
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
211At-labeled immunoconjugate via a one-pot three-component double click strategy: practical access to α-emission cancer radiotherapeutics†
Katsumasa
Fujiki
ab,
Yousuke
Kanayama
c,
Shinya
Yano
d,
Nozomi
Sato
d,
Takuya
Yokokita
d,
Peni
Ahmadi
a,
Yasuyoshi
Watanabe
c,
Hiromitsu
Haba
d and
Katsunori
Tanaka
 *abe
*abe
aBiofunctional Synthetic Chemistry Laboratory, RIKEN Cluster for Pioneering Research, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. E-mail: kotzenori@riken.jp
bGlycoTargeting Research Laboratory, RIKEN Baton Zone Program, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
cLaboratory for Pathophysiological and Health Science, RIKEN Center for Biosystems Dynamics Research, 6-7-3 Minatojima-minamimachi, Chuo-ku, Kobe, Hyogo 650-0047, Japan
dNuclear Chemistry Research Team, RIKEN Nishina Center for Accelerator-Based Science, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
eBiofunctional Chemistry Laboratory, A. Butlerov Institute of Chemistry, Kazan Federal University, 18 Kremlyovskaya Street, Kazan 420008, Russia
First published on 21st December 2018
Abstract
α-Emission radiotherapeutics has potential to be one of most effective cancer therapeutics. Herein, we report a facile synthesis of an 211At-labeled immunoconjugate for use as an α-emission molecular targeting therapy. We synthesized a tetrazine probe modified with closo-decaborate(2-), a prosthetic group that forms a bioavailable stable complex with 211At. Our one-pot three-component double-click labeling method was used to attach decaborate to trastuzumab (anti-HER2 antibody) using decaborate-tetrazine and TCO-aldehyde probes without reducing the antibody binding affinity. Labeling the decaborate-attached trastuzumab with 211At produced in the cyclotron at the RIKEN Nishina Center, at which highly radioactive 211At can be produced, readily furnished the 211At-labeled trastuzumab with a maximum specific activity of 15 MBq μg−1 and retention of the native binding affinity. Intratumor injection of the 211At-labeled trastuzumab in BALB/c nude mice implanted with HER2-expressing epidermoid cancer cells yielded efficient accumulation at the targeted tumor site as well as effective suppression of tumor growth.
Introduction
Radiation therapeutics, including radionuclides, show promise as effective cancer therapeutics. Radiolabeled cancer-targeting molecules have recently attracted attention in the field of radiopharmaceutical development.1–4 Several radioimmunoconjugates5–7 have been developed, including 90Y-labeled rituximab (an anti-CD20 antibody commercialized under the name Zevalin),8131I-labeled tositumomab (an anti-CD20 antibody commercialized under the name Bexxar),9 and 177Lu-labeled anti-CD20 and anti-CD37 antibodies10 for use in β-emitting therapy.α-Emission radiotherapy has attracted attention for its potent cancer therapeutic efficacy. Radionuclides that decay by emitting alpha particles can damage targeted cancer tissue within a few cell diameters, thereby minimizing off-target damage. α-Emitters, including 225Ac, with a half-life of 10 days, 223Ra, with a half-life of 11.4 days, and 227Th, with a half-life of 18.7 days, are favored for use in radiotherapy techniques involving targeted molecules, such as radioimmunoconjugates, because of their long half-lives.11 Efficient chelators of these radionuclides must be developed before such applications may be realized.12,13211At, with a half-life of 7.2 h, is one of the most attractive α-particle nuclides for use in molecularly targeted therapy,14,15 and several approaches to preparing 211At-labeled molecules for cancer targeting have been explored. Astatination may be achieved using prosthetic groups such as stanylphenyl,16–18 silylphenyl,19 or borylphenyl20 moieties. Aryliodonium salt derivatives21 or caged carborane derivatives22–25 tend to be good chelators of 211At as well. The use of these labeling groups has enabled the manufacture of 211At-labeled molecular complexes that are stable to metabolic deastatination in animal models, thereby realizing in vitro and in vivo evaluations of targeted functionalities.26–28 Wilbur et al. developed a variety of 211At-labeling reagents for use with biofunctional molecules, such as biotin,29 streptavidin,30 anti-renal cell carcinoma antibody A6H F(ab′)2,31 and anti-CD45 antibody.32 Li et al. recently reported the use of α-emitting locoregional therapy in human epidermal growth factor receptor 2 (HER2)-expressing gastric cancer in a xenograft mouse model. The stanylphenyl group was used to prepare the 211At-labeled trastuzumab.33 These studies prepared the 211At-labeled molecules by introducing prosthetic groups onto biofunctional molecules using N-succinimidyl ester34 or isothiocyanate derivatives,35 which are reactive toward amino groups on lysine residue side chains, or a maleimide derivative, which is reactive toward thiols on cysteine residue side chains.36 Conjugation was achieved over long reaction times to ensure a high conversion rate, and extra procedures involving reductants were needed to cleave the disulfide bond of antibodies.22 Efficient methods for introducing prosthetic groups into biomolecules for 211At-labeling using highly reactive and/or chemoselective conjugation approaches, such as click chemistry, have not yet been reported. α-Emission therapies using 211At-labeled molecular targeting agents may be extended by developing efficient methods of preparing 211At-labeled molecules.
Our group recently developed a facile method of radiolabeling amino groups on biomolecules using a “one-pot three-component double-click labeling” based on two types of click reactions in combination: a RIKEN click reaction, 6π-azaelectrocyclization,37–42 was combined with tetrazine ligation43 (Scheme 1).44 According to this labeling method, simply by mixing the DOTA (or NOTA)-tetrazine 1 (or 2) as metal chelators and the TCO-aldehyde 3 (DOTA: 1,4,7,10-tetraazadodecane-1,4,7,10-tetraacetic acid, NOTA: 1,4,7-triazacyclononane-1,4,7-triacetic acid, tetrazine: 3,6-di-(2-pyridyl)-s-tetrazine, TCO: trans-cyclooctene) with biomolecules, such as proteins and antibodies, under mild conditions introduced DOTA (or NOTA) to biomolecules without loss of their bioactivities (Scheme 1a). Subsequent treatment of DOTA (or NOTA)-introduced biomolecules with 67Cu, which is an attractive radionuclide due to its diagnostic and therapeutic properties, efficiently produced 67Cu-labeled biomolecules. This radiolabeling method required only Amicon filtration for purification; thus, time-consuming purification by HPLC was not necessary. Note that based on our one-pot three-component double-click labeling, a variety of prosthetic radiolabeling groups could be introduced onto the amino groups of biomolecules without inhibiting their native functions.
