
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of alkyl-substituted pentaphosphido ligands in the coordination sphere of cobalt†‡
Christoph G. P.
Ziegler
 a,
Thomas M.
Maier
a,
Stefan
Pelties
a,
Clemens
Taube
b,
Felix
Hennersdorf
b,
Andreas W.
Ehlers
cd,
Jan J.
Weigand
*b and
Robert
Wolf
*a
a,
Thomas M.
Maier
a,
Stefan
Pelties
a,
Clemens
Taube
b,
Felix
Hennersdorf
b,
Andreas W.
Ehlers
cd,
Jan J.
Weigand
*b and
Robert
Wolf
*a
aUniversity of Regensburg, Institute of Inorganic Chemistry, 93040 Regensburg, Germany. E-mail: robert.wolf@ur.de
bTU Dresden, Faculty of Chemistry and Food Chemistry, 01062 Dresden, Germany. E-mail: jan.weigand@tu-dresden.de
cUniversity of Amsterdam, Faculty of Science, van't Hoff Institute for Molecular Sciences, Science Park 904, 1090 GS Amsterdam, The Netherlands
dUniversity of Johannesburg, Department of Chemistry, Auckland Park, Johannesburg, 2006, South Africa
First published on 4th December 2018
Abstract
Rare mono- and diorganopentaphosphido cobalt complexes are accessible by P–P condensation using the unprecedented, reactive cobalt-gallium tetraphosphido complex [K(dme)2{(MesBIAN)Co(μ-η4:η2-P4)Ga(nacnac)}] (2). Compound 2 was prepared in good yield by reaction of [K(Et2O){(MesBIAN)Co(η4-1,5-cod)}] [1, BIAN = bis(mesitylimino)acenaphthene diimine, cod = 1,5-cyclooctadiene] with [Ga(nacnac)(η2-P4)] (nacnac = CH[CMeN(2,6-iPr2C6H3)]2). Reactions with R2PCl (R = iPr, tBu, and Cy) selectively afford [(MesBIAN)Co(cyclo-P5R2)] (3a–c), which feature η4-coordinated 1,1-diorganopentaphosphido ligands. The mechanism of formation of these species has been studied by 31P{1H} NMR spectroscopy and DFT calculations. In the case of 3a (R = iPr), it was possible to identify the intermediate [(MesBIAN)Co(μ-η4:η2-P5iPr2)Ga(nacnac)] (4) by single-crystal X-ray diffraction. A related, monosubstituted organopentaphosphido cobalt complex [(MesBIAN)Co(μ-η4:η1-P5tBu)GaCl(nacnac)] (5) was isolated by reacting dichloroalkylphosphane tBuPCl2 with 2. Heterobimetallic complexes such as 2 thus may enable the targeted construction of a range of new metal-coordinated polyphosphorus frameworks by P–P condensation.
Introduction
Over the past decades, much effort has been invested into the synthesis of transition metal polyphosphido complexes.1,2 An impressive and structurally diverse array of early and late transition metal polyphosphorus species has become accessible. Most commonly, such species have been prepared by reaction of low-valent transition metal precursors with white phosphorus. While the functionalization of the polyphosphorus units derived from P4 and in particular the construction of new polyphosphorus ligands, is an attractive target, successful examples of such transformations are surprisingly scarce. This paucity is largely due to the sluggish reactivity of known complexes with electrophiles such as halophosphanes. Seminal reports by Cummins and co-workers have demonstrated the synthetic potential of some early polyphosphido transition metalate anions.2–4 The [Nb(OAr)3(η3-P3)]− anion (A, Ar = 2,6-iPr2C6H3) was used for generating a coordinated diphosphorus molecule in situ under mild conditions to access diphosphanes (Scheme 1a).5 Cummins and co-workers also devised a synthetic cycle to yield useful phosphaalkynes, and they have been using their niobium phosphido complexes to access several further unprecedented P compounds, including the previously unknown AsP3 molecule obtained by reacting A with arsenic trichloride.3,4,6,7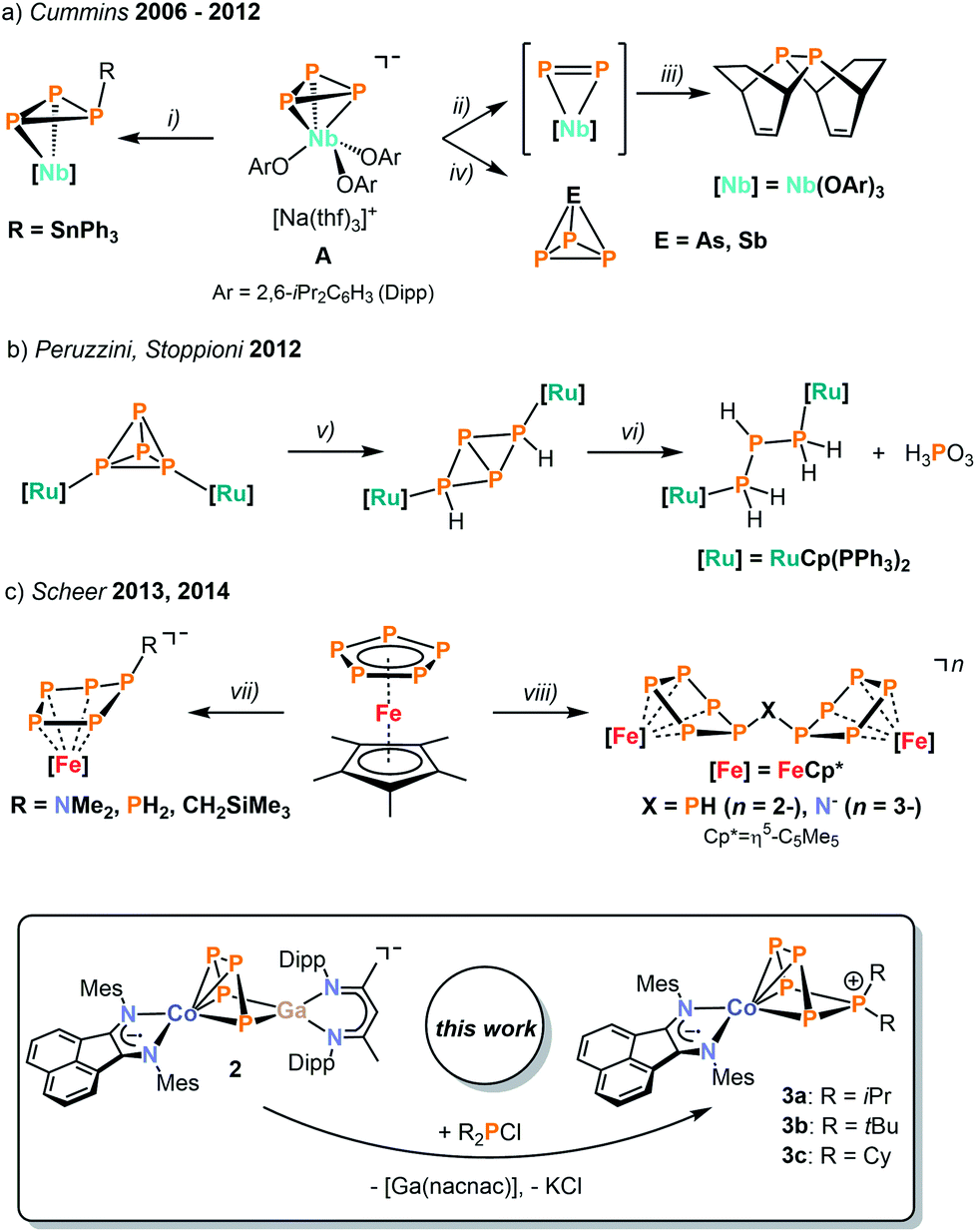 | ||
| Scheme 1 Functionalization of activated phosphorus units; reagents and products: (i) +Ph3SnCl/−NaCl; (ii) +IMo(N[tBu]Ar)3/−NaI, −PMo(N{tBu}Ar)3; (iii) +1,3-cyclohexadiene, +C5H5NO/−C5H5N, −([Nb]O)2; (iv) +ECl3/−NaCl, −[Nb]Cl2; (v) +I2, +H2O, +NaOTf; (vi) +H2O; (vii) +LiCH2SiMe3 or +LiNMe2 or +LiPH2; (viii) +NaNH2; +LiPH2. (top). Functionalization of anionic heterodinuclear tetraphosphido complexes by P–P condensation (bottom). | ||
