalysis. Electron transfer and mass transfer on/inside PdAgCu MNSs thus possessed completely opposite size effects in EOR electrocatalysis. Therefore, we deduced that the size-dependent activity of PdAgCu MNSs may have originated from enhanced electrocatalytic kinetics by the optimization and balance of electron transfer and mass transfer processes. In the current study, PdAgCu MNS-36 exhibited the best electrocatalytic activity and stability, indicating the optimized electron and mass transfer ability.
To reveal the nature of the size-dependent electrocatalytic activity more clearly, the electrocatalytic kinetics were further investigated by comparing their activities at different ethanol concentrations and scan rates, and CO anti-poisoning abilities. First, ethanol concentrations in the range of 0–4.0 mol mL−1 were investigated. As shown in Fig. S18,† with the increase of ethanol concentration in the electrocatalytic solution, the mass activities of all the PdAgCu MNSs increased linearly from initially almost 0 A mgPd−1 at low ethanol concentration (<1.0 mol mL−1), and the increased tendency of mass activity was slowed down at high ethanol concentration (>2.0 mol mL−1). We further figured out the relationship between peak mass activity and ethanol concentration (Fig. S19†). It is clear that MNS-36 possessed the quickest activity enhancement in the electrocatalysis of the EOR, compared to the MNSs with other sizes and PdB. This suggested that MNS-36 had faster electrocatalytic kinetics in ethanol adsorption and desorption processes. Second, gradually increased mass activity in the electrocatalytic EOR was also seen with increasing the scan rates from 10 to 200 mV s−1 (Fig. S20†). We also plotted the mass activity against the square root of the scan rate (ν1/2) (Fig. S21†). All the MNSs with different sizes and PdB exhibited a nearly linear relationship, evidencing a diffusion-driven process.53 Similarly, MNS-36 displayed the steepest slope, distinguishing the enhanced electrocatalytic kinetics among all the nanocatalysts. The above results further indicated that the optimized electron and mass transfers of MNS-36 accelerated the EOR electrocatalysis, and thus resulted in the enhanced electrocatalytic kinetics.
Lastly, CO anti-poisoning tests were also evaluated for all PdAgCu MNSs with different sizes (Fig. 6), to imitate the further oxidation/removal of the intermediates on the nanocatalysts. Two consecutive CV curves of PdAgCu MNSs with different sizes were recorded in the range of −0.9 to 0.2 V. Complete elimination of COads could be obviously discerned after the 2nd forward scan, indicating the better CO anti-poisoning ability of these PdAgCu electrocatalysts. Both the lowest onset potential and largest active area of the CO oxidation peak were seen for MNS-36, compared to its counterparts with different sizes. The results identified the best CO oxidation/removal activity of MNS-36, greatly agreeing with the tendency of EOR activity. These kinetics studies further implied that PdAgCu MNS-36 possessed the optimum electron transfer and mass transfer abilities in EOR electrocatalysis.