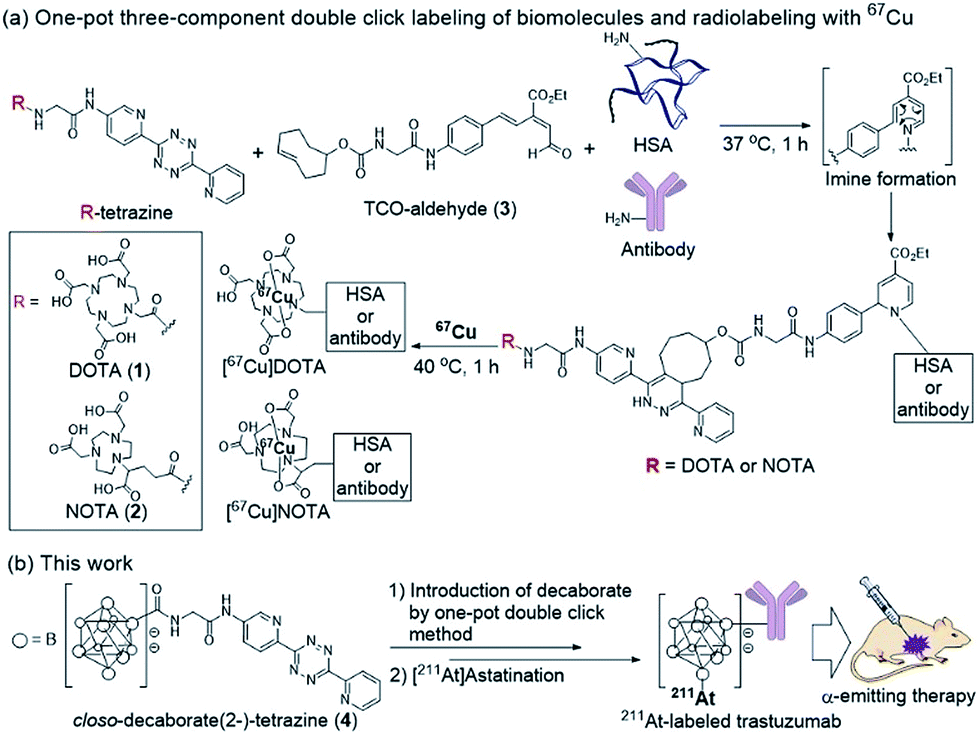 | ||
| Scheme 1 (a) Radiolabeling of human serum albumin (HSA) and an antibody using 67Cu via the one-pot three-component double-click method. (b) Design of the closo-decaborate(2-)-tetrazine probe and one-pot three-component double-click production of the 211At-labeled trastuzumab. Application to an α-emitting cancer therapeutic in a xenograft mouse model. DOTA: 1,4,7,10-tetraazadodecane-1,4,7,10-tetraacetic acid; NOTA: 1,4,7-triazacyclononane-1,4,7-triacetic acid; TCO: trans-cyclooctene. | ||
The clinical use of 211At-labeled molecules would require methods of efficiently producing 211At. To date, only one report has addressed this obstacle.45 We envisioned that the world-class cyclotron at RIKEN, Japan, could be used to produce radioactive 211At in sufficient GBq units to reproducibly prepare α-emission cancer therapies for use in humans. In this context, we designed and synthesized a tetrazine probe 4 having closo-decaborate(2-) as the prosthetic group46 for binding 211At. We developed a facile non-invasive synthetic method of preparing 211At-labeled trastuzumab as an α-emitting therapeutic HER2-recognizing antibody using a double-click labeling strategy (Scheme 1b). Intratumor injection was found to provide excellent α-emitting therapeutic efficacy of the 211At-labeled trastuzumab against HER2-positive epidermoid cancer in a xenograft mouse model.
Results and discussion
Synthesis of the decaborate-tetrazine 4
We previously developed a concise method of labeling biofunctional molecules with DOTA and NOTA as conventional metal chelators without reducing their bioactivities using a one-pot three-component double-click method. Subsequent treatment of the DOTA/NOTA-labeled molecules with 67Cu enabled the synthesis of 67Cu-labeled biomolecules.44 This double-click method was applied to 211At-labeling by designing a tetrazine probe 4 bearing closo-decaborate(2-), which is stable in vivo against deastatination47 (Scheme 2). According to our previous synthesis,44 the glycine-labeled tetrazine 5 was deprotected to give the amine 6 as a hydrochloride salt. 6 was then coupled with the oxocarbenium derivative of closo-decaborate(2-), 7, prepared according to the procedure reported by Wilbur,24 and the decaborate-tetrazine 4 was obtained in good yield over 2 steps.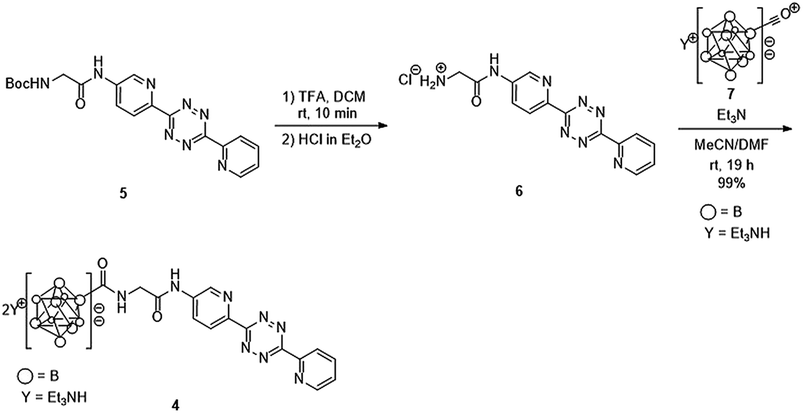 | ||
| Scheme 2 Synthesis of the closo-decaborate-tetrazine 4. TFA = trifluoroacetic acid, DCM = dichloromethane, DMF = N,N-dimethylformamide. | ||
Synthesis of decaborate-trastuzumab via a one-pot three-component double-click reaction
With the decaborate-tetrazine 4 and TCO-aldehyde 3 in hand, we next examined the introduction of the decaborate moiety to biomolecules using a one-pot three-component double-click strategy. A model protein, HSA (human serum albumin), was modified with the decaborate moiety in the presence of 100 μM 3, 100 μM 4, and 10 μM HSA in a 5% DMSO aqueous solution at 37 °C for 1 h (Scheme 3). In our previous investigation of one-pot double click labeling, we established the conditions, e.g., reagent concentrations, in which the tetrazine ligation between TCO-aldehyde 3 and tetrazine was completed within a few minutes, while the RIKEN click reaction proceeds over an hour to label a few lysines. In order to further ensure the completion of the tetrazine ligation, we initially mixed the TCO-aldehyde 3 and decaborate-tetrazine 4 prior to adding the biomolecules in one-pot manner. The reaction mixture was briefly purified by Amicon ultrafiltration to remove unreacted labeling probes. MALDI-TOF mass spectroscopic analysis of the obtained decaborate-HSA 8 revealed that an average of 2.3 decaborate moieties were attached to each HSA (Scheme 3). The number of attached molecules (a 3 + 4 molecule underwent a 1065 MW increase) was determined in comparison to the intact HSA molecular weight. Under the conditions used to label HSA, trastuzumab, which is commercially available and a promising anti-cancer antibody drug, was treated with the TCO-aldehyde 3 and decaborate-tetrazine 4 to prepare the decaborate-labeled trastuzumab 9 in a one-pot double click manner (Scheme 3). Although the exact molecular weight of the decaborate-trastuzumab 9 could not be detected using MALDI-TOF mass analysis (presumably due to the highly hydrophobic nature of the decaborate moiety), the number of prosthetic group introduced was found to be similar to the number introduced into the decaborate-HSA 8, as reported previously.44 This was confirmed using radiolabeling experiments, as discussed below.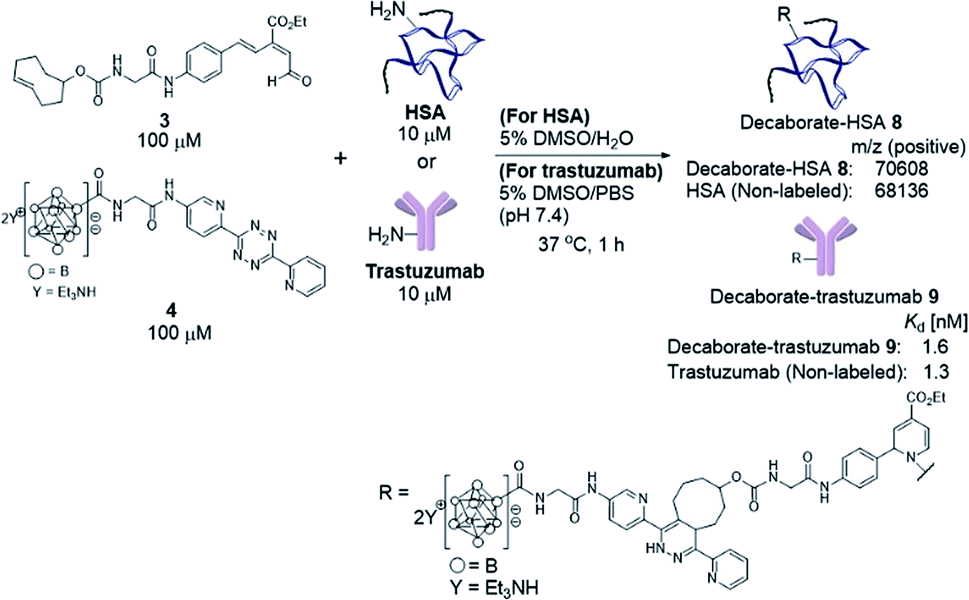 | ||