While these results indicate that anionic polyphosphido complexes are attractive precursors for functionalisation reactions, there have only been scattered examples with other metals.8–11 Peruzzini and Stoppioni reported the alkylation and hydrolysis of group 8 and 9 complexes, for example using ruthenium phosphido compounds (Scheme 1b).8 Moreover, Scheer described the functionalization of cyclo-P5 and cyclo-P3 units in iron and nickel complexes by main group element nucleophiles such as amides, phosphanides, and hydrocarbyl anions (Scheme 1c).9,10 The same group recently reported the synthesis of the first triarsa- and the triphosphatrisilabenzenes by a successful metathesis reaction of a tetraphosphido zirconium complex with a chlorosilylene.11
Here, we describe a new strategy for the synthesis of unprecedented polyphosphido complexes, which uses heterobimetallic complexes derived from P4 as a tool for the construction of more extended Pn units. As a proof of principle, we have synthesized the new CoGaP4 complex [K(dme)2{(MesBIAN)Co(μ-η4:η2-P4)Ga(nacnac)}] (2, MesBIAN = 1,2-bis(2,4,6-dimethylphenylimino)acenaphthene, nacnac = CH[CMeN(2,6-iPr2C6H3)]2). This complex is a useful precursor for the targeted synthesis of the first diorganopentaphosphido complexes [(MesBIAN)Co(cyclo-P5R2)] (3a, R = iPr; 3b, R = tBu; 3c, R = Cy). 31P NMR monitoring and the structural characterization of a presumed intermediate [(MesBIAN)Co(μ-η4:η2-P5iPr2)-Ga(nacnac)] (4) shed light on the reaction mechanism. Moreover, we report the synthesis of [(MesBIAN)Co(μ-η4:η1-P5tBu)GaCl(nacnac)] (5). The molecular structure of 5 is unusual in that it contains a disubstituted P5 ligand with a single tBu moiety and a gallyl-substituent GaCl(nacnac).
Results
Reaction of [K(Et2O){(MesBIAN)Co(η4-1,5-cod)}] (1)12,13 with [(nacnac)Ga(η2-P4)], obtained from white phosphorus and [Ga(nacnac)] according to a literature procedure,14 in THF affords the heterodinuclear complex 2 (Scheme 2). 31P{1H} NMR monitoring indicates that the reaction is very selective and affords 2 as the sole P-containing product. After work-up, 2 was isolated in 59% yield by crystallization from DME/n-hexane. It is noteworthy that the 2,6-diisopropylphenyl-substituted complex [K(thf)2{(DippBIAN)Co(μ-η4:η2-P4)Ga(nacnac)}] (2′) can be synthesized and isolated in an analogous manner in 53% yield by recrystallization from THF/n-hexane.13 Both complexes are very similar, therefore subsequent reactivity studies focused exclusively on the Mes-substituted complex 2. The molecular structure of 2 (Fig. 1) shows a chain of four P atoms sandwiched between cobalt and gallium. The terminal P–P bonds (P1–P2 = 2.1198(7) Å, P3–P4 = 2.1286(7) Å) are shorter than the internal P–P bond (P2–P3 = 2.1755(8) Å) and the distance between the terminal P atoms (P1–P4 = 3.3073(6) Å) is large. The dihedral angle P1, P2, P3, P4 with a value of 1.3° indicates that the P4 chain is nearly planar. The C–C and C–N bond distances of the BIAN moiety suggests that it is present in its radical anionic form.12 The coordination sphere of the potassium cation contains two DME molecules and two P atoms of the P4 chain (K1–P4 = 3.3430(6) Å, K1–P3 = 3.7168(7) Å, cf. the sum of the van-der-Waals radii of K and P: 4.63 Å).15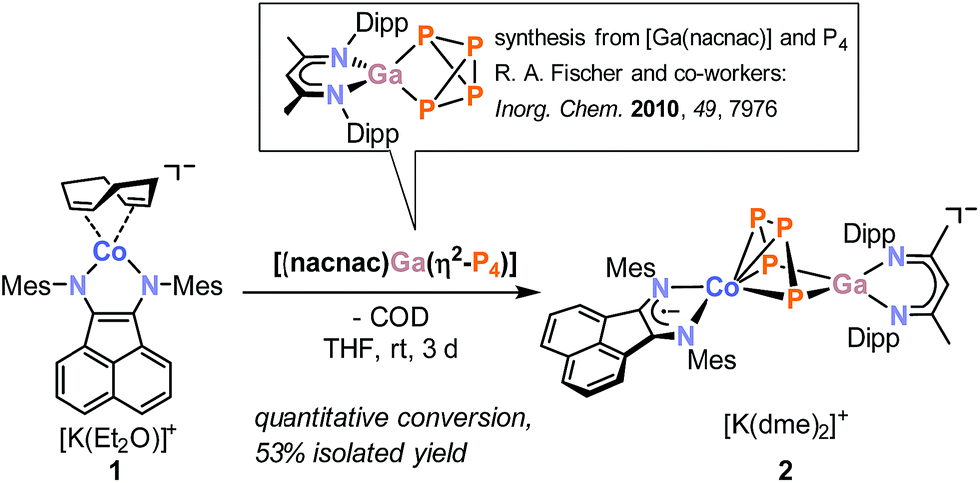 | ||
| Scheme 2 Synthesis of [K(dme)2{(MesBIAN)Co(μ-η4:η2-P4)Ga(nacnac)}] (2). | ||
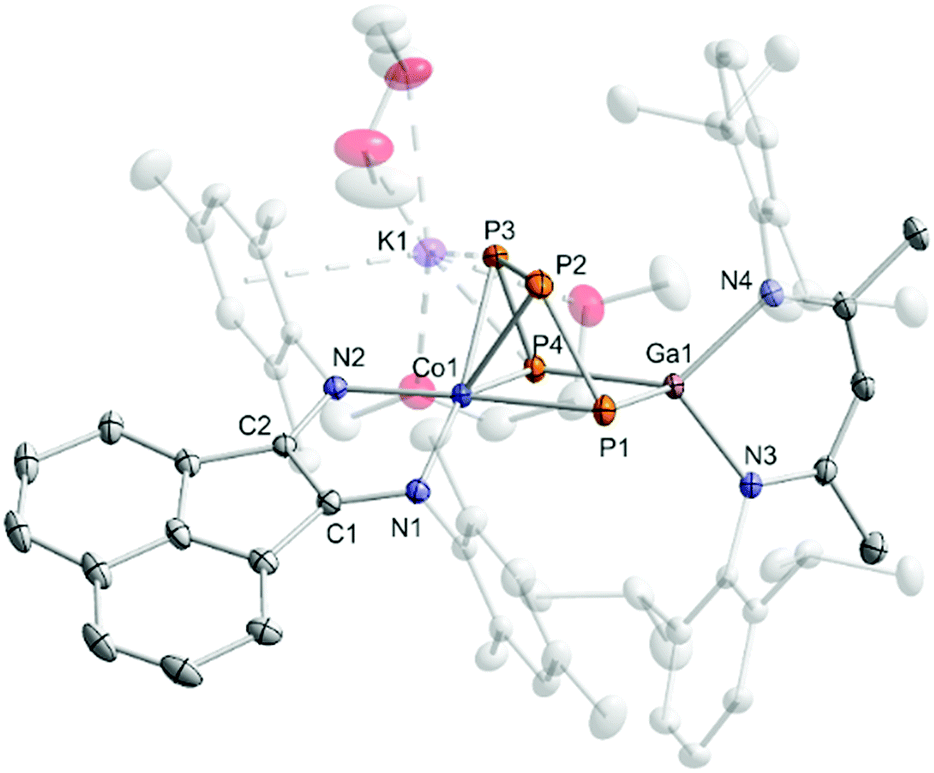 | ||
| Fig. 1 Solid-state molecular structure of [K(dme)2{(MesBIAN)Co(μ-η4:η2-P4)Ga(nacnac)}] (2) hydrogen atoms are omitted for clarity and thermal ellipsoids are drawn at the 40% probability level; selected bond lengths [Å] and angles [°]: P1–P2 2.1198(7), P2–P3 2.1755(8), P3–P4 2.1286(7), P1⋯P4 = 3.3073(6), P4–Ga1 2.3328(5), P1–Ga1 2.3179(5), Ga1–N3 1.991(1), Ga1–N4 2.014(2), Co1-P1 2.3514(6), Co1-P2 2.3098(6), Co1–P3 2.3117(5), Co1–P4 2.3961(6), Co1–N1 1.918(2), Co1–N2 1.948(2), N1–C1 1.337(2), N2–C2 1.334(2), C1–C2 1.411(3), K1–P3 3.7168(7); K1–P4 3.33073(6), Ga1–P4–P3 94.85(2), P4–P3–P2 106.97(3), P3–P2–P1 103.92(3), P2–P1–Ga1 97.31(2), P1–Ga1–P4 90.66(2); bond distances and angles of derivative 2′ are presented in the ESI (see Fig. S39, ESI‡).13 | ||