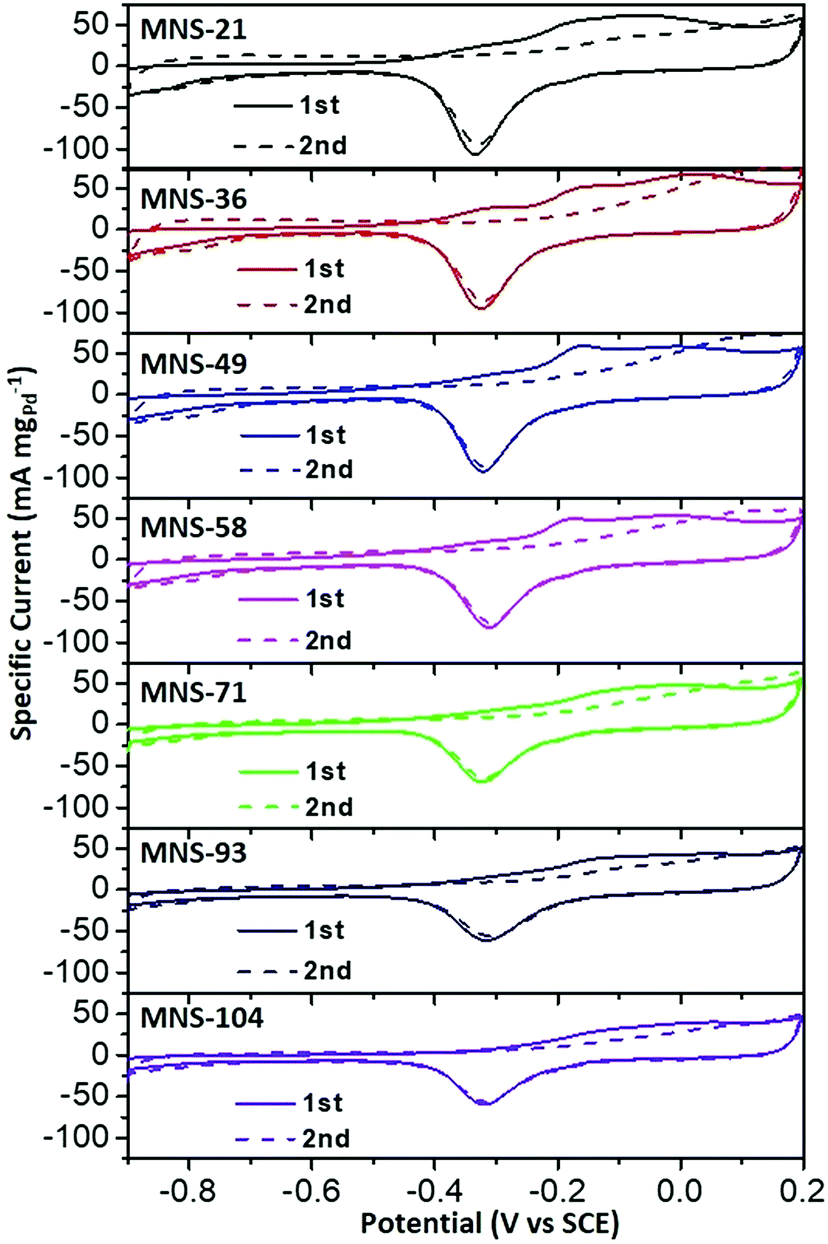 | ||
| Fig. 6 CO antipoisoning experiments. CO stripping voltammograms of trimetallic PdAgCu MNSs with different sizes in 1.0 M KOH at a scan rate of 50 mV s−1. | ||
3. Conclusion
In summary, we have developed a facile and high-yielding solution-phase method to synthesize trimetallic Pd-based MNSs with continuous mesoporous frameworks and cylindrically open mesochannels using DODAC as the template and ethanol/H2O as the mixed solvent. Through carefully tuning the reduction kinetics by varying the reaction conditions (pH, ethanol content, surfactant, and precursor concentrations), the sizes and elemental compositions of PdAgCu MNSs as an example were precisely tailorable. Moreover, the synthetic protocol was easily adopted to synthesize other nanosized Pd-based trimetallic MNSs, for example PdAgPt, PdAgFe, PdPtCu, and PdCuRu MNSs. We further demonstrated a typical size effect of PdAgCu MNSs on electrocatalytic activity and stability toward the EOR in alkaline media. Electrocatalytic kinetics studies indicated that the size effect of PdAgCu MNSs was largely related to the size-dependent electron and mass transfer abilities. This size effect of PdAgCu MNSs was also thought to be extendable to other electrocatalytic reactions, for example the methanol oxidation reaction and hydrogen evolution reaction. We thus expect that the size-dependent synthetic strategy and electrocatalytic performance could readily provide some new opportunities for rational design of electrocatalysts for a broad range of applications, especially in electrochemical catalysis and sensors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for the financial support from the Jiangsu Specially Appointed Professor Plan, National Natural Science Foundation of China (No. 21501095 and 21665021), and Natural Science Foundation of Jiangsu Province (No. BK20150969 and BK20180723). This work is also supported by the Priority Academic Program Development of Jiangsu Higher Education Institutions, National and Local Joint Engineering Research Center of Biomedical Functional Materials.Notes and references
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS.
- S. Hu and X. Wang, Chem. Soc. Rev., 2013, 42, 5577–5594 RSC.
- H. L. Liu, F. Nosheen and X. Wang, Chem. Soc. Rev., 2015, 44, 3056–3078 RSC.
- Z. Fan, X. Huang, C. Tan and H. Zhang, Chem. Sci., 2015, 6, 95–111 RSC.
- J. N. Tiwari, R. N. Tiwari and K. S. Kim, Prog. Mater. Sci., 2012, 57, 724–803 CrossRef CAS.
- D. Wang, H. L. Xin, R. Hovden, H. Wang, Y. Yu, D. A. Muller, F. J. DiSalvo and H. D. Abruna, Nat. Mater., 2013, 12, 81–87 CrossRef CAS PubMed.