Scheme 3 Preparation of the decaborate-albumin 8 and trastuzumab 9via the one-pot three-component double-click labeling method. Purification was performed by ultrafiltration using an Amicon filter, 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 molecular weight. Mass analysis of the decaborate-HSA 8 by MALDI-TOF. Dissociation constants (Kd) of the decaborate-trastuzumab 9 measured by the QCM method. DMSO = dimethylsulfoxide. QCM = quartz-crystal microbalance. 000 molecular weight. Mass analysis of the decaborate-HSA 8 by MALDI-TOF. Dissociation constants (Kd) of the decaborate-trastuzumab 9 measured by the QCM method. DMSO = dimethylsulfoxide. QCM = quartz-crystal microbalance. | ||
The recognition activity of 9 was investigated by measuring the dissociation constant (Kd) of 9 using the quartz crystal microbalance (QCM) method (as described in the ESI†). As shown in Scheme 3, the dissociation constant of 9 was determined to be 1.6 nM, comparable to the dissociation constant of the non-labeled trastuzumab, 1.3 nM. The double click method introduced a decaborate moiety into trastuzumab without diminishing its binding affinity.
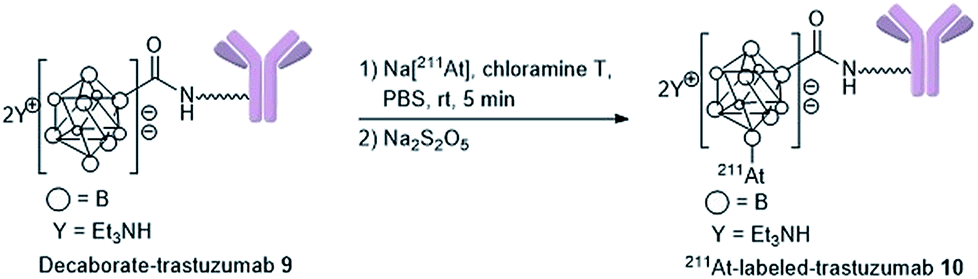
Preparation of 211At-labeled trastuzumab
With the biologically functionally intact decaborate-trastuzumab 9 in hand, we next optimized the astatination of 9 under mild conditions. The astatination of 9 was conducted by treating solutions of 9 (at various concentrations) with Na[211At] in the presence of chloramine T as an oxidant24,48 over 5 min at room temperature (Table 1). We optimized the 211At labeling by treating 10 μM 9 with 5.8 MBq Na[211At] in phosphate buffer saline (PBS). The 211At-labeled trastuzumab 10 was successfully obtained in 78% RCY with a specific activity of 0.050 MBq μg−1 (Table 1, entry 1). The absence of non-specific binding between 211At and trastuzumab was confirmed by mixing a 10 μM PBS solution of the intact trastuzumab with Na[211At], 5.2 MBq (Table 1, entry 2). After ultrafiltration of the reaction mixture, most of the loaded Na[211At] was washed away, and very weak radioactivity from 211At, attributed to residual Na[211At], was observed in the purified trastuzumab. To reduce the administered quantity and to maximize the radiotherapeutic efficacy of the 211At-labeled trastuzumab in in vivo experiments (with an eye for future human applications), we prepared the 211At-labeled trastuzumab 10 with a higher specific activity by increasing the radioactivity of Na[211At] and by reducing the loading of trastuzumab. The labeling was performed using 1 μM 9 in 0.05% PBS-T and Na[211At], 75 MBq, in PBS to furnish 10 with a specific activity of 1.7 MBq μg−1 in 49% RCY (Table 1, entry 3). The potential loss of antigen recognition activity in the 211At-labeled trastuzumab with a high specific activity was assessed by measuring the dissociation constant Kd of the obtained 10. This value was found to be 1.0 nM, indicating no impairment to the affinity. Reacting 0.1 μM 9 with Na[211At], 104 MBq, in PBS, provided 10 in 30% RCY with a very high specific activity of 15 MBq μg−1 (Table 1, entry 4). In this case, the dissociation constant of 10 could not be measured exactly due to the very low concentration of the antibody in the product. We also conducted a TLC analysis of 10 after Amicon ultrafiltration to confirm the formation of the 211At-trastuzumab complex (see Table S1 in the ESI†).|
|
|||||
|---|---|---|---|---|---|
| Entry | Conc. of 9 (μM) | Na[211At] (MBq) | RCY (%) | Specific activity (MBq μg−1) | K d (nM) |
| a Dissociation constants were estimated using the QCM method. b Instead of 9, trastuzumab without the decaborate label was used as a negative control. c The dissociation constant could not be measured accurately due to the very low amount of antibody present. Conc. = concentration. RCY = radiochemical yield. | |||||
| 1 | 10 | 5.8 | 78 | 0.050 | — |
| 2 | 10b | 5.2 | 5 | — | — |
| 3 | 1 | 75 | 49 | 1.7 | 1.0 |
| 4 | 0.1 | 104 | 30 | 15 | —c |
The stability of the 211At-labeled trastuzumab 10 was investigated by storing PBS solutions of 10 with different specific activities for 24 h at room temperature. The stability tests were conducted by preparing the 211At-labeled trastuzumab 10 with specific activities of 0.64, 1.3, or 2.4 MBq μg−1 under the following labeling conditions: 1, 0.5, or 0.25 μM 9 were treated with Na[211At], 22 MBq, respectively. The stock solutions in PBS were subjected to ultrafiltration, and the radioactivities were measured. As shown in Fig. 1, the 211At-labeled trastuzumab 10 solutions prepared with specific activities of 0.64, 1.3, and 2.4 MBq μg−1 displayed radioactive 211At corresponding to 0.82, 0.85, and 0.81 MBq, respectively, after 24 h and before Amicon ultrafiltration. After re-purification by Amicon ultrafiltration, 211At solutions of 0.72, 0.75, and 0.69 MBq were detected in the re-purified 10; thus, the 211At-labeled trastuzumab synthesized via double clicks was robust, and degradation to small peptide fragments under proximal α-ray radiation produced by 211At was minimal.