A few transition metal complexes with structures related to 2 are known. For example, Scherer's dirhodium complex [(CpRRh)(μ-η4:η2-P4){Rh(CO)CpR}] (CpR = η5-C5Me4Et) shows a very similar motif with a P4 chain (P–P 2.150(3) Å – 2.160(3) Å) coordinating to two rhodium centers in an η2- and an η4-fashion, respectively.16 [LSi(μ-η2:η2-P4)Ni(nacnac)] (L = CH[(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2)CMe][N(2,6-iPr2C6H3)]2), reported by Driess and co-workers, is also similar, but this complex features a more weakly activated, “butterfly”-P4 ligand (P1–P4 = 2.335(4) Å).17 Roesky, Konchenko, Scheer, and co-workers synthesized [(Cp′′′Co)2(μ3-η2:η2:η2-P4)SmCp*2] (Cp* = η5-C5Me5, Cp′′′ = η5-1,2,4-tBu3C5H2) via an unusual intramolecular P–P coupling process.18 The P4 chain observed in this trimetallic complex shows even somewhat longer P–P bonds (2.154(4) Å – 2.251(4) Å) than 2. Further examples of transition metal complexes with bridging catena-P4 units are diiron species reported by Scherer,19 Miluykov,20 and Walter,21 as well as a dizirconium complex described by Fryzuk.22
CH2)CMe][N(2,6-iPr2C6H3)]2), reported by Driess and co-workers, is also similar, but this complex features a more weakly activated, “butterfly”-P4 ligand (P1–P4 = 2.335(4) Å).17 Roesky, Konchenko, Scheer, and co-workers synthesized [(Cp′′′Co)2(μ3-η2:η2:η2-P4)SmCp*2] (Cp* = η5-C5Me5, Cp′′′ = η5-1,2,4-tBu3C5H2) via an unusual intramolecular P–P coupling process.18 The P4 chain observed in this trimetallic complex shows even somewhat longer P–P bonds (2.154(4) Å – 2.251(4) Å) than 2. Further examples of transition metal complexes with bridging catena-P4 units are diiron species reported by Scherer,19 Miluykov,20 and Walter,21 as well as a dizirconium complex described by Fryzuk.22
In agreement with the solid-state structure, the 31P{1H} NMR spectrum of 2 in THF-d8 (Fig. 2 and S6, ESI‡) shows an AA′XX′ spin systems.13 DFT calculations indicate that the multiplet at low frequency (−125.4 ppm) can be assigned to the terminal P atoms (Pxx′), while the multiplet at high frequency can be assigned to the internal P atoms (PAA′, see the ESI‡ for details).13 Iterative fitting of the 31P{1H} NMR spectrum revealed a 1JAX′ coupling constant of −450.5 Hz, which is 70 Hz larger in magnitude than the 1JAA coupling constant (−380.5 Hz). The 2JAX′ (6.6 Hz) and 3JXX′ (−7.2 Hz) couplings are rather small which is in line with the constrained alignment of the P atoms in the P4 chain observed in the solid-state structure, causing an antiparallel orientation of the lone pairs.23
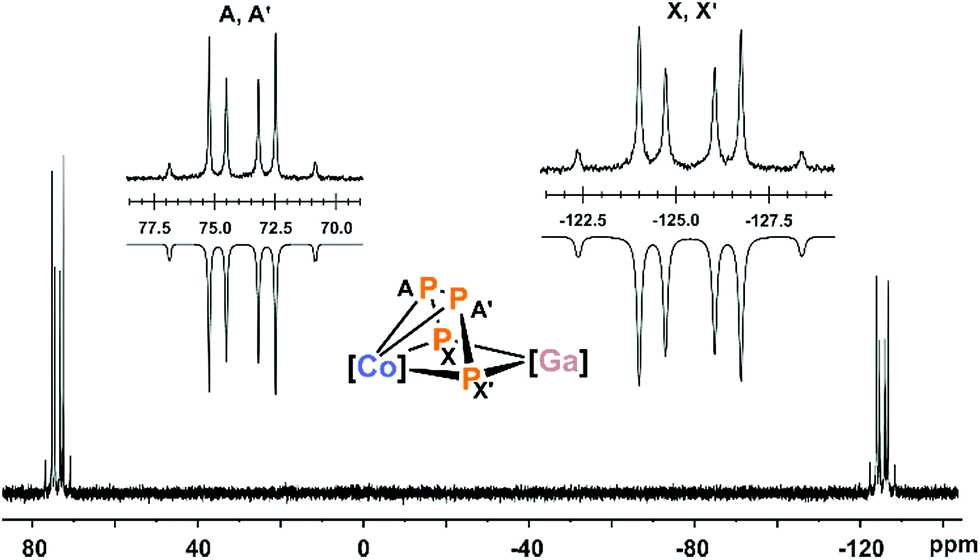 | ||
| Fig. 2 31P{1H} NMR spectrum of compound 2 with nuclei assigned to an AA′XX′ spin system; insets: extended signals (upwards) and simulations (downwards); δ(PAA′) = 74.0 ppm, δ(PXX′) = −125.4 ppm, 1JAA′ = 1JA′A = −380.5 Hz, 1JAX = 1JA′X′ = −450.5 Hz, 2JAX′ = 2JA′X = 6.6 Hz, 3JXX′ = 3JX′X = −7.2 Hz; the spectrum of 2′ is very similar (see Fig. S11 and S12, ESI‡),13 [Co] = (MesBIAN)Co, [Ga] = (nacnac)Ga. | ||
Initial reactivity studies of 2 focused on reactions with dialkylchlorophosphanes. 31P{1H} NMR monitoring of the reactions of 2 with R2PCl (R = iPr, tBu, and Cy) suggests the formation of pentaphosphido complexes [(MesBIAN)Co(η4-P5R2)] (3a, R = iPr; 3b, R = tBu; 3c, R = Cy, Scheme 3). Chromatographic work-up is necessary to remove the by-product [Ga(nacnac)]. Recrystallization from n-hexane (3a and b) or n-hexane/toluene (3c) gave analytically pure, cyan-colored crystals of the products 3a–c in moderate yields (26% to 31%).