- B. Lim, M. Jiang, P. H. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1305 CrossRef CAS PubMed.
- V. Malgras, H. Ataee-Esfahani, H. Wang, B. Jiang, C. Li, K. C. W. Wu, J. H. Kim and Y. Yamauchi, Adv. Mater., 2016, 28, 993–1010 CrossRef CAS PubMed.
- C. Zhu, D. Du, A. Eychmüller and Y. Lin, Chem. Rev., 2015, 115, 8896–8943 CrossRef CAS PubMed.
- Y. Xu and B. Zhang, Chem. Soc. Rev., 2014, 43, 2439–2450 RSC.
- N. Su, X. Chen, B. Yue and H. He, Catal. Sci. Technol., 2015, 5, 638–649 RSC.
- J. Zhang and C. M. Li, Chem. Soc. Rev., 2012, 41, 7016–7031 RSC.
- S. Yang and X. Luo, Nanoscale, 2014, 6, 4438–4457 RSC.
- B. Jiang, C. Li, O. Dag, H. Abe, T. Takei, T. Imai, M. S. A. Hossain, M. T. Islam, K. Wood, J. Henzie and Y. Yamauchi, Nat. Commun., 2017, 8, 15581 CrossRef CAS.
- L. Zhang, Q. Fan, X. Sha, P. Zhong, J. Zhang, Y. Yin and C. Gao, Chem. Sci., 2017, 8, 6103–6110 RSC.
- X. Huang, Y. Li, Y. Chen, E. Zhou, Y. Xu, H. Zhou, X. Duan and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 2520–2524 CrossRef CAS.
- C. Li, B. Jiang, N. Miyamoto, J. H. Kim, V. Malgras and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 11558–11561 CrossRef CAS.
- H. Lv, A. Lopes, D. Xu and B. Liu, ACS Cent. Sci., 2018, 4, 1412–1419 CrossRef CAS PubMed.
- Y. Kuang, Y. Zhang, Z. Cai, G. Feng, Y. Jiang, C. Jin, J. Luo and X. Sun, Chem. Sci., 2015, 6, 7122–7129 RSC.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron and M. Mavrikakis, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- J. Fang, L. Zhang, J. Li, L. Lu, C. Ma, S. Cheng, Z. Li, Q. Xiong and H. You, Nat. Commun., 2018, 9, 521 CrossRef PubMed.
- Y. Kuroda and K. Kuroda, Angew. Chem., Int. Ed., 2010, 49, 6993–6997 CrossRef CAS.
- H. Wang, H. Y. Jeong, M. Imura, L. Wang, L. Radhakrishnan, N. Fujita, T. Castle, O. Terasaki and Y. Yamauchi, J. Am. Chem. Soc., 2011, 133, 14526–14529 CrossRef CAS PubMed.
- A. Mohanty, N. Garg and R. Jin, Angew. Chem., Int. Ed., 2010, 49, 4962–4966 CrossRef CAS PubMed.
- Y. Song, Y. Yang, C. J. Medforth, E. Pereira, A. K. Singh, H. Xu, Y. Jiang, C. J. Brinker, F. van Swol and J. A. Shelnutt, J. Am. Chem. Soc., 2004, 126, 635–645 CrossRef CAS.
- C. Li, Ö. Dag, T. D. Dao, T. Nagao, Y. Sakamoto, T. Kimura, O. Terasaki and Y. Yamauchi, Nat. Commun., 2015, 6, 6608 CrossRef CAS.
- A. Lehoux, L. Ramos, P. Beaunier, D. B. Uribe, P. Dieudonné, F. Audonnet, A. Etcheberry, M. José-Yacaman and H. Remita, Adv. Funct. Mater., 2012, 22, 4900–4908 CrossRef CAS.
- J. Ying, X. Y. Yang, G. Tian, C. Janiak and B. L. Su, Nanoscale, 2014, 6, 13370–13382 RSC.
- F. Wang, C. Li, L. D. Sun, C. H. Xu, J. Wang, J. C. Yu and C. H. Yan, Angew. Chem., Int. Ed., 2012, 51, 4872–4876 CrossRef CAS PubMed.
- B. Jiang, C. Li, V. Malgras, M. Imura, S. Tominaka and Y. Yamauchi, Chem. Sci., 2016, 7, 1575–1581 RSC.
- K. Eid, Y. H. Ahmad, H. Yu, Y. Li, X. Li, S. Y. AlQaradawi, H. Wang and L. Wang, Nanoscale, 2017, 9, 18881–18889 RSC.