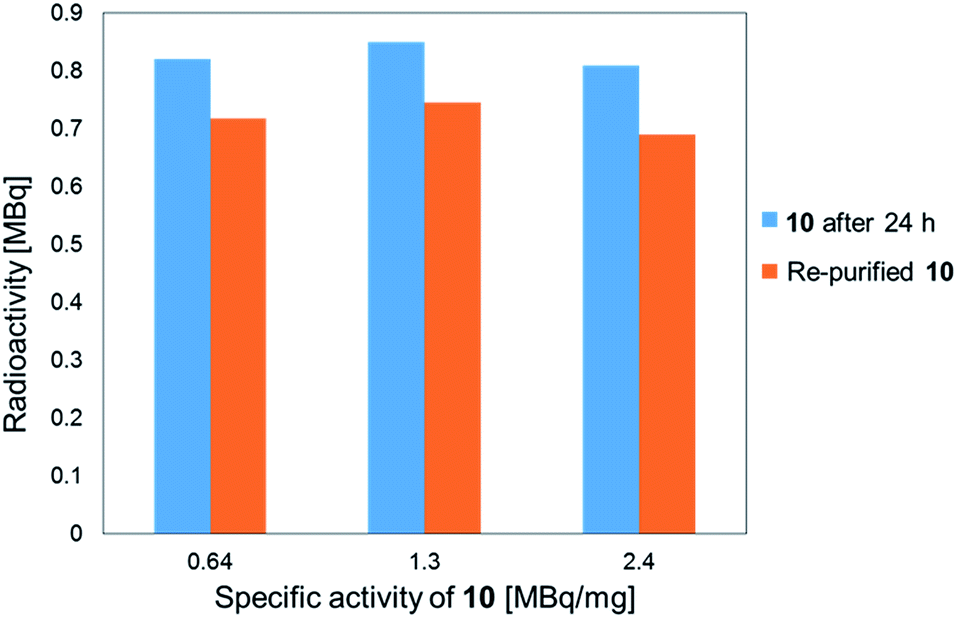 | ||
| Fig. 1 Stability analysis of the 211At-labeled trastuzumab 10. The radioactivity of 10 and the specific activity was measured for each sample 24 h after sample preparation (light blue bar) and after re-purification of the samples (orange bar). | ||
Biodistribution and α-emission therapeutic efficacy of 211At-labeled trastuzumab
In addition to efficiently synthesizing 211At-labeled trastuzumab, 10, without degrading the HER2 recognition activity, we evaluated the biological function as an α-emitting cancer therapy in vivo using tumor-bearing mice. We first performed a biodistribution study of the 211At-labeled trastuzumab 10 in a xenograft model mouse using A431 human epidermoid carcinoma cells as HER2-positive cancer cells to examine the specific accumulation of 10 at the tumor. We previously reported the PET imaging of trastuzumab labeled with 64Cu-DOTA in the A431 xenografted mouse model, the same model as used in this study.49 The HER2 specific accumulation of 64Cu-labeled trastuzumab (20% ID/g of trastuzumab in tumor) was clearly visualized after intravenous injection.We therefore prepared 10 with a specific activity of 0.030 MBq μg−1 after adjusting the appropriate volume of 0.05% Tween 20 – containing PBS (PBS-T) solution for injection into the mice (see the ESI† for protocol details). Next, 150 μL of a 0.05% PBS-T solution containing 20 μg 10 labeled with 211At, 0.60 MBq, was intravenously injected into each tumor-bearing mouse. The radioactivity in each organ dissected 16, 24, and 40 h after intravenous injection was measured, and high radioactivity was detected only in the liver and kidney, and the selective accumulation of 10 was not observed in the tumor (Fig. 2). Specific accumulation in the tumor did not improve upon intravenously injecting a reduced amount of 10 with a higher specific activity (see Fig. S3 in the ESI†).
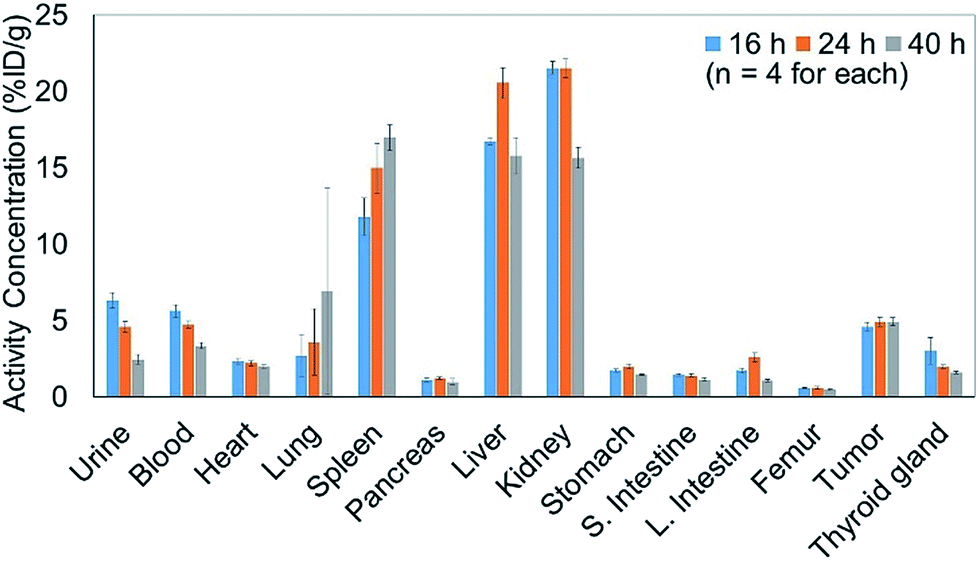 | ||
| Fig. 2 Concentration of the 211At-labeled trastuzumab 10 in each organ of the A431 cell xenograft mouse model. A 150 μL 0.05% PBS-T solution containing 20 μg 10 labeled with 211At (0.60 MBq) was intravenously injected into the A431 xenograft mice (n = 4). The radioactivity of each organ was measured after dissection at 16 h (light blue bar), 24 h (orange bar), or 40 h (gray bar) post injection. | ||
Since the stability of the “RIKEN click” linkage was concerned in serum after intravenous injection, we performed the PET imaging (intravenous injection in A431 xenografted mouse) of 64Cu-DOTA labeled trastuzumab, which was similarly prepared via one-pot double click reaction, using DOTA-tetrazine and TCO-aldehyde 3 (see Fig. S5 in the ESI†). The PET image of 64Cu-labeled trastuzumab in Fig. S5a† clearly showed the accumulation in A431 tumor. In addition, the biodistribution studies after 2 days (Fig. S5b†) found sufficient radioactivity in tumor and blood, and less radioactivity in kidney in comparison with those obtained by intravenously injected 211At-labeled trastuzumab (Fig. 2). Alternatively, biodistribution experiments using the [211At]decaborate moiety provided similar results to 211At-labeled trastuzumab (Fig. S4 in the ESI†), i.e., uptake in the liver followed by clearance through the kidneys. These results indicate that the low tumor uptake under intravenous injection of antibody is not due to the instability of “RIKEN click” linkage in serum, but rather introduction of a large hydrophobic decaborate moiety into the antibody significantly altered the in vivo kinetics. Namely, the hydrophobic trastuzumab conjugate could be captured and degradated by the liver for the renal clearance.