 | ||
| Scheme 3 Synthesis of [(MesBIAN)Co(η4-P5R2)] [R = iPr (3a), R = tBu (3b), R = Cy (3c)]. | ||
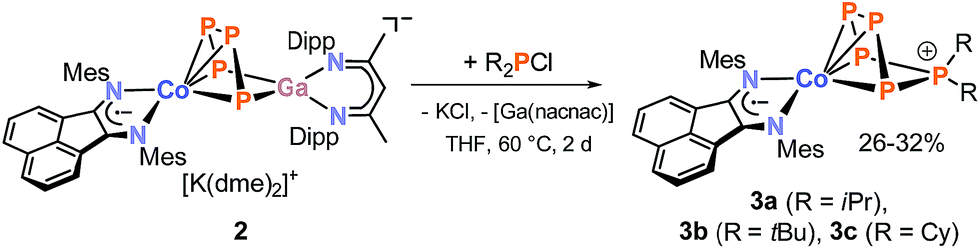
According to single-crystal XRD studies, compounds 3a–c are isostructural and feature an unprecedented η4-coordinated cyclo-P5R2 ligand in an envelope conformation. Interestingly, 3a–c may be regarded as transition metal complexes of the corresponding [P5R2]+ cage cations,24–27 but in fact they show structural isomers of these cations previously prepared by phosphenium ion insertion into P4. The molecular structure of 3a is shown in Fig. 3, while those of 3b and 3c are presented in the ESI (Fig. S40 and S41‡).13 The structural parameters of the BIAN ligand are similar to those of 2 (vide supra). The coordinating phosphorus atoms P1, P2, P3, and P4 form an almost planar arrangement (Co–P = 2.3442(1) Å − 2.3720(1) Å for 3a). The coordinated P–P bonds show a short-long-short pattern (3a: P1–P2 2.12969(2) Å, P2–P3 2.1576(2) Å, P3–P4 2.1297(2) Å), which indicates a diene-like arrangement. Scheer's salts [Li(Et2O)][Cp*Fe(η4-P5CH2SiMe3)], [Na3(dme)5][{Cp*Fe(η4-P5)}2N] and [Li2(dme)6][{Cp*Fe(η4-P5)}2PH] (vide supra, Scheme 1c) display a similar envelope conformation of the monosubstituted cyclo-P5R units.10
 | ||
| Fig. 3 Solid-state molecular structure of [(MesBIAN)Co(η4-P5iPr2)] (3a); hydrogen atoms are omitted for clarity and thermal ellipsoids are drawn at the 40% probability level; selected bond lengths [Å] and angles [°]: P1–P2 2.12969(2), P2–P3 2.1576(2), P3–P4 2.1297(2), P4–P5 2.1347(2), P5–P1 2.1506(1), P5–C1 1.8423(1), P5–C2 1.8458(1), Co1–P1 2.3720(1), Co1–P2 2.3442(1), Co1–P3 2.3447(2), Co1–P4 2.3595(2), Co1–N1 1.9104(1), Co1–N2 1.9480(1), N1–C3 1.32559(8), N2–C4 1.32069(8), C3–C4 1.4366(1); P1–P2–P3 103.926(5), P2–P3–P4 107.049(5), P3–P4–P5 98.104(6), P4–P5–P1 99.966(5), P5–P1–P2 100.689(5), C1–P5–C2 113.0644(5); bond distances and angles of derivatives 3b and c are presented in the ESI (see Fig. S40 and S41, ESI‡).13 | ||
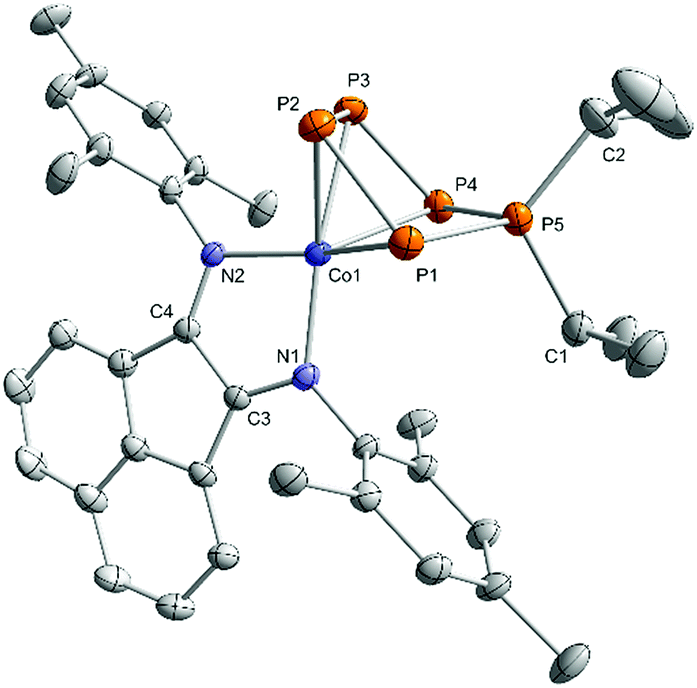
The 31P{1H} NMR spectra of 3a–c in C6D6 (Fig. 4 and S16, S21, and S26, ESI‡)13 show very similar AMM′XX′ spin systems that are consistent with molecular structures observed in the solid state. The spectrum of 3a will be discussed in more detail here. The tetracoordinate, diorganosubstituted phosphorus nucleus (PA) resonates at higher frequency (161.0 ppm for 3a) compared to the resonances of the metal-coordinated P atoms (88.6 ppm and 111.4 ppm, respectively, for 3a). The JPP coupling constants for the derivatives 3a–c were obtained by the iterative simulation of the 31P{1H} NMR spectra.13 The one-bond P–P coupling constants of the coordinated P atoms (1JMM′ = −380.6 Hz and 1JMX = −414.2 Hz for 3a) are comparable to those reported for [Li(Et2O)][Cp*Fe(η4-P5CH2SiMe3)] (1JMM′ = −409.7 Hz, 1JMX = −382.6 Hz), but the 1JAX coupling constant (−392.9 Hz) is substantially larger (−275.3 Hz for [Li(Et2O)][Cp*Fe(η4-P5CH2SiMe3)].10 The values of the 2JPP (2JMX′ = 39.9 Hz, 2JAM = 10.4 Hz, and 2JXX′ = 9.2 Hz for 3a) are in the usual range.