- B. Jiang, C. Li, M. Imura, J. Tang and Y. Yamauchi, Adv. Sci., 2015, 2, 1500112 CrossRef PubMed.
- L. Bai, X. Wang, Q. Chen, Y. Ye, H. Zheng, J. Guo, Y. Yin and C. Gao, Angew. Chem., Int. Ed., 2016, 55, 15656–15661 CrossRef CAS PubMed.
- B. Liu, C. H. Kuo, J. Chen, Z. Luo, S. Thanneeru, W. Li, W. Song, S. Biswas, S. L. Suib and J. He, Angew. Chem., Int. Ed., 2015, 54, 9061–9065 CrossRef CAS PubMed.
- C. Li, M. Imura and Y. Yamauchi, Phys. Chem. Chem. Phys., 2014, 16, 8787–8790 RSC.
- Y. N. Xia, Y. J. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- L. Bu, S. Guo, X. Zhang, X. Shen, D. Su, G. Lu, X. Zhu, J. Yao, J. Guo and X. Huang, Nat. Commun., 2016, 7, 11850 CrossRef CAS PubMed.
- H. Zhu, G. Gao, M. Du, J. Zhou, K. Wang, W. Wu, X. Chen, Y. Li, P. Ma, W. Dong, F. Duan, M. Chen, G. Wu, H. Yang and S. Guo, Adv. Mater., 2018, 30, 1707301 CrossRef PubMed.
- L. Chen, L. Lu, H. Zhu, Y. Chen, Y. Huang, Y. Li and L. Wang, Nat. Commun., 2017, 8, 14136 CrossRef CAS PubMed.
- Y. Wang, S.-I. Choi, X. Zhao, S. Xie, H.-C. Peng, M. Chi, C. Z. Huang and Y. Xia, Adv. Funct. Mater., 2014, 24, 131–139 CrossRef CAS.
- M. Nesselberger, S. Ashton, J. C. Meier, I. Katsounaros, K. J. Mayrhofer and M. Arenz, J. Am. Chem. Soc., 2011, 133, 17428–17433 CrossRef CAS PubMed.
- W. P. Zhou, A. Lewera, R. Larsen, R. I. Masel, P. S. Bagus and A. Wieckowski, J. Phys. Chem. B, 2006, 110, 13393–13398 CrossRef CAS PubMed.
- S. Pedireddy, H. K. Lee, W. W. Tjiu, I. Y. Phang, H. R. Tan, S. Q. Chua, C. Troadec and X. Y. Ling, Nat. Commun., 2014, 5, 4947 CrossRef CAS PubMed.
- H. Lv, X. Chen, D. Xu, Y. Hu, H. Zheng, S. L. Suib and B. Liu, Appl. Catal., B, 2018, 238, 525–532 CrossRef CAS.
- D. Xu, X. Liu, M. Han and J. Bao, Chem. Commun., 2016, 52, 12996–12999 RSC.
- D. Xu, X. Liu, H. Lv, Y. Liu, S. Zhao, M. Han, J. Bao, J. He and B. Liu, Chem. Sci., 2018, 9, 4451–4455 RSC.
- A. Carmona-Ribeiro, Chem. Soc. Rev., 1992, 21, 209–214 RSC.
- E. Feitosa and W. Brown, Langmuir, 1997, 13, 4810–4816 CrossRef CAS.
- S. Fu, C. Zhu, D. Du and Y. Lin, ACS Appl. Mater. Interfaces, 2015, 7, 13842–13848 CrossRef CAS PubMed.
- S. H. Han, H. M. Liu, P. Chen, J. X. Jiang and Y. Chen, Adv. Energy Mater., 2018, 1801326 CrossRef.
- W. Huang, X. Kang, C. Xu, J. Zhou, J. Deng, Y. Li and S. Cheng, Adv. Mater., 2018, 30, 1706962 CrossRef PubMed.
- Z. Liang, T. Zhao, J. Xu and L. Zhu, Electrochim. Acta, 2009, 54, 2203–2208 CrossRef CAS.
- W. Hong, C. Shang, J. Wang and E. Wang, Energy Environ. Sci., 2015, 8, 2910–2915 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details of the mesoporous multimetallic spheres, electrochemical measurements and structural characterization as well as additional TEM images and electrocatalytic EOR performances. See DOI: 10.1039/c8sc04696d |
| This journal is © The Royal Society of Chemistry 2019 |