The antibody valency of the 211At-labeled trastuzumab 10 was exploited to achieve selective therapeutic effects in tumor regions by applying intratumor injections of 10 to the tumor-bearing mice. Intratumor injections of 10 with a specific activity of 0.41 MBq μg−1 were conducted by preparing 5 μL of a 0.05% PBS-T solution containing 6.3 μg 10 labeled with 211At, 1.4 MBq, and administering this solution to each tumor xenograft mouse. The radioactivity in each organ of the mouse dissected after 1 day post-injection was measured, revealing that most of the administered 10 remained in the tumor (Fig. 3a). Furthermore, even after 2 days post-injection, a sufficient quantity of the radioactive 10 could be observed in the tumor. By contrast, administration of Na[211At], 1.0 MBq, in 5 μL PBS to the tumors of xenograft mice via intratumor injection did not result in 211At radioactivity in the tumor, apparently because the salt leached out of the blood vessels (Fig. 3b). Although a relatively high level of 211At radioactivity was detected in the thyroid gland, in agreement with the literature,50,51 most 211At administered had been excreted from the bodies of the mice after 1 day post-injection. Thus, even under intratumor injection conditions, the tumor-anchoring effects of the 211At by the antibody were notable, highlighting the utility of the 211At antibody conjugate.
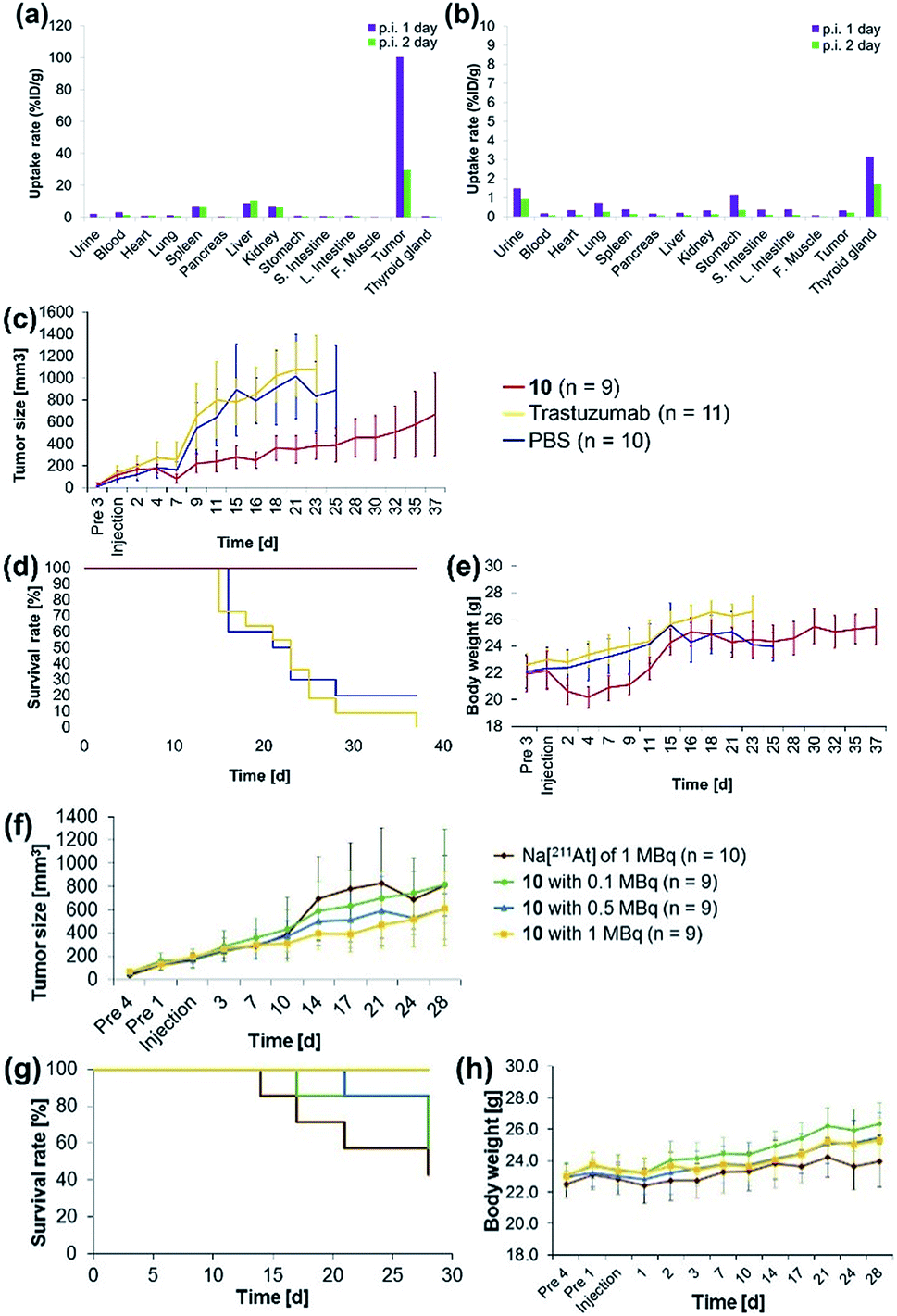 | ||
| Fig. 3 Concentrations of (a) the 211At-labeled trastuzumab 10 or (b) a 0.05% PBS-T solution of Na[211At] in each organ of the A431 cells xenograft mouse. (a) 6.3 μg 10 labeled with 211At (1.4 MBq) in 5 μL 0.05% PBS-T or (b) 211At (1.0 MBq) in 5 μL PBS were administered to the A431 xenograft mouse by intratumor injection. The radioactivity of each organ was measured after dissection 1 day (purple bar) or 2 days (green bar) post-injection. Therapeutic efficacies of the α-emitting 10 (red line), trastuzumab (non-labeled, yellow line), or PBS (blue line) after intratumor injection: (c) tumor size, (d) survival curve, and (e) body weight of the xenograft mice after intratumor injection. Dose dependence of the α-emitting therapeutic efficacy of 211At after intratumor injection of Na[211At] (brown line), 10 with 0.1 MBq (green line), 0.5 MBq (blue line), and 1 MBq (yellow line). (f) Time course analysis of the tumor size. (g) Survival curves of the tumor-bearing mice. (h) Body weight of the tumor-bearing mice after intratumor injection. The tumor size was measured in tumor xenograft mice with tumor sizes exceeding a volume (V) of 1200 mm3, calculated using the formula V = Rr2/2. Here, R and r indicate the long and short axes of the tumor in the xenograft mouse, respectively. | ||
Encouraged by the success of the specific accumulation of 10 in the tumor, we evaluated tumor growth in A431 cell xenograft mice after intratumoral injection of 10. A 0.05% PBS-T solution of 10, the same concentration as was synthesized for the biodistribution experiments, was directly administered to the tumors of A431 cell xenograft mice. The long and short axes of the tumors in the xenograft mice were measured and the size was calculated every couple of days according to Battelle Columbus Laboratories Protocol52 (Fig. 3c). In mice administered 10 labeled with 211At, 1.4 MBq, the average tumor size in the xenograft mice decreased 7 days post-injection, as indicated by the red line. Successive tumor measurements over one month revealed that tumor growth was inhibited by the administration of 10. By contrast, the xenograft mice injected with non-labeled trastuzumab (6.3 μg per mouse) or PBS, used as negative controls, displayed no therapeutic effect, and the average tumor size in the xenograft mice increased dramatically (Fig. 3c, yellow and blue lines). The survival curve revealed that the mice administered 10 did not die (Fig. 3d, red line). The faster tumor growth observed in the two groups of mice to which non-labeled trastuzumab and PBS were administered revealed that most of the mice died by 25 days post-injection (Fig. 3d, yellow and blue lines). Trastuzumab (Herceptin) is a clinically used drug, which is available from Chugai Pharmaceutical Co., Ltd. According to regulated information from electronic Medicines Compendium (UK),53 8 mg kg−1 body weight is suggested for the initial loading dose, and 6 mg kg−1 body weight for the maintenance dose at three-week intervals. When this therapeutically effective dose to human patient was converted to the xenografted mouse (calculated as 25 g of mouse weight), the 200 μg of initial loading and the 150 μg of maintenance dose of trastuzumab are required. In our experiments, only 6.3 μg of trastuzumab was initially administered and no maintenance injections were performed. Therefore, the therapeutic effects could not be observed without labeling with 211At.