 | ||
| Fig. 4 31P{1H} NMR spectrum of compound 3a with nuclei assigned to AMM′XX′ spin system; insets: extended signals (upwards) and simulations (downwards); δ(PA) = 161.0 ppm, δ(PMM′) = 88.6 ppm, δ(PXX′) = −111.4 ppm, 1JAX = 1JAX′ = −392.9 Hz, 1JMM′ = −380.6 Hz, 1JMX = 1JM′X′ = −414.2 Hz, 2JMX′ = 2JM′X = 39.9 Hz, 2JAM = 2JAM′ = 10.4 Hz, 2JXX′ = 9.2 Hz; the spectra of 3b and c are very similar (see Fig. S21 and S26, ESI‡);13 [Co] = (MesBIAN)Co. | ||
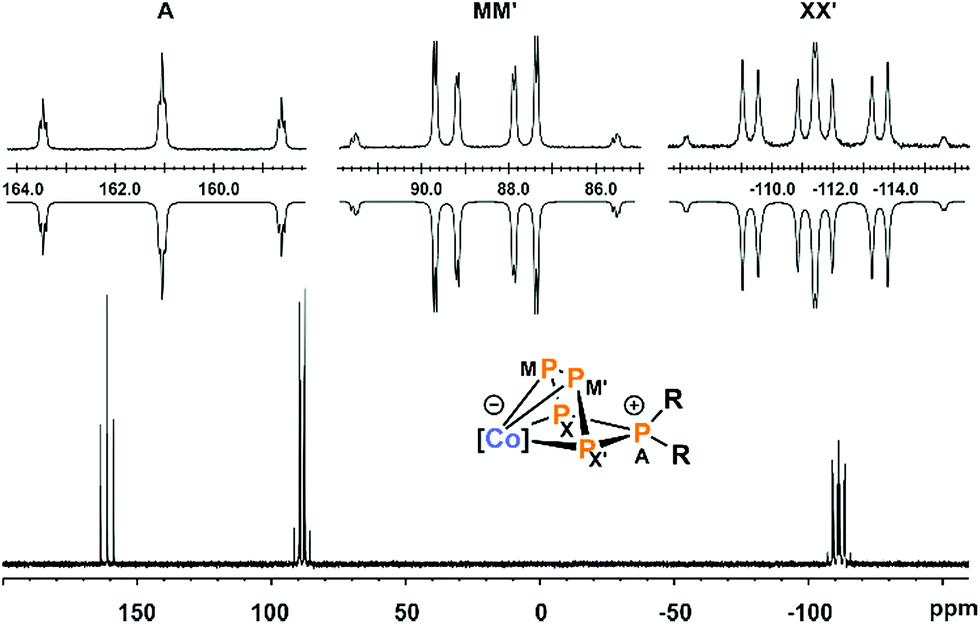
In order to gain more insight in the mechanism of formation of the diorganopentaphosphido ligands in 3a–c, we studied the reactions of 2 with R2PCl (R = tBu and iPr) by 31P{1H} NMR spectroscopy. While we did not detect any intermediate in the reaction with tBu2PCl, we observed two intermediate species in case of iPr2PCl (Fig. 5). The starting materials are consumed within ten minutes, while two similar ABCDE spin systems arise that are presumably assigned to the two intermediates Int-A and Int-B. Monitoring the reaction by 31P{1H} VT NMR spectroscopy initially shows the exclusive formation of intermediate Int-A at −30 °C. Upon warming the reaction mixture above 0 °C, the signals of Int-B arise in the 31P{1H} NMR spectra. According to 31P{1H} NMR integration, Int-A and Int-B are present in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at room temperature independent of the reaction time (see Fig. S36, ESI‡).13 Given the fairly similar 31P NMR patterns, it seems probable that Int-A and Int-B are constitutional isomers.
:1 ratio at room temperature independent of the reaction time (see Fig. S36, ESI‡).13 Given the fairly similar 31P NMR patterns, it seems probable that Int-A and Int-B are constitutional isomers.
 | ||
Fig. 5
31P{1H} NMR monitoring of the reaction of 2 dissolved in THF-d8 with iPr2PCl in a 1:1 ratio at room temperature over seven days; resonances marked with  are assigned to product [(MesBIAN)Co(η4-P5iPr2)] (3a) whereas those marked with are assigned to product [(MesBIAN)Co(η4-P5iPr2)] (3a) whereas those marked with  (Int-A) and (Int-A) and  (Int-B) are assigned to intermediates; inset: section from 25 ppm to −25 ppm. (Int-B) are assigned to intermediates; inset: section from 25 ppm to −25 ppm. | ||
It is difficult to determine the precise molecular structures of Int-A and Int-B only from 31P NMR investigation, but fortunately one of the intermediates crystallized from the reaction mixture and was characterized as [(MesBIAN)Co(μ-η4:η2-P5iPr2)Ga(nacnac)] (4, Fig. 6).13 The 31P{1H} NMR spectrum of crystalline 4 in C6D6 recorded at room temperature after 10 min showed two sets of resonances which were identified as Int-A and Int-B in the same ratio as observed in the reaction mixture. Based on our calculated 31P NMR chemical shieldings of 4, it seems plausible that intermediate Int-A can be assigned to 4 (see the ESI‡ for details).13 Solid 4, when stored under an inert atmosphere, is stable for several weeks without decomposition, but it irreversibly converts to 3a when dissolved in C6D6 at ambient temperature over the course of five days as indicated by 31P{1H} NMR monitoring.
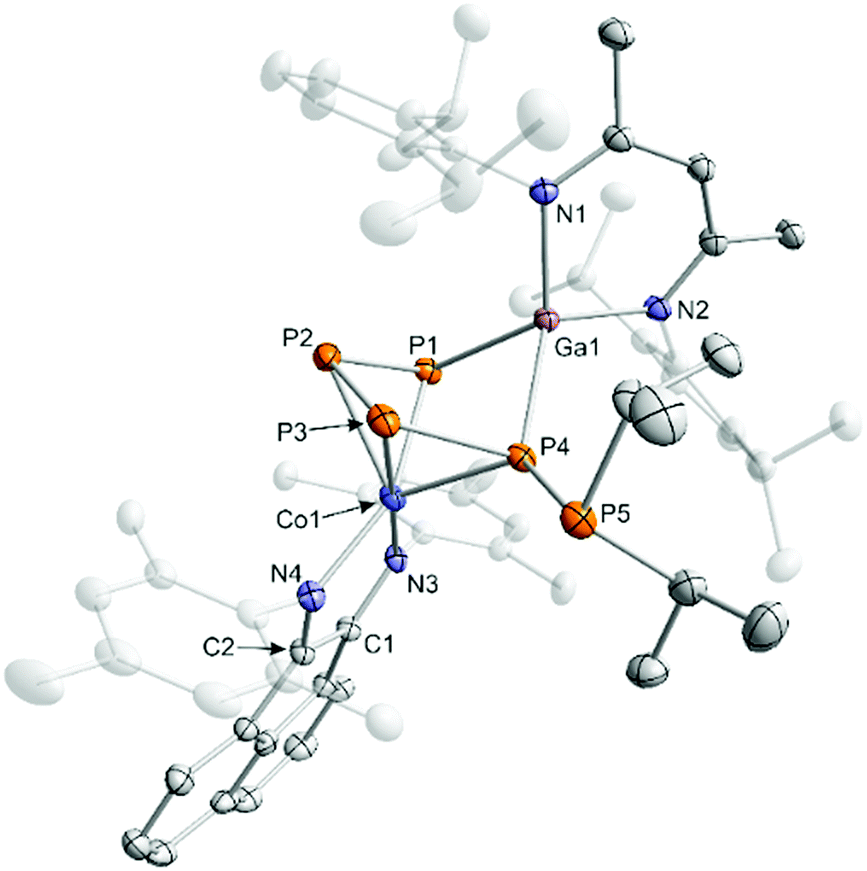 | ||
| Fig. 6 Solid-state molecular structure of [(MesBIAN)Co(μ-η4:η2-P5iPr2)Ga(nacnac)] (4); hydrogen atoms are omitted for clarity and thermal ellipsoids are drawn at the 40% probability level; selected bond lengths [Å] and angles [°]: P1-P2 2.122(1), P2-P3 2.159(2), P3-P4 2.164(1), P4-P5 2.239(1), P1–Ga1 2.3320(9), P4–Ga1 2.418(1), Ga1–N1 1.992(3), Ga1–N2 1.974(3), Co1–P1 2.348(2), Co1–P2 2.37(1), Co1–P3 2.306(1), Co1–P4 2.353(1), Co1–N3 1.919(3), Co1–N4 1.963(3), C1–N3 1.335(4), C2–N4 1.323(4), C1–C2 1.422(5), P1–P2–P3 103.59(5); P2–P3–P4 102.46(5), P3–P4–P5 93.59(5), P5–P4–Ga1 133.22(5), P1–Ga1–P5 82.28(3), Ga1–P1–P2 97.13(4). | ||
Based on these data, a mechanism of formation can be proposed for 3a (Scheme 4), which involves a pre-equilibrium between Int-A and Int-B. The latter species slowly converts into 3a by dissociation of [Ga(nacnac)]; this process appears to be irreversible.
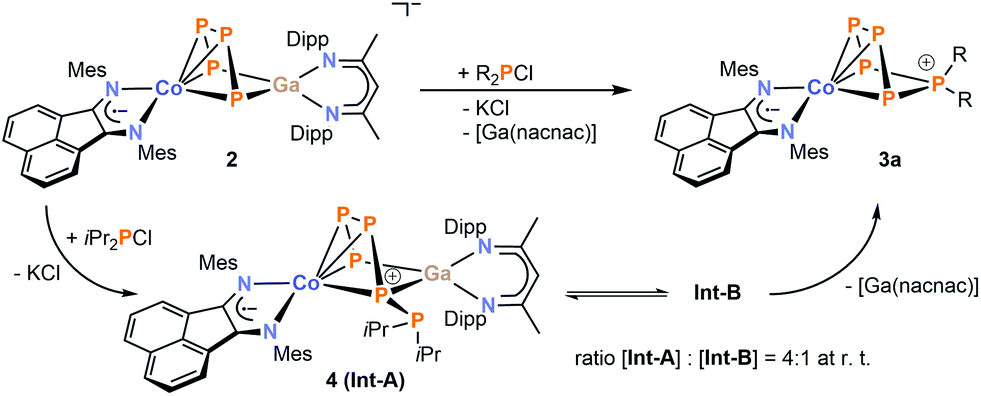 | ||
| Scheme 4 Proposed mechanism of the condensation of iPr2PCl with 2 leading to product 3a. 31P{1H} NMR monitoring revealed two intermediates Int-A and Int-B in a 4:1 integral ratio at room temperature. | ||
The molecular structure of 4 confirms that P–P bond formation has already occurred in this intermediate (Fig. 6). The structure is in line with the 31P{1H} NMR data (ABCDE spin system, vide infra) and shows an almost planar P5 chain which coordinates to cobalt via the four unsubstituted P atoms (P1–P2 = 2.122(1) Å, P2–P3 = 2.159(2) Å, and P3–P4 = 2.164(1) Å). The PiPr2 unit adopts the terminal position (P4–P5 2.239(1) Å) and does not show close contacts to cobalt or gallium. The [Ga(nacnac)] moiety is η2-coordinated to the 1,4-positions of the P5 chain (P1–Ga1 = 2.3320(9) Å, P4–Ga1 = 2.418(1) Å) and the C–N and C–C bond lengths of the BIAN moiety are again comparable to 2 and 3a–c, suggesting that the ligand is present in its radical monoanionic form.
Dialkylchlorophosphanes smoothly reacted with 2 to form a pentaphosphido framework, however, mono- and diarylchlorophosphanes gave intractable products. By contrast, tBuPCl2 (Scheme 5) readily affords tBu-substituted [(MesBIAN)Co(μ-η4:η1-P5tBu)GaCl(nacnac)] (5) as the sole P-containing species detected by 31P{1H} NMR. An unidentified paramagnetic by-product was detected in the 1H NMR spectrum of the crude reaction mixture. This undesired species can be completely removed by several recrystallization steps from toluene. This work-up procedure is the reason for the relatively low yield (6%) for the spectroscopically and analytically pure isolated compound 5. The synthesis of 5 nevertheless is remarkable because it shows that P–P condensations also occur with monoalkyldichlorophanes. The molecular structure of 5 determined by single-crystal XRD (Fig. 7) features an η4:η1-coordinated cyclo-P5tBu ligand similar to the dialkyl-substituted ligands in 3a–c. The phosphorus atoms P2, P3, P4, and P5 coordinated to cobalt form an almost planar arrangement (Co–P = 2.33156(7) Å − 2.37439(6) Å). Notably, the P atom at the tip of the cyclo-P5 envelope is coordinated to the gallium atom of GaCl(nacnac) (P1–Ga1–P5 82.28(3)°, Ga1–P1–P5 115.834(2)°, and Ga1–P1–P2 112.634(2)°). The structural parameters of the BIAN ligand are close to 2 and 3a–c (vide supra). The P–P bond distances (P1–P5 = 2.14148(6) Å, P2–P3 = 2.14129(5) Å, P3–P4 = 2.13102(7) Å, and P4–P5 = 2.13310(5) Å) are in a very close range except for the P1–P2 bond (2.19903(6) Å).
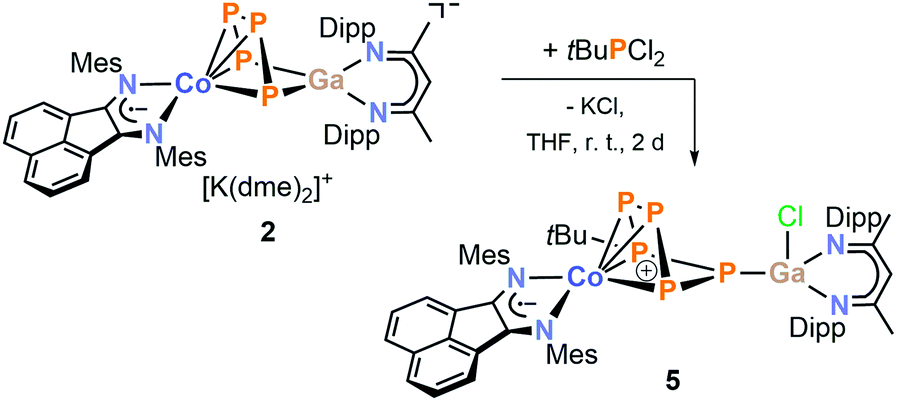 | ||
| Scheme 5 Synthesis of [(MesBIAN)Co(μ-η4:η1-P5tBu)GaCl(nacnac)] (5). | ||
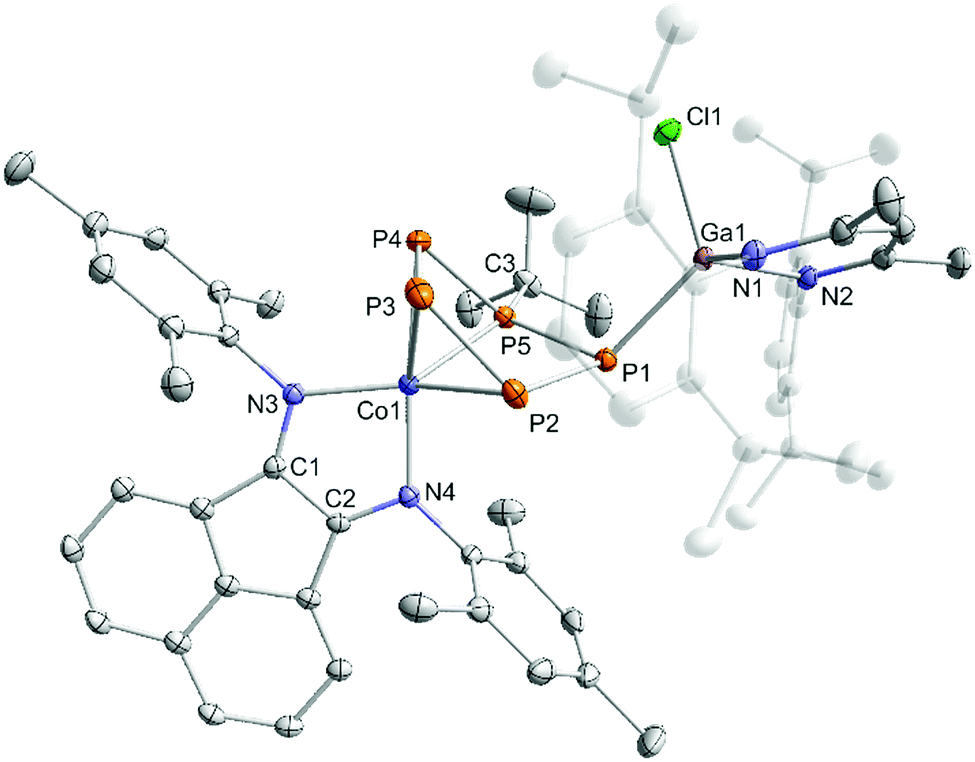 | ||
| Fig. 7 Solid-state molecular structure of [(MesBIAN)Co(μ-η4:η1-P5tBu)GaCl(nacnac)] (5); hydrogen atoms are omitted for clarity and thermal ellipsoids are drawn at the 40% probability level; selected bond lengths [Å] and angles [°]: P1–P2 2.19903(6), P2–P3 2.14129(5), P3–P4 2.13102(7), P4–P5 2.13310(5), P5–P1 2.14148(6), P1–Ga1 2.34200(6), P5–C3 1.89094(6), Ga1–N1 1.94202(6), Ga1–N2 1.96271(5), Ga1–Cl1 2.20080(5), Co1–P2 2.33156(7), Co1–P3 2.33788(6), Co1–P4 2.37439(6), Co1–P5 2.23375(6), Co1–N3 1.95761(5), Co1–N4 1.90519(4), C1–N3 1.31832(3), C2–N4 1.32622(3), C1–C2 1.42740(3); P1–P2–P3 113.062(2), P2–P3–P4 103.661(2), P3–P4–P5 97.566(2), P4–P5–P1 118.046(2), P5–P1–P2 84.471(2), P4–P5–C3 112.065(2), P4–P5–C3 115.399(2), Ga1–P1–P5 115.834(2), Ga1–P1–P2 112.634(2). | ||