Importantly, although a 9% body weight loss (relative to the initial weight) was observed 4 days post-injection in the mice administered 10, the body weight recovered its initial value over the following 8 days. Serious toxicity associated with 211At was not, therefore, observed (Fig. 3e, see below for detailed discussion for the toxicity).
The α-emission therapeutic efficacy of 211At was assessed using 10 by investigating the dose-dependent therapeutic effects of 211At. We prepared a series of 10 labeled with 211At, 1, 0.5, or 0.1 MBq using 6.3 μg antibody in 5 μL 0.05% PBS-T. These solutions were prepared from a 0.05% PBS-T solution of 10 labeled with 211At of 63 MBq (see the ESI† for protocol details). Intratumoral injection of 5 μL of each 0.05% PBS-T solution containing the radioactive 10, or containing Na[211At], 1 MBq, as a free 211At sample, was administered to A431 xenograft mice, and the tumor growth in each mouse was monitored daily over one month. Tumor growth in the three groups of the mice did not differ until 10 days post injection. After two weeks post-injection, the tumors grew more slowly in the mice administered higher doses of 10 (Fig. 3f, green, blue, and yellow lines). In the absence of trastuzumab as a cancer-targeting molecule, no radiotherapeutic effect was observed, even though Na[211At], 1 MBq, should have been sufficient to provide effective therapy (vide infra, Fig. 3f, brown line). The survival curve revealed that all mice in the group administered 10 labeled with 211At, 1 MBq, remained alive during the monitoring period (survival rate of 100%, Fig. 3g, yellow line). On the other hand, the first death was observed after 14 days post-injection among the mice administered Na[211At]. Initial deaths occurred 17 or 21 days post-injection among mice administered 10 labeled with 211At, 0.1 or 0.5 MBq, respectively (Fig. 3g, brown, green, and blue lines). After 28 days post-injection, survival rates reached 42% among mice administered Na[211At], 57% among mice administered 10 labeled with 211At, 0.1 MBq, and 85% among mice administered 10 labeled with 211At, 0.5 MBq (Fig. 3g, brown, green, and blue lines). The intratumoral injection of 10 labeled with 211At, 1 MBq, was found to be quite effective in prolonging survival in this mouse model.
Conclusions
We described the facile synthesis of 211At-labeled trastuzumab, a potentially useful α-emitting cancer-targeting therapy, based on a one-pot three-component double-click labeling strategy. The 211At-labeled trastuzumab was prepared for in vivo use by employing closo-decaborate as a prosthetic group to create a stable complex with halogens, including astatine, and by developing the closo-decaborate-tetrazine 4, synthesized in only two steps from the intermediate 5, as reported previously, as a key labeling probe for the one-pot double-click labeling reaction. Using the decaborate-tetrazine 4 and TCO-aldehyde 3 as the RIKEN click probes and conditions under which an average of 2.3 decaborate moieties were attached to each HSA model protein, trastuzumab as an anti-HER2 antibody could be readily modified with decaborate in a little over 1 h using Amicon ultrafiltration. The decaborate-trastuzumab 9 was efficiently labeled with 211At within 5 min and without reducing the antigen recognition activity of the antibody. We developed the 211At-labeled trastuzumab 10 with a high specific activity (15 MBq μg−1) using highly radioactive 211At, >100 MBq, produced at the RIKEN Nishina Center.Intratumor injection revealed that the 211At-labeled trastuzumab 10 specifically accumulated in the A431 tumor cells of a xenograft mouse model. The therapeutic efficacy of this construct via proximal α-emission of 211At was found to be high, especially upon administration of 10 labeled with 211At, >1 MBq. Significantly, therapeutic effects were observed even in the presence of small quantities of the antibody (6 μg antibody per mouse).
The toxicity by astatine radioactivity under the experimental conditions was not significant or even negligible. Hasegawa and co-workers recently reported that the intravenous injection of 0.5 MBq of 211At-labeled trastuzumab caused the temporal decrease in the number of leukocytes and slightly reduced the body weight.33 Our intratumor injection date cannot simply be compared with those of the intravenous injection, but the toxicity caused by intratumoral injection of 1 MBq antibody is less than that by 0.5 MBq of intravenous injection. In fact, we cannot observe the significant decrease in body weight when injected 1 MBq of antibody (Fig. 3h). We found the slight reduction of the body weight when injected the 1.4 MBq of antibody (Fig. 3e), but that is temporal, and the weight has recovered and kept normal after 2 weeks.
Finally, we found that the [211At] decaborate complex was not appropriate as an antibody-based therapeutic delivered by intravenous injection because the notable hydrophobic properties of the labeling agent may have altered the native biodistribution of the antibody. Intratumor injection of the 211At-labeled antibody may be performed efficiently after diagnosis, e.g., by positron emission tomography. Our 211At-labeling strategy should enable access to antibodies as well as to a variety of cancer-targeting molecules, thereby providing one of the most practical 211At-labeling methods for developing molecular cancer radiotherapeutics, especially against micrometastases, blood cancer, or disseminated cancer cells, which are easily accessible from the blood vessels. In fact, 211At-labeled antibodies have recently been tested for the clinical trials.54 In combination with the pretargeted radioimmunotherapy,55 future translational research of the targeted α-therapy becomes more significant.
Experimental
All commercially available reagents were used without further purification. Distilled water was purchased from Nacalai Tesque. All organic solvents were purchased from Wako Pure Chemicals Industries. All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of RIKEN and approved by the Animal Ethics Committee of RIKEN (H29-2-103).One-pot three-component double-click reactions to attach decaborate to HSA (8)
DMSO solutions of the decaborate-tetrazine 4 (4 × 10−3 M, 10 μL) and TCO-aldehyde 3 (4 × 10−3 M, 10 μL) were mixed in distilled water (180 μL). To the solution was added an aqueous solution of HSA (2 × 10−5 M, 200 μL). The solution was heated to 37 °C. After 1 h, the reaction mixture was transferred into an Amicon molecular weight 10k filtration unit and centrifuged under 14000 × g for 10 min. To the filter was added distilled water (400 μL), and the solution was centrifuged under 14000 × g for 10 min. This wash was repeated 3 more times. The residue on the filter was collected to give 8. The aqueous solution of 8 was adjusted to a concentration of 2 × 10−5 M for use. Mass spectral analysis of the decaborate-HSA was performed to estimate the number of decaborate moieties attached to each HSA protein. MALDI-TOF MS (positive): m/z 70608 (68136 for non-labeled HSA).
One-pot three-component double-click reaction to attach decaborate to trastuzumab (9)
DMSO solutions of the decaborate-tetrazine 4 (4 × 10−3 M, 10 μL) and TCO-aldehyde 3 (4 × 10−3 M, 10 μL) were mixed in PBS (180 μL). To the solution were added a PBS solution of trastuzumab (2 × 10−5 M, 200 μL). The solution was heated to 37 °C. After 1 h, the reaction mixture was transferred into an Amicon molecular weight 50000 filtration unit and centrifuged under 14000 × g for 5 min. To the filter was added distilled water (400 μL), and the solution was centrifuged under 14000 × g for 5 min. This wash was repeated 3 more times. The residue on the filter was collected to give 9 as a PBS solution.
General procedure for 211At-labeling decaborate-trastuzumab (10)
000 filtration unit, 0.05% PBS-T was added to wash the reaction tube, and the solution was centrifuged under 14000 × g for 5 min. Four hundred milliliters of 0.05% PBS-T were added, and the solution was centrifuged at 14000 × g for 5 min. α-Ray doses of the residue on the filter and filtrate were measured using a germanium semiconductor detector. The product 10, labeled with 211At (4.5 MBq, 78% RCY) and having a specific activity (0.050 MBq μg−1), was obtained in a 0.05% PBS-T solution.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the JSPS KAKENHI Grant Numbers JP16H03287, JP18K19154, JP17K10382, and JP15H05843 in Middle Molecular Strategy, by the AMED Grant Number JP18am0301006, and by RIKEN Incentive Research Projects 2016. This work was also performed with the support of the Russian Government Program for Competitive Growth, granted to Kazan Federal University. A part of this work was performed at the RI Beam Factory operated by the RIKEN Nishina Center. We are grateful to Dr Yukiko Komori (RIKEN) for help in producing the 211At radiotracer, and to Etsuko Ebisui, Dr Soichi Kojima, Yasuko Egawa, and Motoko Fujii (RIKEN) for their helpful support in animal experiments conducted in tumor xenograft mouse models.References
- S. Liu, Adv. Drug Delivery Rev., 2008, 60, 1347–1370 CrossRef CAS PubMed.
- M. L. Janssen, W. J. Oyen, I. Dijkgraaf, L. F. Massuger, C. Frielink, D. S. Edwards, M. Rajopadhye, H. Boonstra, F. H. Corstens and O. C. Boerman, Cancer Res., 2002, 62, 6146–6151 CAS.
- S. Liu, Chem. Soc. Rev., 2004, 33, 445–461 RSC.
- T. M. Behr, N. Jenner, M. Behe, C. Angerstein, S. Gratz, F. Raue and W. Becker, J. Nucl. Med., 1999, 40, 1029–1044 CAS.
- D. M. Goldenberg, J. Nucl. Med., 2002, 43, 693–713 CAS.
- D. M. Goldenberg, Arch. Pathol. Lab. Med., 1988, 112, 580–587 CAS.
- D. E. Milenic, E. D. Brady and M. W. Brechbiel, Nat. Rev. Drug Discovery, 2004, 3, 488–498 CrossRef CAS PubMed.
- S. J. Knox, M. L. Goris, K. Trisler, R. Negrin, T. Davis, T. M. Liles, A. Grillo-Lopez, P. Chinn, C. Varns, S. C. Ning, S. Fowler, N. Deb, M. Becker, C. Marquez and R. Levy, Clin. Cancer Res., 1996, 2, 457–470 CAS.
- M. S. Kaminski, M. Tuck, J. Estes, A. Kolstad, C. W. Ross, K. Zasadny, D. Regan, P. Kison, S. Fisher, S. Kroll and R. L. Wahl, N. Engl. J. Med., 2005, 352, 441–449 CrossRef CAS.
- A. H. Repetto-Llamazares, R. H. Larsen, S. Patzke, K. G. Fleten, D. Didierlaurent, A. Pichard, J. P. Pouget and J. Dahle, PLoS One, 2015, 10, e0128816 CrossRef.
- D. E. Milenic, K. E. Baidoo, Y. S. Kim, R. Barkley and M. W. Brechbiel, Dalton Trans., 2017, 46, 14591–14601 RSC.
- N. A. Thiele, V. Brown, J. M. Kelly, A. Amor-Coarasa, U. Jermilova, S. N. MacMillan, A. Nikolopoulou, S. Ponnala, C. F. Ramogida, A. K. H. Robertson, C. Rodriguez-Rodriguez, P. Schaffer, C. Williams Jr, J. W. Babich, V. Radchenko and J. J. Wilson, Angew. Chem., Int. Ed., 2017, 56, 14712–14717 CrossRef CAS.
- I. Captain, G. J. Deblonde, P. B. Rupert, D. D. An, M. C. Illy, E. Rostan, C. Y. Ralston, R. K. Strong and R. J. Abergel, Inorg. Chem., 2016, 55, 11930–11936 CrossRef CAS.
- B. Molina, J. R. Soto and J. J. Castro, Phys. Chem. Chem. Phys., 2018, 20, 11549–11553 RSC.
- O. R. Pozzi and M. R. Zalutsky, J. Nucl. Med., 2007, 48, 1190–1196 CrossRef CAS PubMed.
- R. A. Milius, W. H. McLaughlin, R. M. Lambrecht, A. P. Wolf, J. J. Carroll, S. J. Adelstein and W. D. Bloomer, Int. J. Radiat. Appl. Instrum., Part A, 1986, 37, 799–802 CrossRef CAS.
- D. S. Wilbur, M. K. Chyan, D. K. Hamlin, B. B. Kegley, R. Risler, P. M. Pathare, J. Quinn, R. L. Vessella, C. Foulon, M. Zalutsky, T. J. Wedge and M. F. Hawthorne, Bioconjugate Chem., 2004, 15, 203–223 CrossRef CAS PubMed.
- S. Lindegren, S. Frost, T. Back, E. Haglund, J. Elgqvist and H. Jensen, J. Nucl. Med., 2008, 49, 1537–1545 CrossRef CAS PubMed.
- G. Vaidyanathan, D. J. Affleck and M. R. Zalutsky, Bioconjugate Chem., 1996, 7, 102–107 CrossRef CAS.
- S. W. Reilly, M. Makvandi, K. Xu and R. H. Mach, Org. Lett., 2018, 20, 1752–1755 CrossRef CAS PubMed.
- F. Guerard, L. Navarro, Y. S. Lee, A. Roumesy, C. Alliot, M. Cherel, M. W. Brechbiel and J. F. Gestin, Bioorg. Med. Chem., 2017, 25, 5975–5980 CrossRef CAS PubMed.
- D. S. Wilbur, M. K. Chyan, H. Nakamae, Y. Chen, D. K. Hamlin, E. B. Santos, B. T. Kornblit and B. M. Sandmaier, Bioconjugate Chem., 2012, 23, 409–420 CrossRef CAS PubMed.
- D. S. Wilbur, M. K. Chyan, D. K. Hamlin, H. Nguyen and R. L. Vessella, Bioconjugate Chem., 2011, 22, 1089–1102 CrossRef CAS PubMed.
- D. S. Wilbur, M. K. Chyan, D. K. Hamlin and M. A. Perry, Bioconjugate Chem., 2009, 20, 591–602 CrossRef CAS.
- D. S. Wilbur, M. K. Chyan, D. K. Hamlin, R. L. Vessella, T. J. Wedge and M. F. Hawthorne, Bioconjugate Chem., 2007, 18, 1226–1240 CrossRef CAS PubMed.
- J. J. Orozco, T. Back, A. Kenoyer, E. R. Balkin, D. K. Hamlin, D. S. Wilbur, D. R. Fisher, S. L. Frayo, M. D. Hylarides, D. J. Green, A. K. Gopal, O. W. Press and J. M. Pagel, Blood, 2013, 121, 3759–3767 CrossRef CAS PubMed.