Compound 5 gives rise to an ABEMX spin system in the 31P{1H} NMR spectrum, which was simulated using an iterative fitting procedure (Fig. 8). Based on the observed P–P couplings and an additional 31P–1H HMBC spectrum, the resonance at 70.3 ppm (PA) can be assigned to tBu-substituted phosphorus atom. At room temperature, this resonance is broad; hence, the simulation was carried out for the spectrum recorded at −60 °C. The resulting 31P NMR data suggest that the signals at 12.0 ppm (PM) and −66.8 ppm (PX) can be assigned to the P atoms adjacent to the tBu-substituted P atom (PA, Fig. S34, ESI‡).13
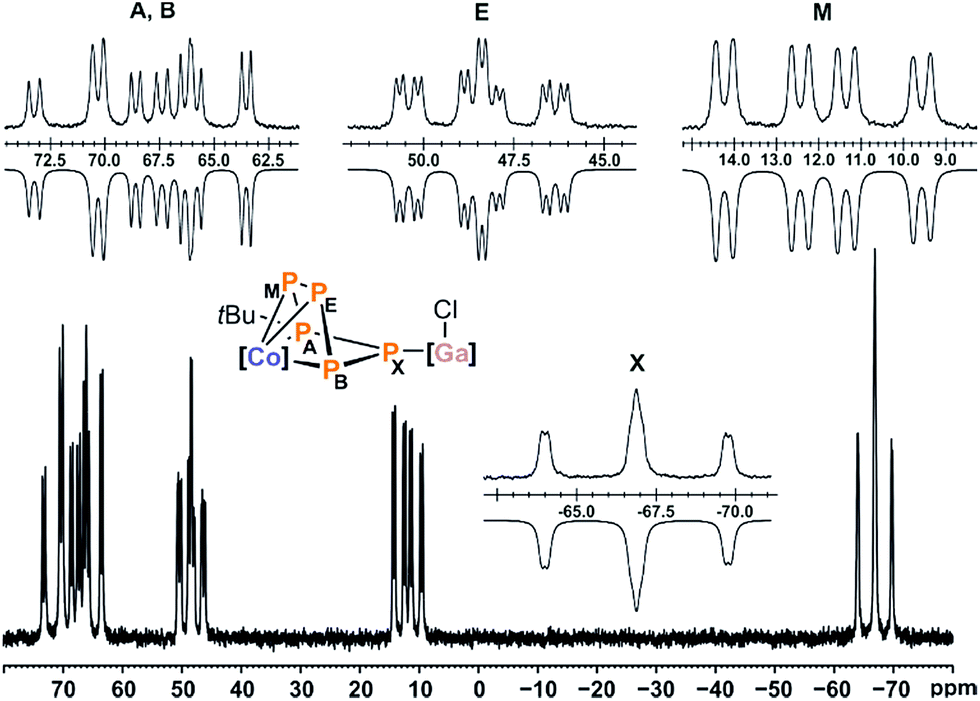 | ||
| Fig. 8 31P{1H} NMR spectrum of compound 5 at −60 °C with nuclei assigned to ABEMX spin system; insets: extended signals (upwards) and simulations (downwards); δ(PA) = 70.3 ppm, δ(PB) = 66.0 ppm, δ(PE) = 48.4 ppm, δ(PM) = 12.0 ppm, δ(PX) = −66.8 ppm, 1JAM = −466.3 Hz, 1JAX = −481.4 Hz, 1JBX = −455.1 ppm, 1JBE = −367.0 Hz, 1JEM = −291.6 Hz, 2JAE = 81.8 Hz, 2JAB = 8.2 Hz, 2JBM = 67.7 Hz, 2JEX = 33.7 Hz, 2JMX = 12.2 Hz; [Co] = (MesBIAN)Co, [Ga] = (nacnac)Ga. | ||
Conclusions
The anionic cobalt-gallium tetraphosphido complex 2 is readily accessible by reaction of [K(Et2O){(MesBIAN)Co(η4-1,5-cod)}] (1) with [Ga(nacnac)(η2-P4)]. This unique heterobimetallic complex features an activated catena-P4 unit amenable to P–P condensation reactions. Remarkably, 2 readily forms unprecedented organosubstituted pentaphosphido complexes 3a–c, 4, and 5 with R2PCl (R = iPr, tBu, and Cy) and tBuPCl2. Related metal-free [P5R2]+, [P6R4]2+, and [P7R6]2+ cations have been prepared by phosphenium cation insertion into P4;24–27 metal complexes with monosubstituted PnR ligands were accessed by reaction of nucleophiles, e.g. alkali metal alkyls, amides and phosphides, on pentaphosphaferrocene and related complexes.9,10 Notwithstanding these previous examples, the results reported in this study show that P–P condensation reactions of anionic polyphosphido complexes and halophosphanes are a potentially powerful synthetic approach which can give rise to unusual new polyphosphorus species. NMR and single-crystal XRD experiments have revealed that P–P bond formation is facile as shown by the formation of intermediate 4 characterized by X-ray crystallography. By contrast, the subsequent elimination of the [Ga(nacnac)] building block from 4 seems to be associated with a considerable barrier. The reaction properties of 3a–c and 5 are currently under investigation. Moreover, efforts are underway to extend and fine tune the P–P condensation approach for the synthesis of further unprecedented polyphosphorus compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Stefanie Gärtner (Central Analytical Services, University of Regensburg) for crystallographic assistance and Kerstin Rothermel (Gschwind group, University of Regensburg) for assistance with NMR measurements. Generous funding by the Deutsche Forschungsgemeinschaft (WE4621/3-1 and WO1496/7-1) and the Fonds der Chemischen Industrie (fellowship to T. M. Maier) is gratefully acknowledged.Notes and references
- (a) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178 CrossRef CAS PubMed; (b) M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236 CrossRef CAS PubMed; (c) M. Peruzzini, L. Gonsalvi and A. Romerosa, Chem. Soc. Rev., 2005, 34, 1038 RSC; (d) N. A. Giffin and J. D. Masuda, Coord. Chem. Rev., 2011, 255, 1342 CrossRef CAS.