- H. Nakamae, D. S. Wilbur, D. K. Hamlin, M. S. Thakar, E. B. Santos, D. R. Fisher, A. L. Kenoyer, J. M. Pagel, O. W. Press, R. Storb and B. M. Sandmaier, Cancer Res., 2009, 69, 2408–2415 CrossRef CAS PubMed.
- A. C. Steffen, Y. Almqvist, M. K. Chyan, H. Lundqvist, V. Tolmachev, D. S. Wilbur and J. Carlsson, Oncol. Rep., 2007, 17, 1141–1147 CAS.
- D. S. Wilbur, D. K. Hamlin, M. K. Chyan, B. B. Kegley, J. Quinn and R. L. Vessella, Bioconjugate Chem., 2004, 15, 601–616 CrossRef CAS PubMed.
- D. S. Wilbur, D. K. Hamlin, M. K. Chyan and M. W. Brechbiel, Bioconjugate Chem., 2008, 19, 158–170 CrossRef CAS PubMed.
- D. S. Wilbur, R. L. Vessella, J. E. Stray, D. K. Goffe, K. A. Blouke and R. W. Atcher, Nucl. Med. Biol., 1993, 20, 917–927 CrossRef CAS.
- D. S. Wilbur, M. S. Thakar, D. K. Hamlin, E. B. Santos, M. K. Chyan, H. Nakamae, J. M. Pagel, O. W. Press and B. M. Sandmaier, Bioconjugate Chem., 2009, 20, 1983–1991 CrossRef CAS PubMed.
- H. K. Li, Y. Morokoshi, K. Nagatsu, T. Kamada and S. Hasegawa, Cancer Sci., 2017, 108, 1648–1656 CrossRef CAS PubMed.
- J. Choi, G. Vaidyanathan, E. Koumarianou, D. McDougald, M. Pruszynski, T. Osada, T. Lahoutte, H. K. Lyerly and M. R. Zalutsky, Nucl. Med. Biol., 2014, 41, 802–812 CrossRef CAS PubMed.
- D. J. Green, M. Shadman, J. C. Jones, S. L. Frayo, A. L. Kenoyer, M. D. Hylarides, D. K. Hamlin, D. S. Wilbur, E. R. Balkan, Y. Lin, B. W. Miller, S. H. Frost, A. K. Gopal, J. J. Orozco, T. A. Gooley, K. L. Laird, B. G. Till, T. Back, B. M. Sandmaier, J. M. Pagel and O. W. Press, Blood, 2015, 125, 2111–2119 CrossRef CAS PubMed.
- E. Aneheim, A. Gustafsson, P. Albertsson, T. Back, H. Jensen, S. Palm, S. Svedhem and S. Lindegren, Bioconjugate Chem., 2016, 27, 688–697 CrossRef CAS PubMed.
- K. Tanaka, T. Masuyama, K. Hasegawa, T. Tahara, H. Mizuma, Y. Wada, Y. Watanabe and K. Fukase, Angew. Chem., Int. Ed., 2008, 47, 102–105 CrossRef CAS PubMed.
- K. Tanaka, E. R. Siwu, K. Minami, K. Hasegawa, S. Nozaki, Y. Kanayama, K. Koyama, W. C. Chen, J. C. Paulson, Y. Watanabe and K. Fukase, Angew. Chem., Int. Ed., 2010, 49, 8195–8200 CrossRef CAS PubMed.
- L. Latypova, R. Sibgatullina, A. Ogura, K. Fujiki, A. Khabibrakhmanova, T. Tahara, S. Nozaki, S. Urano, K. Tsubokura, H. Onoe, Y. Watanabe, A. Kurbangalieva and K. Tanaka, Adv. Sci., 2017, 4, 1600394 CrossRef PubMed.
- A. Ogura, T. Tahara, S. Nozaki, K. Morimoto, Y. Kizuka, S. Kitazume, M. Hara, S. Kojima, H. Onoe, A. Kurbangalieva, N. Taniguchi, Y. Watanabe and K. Tanaka, Sci. Rep., 2016, 6, 21797 CrossRef CAS PubMed.
- K. Tanaka, H. Mori, M. Yamamoto and S. Katsumura, J. Org. Chem., 2001, 66, 3099–3110 CrossRef CAS PubMed.
- K. Fujiki and K. Tanaka, RIKEN click reagent for protein labeling in e-EROS Encyclopedia of reagents for organic synthesis, 2018, DOI:10.1002/047084289X.rn02050.
- M. L. Blackman, M. Royzen and J. M. Fox, J. Am. Chem. Soc., 2008, 130, 13518–13519 CrossRef CAS PubMed.
- K. Fujiki, S. Yano, T. Ito, Y. Kumagai, Y. Murakami, O. Kamigaito, H. Haba and K. Tanaka, Sci. Rep., 2017, 7, 1912 CrossRef PubMed.
- M. R. Zalutsky, X. G. Zhao, K. L. Alston and D. Bigner, J. Nucl. Med., 2001, 42, 1508–1515 CAS.
- D. S. Wilbur, M. K. Chyan, D. K. Hamlin and M. A. Perry, Nucl. Med. Biol., 2010, 37, 167–178 CrossRef CAS PubMed.
- D. Teze, D. C. Sergentu, V. Kalichuk, J. Barbet, D. Deniaud, N. Galland, R. Maurice and G. Montavon, Sci. Rep., 2017, 7, 2579 CrossRef PubMed.
- A. Orlova, O. Lebeda, V. Tolmachev, S. Sjoberg, J. Carlsson and H. Lundqvist, J. Labelled Compd. Radiopharm., 2000, 43, 251–260 CrossRef CAS.
- K. Tamura, H. Kurihara, K. Yonemori, H. Tsuda, J. Suzuki, Y. Kono, N. Honda, M. Kodaira, H. Yamamoto, M. Yunokawa, C. Shimizu, K. Hasegawa, Y. Kanayama, S. Nozaki, T. Kinoshita, Y. Wada, S. Tazawa, K. Takahashi, Y. Watanabe and Y. Fujiwara, J. Nucl. Med., 2013, 54, 1869–1875 CrossRef CAS PubMed.
- B. Langen, N. Rudqvist, K. Helou and E. Forssell-Aronsson, J. Nucl. Med., 2017, 58, 346–353 CrossRef CAS PubMed.
- N. Rudqvist, J. Spetz, E. Schuler, B. Langen, T. Z. Parris, K. Helou and E. Forssell-Aronsson, EJNMMI Res., 2015, 5, 59 CrossRef PubMed.
- A. A. Ovejera, D. P. Houchens and A. D. Barker, Ann. Clin. Lab. Sci., 1978, 8, 50–56 CAS.
- The information for intended use of trastuzumab by health professionals: https://www.medicines.org.uk/emc/product/3856/smpc.
- M. R. Zalutsky, D. A. Reardon, G. Akabani, R. E. Coleman, A. H. Friedman, H. S. Friedman, R. E. McLendon, T. Z. Wong and D. D. Bigner, J. Nucl. Med., 2008, 49, 30–38 CrossRef CAS.
- E. J. L. Steen, P. E. Edem, K. Norregaard, J. T. Jorgensen, V. Shalgunov, A. Kjaer and M. M. Herth, Biomaterials, 2018, 179, 209–245 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc04747b |
| This journal is © The Royal Society of Chemistry 2019 |