- B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164 CrossRef CAS PubMed.
- J. S. Figueroa and C. C. Cummins, Dalton Trans., 2006, 2161 RSC.
- J. S. Figueroa and C. C. Cummins, J. Am. Chem. Soc., 2004, 126, 13916 CrossRef CAS PubMed.
- (a) N. A. Piro, J. S. Figueroa, J. T. McKellar and C. C. Cummins, Science, 2006, 313, 1276 CrossRef CAS PubMed; (b) B. M. Cossairt and C. C. Cummins, Angew. Chem., Int. Ed., 2010, 49, 1595 CrossRef CAS PubMed; (c) A. Velian and C. C. Cummins, Chem. Sci., 2012, 3, 1003 RSC.
- B. M. Cossairt, M.-C. Diawara and C. C. Cummins, Science, 2009, 323, 602 CrossRef CAS.
- (a) J. S. Figueroa and C. C. Cummins, Angew. Chem., Int. Ed., 2004, 43, 984 CrossRef CAS PubMed; (b) D. Tofan, B. M. Cossairt and C. C. Cummins, Inorg. Chem., 2011, 50, 12349 CrossRef CAS PubMed.
- (a) P. Barbaro, C. Bazzicalupi, M. Peruzzini, S. Seniori Costantini and P. Stoppioni, Angew. Chem., Int. Ed., 2012, 51, 8628 CrossRef CAS PubMed; (b) P. Barbaro, M. Di Vaira, M. Peruzzini, S. Seniori Costantini and P. Stoppioni, Chem. - Eur. J., 2007, 13, 6682 CrossRef CAS PubMed; (c) M. Di Vaira, P. Frediani, S. S. Costantini, M. Peruzzini and P. Stoppioni, Dalton Trans., 2005, 2234 RSC; (d) M. Di Vaira, M. Peruzzini, S. Seniori Costantini and P. Stoppioni, J. Organomet. Chem., 2006, 691, 3931 CrossRef CAS; (e) P. Barbaro, M. Peruzzini, J. A. Ramirez and F. Vizza, Organometallics, 1999, 18, 4237 CrossRef CAS; (f) P. Barbaro, A. Ienco, C. Mealli, M. Peruzzini, O. J. Scherer, G. Schmitt, F. Vizza and G. Wolmershäuser, Chem.–Eur. J., 2003, 9, 5196 CrossRef; (g) M. Peruzzini, J. A. Ramirez and F. Vizza, Angew. Chem., Int. Ed., 1998, 37, 2255 CrossRef CAS.
- (a) R. F. Winter and W. E. Geiger, Organometallics, 1999, 18, 1827 CrossRef CAS; (b) M. V. Butovskiy, G. Balázs, M. Bodensteiner, E. V. Peresypkina, A. V. Virovets, J. Sutter and M. Scheer, Angew. Chem., Int. Ed., 2013, 52, 2972 CrossRef CAS PubMed; (c) E. Mädl, G. Balázs, E. V. Peresypkina and M. Scheer, Angew. Chem., Int. Ed., 2016, 55, 7702 CrossRef.
- (a) E. Mädl, M. V. Butovskii, G. Balázs, E. V. Peresypkina, A. V. Virovets, M. Seidl and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 7643 CrossRef PubMed; (b) E. Mädl, G. Balázs, E. V. Peresypkina and M. Scheer, Angew. Chem., Int. Ed., 2016, 55, 7702 CrossRef.
- A. E. Seitz, M. Eckhardt, A. Erlebach, E. V. Peresypkina, M. Sierka and M. Scheer, J. Am. Chem. Soc., 2016, 138, 10433 CrossRef CAS PubMed.
- S. Pelties, T. Maier, D. Herrmann, B. d. Bruin, C. Rebreyend, S. Gärtner, I. G. Shenderovich and R. Wolf, Chem. - Eur. J., 2017, 23, 6094 CrossRef CAS.
- See the ESI‡ for further details..
- (a) G. Prabusankar, A. Doddi, C. Gemel, M. Winter and R. A. Fischer, Inorg. Chem., 2010, 49, 7976 CrossRef CAS PubMed; (b) F. Hennersdorf, J. Frötschel and J. J. Weigand, J. Am. Chem. Soc., 2017, 139, 14592 CrossRef CAS PubMed; (c) F. Hennersdorf and J. J. Weigand, Angew. Chem., Int. Ed., 2017, 56, 7858 CrossRef CAS PubMed.
- (a) S. Alvarez, Dalton Trans., 2013, 42, 8617 RSC; (b) B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC.
- O. J. Scherer, M. Swarowsky, H. Swarowsky and G. Wolmershäuser, Angew. Chem., Int. Ed. Engl., 1988, 27, 694 CrossRef.
- Y. Xiong, S. Yao, E. Bill and M. Driess, Inorg. Chem., 2009, 8, 7522 CrossRef PubMed.
- T. Li, N. Arleth, M. T. Gamer, R. Köppe, T. Augenstein, F. Dielmann, M. Scheer, S. N. Konchenko and P. W. Roesky, Inorg. Chem., 2013, 52, 14231 CrossRef CAS PubMed.
- O. J. Scherer, T. Hilt and G. Wolmershäuser, Organometallics, 1998, 17, 4110 CrossRef CAS.
- V. A. Miluykov, O. G. Sinyashin, P. Lönnecke and E. Hey-Hawkins, Mendeleev Commun., 2003, 13, 212 CrossRef.
- M. D. Walter, J. Grunenberg and P. S. White, Chem. Sci., 2011, 2, 2120 RSC.
- W. W. Seidel, O. T. Summerscales, B. O. Patrick and M. D. Fryzuk, Angew. Chem., Int. Ed., 2009, 48, 115 CrossRef CAS.
- M. Baudler, Y. Aktalay, K.-F. Tebbe and T. Heinlein, Angew. Chem., Int. Ed. Engl., 1981, 20, 967 CrossRef.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2001, 40, 4406 CrossRef CAS.
- M. H. Holthausen, S. K. Surmiak, P. Jerabek, G. Frenking and J. J. Weigand, Angew. Chem., Int. Ed., 2013, 52, 11078 CrossRef CAS.
- M. H. Holthausen, A. Hepp and J. J. Weigand, Chem. - Eur. J., 2013, 19, 9895 CrossRef CAS PubMed.
- M. H. Holthausen, K.-O. Feldmann, S. Schulz, A. Hepp and J. J. Weigand, Inorg. Chem., 2012, 51, 3374 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Professor Dietmar Stalke on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 1861829–1861837 and 1874059. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc04745f |
| This journal is © The Royal Society of Chemistry 2019 |